
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproved ClickTags enable live-cell barcoding for highly multiplexed single cell sequencing†
Xinlu
Zhao‡
a,
Shiming
Sun‡
a,
Wenhao
Yu‡
a,
Wenqi
Zhu
b,
Zihan
Zhao
c,
Yiqi
Zhou
b,
Xiuheng
Ding
b,
Nan
Fang
b,
Rong
Yang
c and
Jie P.
Li
 *a
*a
aState Key Laboratory of Coordination Chemistry, Chemistry and Biomedicine Innovation Center (ChemBIC), School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, Jiangsu, China. E-mail: jieli@nju.edu.cn
bSingleron Biotechnologies, Nanjing, Jiangsu, China
cDepartment of Urology, Affiliated Drum Tower Hospital, Medical School of Nanjing University, Nanjing, Jiangsu, China
First published on 31st May 2022
Abstract
Click chemistry-enabled DNA barcoding of cells provides a universal strategy for sample multiplexing in single-cell RNA-seq (scRNA-seq). However, current ClickTags are limited to fixed samples as they only label cells efficiently in methanol. Herein, we report the development of a new protocol for barcoding live cells with improved ClickTags. The optimized reactions barcoded live cells without perturbing their physiological states, which allowed sample multiplexing of live cells in scRNA-seq. The general applicability of this protocol is demonstrated in diversified types of samples, including murine and human primary samples. Up to 16 samples across these two species are successfully multiplexed and demultiplexed with high consistency. The wide applications of this method could help to increase throughput, reduce cost and remove the batch effect in scRNA-seq, which is especially valuable for studying clinical samples from a large cohort.
 Jie P. Li | Dr Jie P. Li is a Group Leader at the Chemistry and Biomedicine Innovation Center (ChemBIC), School of Chemistry and Chemical Engineering, Nanjing University, China. After completing his DPhil degree in 2015 at Peking University, China, he undertook postdoctoral work at the Scripps Research Institute, USA. He started his independent research career in 2018 and his research group interest is focused on the topic of “chemical single-cell omics”, which utilizes chemical tools to study intracellular and intercellular signaling at the single-cell level for understanding the immune system. |
High-throughput single-cell RNA sequencing (scRNA-seq) can efficiently provide transcriptomic analysis of heterogeneous multicellular living systems, such as complex tissues and the immune system.1–14 With the advancement of single-cell multi-omics techniques and bioinformatics tools, multimodal scRNA-seq allows deep analysis of transcriptomes and other cell identities simultaneously at a single-cell level, which offers new insights into the understanding of life from a single-cell perspective.15–23 To characterize the single-cell profiles of whole organs or even larger organisms, the pursuit of ultra-high-throughput scRNA-seq has been realized such that one million cells can be analyzed in a single run.24 The increase of throughput in scRNA-seq platforms also provides opportunities for the massively parallel analysis of multiple samples in a single run, which reduces the reagent cost per cell and removes the batch effect physically.20,22
The technique called sample multiplexing is usually used in single-cell techniques to achieve massively parallel analysis in single experiments, in which cells from independent samples were barcoded (hashed) with DNA oligonucleotides (oligos) before pooling together for single-cell analysis.25–31 DNA-labelled antibodies and lectins,19,28,31 lipid-tagged DNA,32 chemical reactive DNA30 and expressed genetic barcodes29 are representative methods to introduce the barcodes on the samples. Among these, chemical barcoding of cell surface proteins with DNA oligos might be the most universal method due to the following facts: (1) it uses the same reagent for all species; (2) it is of low cost per sample; (3) it has minimal mRNA expression perturbations; (4) it labels cells at a high intensity of DNA barcodes. “ClickTag” has been reported to barcode methanol fixed cells for sample multiplexing in scRNA-seq through a one-pot, two-step chemical cross-linking reaction that combined NHS–amine coupling and inverse electron-demand Diels–Alder (IEDDA)33–35 reaction (Scheme 1). However, this reported protocol failed to be applied in live-cell barcoding and demultiplexing. Although methanol fixed samples have been widely used in scRNA-seq, live-cell samples are more preferred since the methanol fixation introduces more steps and may cause the loss of rare cell populations. According to the wide applications of “click chemistry”36 or “bioorthogonal chemistry”37 in live cell engineering,34,38,39 we believe that the optimization of reactions should be able to barcode live cells with DNA oligos.
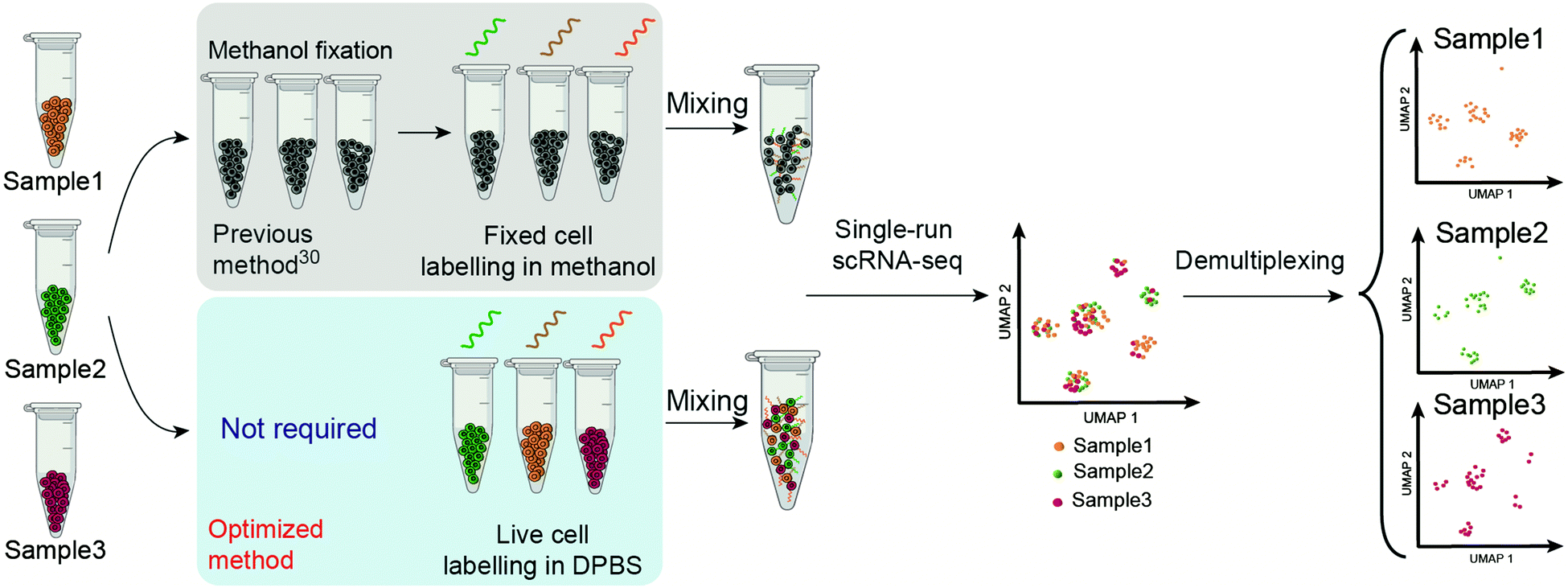 | ||
| Scheme 1 Schematic overview of the workflow of ClickTag-enabled sample multiplexing in scRNA-seq. The previous method30 and the optimized method reported here in this work are shown in parallel. | ||
Here we report the improved “ClickTag” for DNA oligos barcoding on live cells. Our method adds to a growing family of complementary multiplexing technologies for live-cell samples that could be easily adapted into current scRNA-seq platforms (Scheme 1). This method allows accurate sample multiplexing and demultiplexing regardless of genetic background and sample type, and has been successfully applied in patient samples of bladder cancer.
Firstly, to label live cells for multiplexed scRNA-seq, we prepared tetrazine (Tz)-modified DNA oligos (bearing a poly-A tag) according to the previous protocol (Fig. 1A). The subsequent labelling of Tz-oligos to cellular proteins on the cell surface is enabled by the combination of the IEDDA chemistry and NHS ester–amine coupling using the heterobifunctional molecule, NHS ester–trans-cyclooctene (NHS–TCO) (Fig. 2). Accordingly, Tz-oligos and NHS–TCO were premixed as ClickTags and then diluted in an aqueous buffer to label live Jurkat cells. After that, to characterize the labelling efficiency by flow cytometry, a fluorophore-conjugated complementary ssDNA oligo (5′ Cy5–poly-T) was used to detect the ClickTags labelled on the cell surface (Fig. S1A, ESI†). As expected, ClickTags made by the previously reported protocol could not be detected on the cell surface after labelling (Fig. S1B, ESI†). To make the chemistry compatible with live cells, we double checked the procedure of Tz-oligo synthesis and found that the purification by ethanol precipitation might lead to the contamination of Tz-oligos by unreacted Tz. More importantly, the following characterization of Tz-oligos by TCO–Cy5 labelling and PAGE–gel imaging is semi-quantitative and not sufficient to characterize the purity (Fig. 1A). Both issues of contamination and a low reaction yield might lead to the insufficient labelling of live cells in aqueous buffer. Thus, we decided to upgrade the synthesis procedures through strict quality control to obtain pure Tz-oligos, which is the molecular basis to precisely control the cell surface labelling chemistry. To improve the procedure, the reaction between NHS–Tz and 5′ NH2-oligo was optimized and monitored by LC-MS analysis (Methods, ESI†). After completion of the reactions, we introduced column purification to purify Tz-oligos, in which unreacted NHS–Tz could be washed away, while the negatively charged DNA oligos functionalized with Tz groups remained on the column (Fig. 1A). Moreover, the purity of the ClickTags was further characterized by LC-MS after elution (Fig. 1B, C). Through these improvements, pure Tz-oligos could be readily prepared in our lab to make improved ClickTags.
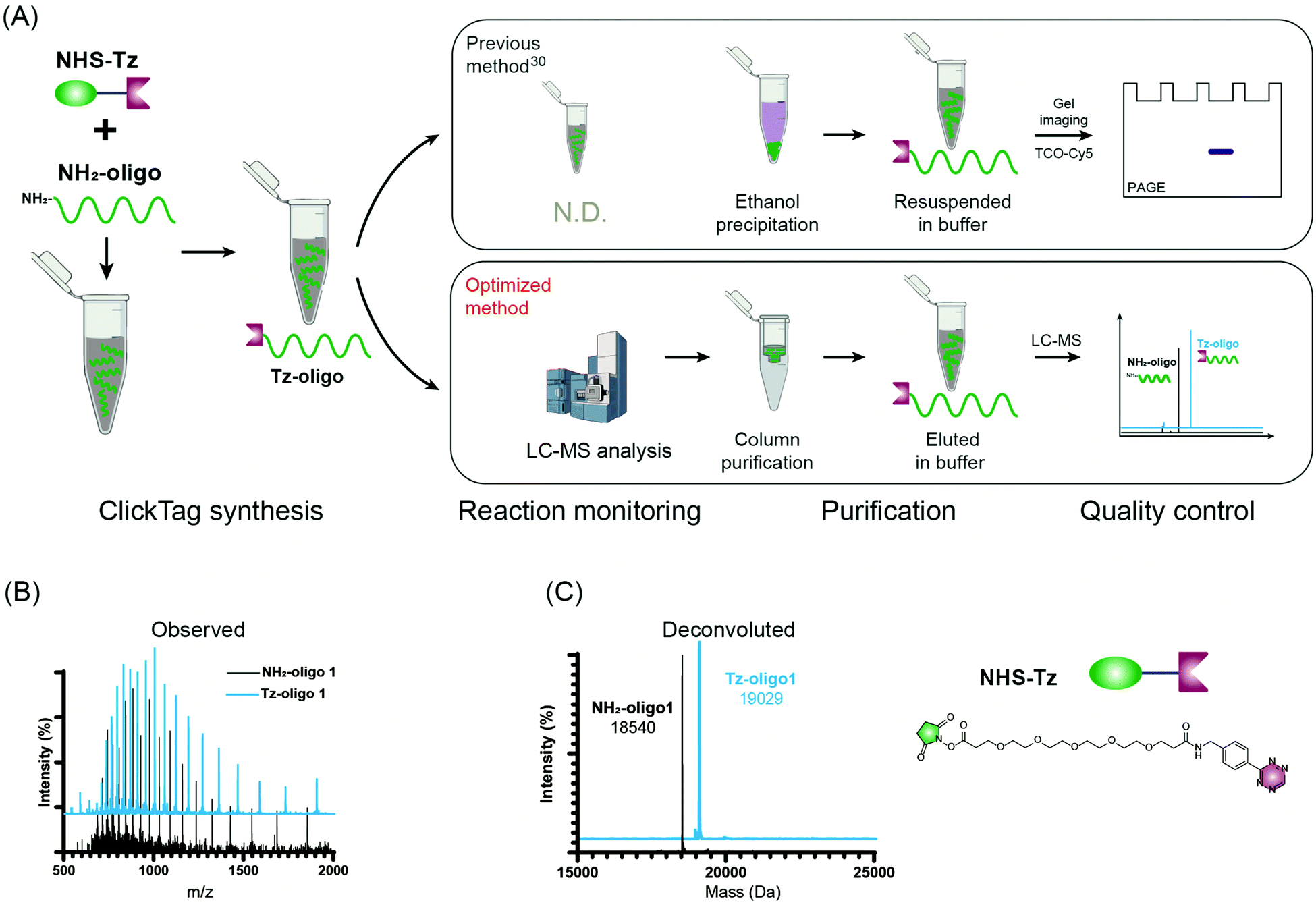 | ||
| Fig. 1 The optimized synthesis procedure for Tz-oligos. (A) Schematic procedures of ClickTag synthesis. Previous method: top; optimized method: bottom. Observed (B) and deconvoluted (C) mass spectra of NH2-oligo 1 and final product Tz-oligo 1 (related to ClickTag1). | ||
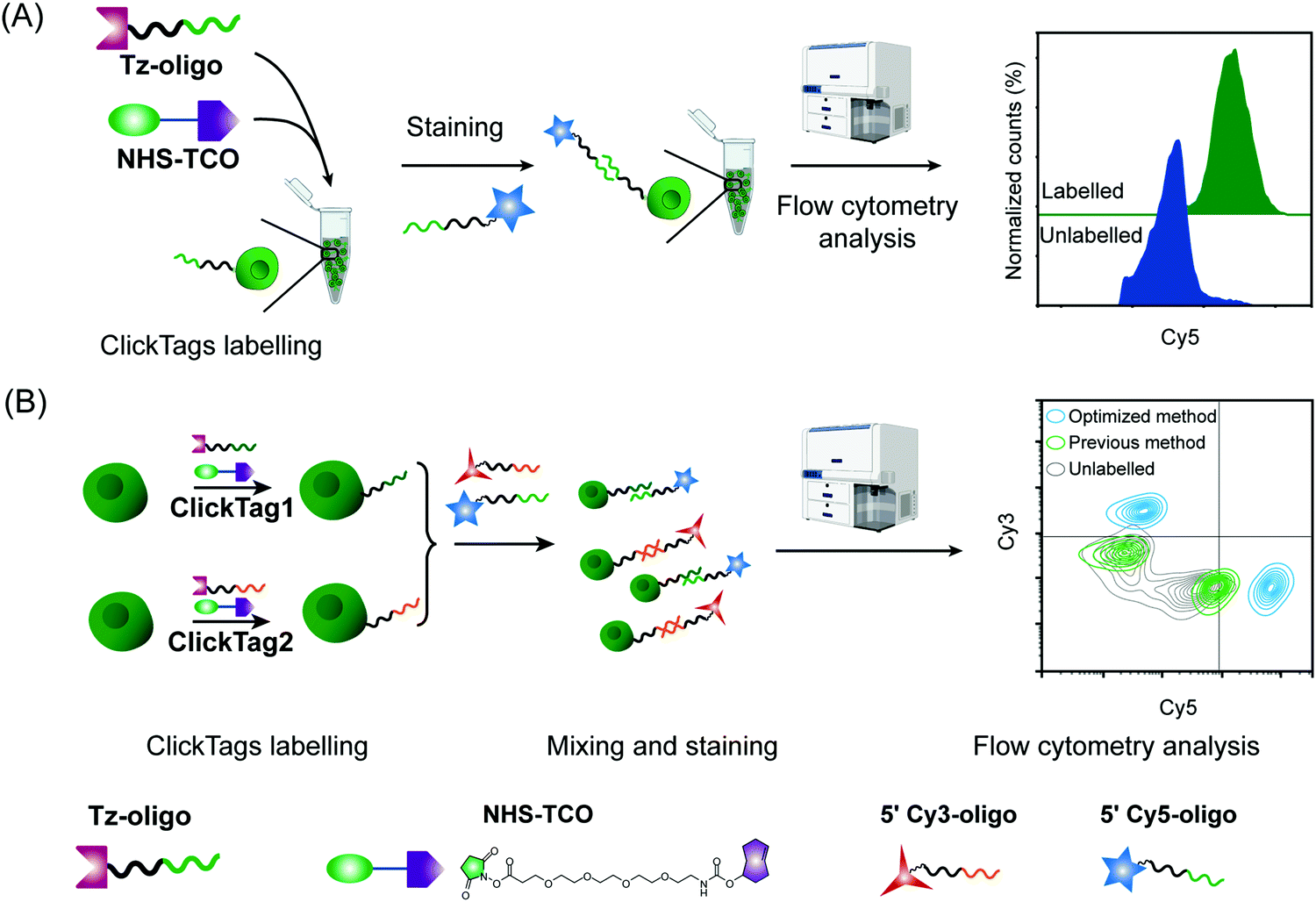 | ||
| Fig. 2 ClickTag labelling and sample multiplexing detected by flow cytometry. (A) Jurkat cells were labelled by optimized ClickTags and detected by flow cytometry via Cy5 conjugated complementary oligos. (B) Two cell samples were labelled with ClickTag1 and ClickTag2 following different protocols and analyzed by flow cytometry using complementary fluorophore-conjugated oligos. Optimized method: blue; previous method: green; unlabelled: grey. | ||
With pure Tz-oligos in hand, we optimized the concentrations and reaction time between Tz-oligos and NHS–TCO to make ClickTags capable of effective and non-cytotoxic labelling of live cells (Fig. 2A). ClickTag-labelled cells were probed by 5′ Cy5–poly-T and analyzed by flow cytometry. To our delight, a dose-dependent labelling of ClickTags was observed on live Jurkat cells (Fig. S1C, ESI†). Specifically, a significantly high level of DNA barcoding was achieved at 25 μM of NHS–TCO and 25 μM of Tz-oligos (Fig. 2A). Under these concentrations, the intensity of ClickTags coupled to the surface of Jurkat cells reached a plateau after 30 minutes (Fig. S1D, ESI†). As the reactivity of different cells varied, reaction times of 15–30 minutes were usually used for the following experiments. To test the ClickTag-enabled multiplexing and demultiplexing, we further labelled two samples with different ClickTags (ClickTag1 and ClickTag2, Table S1, ESI†), respectively, and mixed them as one sample. Complementary ssDNA oligos of ClickTag1 (5′ Cy3-oligo) and ClickTag2 (5′ Cy5-oligo) were added to detect the barcodes of the two samples and their exchange levels. As shown in Fig. 2B, the two samples were successfully separated by individually labelled Cy3 or Cy5 probe with minimal double-positive population. In contrast, ClickTags made by the previous protocol showed unclear separation of labelled and unlabelled cells (Fig. 2B). Taken together, these results demonstrate that ClickTag barcodes are stable on the cell surface and could be demultiplexed by complementary oligo probes, which is potentially applicable in scRNA-seq.
To make ClickTags compatible with the commercial scRNA-seq platform, as shown in Fig. 3A, each sample barcode was designed to include a 21-bp 3′ poly-A capture sequence, a 15-bp sample barcode and a 5′ PCR handle that are necessary for library preparation and sequencing. Live cells labelled with ClickTag barcodes were loaded to a scRNA-seq platform for single cell isolation. In a single droplet/microwell, a single bead anchored with primers consisting of a cell barcode sequence, a unique molecular identifier (UMI) sequence, and a poly-T sequence could capture the poly-A tail of endogenous transcripts and ClickTags from one cell simultaneously. Cell-specific barcodes were then attached to the cDNA or ClickTags during reverse transcription (RT), which allows sample demultiplexing after the standard scRNA-seq workflow of library preparation and next-generation sequencing (NGS). We tested the capacity of ClickTags to perform a proof-of-concept sample multiplexing experiment using four human cell lines, including the AML cell line U937, the promyelocytic cell line NB4, the T lymphoblastoid cell line CCRF-CEM, and the acute monocytic leukemia cell line THP-1. The four types of cells were barcoded with ClickTags (ClickTag A, ClickTag B, ClickTag C, and ClickTag D, Table S1, ESI†) and pooled for analysis.
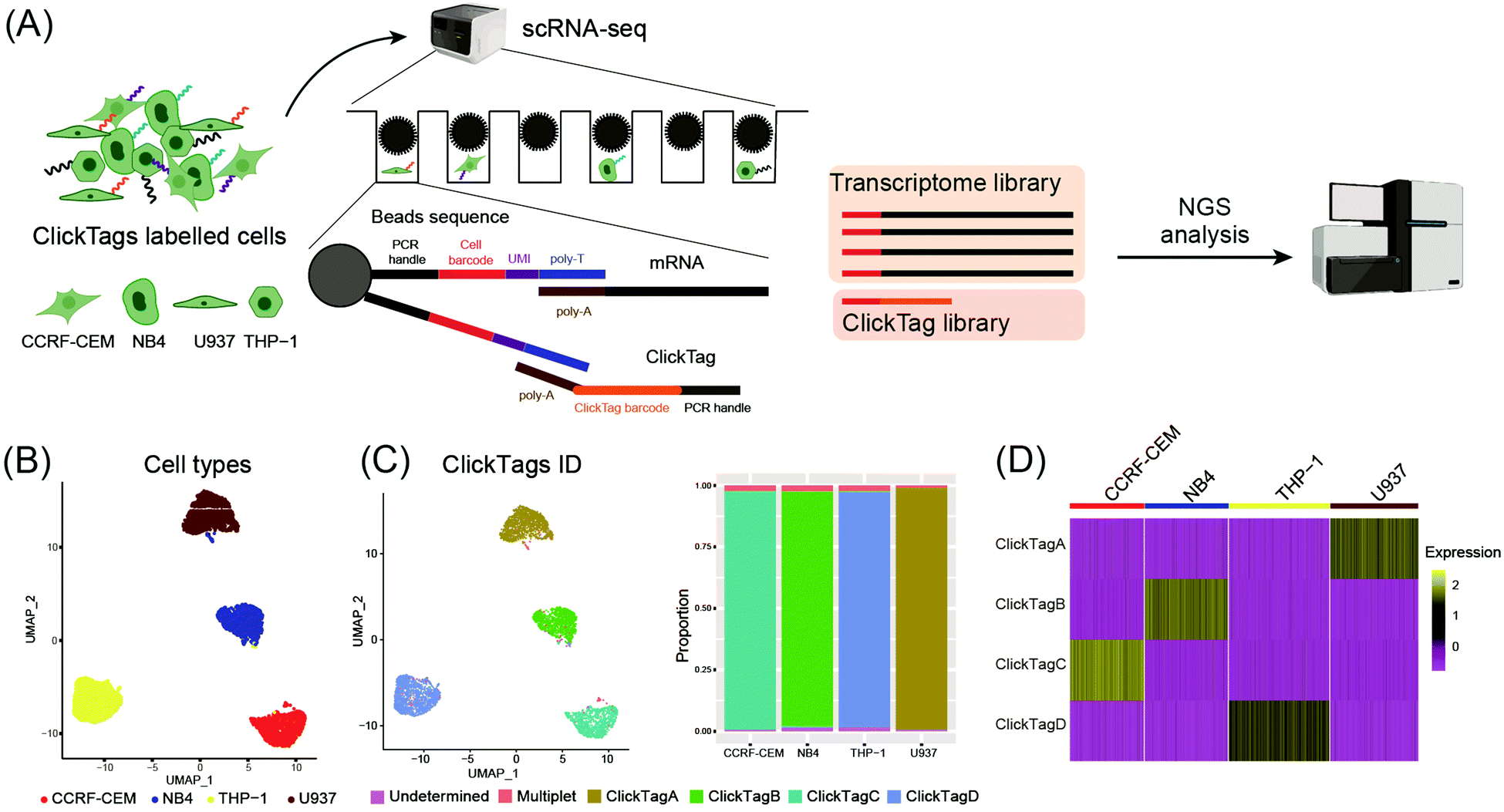 | ||
| Fig. 3 Application of ClickTags to multiplexing of four cell lines in scRNA-seq. (A) Schematic overview of a proof-of-concept sample multiplexing scRNA-seq experiment using ClickTags. Four cell-line samples (U937, NB4, CCRF-CEM, and THP-1) were barcoded with ClickTags A–D and then pooled together before subjecting to the scRNA-seq platform. (B) Transcriptome-based clustering of single-cell expression profiles reveals distinct cell types: CCRF-CEM (red), NB4 (blue), THP-1 (yellow), and U937 (dark red). (C) Projection of the ClickTag dataset onto cell types (on the left). ClickTag A: golden yellow; ClickTag B: green; ClickTag C: cyan; ClickTag D: pale blue. Bar graphs describing the proportion of tags attached to each cell type (on the right). Multiplets: cells labelled with more than one ClickTag. Undetermined: cells barcoded with undetected amounts of ClickTags. (D) The heat map of scaled (z-scores) normalized ClickTag values on the four cell lines. | ||
Following data pre-processing, we obtained a final scRNA-seq dataset containing 4869 total cells. We identified clusters in gene expression space according to known markers for U937, NB4, CCRF-CEM and THP-1 (Fig. 3B; marker genes for the four cell lines are presented in Fig. S2A, ESI†). Projection of ClickTag barcode classifications onto the gene expression space was performed according to the UMI detected for endogenous transcripts and ClickTags in the same cell (Fig. 3C and 3D). The result shows that ClickTag classifications successfully demultiplexed each sample, in which the ClickTags significantly correlate with cell type. The range of ratios of undetermined and multiplet populations are 0.48–1.5% and 1.0–2.3%, respectively (Fig. 3C). Indeed, the multiplets identified based on the mixture of ClickTags A and B consist of both CCRF and NB4 cells, which could be unambiguously detected based on their clustering results (Fig. 3C). Meanwhile, the number of detected UMIs by ClickTags per cell ranged from 112 to 198 (3.0–5.8 percentile), suggesting intensive barcoding of cells (Fig. 3D, Fig. S2B and C, ESI†). These results together showed the success of sample multiplexing and demultiplexing of live cells by ClickTags in scRNA-seq. We also explored the combinatorial barcoding capacity of ClickTags in an experiment where HeLa cells and mouse bone marrow cells were dual-labelled with two unique pairs of ClickTags, respectively (Fig. S3A, ESI†). According to the clustering and classifications, both HeLa cells and mouse marrow cells were labelled with a pair of sample-specific ClickTags. In contrast, when following the previous labelling protocol of ClickTags, the ClickTag classifications failed to generate readily demultiplexed samples, likely owing to the competitive NHS–ester hydrolysis in the aqueous buffer (Fig. S3B, ESI†).
Barcoding of primary samples has been little studied in the development of sample multiplexing techniques due to problems of heterogeneous labelling efficiency, labelling-induced loss of rare cell types, and potential alteration of transcriptomes.28–32 Mouse bone marrow cells that contain various immune cell subtypes were chosen for assessing whether our ClickTags could be applied to primary cells. Mouse bone marrow cells were harvested from C57BL/6J mice. After lysis of erythrocytes and cell counting, four aliquots of mouse bone marrow were barcoded with four ClickTags and pooled together for sequencing. In parallel, we prepared an unbarcoded replicate as a transcriptome reference to test whether ClickTag barcoding influenced gene expression or mRNA capture efficiency. Barcoded and unbarcoded samples were subjected to scRNA-seq platforms, respectively, and then analyzed following the data processing procedure. As a result, a total of 2552 cells were captured with sufficient reads for transcriptome analysis in the barcoded sample, in which 2351 cells were detected as barcoded cells. Through sample demultiplexing, 606, 577, 546, and 622 cells were assigned to ClickTag1-, ClickTag2-, ClickTag3-, and ClickTag4-labelled samples, respectively. Transcriptome-based clustering was used to map the cell subtypes, which showed similar results in demultiplexed samples and the unbarcoded replicates (Fig. S4A, ESI;† marker gene expression is shown as a heat map in Fig. S4D, ESI†). Through analysis of the results, we further confirmed that most of the cell types in each sample could be accurately assigned and the proportion of the cell subtypes were highly consistent in each sample derived from the same parent sample of marrow cells (Fig. S4B, ESI†), including some important immune cell subtypes that account for a very small amount of marrow cells, e.g., basophils, T cells, plasmacytoid dendritic cells (pDCs) and mature B cells. Moreover, Pearson correlation of gene expression from all the samples displayed a strong correlation of the top two thousand highly variable gene expressions across all the samples (Fig. S4C, ESI†). Taken together, our method showed high reproducibility in sample multiplexing of primary cells and had negligible consequences on cell transcriptional states.
Using scRNA-seq to profile samples from high throughput screening is often difficult due to batch effects and high cost.40–42 We next investigated whether our method could enable multiplexing of multiple samples (>10) via rapid and species-unbiased barcoding. Since the scRNA-seq platform we used could allow about 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per run, we designed 16 ClickTags to barcode 16 samples that allow thousands of cells to be captured in each sample. HeLa cells and mouse testicle cells were each divided into eight aliquots and labelled with sixteen distinct ClickTags (Table S1, ESI†). We then pooled these barcoded samples together before scRNA-seq. After quality-control filtering, sample classification and doublet removal, we obtained a final scRNA-seq dataset of 45704 mouse and human single cells spanning all 16 samples. Out of 45704 cells, 16 undetermined cells and 6065 multiplets were detected. As shown in Fig. 4A, the cells from the 16 samples showed highly accurate sample assignment despite major differences in transcriptomes between the two species, demonstrating that improved ClickTags barcoding is a universal sample multiplexing technique. Gene expression of the eight HeLa cell samples showed strong correlation with each other, as was the case for mouse testicle cell samples (Fig. 4B). These data support that improved ClickTags are potentially applicable in drug screening where millions of cells could be analyzed per run in scRNA-seq.
000 cells per run, we designed 16 ClickTags to barcode 16 samples that allow thousands of cells to be captured in each sample. HeLa cells and mouse testicle cells were each divided into eight aliquots and labelled with sixteen distinct ClickTags (Table S1, ESI†). We then pooled these barcoded samples together before scRNA-seq. After quality-control filtering, sample classification and doublet removal, we obtained a final scRNA-seq dataset of 45704 mouse and human single cells spanning all 16 samples. Out of 45704 cells, 16 undetermined cells and 6065 multiplets were detected. As shown in Fig. 4A, the cells from the 16 samples showed highly accurate sample assignment despite major differences in transcriptomes between the two species, demonstrating that improved ClickTags barcoding is a universal sample multiplexing technique. Gene expression of the eight HeLa cell samples showed strong correlation with each other, as was the case for mouse testicle cell samples (Fig. 4B). These data support that improved ClickTags are potentially applicable in drug screening where millions of cells could be analyzed per run in scRNA-seq.
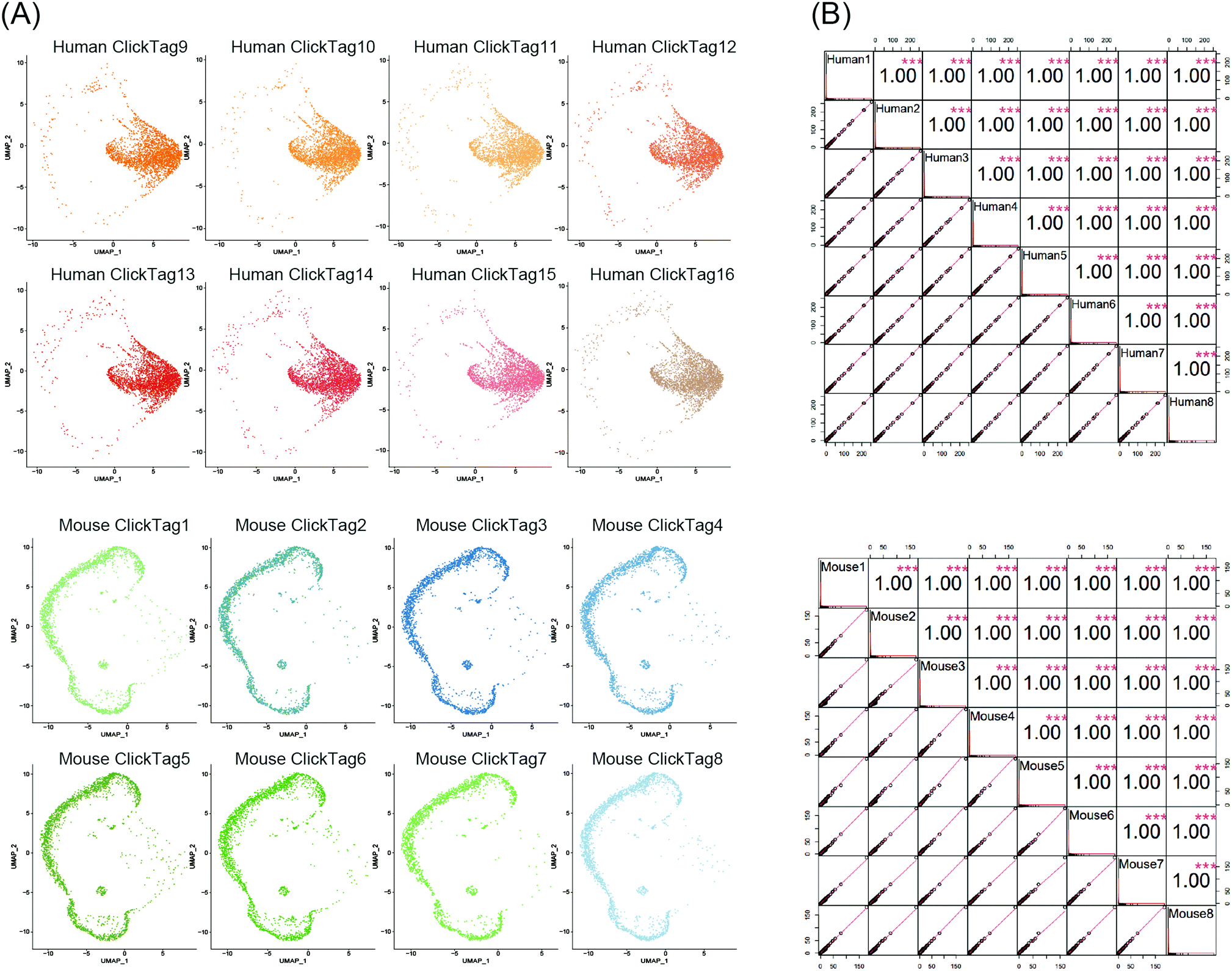 | ||
| Fig. 4 Sample multiplexing of 16 samples across species. Eight HeLa cell samples and eight mouse testicle cell samples were individually barcoded with distinct barcodes. (A) UMAP embedding and demultiplexing of the sixteen samples. (B) Pearson correlation of gene expression from all the samples containing human cells (top) and all samples containing mouse samples (bottom). Gene expression is shown as average counts per sample for each of n = 26098 human genes and 31713 mouse genes. | ||
Archival primary tissue samples from a cohort are rich resources for clinical study, which could be mined by scRNA-seq to generate novel insights at the single-cell level.43,44 However, these samples are usually fragile during the cycle of cryopreservation, thawing, enzymatic digestion and scRNA-seq sample preparation. As a consequence, the application of scRNA-seq in analysis of cryopreserved clinical samples has long been hindered by a lack of rapid, low-cost and nonperturbative sample multiplexing methods. As we have already demonstrated that the improved ClickTag-enabled cell barcoding is a robust multiplexing strategy, we decided to apply ClickTag-based sample multiplexing in cryopreserved clinical samples to expand its potential clinical usage (Fig. 5A). Three cryopreserved bladder tumor clinical samples (single cell suspension) were randomly picked from a cohort of bladder cancer patients (numbered patient #1, #2, and #3). Samples were thawed and CD45+ immune cells were sorted using fluorescence activated cell sorting (FACS) for scRNA-seq. After barcoding by ClickTags, tumor infiltrating lymphocytes (TILs) from three patients were pooled together and subjected to scRNA-seq, while TILs from patient #1 (TILs-1) were also individually subjected to scRNA-seq as the unbarcoded replicate. Following the data processing, a total of 5153 cells were obtained from the multiplexing samples, while 5722 cells were obtained from the unbarcoded samples. In the barcoded samples, 4864 cells from 5153 cells were detected as barcoded cells. Through sample demultiplexing, 1878, 1281, and 1705 cells were assigned to three TILs samples. Transcriptome-based clustering clearly identified monocytes, T cells, natural killer cells (NKs), dendritic cells (DCs) and a small population of epithelial cells (marker gene expression is shown as a heat map in Fig. S5, ESI†). The proportion of different cell types in TILs-1 revealed by demultiplexing is highly correlated with those in the unbarcoded replicate from the same patient but different from TILs of the other two patients (Fig. 5B and C). Gene expressions of TILs-1 control and TILs-1-ClickTag showed strong correlation after ClickTag barcoding (Fig. 5D). These results together showed the great potential of our ClickTags for accelerating the mining of clinical samples that are cryopreserved in a large cohort via scRNA-seq.
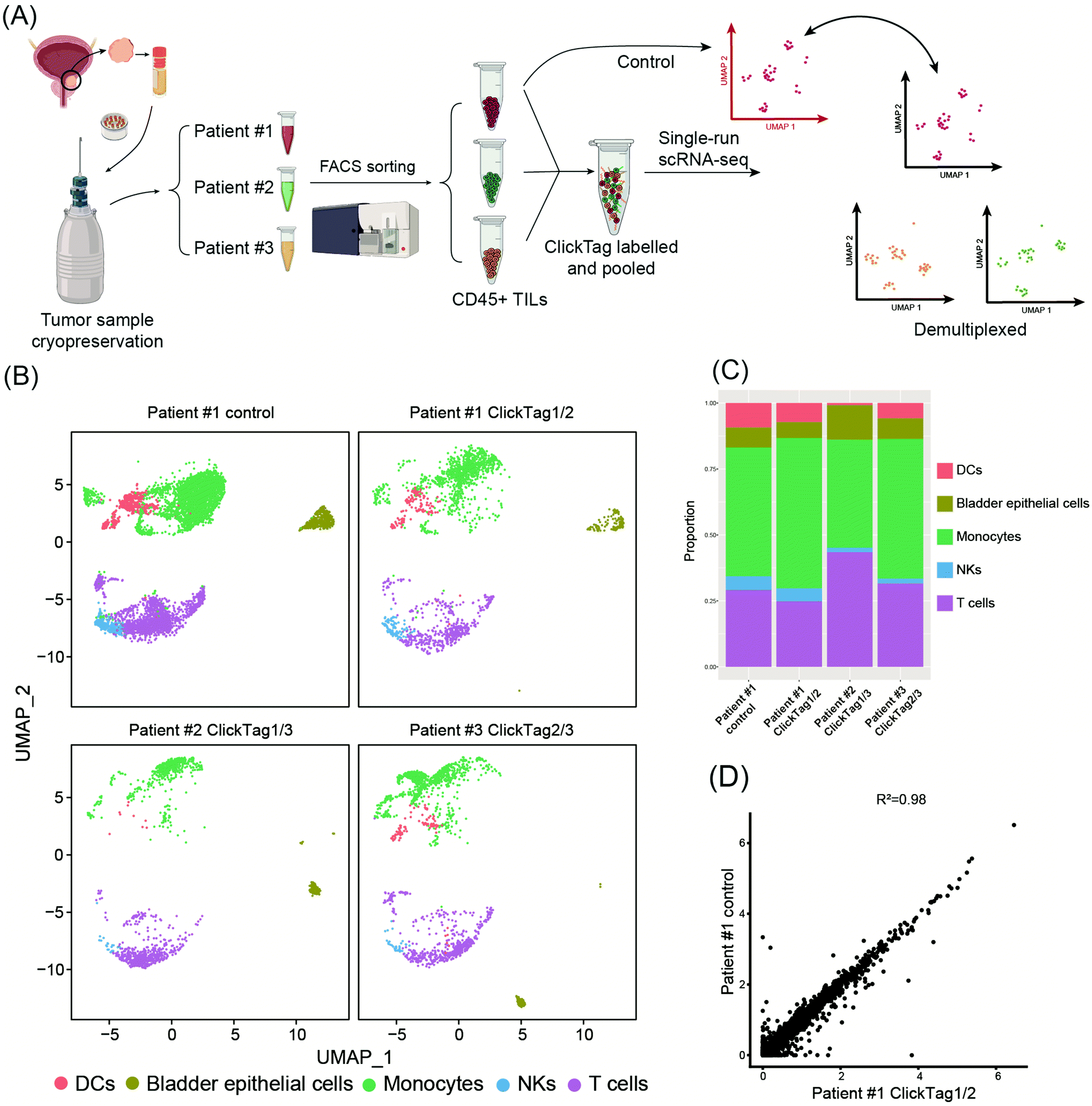 | ||
| Fig. 5 Application of ClickTags to multiplexing of clinical samples from human bladder cancer. (A) Schematic overview of multiplexing TILs from three bladder cancer patients in scRNA-seq. TILs from patient #1 were also analyzed individually as a reference. (B) UMAP analysis and graph visualization of patient TILs samples: patient #1 control (n = 5722); patient #1 ClickTag1/2 (n = 1878), patient #2 ClickTag1/3 (n = 1281), patient #3 ClickTag2/3 (n = 1705). ClickTag1/2 means the sample was dual-labelled with ClickTag1 and ClickTag2; this also applies to the remaining sample designations. (C) Cell type proportions identified by transcriptome-based clustering. (D) Gene expression correlation between unbarcoded control samples of patient #1 and patient #1 ClickTag1/2. | ||
In summary, we have developed live-cell compatible ClickTags that are broadly applicable for multiplexing different types of primary samples in scRNA-seq experiments. As the throughput of single-cell sequencing technologies increases gradually, we anticipate that live-cell compatible ClickTags will become a versatile tool for biological and clinical researchers by incorporating more information in single runs of experiments. For example, sample multiplexing could help in overcoming the obstacles of high cost and batch effects for cohort studies requiring the use of scRNA-seq in the clinic. Among the existing sample multiplexing technologies, the chemical barcoding approach30 possesses several advantages over the widely applied antibody barcoding techniques,28 including cost-effectiveness, versatility across samples with distinct genetic backgrounds, negligible transcriptomic perturbations and significant labelling efficiency. However, clinical samples from a large cohort are usually frozen for storage or transportation purposes. Cells from these samples are very fragile after freeze–thaw cycles and easy to decompose after a long procedure of sample barcoding, e.g., the previously reported ClickTags involve multiple steps of methanol-fixation, labelling and wash. Thus, few of the previously reported methods have been used in these types of clinical studies. By contrast, our live-cell compatible ClickTags are successfully applied to freeze–thaw samples of human TILs, which opens the door to large cohort studies using scRNA-seq. Beyond clinical studies, live-cell compatible ClickTags could make the scRNA-seq accessible to drug screening through sample multiplexing. Through the integration of time points and treatment conditions in a single experiment, one could observe the dynamic drug-perturbed transcription on heterogenous populations of cells. It is noteworthy that the current protocol is not applicable to barcoding of very few cells (<1000 cells) from special biopsy samples. To fulfill this requirement, the development of wash-free probes for barcoding is currently underway in our lab.
Ethics statement
This study was approved by the Ethics Committee of Nanjing Drum Tower Hospital (2021-394-01). Informed consent was obtained from the human participants in this study.Conflicts of interest
The authors declare the following competing financial interest(s): for J. Li, X. Zhao, W. Yu, Y. Yang, W. Zhu, D. Sun, and X. Ding, a patent application for “Chemical sample indexing for high-throughput single-cell analysis”, International application no. PCT/CN2022/080093, filed on 10 March 2022.Acknowledgements
J. P. L. acknowledges support from the National Key R&D Program of China (2019YFA09006600), the National Natural Science Foundation of China (21977048 and 92053111), Beijing National Laboratory for Molecular Sciences (BNLMS202008), Jiangsu Specially-Appointed Professor Plan, Program for Innovative Talents and Entrepreneur in Jiangsu, the Natural Science Foundation of Jiangsu Province (BK20202004) and the Excellent Research Program of Nanjing University (ZYJH004).References
- L. Yan, M. Yang, H. Guo, L. Yang, J. Wu, R. Li, P. Liu, Y. Lian, X. Zheng, J. Yan, J. Huang, M. Li, X. Wu, L. Wen, K. Lao, R. Li, J. Qiao and F. Tang, Nat. Struct. Mol. Biol., 2013, 20, 1131–1139 CrossRef CAS PubMed.
- B. Treutlein, D. G. Brownfield, A. R. Wu, N. F. Neff, G. L. Mantalas, F. H. Espinoza, T. J. Desai, M. A. Krasnow and S. R. Quake, Nature, 2014, 509, 371–375 CrossRef CAS PubMed.
- R. Satija, J. A. Farrell, D. Gennert, A. F. Schier and A. Regev, Nat. Biotechnol., 2015, 33, 495–502 CrossRef CAS PubMed.
- S. Zhong, S. Zhang, X. Fan, Q. Wu, L. Yan, J. Dong, H. Zhang, L. Li, L. Sun, N. Pan, X. Xu, F. Tang, J. Zhang, J. Qiao and X. Wang, Nature, 2018, 555, 524–528 CrossRef CAS PubMed.
- X. Han, Z. Zhou, L. Fei, H. Sun, R. Wang, Y. Chen, H. Chen, J. Wang, H. Tang, W. Ge, Y. Zhou, F. Ye, M. Jiang, J. Wu, Y. Xiao, X. Jia, T. Zhang, X. Ma, Q. Zhang, X. Bai, S. Lai, C. Yu, L. Zhu, R. Lin, Y. Gao, M. Wang, Y. Wu, J. Zhang, R. Zhan, S. Zhu, H. Hu, C. Wang, M. Chen, H. Huang, T. Liang, J. Chen, W. Wang, D. Zhang and G. Guo, Nature, 2020, 581, 303–309 CrossRef CAS PubMed.
- Z. Yao, C. T. J. van Velthoven, T. N. Nguyen, J. Goldy, A. E. Sedeno-Cortes, F. Baftizadeh, D. Bertagnolli, T. Casper, M. Chiang, K. Crichton, S.-L. Ding, O. Fong, E. Garren, A. Glandon, N. W. Gouwens, J. Gray, L. T. Graybuck, M. J. Hawrylycz, D. Hirschstein, M. Kroll, K. Lathia, C. Lee, B. Levi, D. McMillen, S. Mok, T. Pham, Q. Ren, C. Rimorin, N. Shapovalova, J. Sulc, S. M. Sunkin, M. Tieu, A. Torkelson, H. Tung, K. Ward, N. Dee, K. A. Smith, B. Tasic and H. Zeng, Cell, 2021, 184, 3222–3241.e26 CrossRef CAS PubMed.
- E. Tran, P. F. Robbins and S. A. Rosenberg, Nat. Immunol., 2017, 18, 255–262 CrossRef CAS PubMed.
- Y.-C. Lu, Z. Zheng, P. F. Robbins, E. Tran, T. D. Prickett, J. J. Gartner, Y. F. Li, S. Ray, Z. Franco, V. Bliskovsky, P. C. Fitzgerald and S. A. Rosenberg, Mol. Ther., 2018, 26, 379–389 CrossRef CAS PubMed.
- A. A. Tu, T. M. Gierahn, B. Monian, D. M. Morgan, N. K. Mehta, B. Ruiter, W. G. Shreffler, A. K. Shalek and J. C. Love, Nat. Immunol., 2019, 20, 1692–1699 CrossRef CAS PubMed.
- M. Slyper, C. B. M. Porter, O. Ashenberg, J. Waldman, E. Drokhlyansky, I. Wakiro, C. Smillie, G. Smith-Rosario, J. Wu, D. Dionne, S. Vigneau, J. Jané-Valbuena, T. L. Tickle, S. Napolitano, M.-J. Su, A. G. Patel, A. Karlstrom, S. Gritsch, M. Nomura, A. Waghray, S. H. Gohil, A. M. Tsankov, L. Jerby-Arnon, O. Cohen, J. Klughammer, Y. Rosen, J. Gould, L. Nguyen, M. Hofree, P. J. Tramontozzi, B. Li, C. J. Wu, B. Izar, R. Haq, F. S. Hodi, C. H. Yoon, A. N. Hata, S. J. Baker, M. L. Suvà, R. Bueno, E. H. Stover, M. R. Clay, M. A. Dyer, N. B. Collins, U. A. Matulonis, N. Wagle, B. E. Johnson, A. Rotem, O. Rozenblatt-Rosen and A. Regev, Nat. Med., 2020, 26, 792–802 CrossRef CAS PubMed.
- L. Zhang, Z. Li, K. M. Skrzypczynska, Q. Fang, W. Zhang, S. A. O’Brien, Y. He, L. Wang, Q. Zhang, A. Kim, R. Gao, J. Orf, T. Wang, D. Sawant, J. Kang, D. Bhatt, D. Lu, C.-M. Li, A. S. Rapaport, K. Perez, Y. Ye, S. Wang, X. Hu, X. Ren, W. Ouyang, Z. Shen, J. G. Egen, Z. Zhang and X. Yu, Cell, 2020, 181, 442–459.e29 CrossRef CAS PubMed.
- J. Ni, X. Wang, A. Stojanovic, Q. Zhang, M. Wincher, L. Bühler, A. Arnold, M. P. Correia, M. Winkler, P.-S. Koch, V. Sexl, T. Höfer and A. Cerwenka, Immunity, 2020, 52, 1075–1087.e8 CrossRef CAS PubMed.
- S. Cheng, Z. Li, R. Gao, B. Xing, Y. Gao, Y. Yang, S. Qin, L. Zhang, H. Ouyang, P. Du, L. Jiang, B. Zhang, Y. Yang, X. Wang, X. Ren, J.-X. Bei, X. Hu, Z. Bu, J. Ji and Z. Zhang, Cell, 2021, 184, 792–809.e23 CrossRef CAS PubMed.
- L. Zheng and Z. Zhang, Immunity, 2021, 54, 199–201 CrossRef CAS PubMed.
- M. Stoeckius, C. Hafemeister, W. Stephenson, B. Houck-Loomis, P. K. Chattopadhyay, H. Swerdlow, R. Satija and P. Smibert, Nat. Methods, 2017, 14, 865–868 CrossRef CAS PubMed.
- E. P. Mimitou, A. Cheng, A. Montalbano, S. Hao, M. Stoeckius, M. Legut, T. Roush, A. Herrera, E. Papalexi, Z. Ouyang, R. Satija, N. E. Sanjana, S. B. Koralov and P. Smibert, Nat. Methods, 2019, 16, 409–412 CrossRef CAS PubMed.
- J. Lee, D. Y. Hyeon and D. Hwang, Exp. Mol. Med., 2020, 52, 1428–1442 CrossRef CAS PubMed.
- C. Zhu, S. Preissl and B. Ren, Nat. Methods, 2020, 17, 11–14 CrossRef CAS PubMed.
- C. J. Kearney, S. J. Vervoort, K. M. Ramsbottom, I. Todorovski, E. J. Lelliott, M. Zethoven, L. Pijpers, B. P. Martin, T. Semple, L. Martelotto, J. A. Trapani, I. A. Parish, N. E. Scott, J. Oliaro and R. W. Johnstone, Sci. Adv., 2021, 7, eabe3610 CrossRef CAS PubMed.
- O. Stegle, S. A. Teichmann and J. C. Marioni, Nat. Rev. Genet., 2015, 16, 133–145 CrossRef CAS PubMed.
- M. D. Luecken and F. J. Theis, Mol. Syst. Biol., 2019, 15, 1–23, DOI:10.15252/msb.20188746.
- S. C. Hicks, F. W. Townes, M. Teng and R. A. Irizarry, Biostatistics, 2018, 19, 562–578 CrossRef PubMed.
- Y. Hao, S. Hao, E. Andersen-Nissen, W. M. Mauck, S. Zheng, A. Butler, M. J. Lee, A. J. Wilk, C. Darby, M. Zager, P. Hoffman, M. Stoeckius, E. Papalexi, E. P. Mimitou, J. Jain, A. Srivastava, T. Stuart, L. M. Fleming, B. Yeung, A. J. Rogers, J. M. McElrath, C. A. Blish, R. Gottardo, P. Smibert and R. Satija, Cell, 2021, 184, 3573–3587.e29 CrossRef CAS PubMed.
- P. Datlinger, A. F. Rendeiro, T. Boenke, M. Senekowitsch, T. Krausgruber, D. Barreca and C. Bock, Nat. Methods, 2021, 18, 635–642 CrossRef CAS PubMed.
- D. A. Cusanovich, R. Daza, A. Adey, H. A. Pliner, L. Christiansen, K. L. Gunderson, F. J. Steemers, C. Trapnell and J. Shendure, Science, 2015, 348, 910–914 CrossRef CAS PubMed.
- A. M. Klein, L. Mazutis, I. Akartuna, N. Tallapragada, A. Veres, V. Li, L. Peshkin, D. A. Weitz and M. W. Kirschner, Cell, 2015, 161, 1187–1201 CrossRef CAS PubMed.
- J. Cao, J. S. Packer, V. Ramani, D. A. Cusanovich, C. Huynh, R. Daza, X. Qiu, C. Lee, S. N. Furlan, F. J. Steemers, A. Adey, R. H. Waterston, C. Trapnell and J. Shendure, Science, 2017, 357, 661–667 CrossRef CAS PubMed.
- M. Stoeckius, S. Zheng, B. Houck-Loomis, S. Hao, B. Z. Yeung, W. M. Mauck, P. Smibert and R. Satija, Genome Biol., 2018, 19, 224 CrossRef CAS PubMed.
- D. Shin, W. Lee, J. H. Lee and D. Bang, Sci. Adv., 2019, 5, eaav2249 CrossRef CAS PubMed.
- J. Gehring, J. Hwee Park, S. Chen, M. Thomson and L. Pachter, Nat. Biotechnol., 2020, 38, 35–38 CrossRef CAS PubMed.
- L. Fang, G. Li, Z. Sun, Q. Zhu, H. Cui, Y. Li, J. Zhang, W. Liang, W. Wei, Y. Hu and W. Chen, Mol. Syst. Biol., 2021, 17, 1–16, DOI:10.15252/msb.202010060.
- C. S. McGinnis, D. M. Patterson, J. Winkler, D. N. Conrad, M. Y. Hein, V. Srivastava, J. L. Hu, L. M. Murrow, J. S. Weissman, Z. Werb, E. D. Chow and Z. J. Gartner, Nat. Methods, 2019, 16, 619–626 CrossRef CAS PubMed.
- M. Blackman, M. Royzen and J. Fox, J. Am. Chem. Soc., 2008, 130, 13518 CrossRef CAS PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- R. Selvaraj and J. Fox, Curr. Opin. Chem. Biol., 2013, 17, 753–760 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- J. Li and P. R. Chen, Nat. Chem. Biol., 2016, 12, 129–137 CrossRef CAS PubMed.
- H. Wu and N. K. Devaraj, Acc. Chem. Res., 2018, 51, 1249–1259 CrossRef CAS PubMed.
- A. Subramanian, R. Narayan, S. M. Corsello, D. D. Peck, T. E. Natoli, X. Lu, J. Gould, J. F. Davis, A. A. Tubelli, J. K. Asiedu, D. L. Lahr, J. E. Hirschman, Z. Liu, M. Donahue, B. Julian, M. Khan, D. Wadden, I. C. Smith, D. Lam, A. Liberzon, C. Toder, M. Bagul, M. Orzechowski, O. M. Enache, F. Piccioni, S. A. Johnson, N. J. Lyons, A. H. Berger, A. F. Shamji, A. N. Brooks, A. Vrcic, C. Flynn, J. Rosains, D. Y. Takeda, R. Hu, D. Davison, J. Lamb, K. Ardlie, L. Hogstrom, P. Greenside, N. S. Gray, P. A. Clemons, S. Silver, X. Wu, W.-N. Zhao, W. Read-Button, X. Wu, S. J. Haggarty, L. V. Ronco, J. S. Boehm, S. L. Schreiber, J. G. Doench, J. A. Bittker, D. E. Root, B. Wong and T. R. Golub, Cell, 2017, 171, 1437–1452.e17 CrossRef CAS PubMed.
- E. C. Bush, F. Ray, M. J. Alvarez, R. Realubit, H. Li, C. Karan, A. Califano and P. A. Sims, Nat. Commun., 2017, 8, 105 CrossRef PubMed.
- C. Ye, D. J. Ho, M. Neri, C. Yang, T. Kulkarni, R. Randhawa, M. Henault, N. Mostacci, P. Farmer, S. Renner, R. Ihry, L. Mansur, C. G. Keller, G. McAllister, M. Hild, J. Jenkins and A. Kaykas, Nat. Commun., 2018, 9, 4307 CrossRef PubMed.
- E. Armingol, A. Officer, O. Harismendy and N. E. Lewis, Nat. Rev. Genet., 2021, 22, 71–88 CrossRef CAS PubMed.
- R. Sklavenitis-Pistofidis, G. Getz and I. Ghobrial, Nat. Med., 2021, 27, 375–376 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00046f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |