
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the intrinsic catalytic activities of oxygen-evolution catalysts by doping: a comprehensive review
Sivasankara Rao
Ede
and
Zhiping
Luo
 *
*
Department of Chemistry, Physics and Materials Science, Fayetteville State University, Fayetteville, North Carolina 28301, USA. E-mail: zluo@uncfsu.edu
First published on 13th August 2021
Abstract
Electrochemical water splitting produces clean hydrogen fuel as one of the pivotal alternative energies to fossil fuels in the near future. However, the anodic oxygen evolution reaction (OER) is a significant bottleneck that curtails large-scale applications of electrochemical water splitting technology, owing to its sluggish reaction kinetics. In the past few decades, various methods have been proposed to improve the OER kinetics. Among them, doping is a simple and efficient method to mold the OER kinetics of a catalyst by incorporating different or hetero atoms into the host lattice. These efforts are vital to design highly efficient OER catalysts for real-world applications. However, the OER mechanism of a doped catalyst varies, depending on the host lattice and the dopant. This review highlights different doping strategies and associated OER mechanisms of state-of-the-art catalysts, including oxides (noble metal oxides, perovskite oxides, spinel oxides, hydroxides and others), non-oxides (metal sulfides, metal selenides, metal phosphides, metal nitrides and metal carbides), and carbon-based catalysts (graphene, carbon nanotubes and others). Fundamental understanding of the doping effects on the OER from combined experimental and theoretical research provides guidelines for designing efficient catalysts.
1. Introduction
The demand for transitioning the world's energy from fossil fuels to eco-friendly renewable energy is rapidly increasing, in order to cope with the depletion of fossil fuels and to curtail the associated CO2 emission.1 Currently, sunlight and wind are the main contributors to renewable energy, but they are highly intermittent.2 To overcome this challenge, researchers have been working on new energy storage techniques that can sustain the erratic behavior of solar, wind and tidal energies.3 Instead, we can store these excess energies in the form of hydrogen through water electrolysis (eqn (1)),4 and the stored hydrogen is readily available as a fuel cell for energy production in a clean way without CO2 emission (eqn (2)).5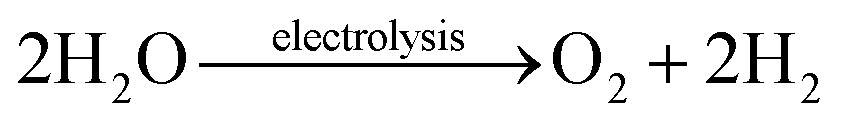 | (1) |
| 2H2 + O2 → 2H2O + heat | (2) |
Hydrogen production through water electrolysis is a thermodynamically infeasible process that requires additional energy to drive this reaction, and theoretically, 1.23 V (pH = 0) electric potential is required.6 However, the oxygen evolution reaction (OER) during the water electrolysis is associated with a sluggish reaction kinetics that comprises O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond formation with a loss of 4 e−/4 H+, requiring an even higher electric potential than the theoretical prediction.7 The excess potential above the theoretical prediction is known as overpotential (η), which is one of the major benchmarks for denoting OER activity, along with high current density and low Tafel slope values.8 Over the past few decades, significant research has been conducted to improve the OER kinetics through various strategies for economically viable hydrogen production.9 So far, RuO2 and IrO2 catalysts have been proven to be two of the most efficient types of electrocatalyst for the OER in both acidic and alkaline media. However, the high cost associated with these catalysts hinders their large-scale water electrolysis application.10 In alkaline media, researchers can replace these expensive catalysts with earth-abundant transition metal (TM)-based catalysts,11 as they are stable in such alkaline environments, while being unstable in acidic media.12,13 The catalyst stability is also an important factor for consideration.
O bond formation with a loss of 4 e−/4 H+, requiring an even higher electric potential than the theoretical prediction.7 The excess potential above the theoretical prediction is known as overpotential (η), which is one of the major benchmarks for denoting OER activity, along with high current density and low Tafel slope values.8 Over the past few decades, significant research has been conducted to improve the OER kinetics through various strategies for economically viable hydrogen production.9 So far, RuO2 and IrO2 catalysts have been proven to be two of the most efficient types of electrocatalyst for the OER in both acidic and alkaline media. However, the high cost associated with these catalysts hinders their large-scale water electrolysis application.10 In alkaline media, researchers can replace these expensive catalysts with earth-abundant transition metal (TM)-based catalysts,11 as they are stable in such alkaline environments, while being unstable in acidic media.12,13 The catalyst stability is also an important factor for consideration.
The OER catalytic activity can be improved by enhancing the number of active sites on the surface of the catalyst, which is termed as the intrinsic activity of the catalyst.14–22 The intrinsic activity of the OER catalyst can be augmented by introducing elemental dopants, creating vacancies, optimizing the morphology, decreasing the particle size, or via surface reconstruction during the OER process.23 Among them, elemental doping is the most efficient way to engineer the activity of OER catalysts.9 However, a fundamental understanding of OER mechanisms is required in catalyst design to shift its efficiency closer to the thermodynamic limit.24 In the past few decades, several reaction mechanisms for the OER have been unveiled to fabricate efficient OER catalysts.25
To date, the current literature on OER catalysts lacks a comprehensive review on the doping effect in the OER mechanisms of catalysts. In this review, by corroborating experimental and theoretical results, we illustrate how particular elemental doping can improve the OER efficiency of a catalyst. In the beginning, OER intermediates by doping are listed. In particular materials, the OER mechanisms are presented, including the influence of dopant on improving the intrinsic activity through optimizing OER descriptors, such as eg orbital occupancy, metal–oxygen covalency, and structural parameters like M–O bond binding energy, length, and angle. Various types of material that have exhibited efficient doping in the OER are reviewed. Understanding of these OER mechanisms will guide the design of new efficient OER catalysts.
2. OER mechanisms
Generally, in the OER, four electrons and protons are generated and combine on the surface of a catalyst. Several mechanisms are proposed for alkaline and acidic electrolytes; however, the research community primarily accepts the oxide26 and electrochemical oxide26 mechanisms. Karsil'shchikov27 and Wade–Hackerman28 pathways are the two known mechanisms in an acidic medium for the OER.28 Here, the symbol * denotes an active catalytic site on the surface of the OER catalyst. In one of the reaction steps below, 2*O → 2* + O2(g) in both oxide mechanisms (Fig. 1a and c) or the electrochemical oxide mechanism (Fig. 1b and d), oxygen gas is generated. In case of recombination of two *O intermediates or generation of oxygen from the *O intermediate, oxygen evolution is impossible because of a substantial thermodynamic barrier. The oxygen gas is generated from the active catalytic site after two steps via the formation of intermediate *O–OH, a step which is the most sluggish one out of all rate-determining steps (RDSs) in the OER.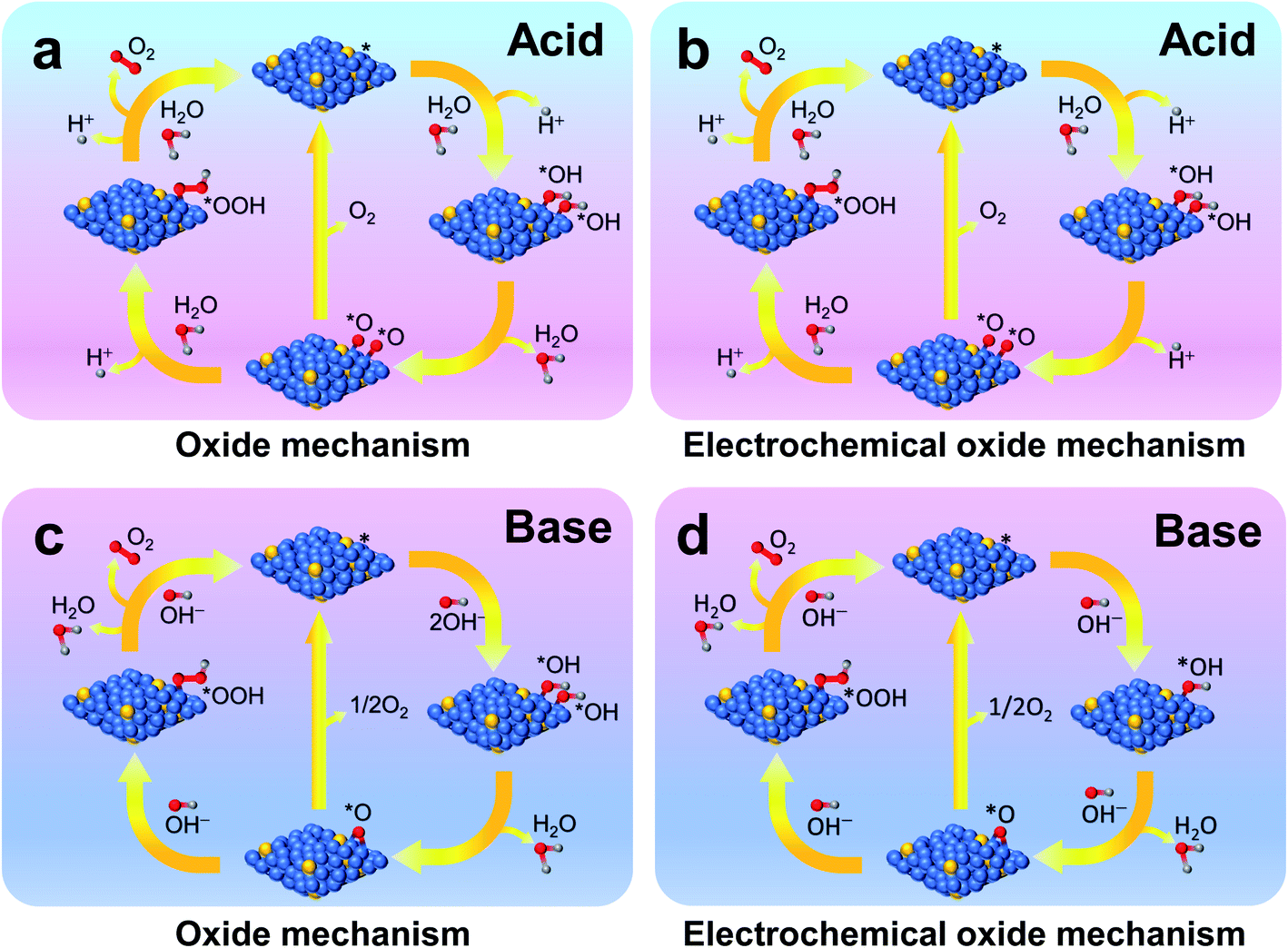 | ||
| Fig. 1 Schematic illustrations of oxide (a and c) and electrochemical oxide (b and d) mechanisms of the OER in acid (a and b) and base (c and d) electrolytes, respectively. | ||
In acidic media, the cathodic reaction is expressed as
| 4H+ + 4e− → 2H2 | (3) |
| 2H2O → O2(g) + 4H+ + 4e− | (4) |
The elementary steps of the oxide and electrochemical oxide mechanisms are described below:26
(a) Oxide mechanism (Fig. 1a), including reaction steps of * + H2O → *OH + H+ (aq) + e−, 2*OH → *O + H2O, 2*O → 2* + O2 (g), 2*O + H2O → *O–OH + H+ + e−, and *O–OH + H2O → * + O2 (g) + H+ + e−.
(b) Electrochemical oxide mechanism (Fig. 1b), including reaction steps of * + H2O → *OH + H+ (aq) + e−, 2*OH → *O + H+ (aq) + e−, 2*O → 2* + O2 (g), 2*O + H2O → *O–OH + H+ + e−, and *O–OH + H2O → * + O2 (g) + H+ + e−.
Likewise, in the alkaline medium, the cathodic reaction is
| 4H2O + 4e− → 2H2 + 4OH− | (5) |
| 4OH− → 2O2 (g) + 2H2O + 4e− | (6) |
The elementary steps of the oxide and electrochemical oxide mechanisms are described below:26
(a) Oxide mechanism (Fig. 1c), including reaction steps of * + OH− → *OH + e−, 2*OH → *O + H2O, 2*O → 2* + O2 (g), *O + OH− → *O–OH + e−, and *O–OH + OH− → * + O2 (g) + H2O.
(b) Electrochemical oxide mechanism (Fig. 1d), including reaction steps of * + OH− → *OH + e−, *OH + OH− → *O + H2O + e−, 2*O → 2* + O2 (g), *O + OH− → *O–OH + e−, and *O–OH + OH− → * + O2 (g) + H2O.
There are three other well-known mechanisms proposed by, namely, Karsil'shchikov,27 Yeager,29 and Bockris,30 in addition to the primarily recognized oxide and electrochemical oxide mechanisms.26,28
3. Why doping?
Besides the electrocatalyst type and chemical stability, the geometry and electronic structure are the key parameters for optimization in developing efficient OER catalysts.31Geometrical surface area is the most fundamental and critical parameter, and it can be improved by synthesizing nano-forms of an electrocatalyst. By improving the specific surface area of the catalyst, the mass transfer rate and density of active catalytic sites can be accelerated.32 However, nanomaterials are often aggregated, resulting in poor stability during long-term service.9
Tuning the electronic structure is another critical factor in improving the OER activities, and elemental doping is one of the best ways of tuning the electronic structure.33 The tuning of the electronic structure by doping influences the binding energy of OER intermediates, such as *O, *OH, and *O–OH. Hence, compared with other approaches for improving OER activities, doping provides an effective way to achieve large-scale activity and long-term stability under harsh applied anodic overpotentials.
The dopant is present in trace or small amounts. Doping generates ionizable species within the host material by incorporating impurities. The ionizable species could lead to the shift of the Fermi level (EF), enabling carrier transport with superior catalytic activity. Doping can be performed through (a) gating for direct carrier injection; (b) antisites and vacancies as structural defects; and (c) photodoping through generation of transient carrier species via photo-excitation. Incorporation of foreign atoms that do not produce free carriers is not considered as doping.34
In the following sections, we will elaborate on the influences of doping in the following common catalytic systems, including various oxides (noble metal oxides, perovskite oxides, spinel oxides, hydroxides and other oxides), non-oxides (metal sulfides, metal selenides, metal phosphides, metal nitrides, and metal carbides), and carbon-based catalysts. Fig. 2 shows the overpotential at 10 mA cm−2 and Tafel slope of the representative types of catalyst (data collected from the following sections). Lower overpotential and lower Tafel slopes are desirable for high catalytic performance. The effects of doping and its impacts on OER activity in the various prescribed electrocatalysts are finely discussed hereafter in terms of accelerating OER kinetics with less energy input.
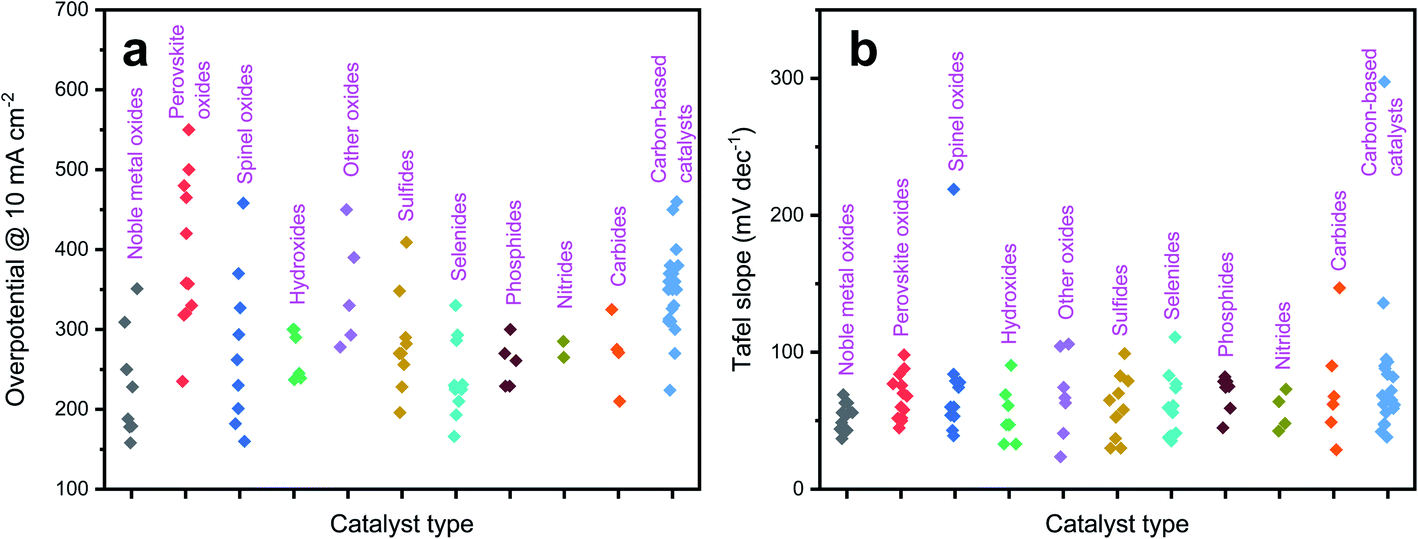 | ||
| Fig. 2 Catalytic performance of representative types of catalyst: (a) overpotential @ 10 mA cm−2; (b) Tafel slope. | ||
4. Doping of oxide electrocatalysts
4.1 Noble metal (Ir and Ru) oxides
Table 1 provides a summary of some representative doped noble metal oxides and their catalytic performances. Both IrO2 and RuO2 are considered as benchmark catalysts for the OER in basic and acidic electrolytes. Under OER conditions, IrO2 is more stable than RuO2, but both oxides are unstable at high anodic potentials. RuO2 leaches out into the electrolyte in the form of RuO4 at high anodic potentials.35 Likewise, IrO2 also dissolves in the electrolyte by forming IrO3 at high anodic potentials.36 However, IrO2 shows considerably higher stability but lower OER performance, compared to RuO2.| Catalyst | Overpotential at a specific current density | Tafel slope (mV dec−1) | Durability | Electrolyte |
|---|---|---|---|---|
| Noble metal oxides | ||||
| Ce0.2–IrO2@NPC45 | 224 mV @ 10 mA cm−2 | 55.9 | 100 h @ 10 mA cm−2 | 0.5 M H2SO4 |
| Co-doped RuO2 (ref. 41) | 169 mV @ 10 mA cm−2 | 63 | 50 h @ 10 mA cm−2 | 0.5 M H2SO4 |
| Cr-doped IrOx (ref. 43) | 250 mV @ 10 mA cm−2 | 69 | 25 h @ 10 mA cm−2 | 0.5 M H2SO4 |
| Cr0.6Ru0.4O2 (ref. 44) | 178 mV @ 10 mA cm−2 | 56 | 10 h @ 10 mA cm−2 | 0.5 M H2SO4 |
| Cu-doped RuO2 (ref. 38) | 188 mV @ 10 mA cm−2 | 43.96 | 8 h @ 10 mA cm−2 | 0.5 M H2SO4 |
| Cu-doped IrO2 (ref. 39) | 351 mV @ 10 mA cm−2 | 63 | 6000 s @ 1.68 V vs. RHE | 0.1 M HClO4 |
| IrCoNi nanocrystals40 | 309 mV @ 10 mA cm−2 | 53.8 | — | 0.5 M H2SO4 |
| Mg-doped RuO2 (ref. 47) | 228 mV @ 10 mA cm−2 | 48.66 | 30 h @ 1.15 V vs. RHE | 0.5 M H2SO4 |
| Mn-doped RuO2 (ref. 42) | 158 mV @ 10 mA cm−2 | 42.94 | 10 h @ 10 mA cm−2 | 0.5 M H2SO4 |
| W-doped IrO2 (ref. 46) | 250 mV @ 10 mA cm−2 | 56.6 | — | 0.1 M HClO4 |
| Zn-doped RuO2 (ref. 48) | 179 mV @ 10 mA cm−2 | 36.9 | 20 h @ 10 mA cm−2 | 0.5 M H2SO4 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Perovskite oxides | ||||
| Ba2CoMo0.5Nb0.5O6−δ49 | 445 mV @ 10 mA cm−2 | 77 | 2 h @ 10 mA cm−2 | 0.1 M KOH |
| Ba0.5Sr0.5Co0.8Fe0.2O3−δ50 | 340 mV @ 10 mA cm−2 | 70 | 10 h @ 10 mA cm−2 | 1 M KOH |
| CaMn0.75Nb0.25O3−δ51 | 550 mV @ 10 mA cm−2 | 98 | 7000 s @ 1.65 V vs. RHE | 0.1 M KOH |
| Co-doped SrIrO3 (ref. 52) | 235 mV @ 10 mA cm−2 | 51.8 | 20 h @ 10 mA cm−2 | 0.1 M HClO4 |
| Pr0.5Ba0.5CoO3−δ53 | 320 mV @ 10 mA cm−2 | 60 | 120 min @ 5 mA cm−2 | 0.1 M KOH |
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ54 | 358 mV @ 10 mA cm−2 | 52 | 12 h @ 10 mA cm−2 | 0.1 M KOH |
| P-doped LaFeO3−δ55 | 465 mV @ 10 mA cm−2 | 50 | — | 0.1 M KOH |
| SCFW0.4-BM56 | 357 mV @ 10 mA cm−2 | 58 | 18 h @ 10 mA cm−2 | 0.1 M KOH |
| Sr2Co1.5Fe0.5O6−δ57 | 318 mV @ 10 mA cm−2 | 44.8 | 10 h @ 10 mA cm−2 | 1 M KOH |
| SrCo0.95P0.05O3−δ58 | 480 mV @ 10 mA cm−2 | 84 | 7000 s @ 1.65 V vs. RHE | 0.1 M KOH |
| Sr-doped PrBaCo2O5+δ59 | 420 mV @ 10 mA cm−2 | 75.8 | 10 h @ 0.1 mA cm−2 | 1 M KOH |
| SrCo0.9Ti0.1O3−δ60 | 500 mV @ 10 mA cm−2 | 88 | 10 h @ 10 mA cm−2 | 0.1 M KOH |
| Yb-doped CaMnO3 (ref. 61) | 330 mV @ 10 mA cm−2 | 68 | 1500 s @ 520 mV | 0.1 M KOH |
Researchers doped RuO2 with a small amount of Ir (RuxIr1−xO2) and observed a significant improvement in stability without sacrificing the OER performance.37 Another way of enhancing the OER activity with lower total noble metal content is doping with earth-abundant metals, such as Cu,38,39 Ni,40 Co,40,41 Mn,42 Cr,43,44 Ce,45 W,46 Mg,47 and Zn.48 For example, Cu-doped IrO2 (Cu0.3Ir0.7Oδ) displays improved OER activity in acidic, neutral and basic media, compared to pristine IrO2.39 The enhanced OER activity of Cu0.3Ir0.7Oδ is primarily imputed to the Jahn–Teller distortion in IrO6 octahedra induced by Cu, as well as oxygen vacancies induced by Cu doping. Density functional theory (DFT) calculations revealed the improved OER activity mechanism by the Cu doping. With the increase in Cu-doping, σ and π bonding regions become broader and shift to a lower energy level because of the occupancy of σ states. The partial density of states suggests that with an increase in Cu doping, the antibonding states of dxy shift up; in contrast, dz2 shifts down. The dxz and dyz orbitals were crossed by the Fermi level EF, while the dx2−y2 orbital is unchanged. Because of electrons hopping from dxz and dyz (t2g) to dz2, the eg level is partially filled as a consequence of enhanced degeneracy in Ir 5d orbitals by Cu doping. The strong interaction of eg and O 2p orbitals facilitates bonding with oxygen intermediates during σ bond formation, which is more advantageous than the π bonding of t2g orbitals.39
Suntivch et al. postulated that the OER catalyst, specifically perovskite oxide with the eg value close to unity, will exhibit superior OER performance.62 Later, other scientists found that the eg filling values in some oxides also have a similar effect.63 Site-specific deposition of a meager amount of Ir on facets of Ni nano-frameworks led to excellent OER activity in an acidic electrolyte and the catalyst even outperformed an Ir/C catalyst.62 The oxygen adsorption of TMs was effectively tuned by doping with Ir. Similarly, Feng et al. reported porous IrCoNi nanocrystals as an efficient OER catalyst in highly corrosive HClO4 electrolyte.40Fig. 3a shows the schematic illustration of IrCoNi porous hollow nanocrystals (PHNCs). A two-step process was followed to synthesize PHNCs. In the first step, IrCo single nanocrystals (SNCs) were prepared in oleylamine; and in the second step, IrCoNi PHNCs were obtained from SNCs by etching with Fe3+ in hexane/ethanol at room temperature. Fig. 3b shows a transmission electron microscopy (TEM) image of the as-synthesized IrCoNi PHNCs, presenting uniform hollow nanocrystals. The polarization curves of IrCo/C, IrCoNi/C, and IrNi/C PHNCs are compared with that of the Ir/C catalyst in Fig. 3c. The IrCoNi/C catalyst shows superior activity with an onset potential of 1.46 V and requires only 303 mV overpotential to achieve 10 mA cm−2 current density. The projected density of states (PDOS) of the d-band of Ir and IrCoNi catalysts is displayed in Fig. 3d, which shows the left side shift of the d-band center of IrCoNi from the Fermi level. The shift in the Fermi level facilitates the desired charge and discharge of OER intermediates. The schematic representation of OER reaction paths, as shown in Fig. 3e, reveals that the adsorption of OER intermediates becomes weaker when Ir is alloyed with Co and Ni, compared to pristine Ir. In another study, porous W-doped IrO2 nanodendrites were found to exhibit superior OER performance under all pH conditions.46 DFT calculations suggested that W doping stabilizes the IrO2 at harsh anodic potentials by lowering the binding energies of oxygen intermediates. Qu et al. synthesized Cr-doped IrOx nanowires and evaluated their OER activity in 0.5 M H2SO4 electrolyte.43 The catalyst exhibits excellent OER activity with 25 h durability without any decay in activity. This high activity was ascribed to an increase in active site number and facile adsorption of OER intermediates via electronic structure tuning.
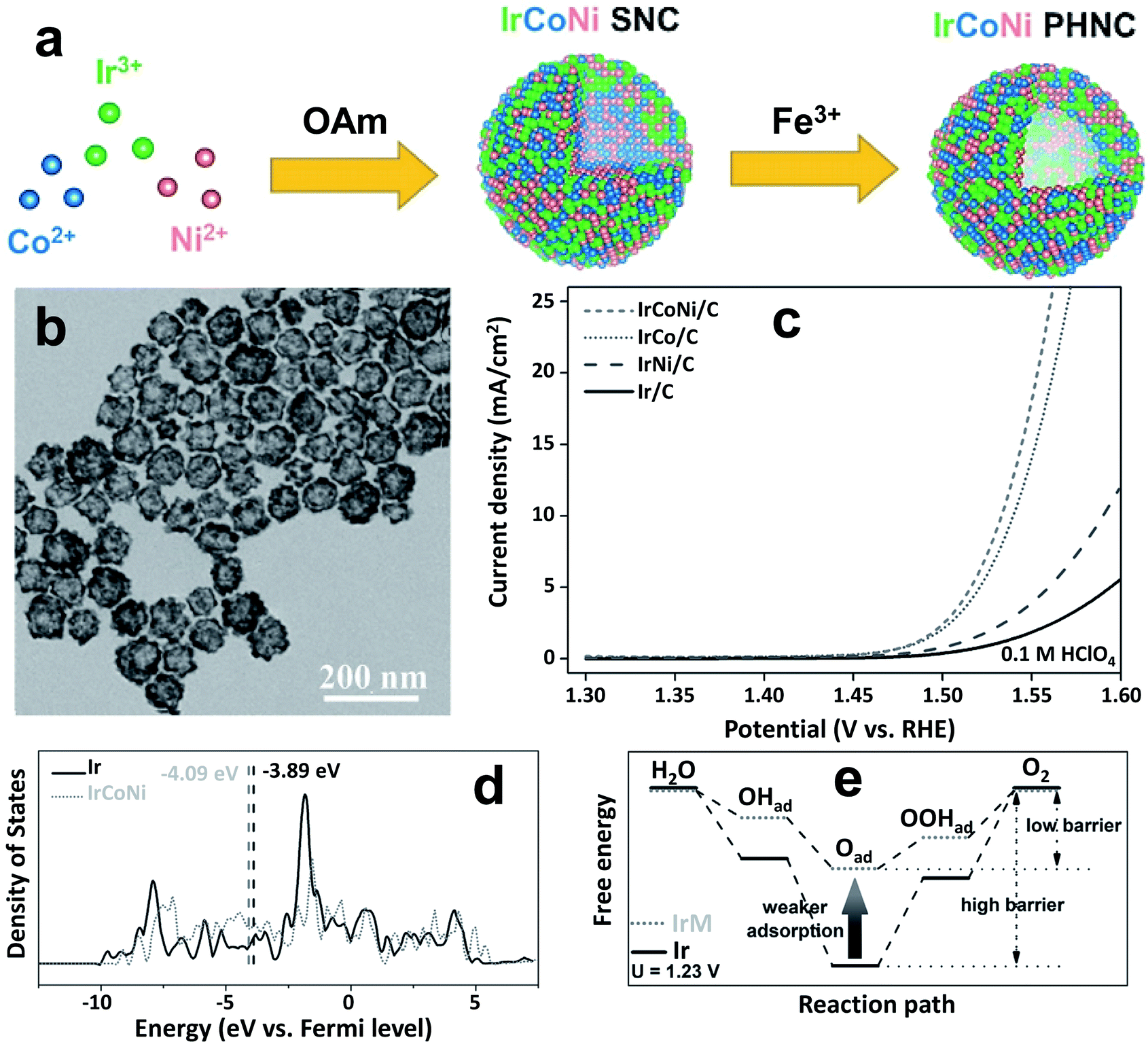 | ||
| Fig. 3 (a) Schematic illustration of the formation process of IrCoNi PHNCs; (b) TEM image of the as-prepared IrCoNi PHNCs; (c) polarization curves of IrCo, IrCoNi, and IrNi for the OER with 95% iR-compensation; (d) projected DOS of d bands of Ir (solid) and IrCoNi models (dotted) with the corresponding d-band center denoted by dashed lines; (e) schematic illustration of reaction paths for the OER, indicating that alloying Ir with M leads to weaker adsorption of oxygen-based intermediates, thus decreasing the barrier in the reaction path.40 Reproduced from ref. 40 with permission, © 2017 John Wiley and Sons. | ||
Like IrO2, the OER performance and stability of RuO2 were also enhanced by doping. Cu-doped RuO2 porous nano-polyhedra, derived from organic frameworks, are probably one of the best examples for enhancing OER activity via the formation of oxygen vacancies.38 Here, most oxygen vacancies formed near Cu, causing the adjacent Ru atom to become more negative, and thus shifting the O 2p band center towards the Fermi level for superior OER activity. Recently, Chen's group reported Cr-doped RuO2 as an efficient OER catalyst in an acidic medium derived from a metal–organic framework (MOF).44 The schematic representation of the synthetic approach of this catalyst is shown in Fig. 4a. Initially, RuCl3 was incorporated into the MOF of Cr. The resulting composite was annealed between 450 and 600 °C for 4 h, and the composite which was annealed at 550 °C showed the formation of Cr0.6Ru0.4O2. Among Cr1−xRuxO2 catalysts, Cr0.6Ru0.4O2 showed superior OER activity with a low overpotential of 178 mV at 10 mA cm−2 current density, and at the same current density, it maintained its stability for 10 h. Chromium incorporation optimizes the DOS for RuO2 and decreases the Fermi energy level from 2.01 to 1.07 eV, which stabilizes the crystal structure of Cr-doped RuO2. Fig. 4b displays the Ru site, which is coordinated with five atoms that act as an adsorption site for OER intermediates, and the formation of *OOH is found to be a RDS. The free energy change in the RDS is calculated to be 1.87 eV for the doped one, which is nearly 0.1 eV less than the free energy on the RuO2 surface.44 It was predicted that Mn doping also has a similar effect on improving the OER activity of RuO2. Very recently, Chen et al. reported Mn-doped RuO2 as an OER catalyst in an acid with a record low overpotential of 158 mV at 10 mA cm−2, which was stable up to 5000 cycles at a scan rate of 100 mV s−1.42 Recently, Chen and co-workers demonstrated considerable improvement of OER activity of RuO2 in an acid medium by doping with Mg in a one-step annealing of a Ru-exchanged MOF.47 DFT calculations indicate the formation of oxygen vacancies on account of different oxidation states of Mg (2+) and Ru (4+), significantly bringing down the energy barrier for the RDS.47
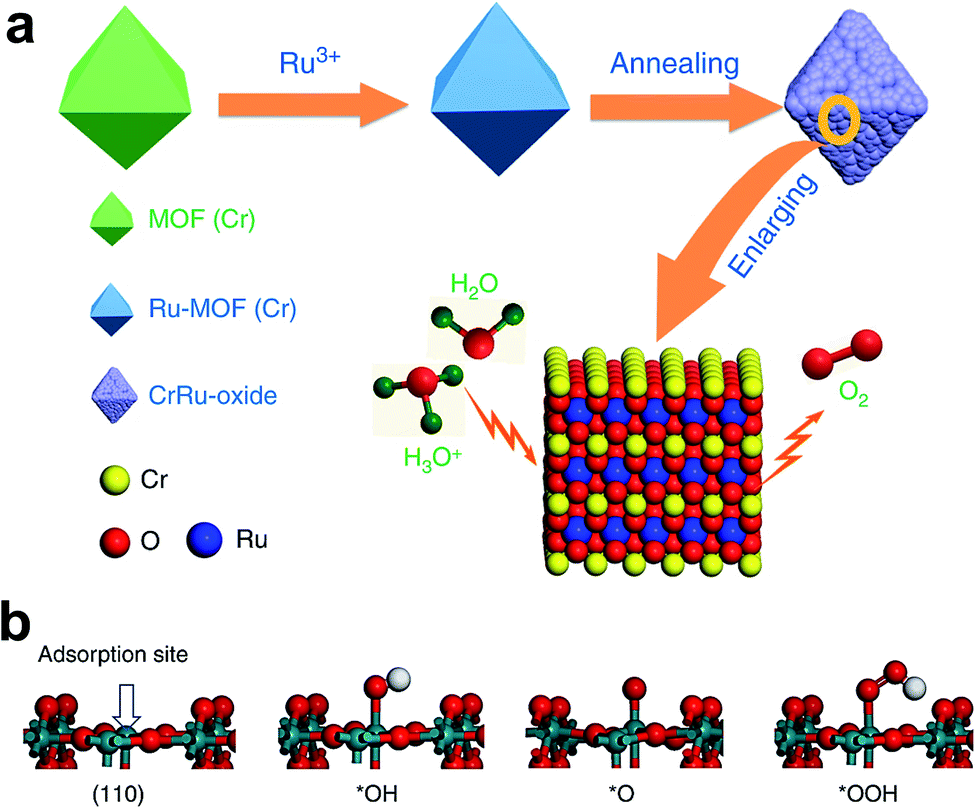 | ||
| Fig. 4 (a) Schematic illustration of the preparation of Cr0.6Ru0.4O2 electrocatalysts for OER application in acid media; (b) four-step OER process.44 Reproduced with open access. | ||
Creating defect-containing RuO2via doping and dopant leaching is another way of improving the OER activity and stability in acidic medium. For example, Tian et al. doped RuO2 with zinc and leached it out by aging in an acid, and the catalyst displayed 179 mV overpotential and 20 h stability at a current density of 10 mA cm−2.48 The high catalytic activity is accredited to the synergetic effect of active sites and tuning of the electronic structure. Recently, an oxygen-defect-containing Ru-based catalyst was synthesized by doping with cobalt, which showed 169 mV overpotential and 50 h endurance at 10 mA cm−2 current density.41 The doping generated a large amount of oxygen defects, following a vacancy-related lattice oxygen oxidation mechanism (LOM) rather than an adsorbate evolution reaction mechanism (AEM). Both computational and experimental studies elucidate this mechanism. All these studies revealed the effectiveness of doping Ir and Ru oxides for enabling higher activity and longer stability.
4.2 Perovskite oxides
Perovskite oxides are a large family of catalysts with the general formula ABO3, where A is either an alkaline earth metal or a rare earth metal, and B is a TM. Table 1 provides a summary of some representative doped perovskite oxides and their catalytic performances. The ideal perovskite oxide displays a cubic crystal structure. The TMs are in the oxidation states of +3, +4, or a mixture of them, governed by the metal on the A-site.18,64 The OER activity can be tailored by doping at either the A-site, B-site, or both sites to create oxygen vacancies in the perovskite.18 The metal–oxygen hybridization depends upon the oxidation state of TMs, and the electronic properties of these perovskites rely on this parameter. The TMs with high electronegativity show high metal–oxygen hybridization, and the d-band center of the TM oxide moves closer to O 2p states as the electronegativity increases. However, besides the metal–oxygen hybridization, octahedral tilting, Jahn–Teller distortion, and displacement of B-site cations also play significant roles in governing the electronic properties of perovskite oxides.18,65,66Grimaud et al. doped a series of lanthanides (Ln = Pr, Sm, Gd and Ho) at the A-site of BaCoO3−δ to optimize the eg filling values, thus improving the OER activity.53 Among Ln-doped perovskites, Pr0.5Ba0.5CoO3−δ shows superior activity. Estimated from X-ray absorption spectroscopy (XAS) and chemical titrations, the eg filling values of all Ln-doped compounds are close to unity. However, DFT calculations suggest that hybridization of Co 3d and O 2p orbitals decreases from Pr to Ho, because of the decreased Co oxidation state with an increase in electronegativity of dopants from Pr to Ho. Similarly, when the A-site of CaMnO3 is doped with Yb (Ca0.9Yb0.1MnO3), the OER activity is found to be 100 times higher than that of the pristine sample.61 The excellent OER performance of Ca0.9Yb0.1MnO3 is attributed to the increased Mn3+/Mn4+ ratio, which provides the double exchange of electrons and optimized eg filling value of ∼0.81. The OER activity of double perovskites was also tuned by doping the A-site; for instance, Sr doping of the Ba site of PrBaCo2O5+δ improves the population of Co4+ ions on the surface.59 The doping enhances the ease of adsorption and deprotonation of *OOH intermediates from the catalyst surface, thus improving the OER performance of the perovskite.
Recently, researchers turned their attention to dope non-metals like phosphorus, sulfur, and silicon at the B-site to stabilize the crystal structure of perovskite oxides. An important reason for doping, specifically phosphorus, for improving electrocatalytic properties is that it has greater affinity towards combining with oxygen. Phosphorus doping at the B-site of SrCoO3−δ (SC) perovskite results in tetragonal perovskite oxide SrCo0.95P0.05O3−δ (SCP), which shows a considerable increase in OER performance in terms of activity and endurance relative to the parent compound.58Fig. 5a and b show linear sweep voltammetry (LSV) curves of SC and SCP before and after the accelerated degradation test (ADT) conducted by repetition of 1000 CV cycles. The ADT results show a decrease in the activity of SC after ADT, while SCP shows an increase in OER activity after the ADT. The enhanced OER activity during the ADT of SCP is due to the presence of P dopant. The high-resolution TEM (HR-TEM) images presented Fig. 5c and d suggest that the decrease in the activity of SC is because of the formation of a 10–15 nm thick amorphous layer after the ADT, which curtails the diffusion by blocking active sites, while SCP shows an only 1–2 nm thick amorphous layer (Fig. 5d). Fig. 5e shows a schematic illustration of the OER mechanism on SCP during the ADT, which indicates improved interaction between active cobalt sites and electrolyte through leaching of either Sr2+ or P5+ from the surface of the catalyst.
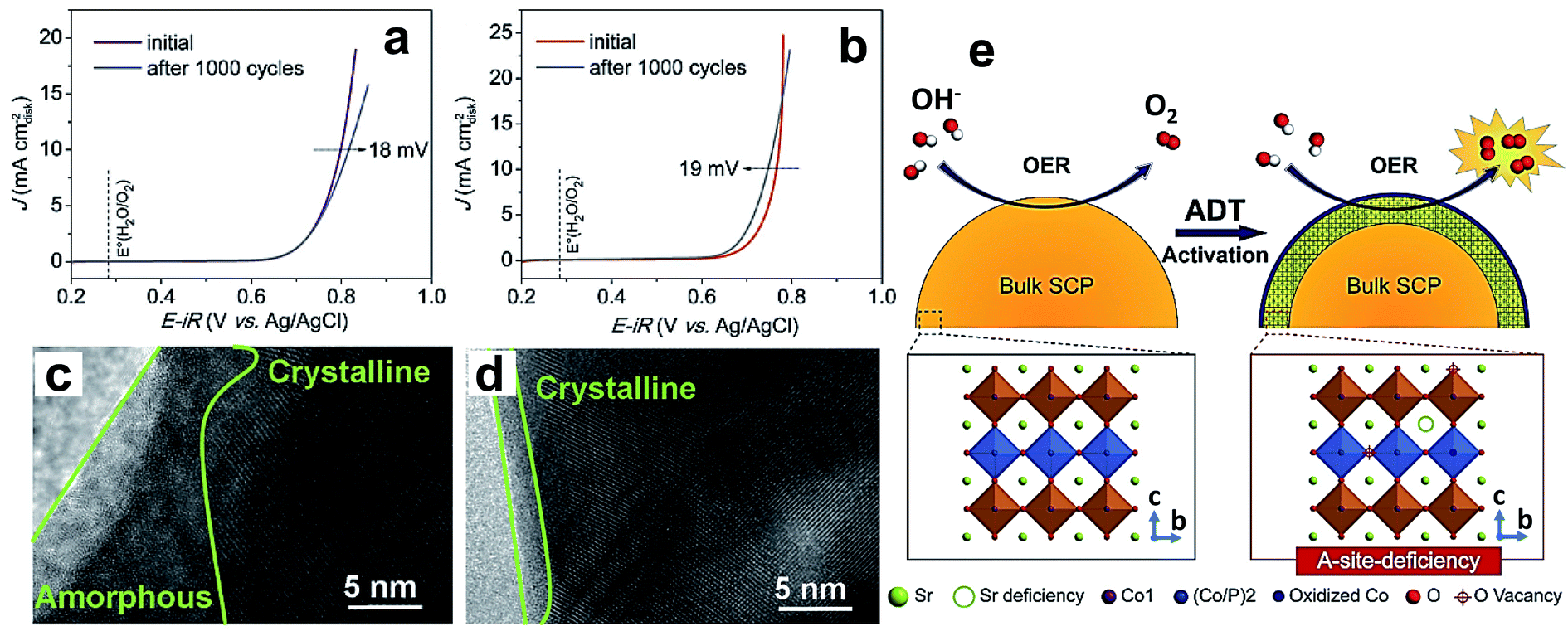 | ||
| Fig. 5 (a and b) LSV curves for the OER on a RDE (1600 rpm) for the (a) SC and (b) SCP samples before and after an ADT of 1000 cycles in O2-saturated 0.1 M KOH solution; HRTEM images of the surface regions of (c) SC and (d) SCP after an ADT of 1000 cycles; (e) schematic illustration of the main origin of the OER activation for the SCP sample during the ADT.58 Reproduced from ref. 58 with permission, © 2017 John Wiley and Sons. | ||
Likewise, Liu's group demonstrated the universality of P doping at the B-site of LaFeO3−δ, LaCoO3−δ and LaNiO3−δ perovskites for improving the electrochemical water oxidation capacity.55 The superior activity is attributed largely to the generation of a large amount of O22−/O− species, and DFT calculation suggests a change in the oxidation state of the B cation that optimizes the eg value (∼1). Zou et al. doped a series of TMs at the B-site of 6H–SrIrO3 for improving its OER activity in an acidic medium.52 Among them, cobalt doping was found to enhance the OER activity of 6H–SrIrO3 significantly. Fig. 6a presents the crystal structure of 6H–SrIrO3 having two types of IrO6 octahedron: one is face sharing (red), and the other one is corner-sharing isolated (olive green). Thus, the doped TMs can replace the Ir present in either the face sharing octahedra or corner-sharing octahedra. Fig. 6b displays the SEM image of Co-doped 6H–SrIrO3 having plate-like morphology and the thickness of these plates decreased after doping. Fig. 6c shows the polarization curves of pristine and Co-doped catalysts in 0.1 M HClO4 electrolyte. The catalyst with a 0.2:1 ratio of Co and Ir shows superior activity with the lowest overpotential of 235 mV at 10 mA cm−2, and the catalytic activity is 2.5 times higher than that of the pristine catalyst. A comparison between the mass activities of pristine and Co-doped catalysts is presented in Fig. 6d, which suggests that the catalyst with the 0.2:1 ratio of Co and Ir has 3 times higher mass activity than 6H–SrIrO3. However, both catalysts show nearly the same OER kinetics with Tafel values of 51.8 mV dec−1 for the catalyst with the 0.2:1 ratio of Co and Ir and 58.7 mV dec−1 for the pristine sample. Computational studies suggest that in the pristine sample, a downshift of the Ir 5d band center away from the Fermi level is observed. In contrast, the O 2p band moves close to the Fermi level in the doped 6H–SrIrO3, which thus results in enhanced covalency and increases the OER activity.52
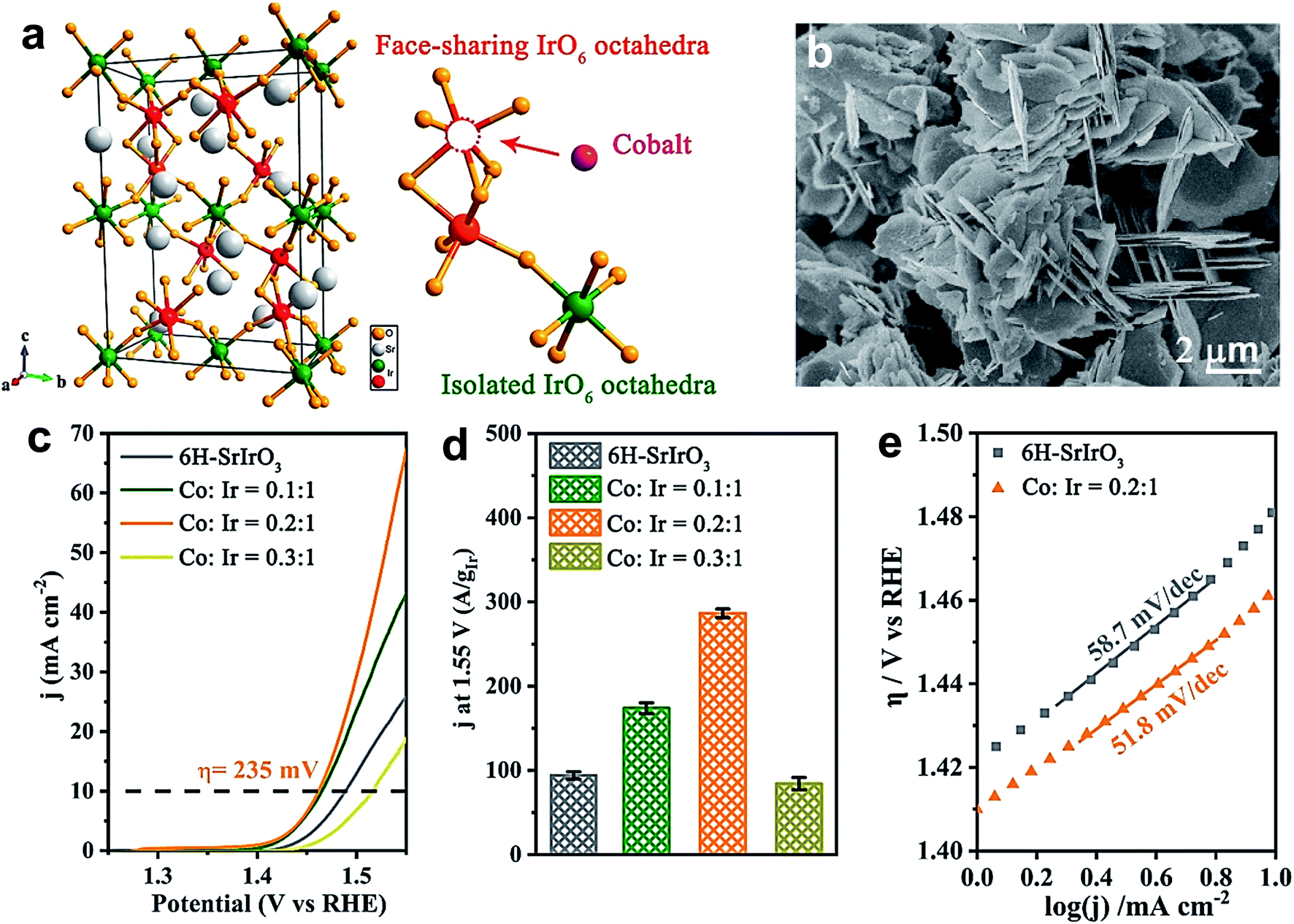 | ||
| Fig. 6 (a) Crystal structure of 6H–SrIrO3, and two kinds of Ir site for Co substitution. The iridium atom at face-sharing IrO6 octahedra is marked by a red color and that at isolated IrO6 octahedra is marked by an olive green color; (b) SEM image of Co doped 6H–SrIrO3; (c) polarization curves of pure 6H–SrIrO3 and Co-doped SrIrO3 (Co:Ir = 0.1:1, 0.2:1, and 0.3:1) in 0.1 M HClO4 solution with 85% iR compensation; (d) iridium amount-normalized mass activity of 6H–SrIrO3 and Co-doped SrIrO3 (Co:Ir = 0.1:1, 0.2:1, and 0.3:1); (e) Tafel slopes of 6H–SrIrO3 and Co-doped 6H–SrIrO3.52 Reproduced from ref. 52 with permission, © 2019 American Chemical Society. | ||
Very recently, our group doped Fe on the B-site of SrCoO3−δ and observed the formation of a tetragonal intermediate structure between perovskite and brownmillerite, with a stoichiometric formula of Sr2Co1.5Fe0.5O6−δ.57Fig. 7a presents the OER activity of Sr–Co–Fe-oxides, which shows that Sr2Co1.5Fe0.5O6−δ exhibits superior OER activity among all doped oxides. The enhanced OER activity is ascribed to the unique stacking of octahedral (oh) and tetrahedral (th) layered units of the tetragonal structure, as shown in Fig. 7b, providing facile interactions between the electrode and electrolyte surface. The stability of the Sr2Co1.5Fe0.5O6−δ catalyst was evaluated at 1.55 V for 10 h as shown in Fig. 7c, and it presents good stability with a slight increase in overpotential of 16 mV as depicted in Fig. 7d. Furthermore, theoretical calculations suggest that the enhanced covalency of Co 3d and O 2p is another prime contributor to the augmented OER performance of Sr2Co1.5Fe0.5O6−δ. This tetragonal intermediate structure presents a model for developing advanced catalysts in other systems.
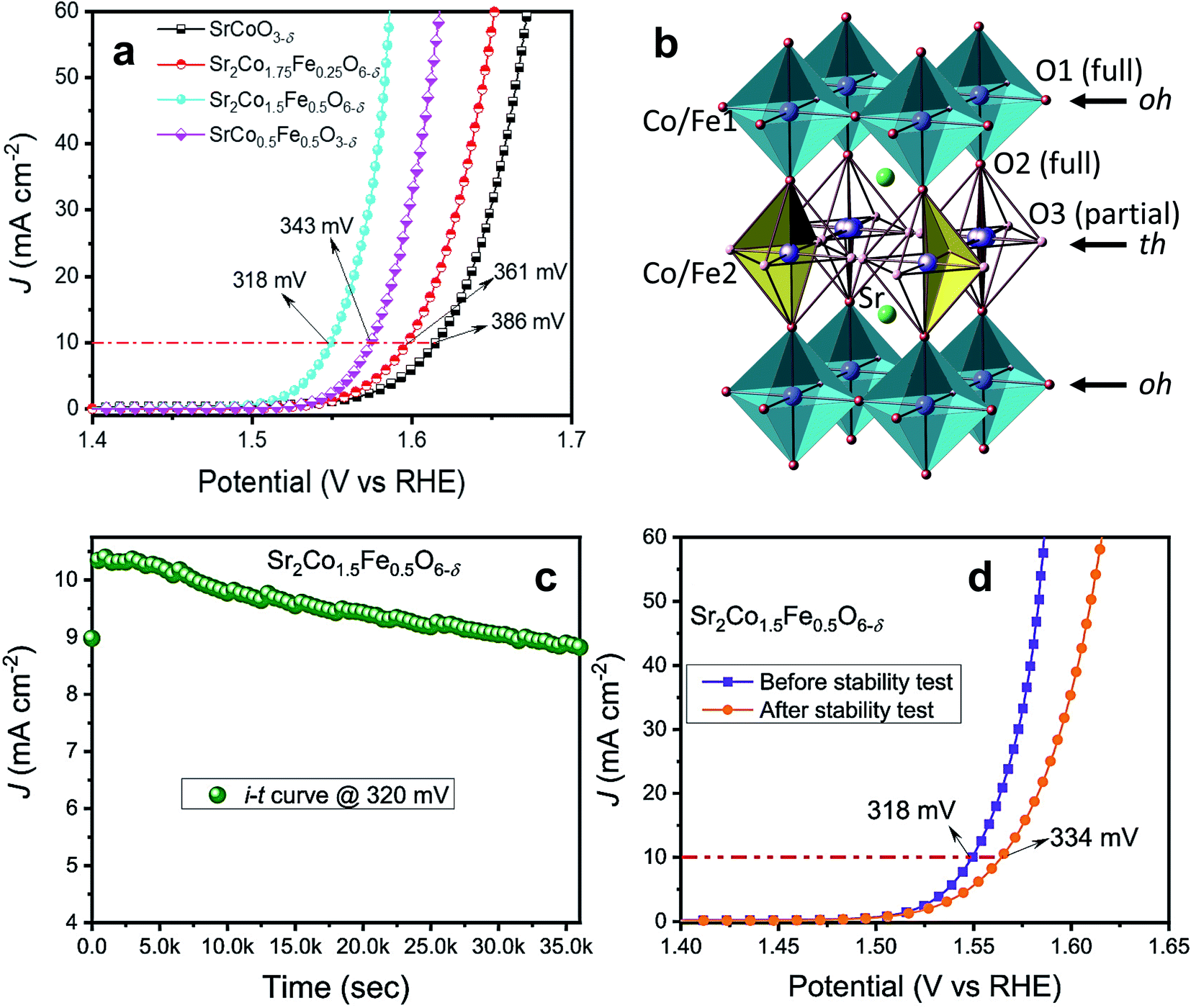 | ||
| Fig. 7 (a) LSV plots of SrCoO3−δ, Sr2Co1.75Fe0.25O6−δ, Sr2Co1.5Fe0.5O6−δ and SrCo0.5Fe0.5O3−δ oxides; (b) structural model of the intermediate tetragonal phase containing a sequence of alternating octahedral (oh) and tetrahedral (th) layers (…oh–th–oh–th…), where only two randomly orientated tetrahedra are shown for clarity; (c) stability test of Sr2Co1.5Fe0.5O6−δ oxide at 1.550 V vs. RHE for 10 h; (d) LSV plots of samples before and after the stability test.57 Reproduced from ref. 57 with permission, © 2021 American Chemical Society. | ||
4.3 Spinel oxides
The general formula of spinel oxides is AB2O4. A and B are metal cations in general, which belong to the first row of TMs, or group 2 or group 13 in the periodic table. Table 2 provides a summary of representative doped spinel oxides and their catalytic performances. The structure of spinel oxides is complicated, because of the presence of two crystallographic sites, namely octahedral oh and tetrahedral th sites, respectively, in a single structure.22 Based on the arrangement of these crystallographic sites, they are classified into two types: one is spinel comprising A2+ on the th site and B3+ on the oh site, and the other one is inverse spinel containing A2+ on the oh site and B3+ on the th site. According to crystal field theory, the TM ions present in the oh and th sites show different d-orbital splitting, i.e., t2geg for oh and egt2g for th. This difference in d-orbital splitting plays a significant role in tuning the electronic properties of spinel oxides and optimizing OER performance.71,72 Spinel oxides are highly stable in a basic medium with good electrical conductivity under harsh OER conditions. Mn-, Fe-, and Co-based spinel oxides are mostly reported as catalysts for the OER.72| Catalyst | Overpotential at a specific current density | Tafel slope (mV dec−1) | Durability | Electrolyte |
|---|---|---|---|---|
| Spinel oxides | ||||
| Ag-doped Co3O4 (ref. 73) | 680 mV @ 10 mA cm−2 | 219 | 10 h @ 370 mV | 0.5 M H2SO4 |
| Co2.25Cr0.75O4 (ref. 74) | 350 mV @ 10 mA cm−2 | 60 | 24 h @ 10 mA cm−2 | 1 M NaOH |
| Cu-doped Co3O4 NAs/NF75 | 230 mV @ 10 mA cm−2 | 84 | 20 h @ 10 mA cm−2 | 1 M KOH |
| Fe-doped Co3O4 (ref. 76) | 262 mV @ 10 mA cm−2 | 43 | 50 h @ 100 mA cm−2 | 1 M KOH |
| Fe-doped Co3O4 (ref. 77) | 160 mV @ 10 mA cm−2 | 78 | ∼160 min @ 1.63 V vs. RHE | 0.1 M KOH |
| Fe-doped NiCo2O4 (ref. 33) | 201 mV @ 10 mA cm−2 | 39 | 12 h @ 10 mA cm−2 | 1 M KOH |
| LiMn0.25Co1.75O4 (ref. 78) | 370 mV @ 10 mA cm−2 | 60 | — | 1 M NaOH |
| Mn1.4Co1.6O4 (ref. 79) | 327 mV @ 10 mA cm−2 | 79 | 25 h @ 1.53 V vs. RHE | 1 M KOH |
| Ni-doped Mn3O4 (ref. 80) | 458 mV @ 10 mA cm−2 | 74.4 | 21 h @ 10 mA cm−2 | 3 M NaCl |
| NPCoO-UCSs81 | 182 mV @ 10 mA cm−2 | 54 | 50000 s @ 1.42 V vs. RHE |
0.1 M KOH |
| P8.6–Co3O4/NF82 | 260 mV @ 20 mA cm−2 | 60 | 25 h @ 1.53 V vs. RHE | 1 M KOH |
| P–Co3O4 (ref. 83) | 280 mV @ 20 mA cm−2 | 51.6 | 40000 s @ 280 mV |
1 M KOH |
| Se/Ni–Co3O4 (ref. 84) | 290 mV @ 20 mA cm−2 | 62.9 | — | 1 M KOH |
| V-doped Co3O4 (ref. 85) | 293.6 mV @ 10 mA cm−2 | 53.3 | 30 h @ 5 mA cm−2 | 1 M KOH |
|
||||
| Hydroxides | ||||
| Ce-doped NiFe LDH/CNT86 | 227 mV @ 10 mA cm−2 | 33 | 30000 s @ 10 mA cm−2 |
1 M KOH |
| Co0.54Fe0.46OOH87 | 290 mV @ 10 mA cm−2 | 47 | — | 0.1 M KOH |
| Cu-doped Co(OH)2 (ref. 88) | 300 mV @ 10 mA cm−2 | 47 | 36 h @ 1.55 V vs. RHE | 1 M KOH |
| Fe-doped Co(OH)2 (ref. 89) | 290 mV @ 10 mA cm−2 | 69 | 12 h @ 10 mA cm−2 | 1 M KOH |
| Fe–NiOOH90 | 181 mV @ 50 mA cm−2 | 43.2 | 56 h @ 20 mA cm−2 | 1 M NaOH |
| Mn and F dual-doped Ni(OH)2 (ref. 91) | 233 mV @ 20 mA cm−2 | 56.9 | 10 h @ 20 mA cm−2 | 1 M KOH |
| Ni-doped Co(OH)2 (ref. 92) | 300 mV @ 10 mA cm−2 | 47 | 36 h @ 1.55 V vs. RHE | 1 M KOH |
| Ni-doped FeOOH93 | 239 mV @ 10 mA cm−2 | 90.4 | — | 1 M KOH |
| Ni0.83Fe0.17(OH)2 (ref. 94) | 245 mV @ 10 mA cm−2 | 61 | 10 h @ 10 mA cm−2 | 1 M KOH |
| Se-doped FeOOH95 | 348 mV @ 500 mA cm−2 | 54 | 100 h @ 100 mA cm−2 | 1 M KOH |
| W-doped Ni(OH)2 (ref. 96) | 237 mV @ 10 mA cm−2 | 33 | — | 1 M KOH |
|
||||
| Other oxides | ||||
| Ce-doped NiO–Au97 | 244 mV @ 100 mA cm−2 | >24 h @ 50 mA cm−2 | 1 M KOH | |
| Co-doped CuO98 | 299 mV @ 50 mA cm−2 | 134 | 15 h @ 300 mV | 1 M KOH |
| Co–MnO2/OV99 | 279 mV @ 20 mA cm−2 | 75 | 12 h @ 10 mA cm−2 | 1 M KOH |
| Co-doped ZnO100 | 450 mV @ 10 mA cm−2 | 106 | 18 h @ 1.68 V vs. RHE | 0.1 M phosphate-buffered saline |
| Fe–CoO/C nanofibers101 | 362 mV @ 10 mA cm−2 | 74.4 | — | 1 M KOH |
| NixCo1−xO102 | 450 mV @ 10 mA cm−2 | 66.8 | — | 1 M KOH |
| Ni-doped MnO2 (ref. 103) | 330 mV @ 10 mA cm−2 | 23.7 | >3 h @ 10 mA cm−2 | 1 M KOH |
| V-, Fe-, Co- and Ni-doped MnO2 (ref. 104) | 390 mV @ 10 mA cm−2 | 104.4 | >27 h @ 20 mA cm−2 | 1 M KOH |
| Y2−xBaxRu2O7 (ref. 105) | 278 mV @ 10 mA cm−2 | 40.8 | 5 h @ 10 mA cm−2 | 0.5 M H2SO4 |
| Zn-doped CoO106 | 293 mV @ 10 mA cm−2 | 63 | 24 h @ 10 mA cm−2 | 1 M KOH |
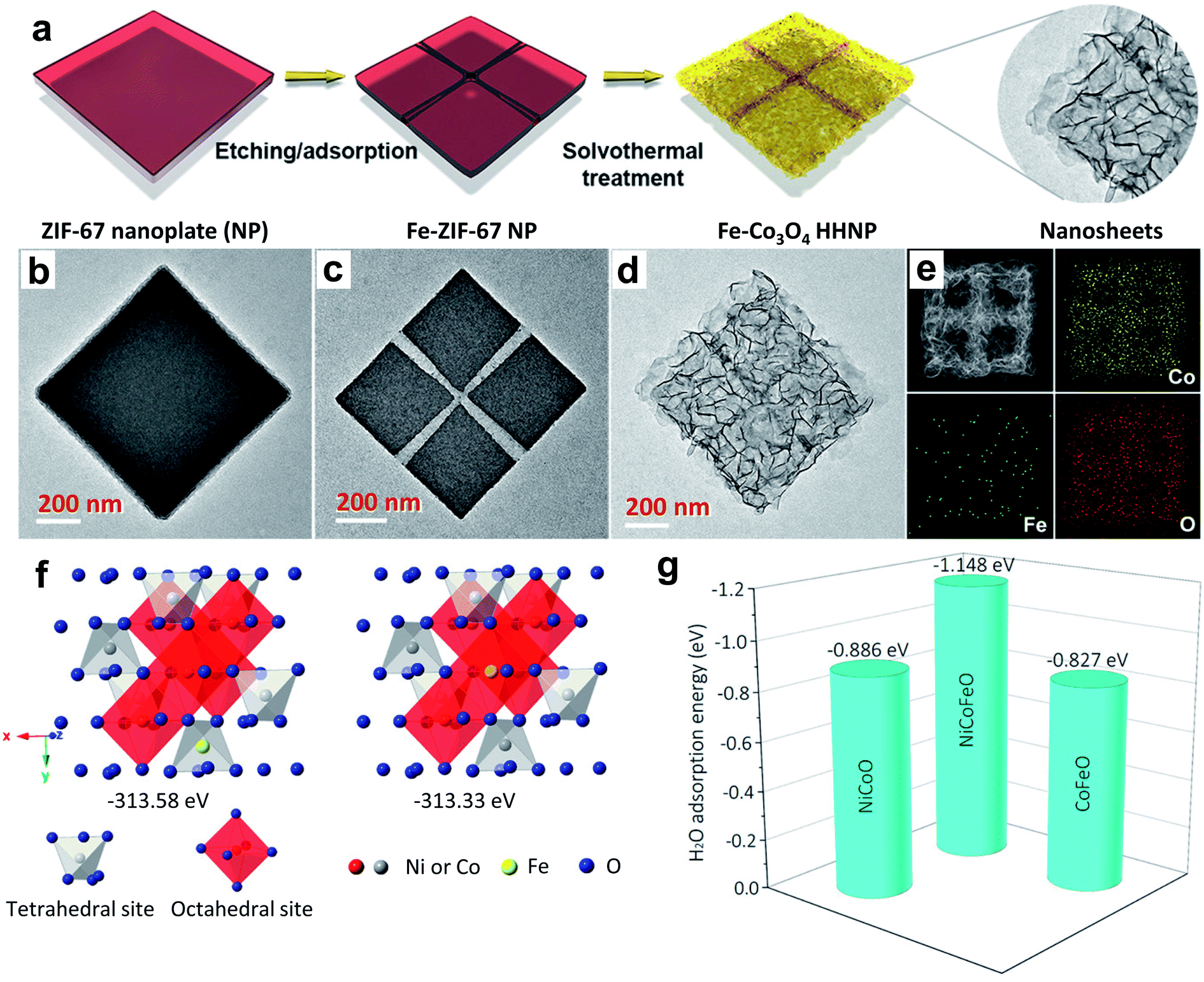 | ||
| Fig. 8 (a) Schematic synthesis process of cross-channeled Fe–Co3O4 hierarchically hollow NPs (HHNPs); (b–d) TEM images of ZIF-67 NPs; (e) HAADF-STEM image and elemental mapping images of one Fe–Co3O4 HHNP.76 Reproduced with open access. (f) Schematic illustration of the preference of Fe doping into tetrahedral sites of Ni–Co–O spinel oxides, and (g) adsorption energy of an H2O molecule on the (001) surface of Ni–Co–O, Co–Fe–O, and Ni–Co–Fe–O spinels.33 Reproduced from ref. 33 with permission, © 2019 American Chemical Society. | ||
In another report, Swaminathan et al. demonstrated how minute changes in the amount of Fe doping influenced the OER activity of Co3O4.77 They doped 3, 4.5, and 6% of Fe into the host lattice and observed that above or below the optimum level of Fe doping, fading of OER activity occurred. The 4.5% Fe doping caused superior activity, and the DFT study identified a considerable downshift of the Fermi energy level, originating from the optimized adsorption energy of reaction intermediates. By comparing Ni, Cu, and Li dopants, Vitanov et al. studied the effectiveness of doping in improving the OER activity of Co3O4 spinel and noticed that doping with Li (LixCo3−xO4) produced a promising activity.113 The raised activity of LixCo3−xO4 spinel is ascribed to faster reaction kinetics owing to the formation of active sites with high transfer coefficients, and a shortened reaction pathway to generate oxygen. Tian et al. revealed the Cu doping effect on improving the OER activity of Co3O4 nanosheets prepared on nickel foam, which outperforms the benchmark catalyst IrO2 on nickel foam.75 The high OER activity is found to have resulted from an increase in Co(III) species in the crystal lattice, along with highly uncovered active sites. Ding et al. demonstrated the improved OER performance of Co3O4 nanosheets by creation of oxygen vacancies upon Zn doping, which even surpassed that of the Ir/C catalyst.81
The stability and activity of Co3O4 spinel could be significantly enhanced in acidic electrolyte through Ag doping. Ag doping plays a crucial role in the growth of Co3O4 nanowire arrays, improving the surface area and stability under harsh acidic conditions and OER activity by providing shortened reaction paths.73 NixCo2−xO4 is an inverse spinel, in which Ni(II/III) is present in 16d oh sites and Co(II/III) is present in both 8a th sites and 16d oh sites. By tuning the Ni doping level, this inverse spinel's OER activity significantly increased, because the Ni2+/Ni3+ ratio plays a key role in governing the conductivity, surface roughness, and density of active sites. For example, Zhang et al. systematically studied the influence of an increase in the Ni dopant level to improve the overall OER performance of NixCo2−xO4.112 When x = 1, it shows superior OER activity. In this composition, Ni is in the +3 oxidation state. The enhanced OER activity is a result of the 0.27 eV upshifting of the valence band maximum toward the Fermi level, which creates a new hole. Such an electronic configuration favors the adsorption of OER intermediates.112 The doping of Fe at the Ni site of NiCo2O4 induces oxygen vacancies, which impacts the OER activity considerably. The Fe0.1Ni0.9Co2O4 composition shows superior activity within the doped series, which is accredited to the presence of an optimum amount of oxygen defects for triggering electrocatalytic activity.114 Huang's group reported doping Fe into the th site of the NiCo2O4 lattice to improve its OER activity through a theoretical prediction.33 As displayed in Fig. 8f, incorporation of Fe into the th site requires a lower energy (313.33 eV) compared to the oh site (313.58 eV) according to DFT calculations. Fig. 8g shows the DFT estimated H2O adsorptive ability where the Fe-doped one is superior to the pristine one and CoFe2O4, which is highly favorable for improving OER performance to increase active sites. Furthermore, X-ray absorption spectroscopy confirms the successful incorporation of Fe into the th site of NiCo2O4, which enhances the OER performance by increasing the population of Ni3+ and Co2+ ions in oh sites. MnxCo2−xO4 is also an inverse spinel, and Mn cations preferably occupy oh sites. Lankauf et al. described MnxCo2−xO4 spinel by adjusting the composition of Mn (x = 0–2) using a soft chemistry method, and the spinel with a moderate content of Mn shows better OER activity.79 The high activity was attributed to the improved oxidizing ability and better electronic conductivity. Chi et al. noticed that the change in copper content in the CuxCo2−xO4 spinel could influence its OER properties.115 When x was increased from 0.7 to 0.9, more Cu2+ ions displaced Co3+ ions from oh sites. They occupy th sites, and thus the Co3+ ion population on th sites was significantly increased, resulting in superior OER performance.
Li and coworkers reported an increase in the OER performance of the NiCo2O4 catalyst through double-exchange interaction (DEI).72 The DEI is induced by synergistic creation of oxygen vacancies (VO) via doping with Fe and creating a heterojunction with MoS2 (MoS2/rFe–NiCo2O4) as shown in Fig. 9a. Fig. 9b and c show the SEM and TEM images of the MoS2/rFe–NiCo2O4 catalyst, which is in the form of nanowires having a diameter of 50–60 nm, decorated with MoS2 on Fe–NiCo2O4. Fig. 9d displays the comparative OER activity of NoCo2O4, Fe–NoCo2O4, MoS2/Fe–NoCo2O4, rFe–NoCo2O4 and MoS2/rFe–NoCo2O4 catalysts, which reveals that DEI has the most powerful effect on enhancing OER activity. Fig. 9e shows the schematic representation of the effect of DEI on enhancing the OER activity of MoS2/rFe–NiCo2O4 through improving electrical conductivity, number of OER active sites, and reactivity of active centers. Recently, non-metal doping into Co3O4 was studied for improving electrochemical activity. For instance, Sun's group reported P-doped Co3O4 nanowires as an efficient OER catalyst in basic electrolyte.82 The P-doped Co3O4 nanowires on nickel foam were prepared by low-temperature annealing using NaH2PO2. First-principles calculations revealed that P-doped Co3O4 nanowires have a much lower free-energy value for the rate-determining step than pristine Co3O4.82 Wang et al. synthesized P-doped Co3O4 spinel by filling the in situ generated oxygen vacancies using an Ar plasma etching technique, and observed excellent OER activities after doping.83 When oxygen vacancies were created in Co3O4, more electrons migrated into oh Co3+ 3d orbitals than th Co2+ 3d orbitals. With P doping, electrons are transferred out of the Co 3d states, resulting in more Co2+ on th sites than Co3+ on oh sites in P–Co3O4. Hence, the superior OER activity of the P-doped Co3O4 is linked with the presence of a greater number of Co2+ ions. Zhang et al. reported doping of Co3O4 with S by using facile solution-combustion and thioacetamide-sulfidation methods.116 The S-doping enhances the OER performance of Co3O4 in terms of activity as well as stability in an alkaline medium. The formation of S-substituted CoO layers on the surface of Co3O4 is a major reason for the superior activity. Theoretical calculations indicate that improving the electronic conductivity by S doping was beneficial for overall electrochemical activity. Dual doping of Co3O4 on both cation and anion sites with Ni and Se, respectively, resulted in a significant increase in its OER performance.84 The Se doping enhances the active sites on the surface of a catalyst by inducing oxygen vacancies, and Ni doping helps in generating highly active Co4+ species.
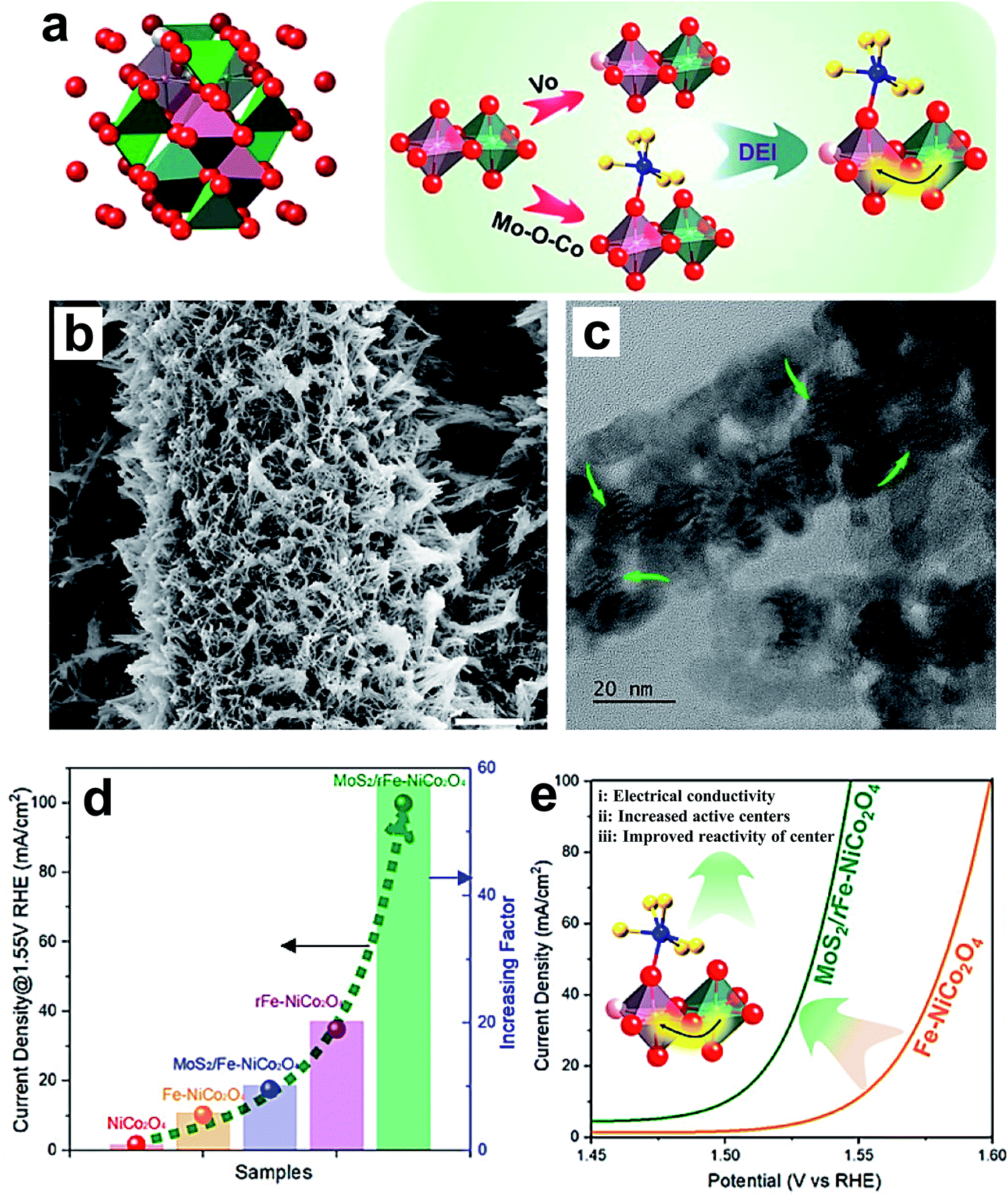 | ||
| Fig. 9 (a) Crystal structure of spinel oxides, and the creation of a double-exchange interaction (DEI); (b) SEM and (c) TEM images of MoS2/rFe–NiCo2O4; (d) OER performance of the electrocatalysts NiCo2O4, Fe–NiCo2O4, MoS2/Fe–NiCo2O4, rFe–NiCo2O4, and MoS2/rFe–NiCo2O4 with respect to current density at 1.55 V RHE and the current density increasing factor; (e) schematic representation of the enhanced OER arising from the DEI effect.72 Reproduced from ref. 72 with permission, © 2020 American Chemical Society. | ||
4.4 Hydroxides
Hydroxides, oxyhydroxides, and layered double hydroxides (LDHs) of oxides of TMs, such as Ni, Co, Fe, Mn, and V, are highly active in the electrocatalytic oxygen evolution reaction.86,117,118 They exhibit typical layered stacking conformation with proton sandwiching between layers, and the metal is centered in the octahedron surrounded by oxygen anions at the corners as MO6.117 Some representative hydroxides and their catalytic performances are listed in Table 2.Fe doping of TM hydroxides is most widely reported in the literature for enhancing their OER performance.119 Specifically, Fe doping in Ni(OH)2 (ref. 94 and 120–123) and Co(OH)2 (ref. 89 and 124–126) has been widely studied. In 1901, Edison,127 for the first time, noticed an increase in the OER activity of Ni(OH)2 because of Fe impurity in the electrolyte in a Ni-based alkaline battery.120,128 Because of oxygen evolution, the capacitance and cycle life of the battery were decreased considerably by the incorporation of Fe into the Ni(OH)2 electrode. Later, researchers revealed that the OER activity of Ni(OH)2 shortened the voltage window of the battery owing to the presence of Fe in the electrolyte, and they also found that the presence of a discrete amount of Fe (<1%) considerably contributed to augmentation of OER activity.129 Corrigan demonstrated that the presence of 0.01% Fe impurity in electrolytes substantially decreased the discharge capacitance.130
Boettcher's group studied the role of Fe doping in enhancing the OER performance of Ni(OH)2 thin films and provided more insights on catalytic reactions at the electrode/electrolyte interface.122,124–126 They witnessed an increase in conductivity of thin films with systematically increasing the doping amount from 5 to 25%. Still, the CV curves show drastic changes, which signify the change in surface reconstruction with varying dopant levels.122 Klaus et al. studied the Fe inclusion by aging the thin films in an electrolyte with or without Fe impurities for 6 days.131 The sample aged in the electrolyte with Fe impurity shows an increase in OER activity gradually from the first day to the 6th day. In contrast, the other one without Fe shows a decrease in activity gradually upon aging, and the enhanced OER activity is attributed to the increase in conductivity. Landon et al. noticed a rise in the coordination number of Fe in the OER environment using an in situ extended X-ray absorption fine structure (EXAFS) method.132 They proposed that forming a new phase (NiFe2O4) through reconstruction increased the coordination number of Fe. Nevertheless, the formation of NiFe2O4 is rejected by other researchers with the aid of in situ Raman spectroscopy.133 They did not notice any signals for NiFe2O4 formation in the in situ Raman spectra (700 cm−1), even after aging in KOH electrolyte with Fe at high OER potentials. In contrast, they observed two peaks which are characteristic of the NiOOH phase at 555 and 475 cm−1, even in the presence of a high percentage of Fe.
Recently, Zhao's group studied the effect of doping Co and Fe into the lattice of Ni(OH)2 for improving the OER performance, and they conducted electron energy-loss spectroscopy (EELS) characterization and Hubbard corrected density functional theory (DFT + U) calculations to understand the activity trend.90 The Fe-doped sample shows superior OER activity to Co and Fe co-doped or Co-doped samples.90 EELS analysis suggests electronic interactions between Ni, Co, and Fe dopants even after surface reconstruction during the OER process. A DFT + U study reveals that the d-band center of the Fe-doped sample is much closer to the Fermi level than that of the others, enhancing the binding energy values of OER intermediates to an optimal value. Wang et al. demonstrated an enhancement in OER activity of Ni(OH)2 by co-doping Mn and F elements.91 A considerable increase in oxygen deficiencies was found after doping, which reduced the energy barrier for the key OER of *OH → *O. Ma's group doped a single atom of W6+ into the lattice of Ni(OH)2 and studied its electrocatalytic oxygen evolution behavior.96 Interestingly, W doping significantly enhances the OER activity compared to the pristine sample, and the superior activity is ascribed to the boosting of carrier migration at the interface of the electrode and electrolyte. Furthermore, a DFT study reveals that low spin W6+ (d0) stabilizes the O* and thus helps in the formation of O–O.
The augmentation of OER activity was also reported for the Co(OH)2 catalyst by Fe doping. Boettcher's group reported the effect of Fe doping on the OER activity of electrodeposited films of Co(OH)2.124 They observed an enhancement of OER performance by 100 fold when the stoichiometric ratio of Co and Fe was 1:0.6–0.7. The reason for the superior OER activity is increased Co3+ population and improved thin-film conductivity with Fe doping. Unlike Ni(OH)2, Co(OH)2 requires a huge amount of Fe doping (60–70%) to display enhanced OER activity, while Ni(OH)2 shows a significant effect even with 0.01% Fe inclusion. Above the 60–70% level, the OER activity started decreasing for Co(OH)2. Researchers also doped other TMs into Co(OH)2 to improve its OER performance. For example, Chen et al. doped Cu into the lattice of Co(OH)2, which significantly enhances its OER activity and outperforms the state-of-the-art catalyst IrO2. The considerable increase in the catalytic performance is attributed to an increase in Co3+ population after doping with Cu.88 Koel's group studied the doping effect on the OER behavior of CoOOH nanowires by doping with Ni and Mn.92 Among them, Ni doping turned out to be fruitful compared to Mn doping because of Ni's ability to stabilize the OER intermediates. Similarly, FeOOH was also doped with different elements such as Ni, Co, and Se to enhance its OER performance. Tolstoy et al. doped Ni2+ into the lattice of FeOOH nanocrystals, and 25% Ni2+ doping was proven to improve the OER activity considerably.93 Zhang et al. doped Co into FeOOH, and the Co0.54Fe0.46OOH composition showed superior activity to other doped samples because Co inclusion created more active sites at the edges of the three-dimensional hierarchical structures.87 Recently, Niu et al. reported doping of Se into FeOOH via electrochemical oxidation of an FeSe/iron foam (IF) pre-catalyst by applying a constant 10 mA cm−2 current density for 4 h.95 XRD patterns, as shown in Fig. 10a, display the complete disappearance of diffraction peaks of FeSe which was converted into Se-doped FeOOH. However, the electrochemical oxidation did not disturb the nanosheet morphology as revealed by SEM and TEM images in Fig. 10b and c, respectively. Fig. 10d shows the polarization curves of the as-prepared catalysts, and among them, FeOOH(Se)/IF delivers industrial scale 500 mA cm−2 current density at 348 mV overpotential. Tafel plots of these catalysts are presented in the inset of Fig. 10d, which shows that the FeOOH(Se)/IF catalyst possesses OER kinetics with the lowest Tafel slope value of 54 mV dec−1. As a unary Fe-based catalyst, FeOOH(Se)/IF shows comparative activity and even outperforms Ni and Co-based OER catalysts as observed in Fig. 10e. Furthermore, DFT calculations revealed a considerable decrease in the energy barrier for the RDS from 3.20 eV to 1.45 eV after Se inclusion, which accounts for the superior OER activity.
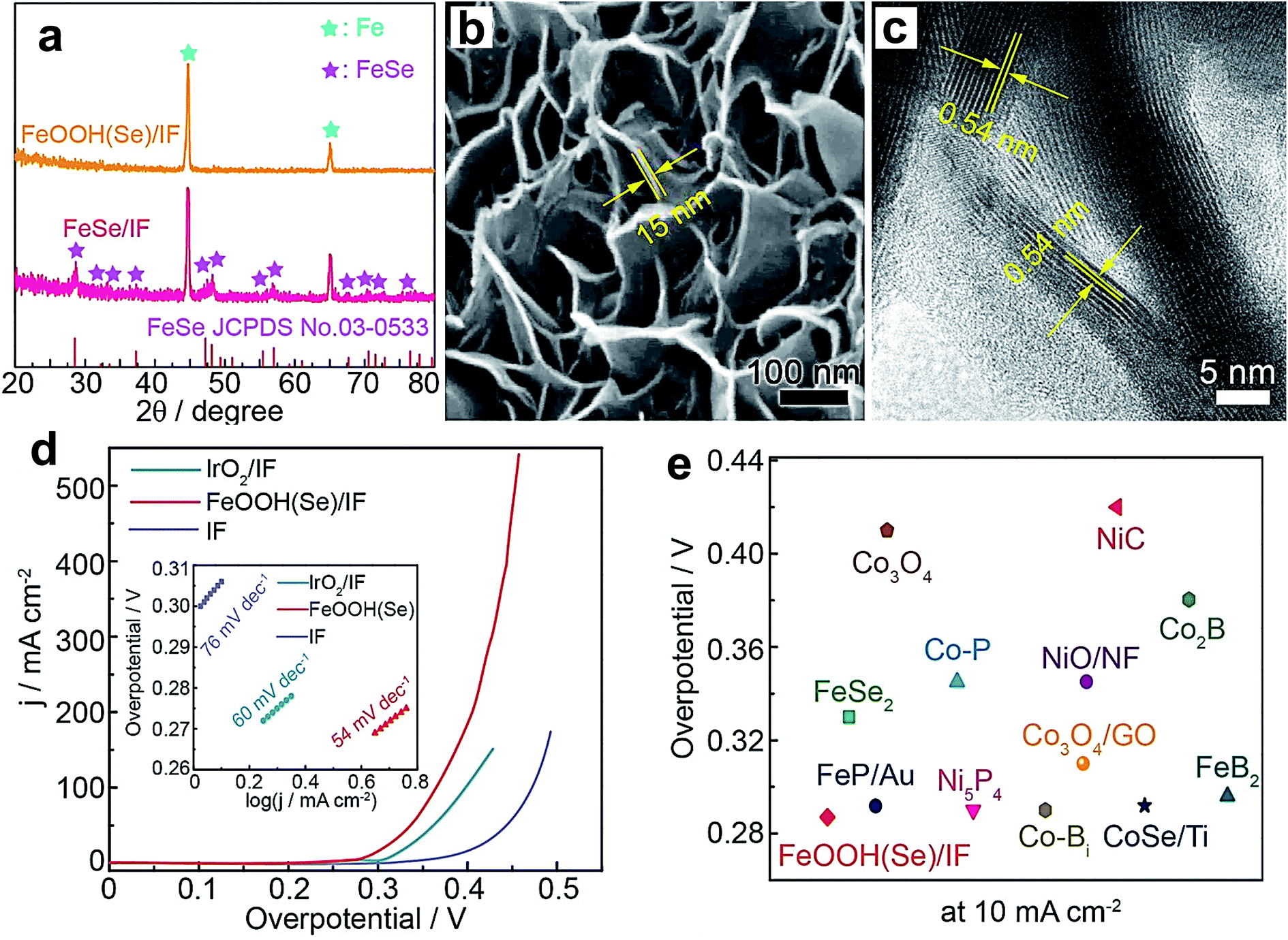 | ||
| Fig. 10 (a) XRD patterns of FeSe/iron foam (IF) and FeOOH(Se)/IF; (b) SEM image of FeSe/IF; (c) HRTEM image of FeSe/IF; (d) LSV curves of the as-prepared catalysts (inset: Tafel plots); (e) overpotential comparison of FeOOH(Se)/IF and state-of-the-art unary metal-based OER electrocatalysts.95 Reproduced from ref. 95 with permission, © 2019 American Chemical Society. | ||
4.5 Other oxides
TM oxides, such as NiO, CoO, and MnO2, are widely reported as catalysts for the OER owing to their high durability and low cost.5 Some other representative oxides and their catalytic performances are listed in Table 2. The electrochemical activity of these oxides was further improved by tuning their electronic structure through the inclusion of various dopants. For instance, lithium-doped NiO (LixNi1−xO) has shown unprecedented OER performance and even outperformed non-noble benchmark perovskite catalysts, such as BSCF and LaNiO3.134 Zhang et al. studied the role of lithium incorporation into the lattice of NiO.134Fig. 11a shows the BET surface area normalized LSV curves of pristine and Li-doped NiO catalysts, and among them, the catalyst with a 1:1 ratio of Li and Ni shows 11 fold higher activity than pristine NiO. The inset of Fig. 11a presents cyclic voltammogram (CV) curves, which further reveals a positive shift in redox potential with an increase in Li content. A combinatorial study of the electronic structure of LixNi1−xO was conducted using X-ray absorption spectroscopy and synchrotron-based photoemission spectroscopy. The schematic illustration of covalency of Ni 3d–O 2p from DFT calculations is shown in Fig. 11b, which reveals the formation of a new hole state near the Fermi level, and the increased covalency of Ni 3d–O 2p that ensured higher OER activity.
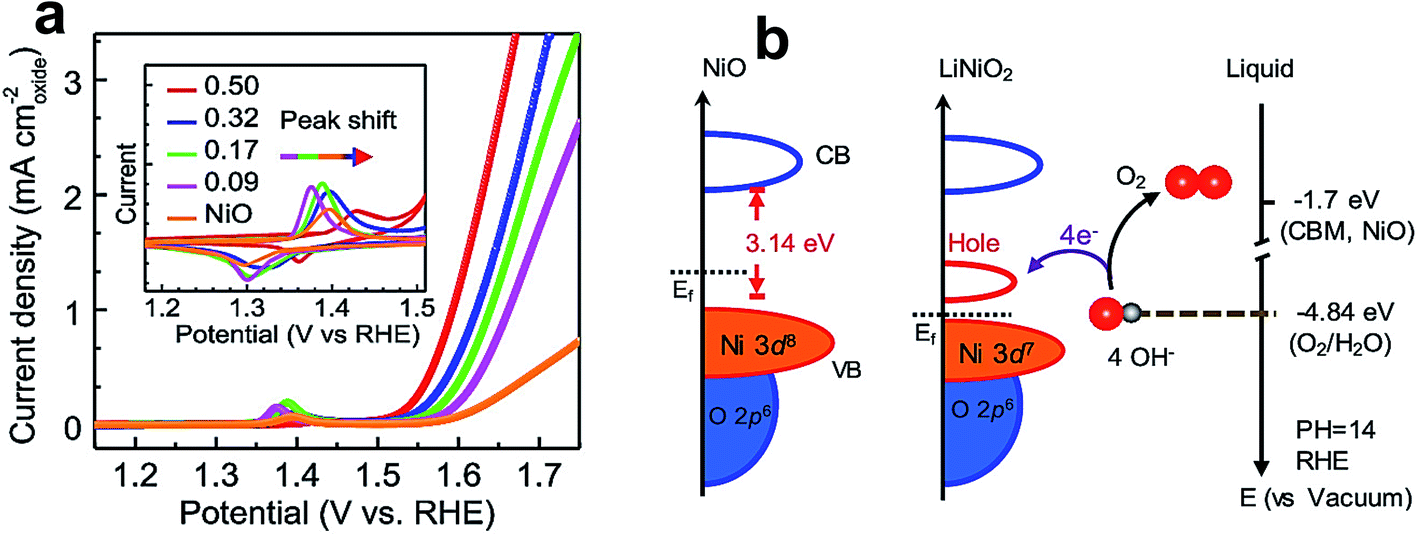 | ||
| Fig. 11 (a) LSV polarization curves of the OER normalized by the BET surface area in 1 M KOH solution at a scan rate of 10 mV s−1; the inset shows cyclic voltammogram curves at a scan rate of 10 mV s−1; (b) schematic diagram for electronic structures of NiO (left) and LiNiO2 (middle), and energy diagram at the oxide–liquid interface on the vacuum level scale at pH = 14 (right); the redox level for O2/H2O (red dashed line) is −4.84 eV, the conduction band minimum (CBM) level for NiO is −1.7 eV according to the formula ECBM = Ebandgap − Eionization (Ebandgap = 3.7 eV, Eionization = 5.4 eV).134 Reproduced from ref. 134 with permission, © 2019 American Chemical Society. | ||
Enhancement in the OER activity of the NiO catalyst with Fe doping was first observed by Corrigan in 1987,130 and Fe doping into the NiO thin film was found to be affected by the impurities present in KOH electrolyte.135 Recently, Nocera et al. identified the essential role of Fe doping in improving the electrocatalytic activity of Ni oxides.136 5–10% doping of Fe increased the population of Ni4+ ions, as revealed by coulometric titrations. Consequently, O and Ni K-edge spectra indicated an increase in hybridization between Ni 3d and O 2p orbitals. Both experimental and computational studies showed that Fe3+ is a Lewis acid with pKa = 2.2, and is the most acidic among TMs. Hence, the incorporation of Fe3+ into Ni oxides helps in enhancing the acidity of OHx moieties coordinated with Ni, which increases the population of Ni4+ ions by lowering the Ni3+/Ni4+ reduction potential.136 Ce-doped NiOx deposited on a Au substrate shows significantly higher OER performance than pristine NiOx.97 The enhanced OER activity is accredited to the modification of the local environment favorable for binding of OER intermediates.
A nickel-doped rock-salt-structured OER catalyst, NixCo1−xO, was synthesized using a solid-state method.102 The inclusion of a discrete amount of Ni into the lattice of CoO stabilizes the crystal structure and tunes its eg value. Within the doped series, the Ni0.3Co0.7O catalyst displays the highest OER activity with an eg value of ∼1.3. Huo et al. reported the improved OER performance of CoO nanoflowers by doping with Zn.106 A DFT study confirmed that OER active sites were increased by Zn doping, and the high surface area of nanoflowers was another contributor to the improved catalytic activity. Like in NiO, Fe doping of CoO could also greatly enhance its electrocatalytic performance by increasing the number of OER active sites.101 Ye et al. prepared V-, Fe-, Co-, and Ni-doped MnO2 ultrathin nanosheets on carbon fiber paper (CFP) using an electrodeposition method as shown in Fig. 12a, and studied their OER activity in a basic medium.104Fig. 12b and c show the doped MnO2 ultrathin nanosheet/CFP composite electrode at low and high magnifications, which shows very dense nanoclusters with 5 nm thick nanosheets. Fig. 12d shows the LSV curves of CFP, pure MnO2/CFP, metal-ion-doped MnO2 ultrathin nanosheet/CFP, and IrO2/CFP composite electrodes in 1 M KOH with a scan rate of 5 mV s−1. The authors noticed that doping with these TM cations vastly enhances MnO2 conductivity, resulting in higher catalytic activity compared to the pristine sample. The electrochemical stability of doped and pristine MnO2 compared with IrO2 evaluated at 20 mA cm−2 is shown in Fig. 12e. Doped MnO2 ultrathin nanosheet/CFP shows significant stability compared to the pristine sample with a slight increase in overpotential.
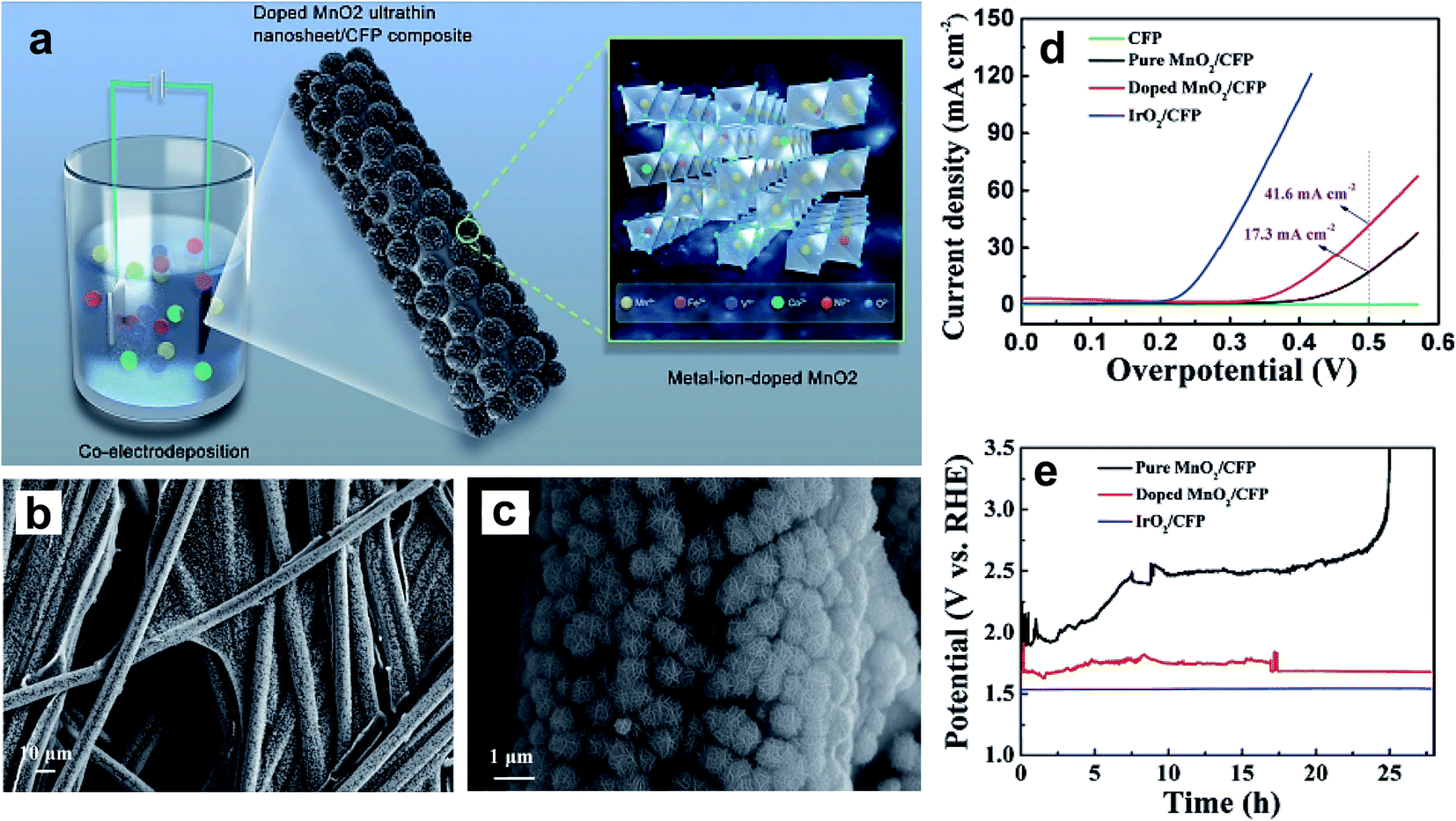 | ||
| Fig. 12 (a) Schematic representation for the preparation of the metal-ion (Fe, V, Co, and Ni)-doped MnO2 ultrathin nanosheet/CFP composite; (b and c) FESEM images showing the morphology of the metal-ion-doped MnO2 ultrathin nanosheet/CFP composite electrode at different magnifications; (d) LSV curves of the CFP, the metal-ion-doped MnO2 ultrathin nanosheet/CFP and the IrO2/CFP composite electrodes measured in 1 M KOH with a scan rate of 5 mV s−1, where the current is normalized by the geometrical area of the carbon fiber paper; (e) galvanostatic measurements at a current density of 20 mA cm−2 for the different composite electrodes.104 Reproduced from ref. 104 with permission, © 2017 John Wiley and Sons. | ||
Yang et al. intercalated 3d block metal cations such as Zn2+, Cu2+, Ni2+, Co2+, and Fe3+ into MnO2 nanosheets to enhance OER activity.103 Among them, the inclusion of Ni2+ ions was more effective in enhancing the OER activity of MnO2 nanosheets. The increased activity is associated with strengthening the Mn–O bond perpendicular to layered chains to promote the catalysis between two Mn sites. Oxygen vacancy formation in MnO2 nanosheets by Co doping is demonstrated by Zhao et al.99 Co doping enhances the OER activity of MnO2 considerably by enhancing its conductivity and decreasing the energy barrier for adsorption of OER intermediates on the catalyst surface. Interestingly, a small amount of cobalt doping (3.47%) could activate the OER activity of ZnO. Theoretical studies suggest that Co activates inherently inactive adjacent Zn sites by increasing conductivity, which act as OER active sites for adsorption of OER intermediates.100 Co doping also activates the OER activity in CuO. Sun et al. demonstrated a multifold enhancement of OER activity in a basic electrolyte after doping with Co on a Cu foam substrate,98 which was related to a change in the electronic structure of CuO by Co doping. Meyer et al. reported the fabrication of an OER catalyst by modifying fluorine-doped tin oxide (FTO) by Co2+ single-atom doping.137 X-ray photoelectron spectroscopy (XPS) and TEM reveal that doping Co2+ into FTO by replacing Sn4+ results in oxygen vacancies. The as-modified FTO electrode shows water oxidation activity at pH 7.2 with a turnover frequency of 29000 per cobalt site, at a current density of 0.16 mA cm−2 during an endurance test for 2 h. Furthermore, the current density was enhanced by 5 fold by utilizing nanoporous FTO in place of planar FTO. Wang et al. reported Ba doping of Y2Ru2O7 pyrochlore oxide on the yttrium position (YBRO) as an efficient catalyst for the OER in an acid electrolyte.105 Among the doped samples, YBRO-0.15 shows superior activity, and the OER activity decreases in the following sequence: YBRO-0.15 > YBRO-0.1 > YBRO-0.25 > YBRO-0.4 > YRO > RuO2. The enhanced activity is attributed to the oxygen vacancy-rich surface because of partial replacement of Ba2+ with Y3+ and simultaneous formation of multivalent Ru (Ru5+/Ru4+) species.
5. Doping non-oxide catalysts
Non-oxide catalysts include metal sulfides, metal selenides, metal phosphides, metal nitrides, and metal carbides. These catalysts are pre-catalysts, and the actual catalysts are the oxides of the corresponding cations generated on the surface during electrochemical water oxidation.5.1 Metal sulfides
Transition metal sulfides are widely explored in electrocatalysis due to their high stability in acidic and alkaline electrolytes and abundant redox chemistry.138 Some doped metal sulfides and their catalytic performances are listed in Table 3. Like in metal oxides and hydroxides, Fe doping is effective in significantly improving the OER activity of TM sulfides.139,140 For example, Zeng et al. synthesized Fe-doped CoS electrocatalysts via a simple hydrothermal method, and 20% Fe doping produced considerably the highest OER activity among them because of the high electrical conductivity by doping, which accelerated the charge transfer process by changing the local Co–S environment for easy adsorption of OER intermediates.141 Dong and coworkers synthesized nano-micro spheres of Fe-doped Co9S8 on nickel foam using a hydrothermal method; for comparison, they also synthesized Ni-doped Co9S8.142 The Fe-doped sample showed superior OER activity to the pristine and Ni-doped samples. It was found that the high activity was contributed by the synergetic effect of tuning the electronic structure of cobalt by an Fe dopant and high surface area induced by the nickel foam. Sun et al. synthesized Fe-doped NiS2 nanoarrays on a Ti mesh through sulfidation of a NiFe layered hydroxide as a precursor, and they found that the catalyst with 10% Fe doping showed superior OER activity with an overpotential of 231 mV at 100 mA cm−2 current density.143 Wu's group reported Fe-doped Ni3S2 nanowires synthesized on a Ni foam and found that Ni3S2 nanowires with a 13.7% dopant level of Fe exhibit superior activity and only require 223 mV overpotential to achieve 200 mA cm−2 in alkaline electrolyte.144 The incorporation of iron into the matrix of Ni3S2 enhances the number of active site edges along with conductivity. Sugahara and coworkers prepared hollow iron-doped bimetallic CoMoS heterostructures, which show enhanced OER activity over the gold standard catalyst RuO2.145| Catalyst | Overpotential at a specific current density | Tafel slope (mV dec−1) | Durability | Electrolyte |
|---|---|---|---|---|
| Metal sulfides | ||||
| Al-doped Ni3S2/NF146 | 223 mV @ 10 mA cm−2 | 37 | 15 h @ 1.53 V vs. RHE | 1 M KOH |
| Ce-doped Ni3S2 (ref. 147) | 257 mV @ 50 mA cm−2 | 81 | 24 h @ 0.6 V vs. Ag/AgCl | 1 M KOH |
| Co-doped WS2 (ref. 148) | 303 mV @ 10 mA cm−2 | 79 | — | 1 M KOH |
| CoFeS/CNT-P 1000 (ref. 149) | 309 mV @ 100 mA cm−2 | 47 | 12 h @ 20 mA cm−2 | 1 M KOH |
| Fe-doped CoS141 | 290 mV @ 10 mA cm−2 | 52.6 | 10 h @ 10 mA cm−2 | 1 M KOH |
| Fe-doped Co9S8 (ref. 142) | 270 mV @ 10 mA cm−2 | 70 | 10 h @ 270 mV | 1 M KOH |
| Fe-doped NiS2 (ref. 143) | 231 mV @ 100 mA cm−2 | 43 | 15 h @ 20 mA cm−2 | 1 M KOH |
| Fe-doped Ni3S2 (ref. 144) | 223 mV @ 200 mA cm−2 | 55.7 | 14 h @ 223 mV | 1 M KOH |
| Fe-doped Ni3S2/NF139 | 249 mV @ 100 mA cm−2 | 42 | 20 h @ 270 mV | 1 M KOH |
| Fe2.1% doped Ni3S2/NF140 | 213 mV @ 100 mA cm−2 | 33.2 | — | 1 M KOH |
| Fe-doped H–CoMoS145 | 282 mV @ 10 mA cm−2 | 58 | 1 M KOH | |
| Ni1.29Co1.49Mn0.22S4 (ref. 150) | 348 mV @ 10 mA cm−2 | 65 | 40000 s @ 10 mA cm−2 |
1 M KOH |
| N-doped Co9S8/G151 | 409 mV @ 10 mA cm−2 | 82.7 | — | 0.1 M KOH |
| Ni-doped FeS152 | 228 mV @ 10 mA cm−2 | 53 | 10 h @ 1.47 V vs. RHE | 1 M KOH |
| N2–NiS2-500 (ref. 153) | 270 mV @ 10 mA cm−2 | 40 h @ 270 mV | 1 M KOH | |
| (N–Ni3S2@C)/NF154 | 310 mV @ 100 mA cm−2 | 75 | 20 h @ 1.70 V vs. RHE | 1 M KOH |
| N-doped NiS/NiS2 (ref. 155) | 270 mV @ 10 mA cm−2 | 99 | 20 h @ 270 mV | 1 M KOH |
| P-doped Co–Ni–S nanosheets156 | 296 mV @ 100 mA cm−2 | 61.1 | 16 h @ 10 mA cm−2 | 1 M KOH |
| P–Ni3S2/NF157 | 256 mV @ 10 mA cm−2 | 30 | 30 h @ 1.525 V vs. RHE | 1 M KOH |
| P–Ni3S2/NF158 | 306 mV @ 100 mA cm−2 | 99 | 10 h @ 1.54 V vs. RHE | 1 M KOH |
| (P-(Ni,Fe)3S2/NF159 | 196 mV @ 10 mA cm−2 | 30 | 15 h @ 295 mV | 1 M KOH |
| Zn-doped Ni3S2 (ref. 160) | 330 mV @ 100 mA cm−2 | 87 | 20 h @ 300 mV | 1 M KOH |
|
||||
| Metal selenides | ||||
| Fe-doped CoSe2/NF161 | 256 mV @ 100 mA cm−2 | 35.6 | 10 h @ 231 mV | 1 M KOH |
| Ag-doped CoSe2 nanobelts162 | 320 mV @ 10 mA cm−2 | 56 | — | 0.1 M KOH |
| B-doped Fe5Co4Ni20Se36 (ref. 163) | 279.8 mV @ 10 mA cm−2 | 59.5 | 10 h @ 10 mA cm−2 | 1 M KOH |
| Co-doped NiSe164 | 380 mV @ 100 mA cm−2 | 111 | >10 h @ 320 mV | 1 M KOH |
| Co-doped nickel selenide165 | 275 mV @ 30 mA cm−2 | 63 | 24 h @ 1.5 V vs. RHE | 1 M KOH |
| Co0.75Fe0.25(S0.2Se0.8)2 (ref. 166) | 293 mV @ 10 mA cm−2 | 77 | — | 1 M KOH |
| Cu-14-Co3Se4/GC167 | 166 mV @ 10 mA cm−2 | 111 | 12000 s @ 166 mV |
1 M KOH |
| Cu-doped FeSe2 (ref. 168) | 193 mV @ 10 mA cm−2 | 59.2 | 10000 s @ 1.526 V vs. RHE |
1 M KOH |
| FCS@N-CT169 | 330 mV @ 10 mA cm−2 | 74 | — | 1 M KOH |
| Fe-doped NiSe170 | 291 mV @ 1000 mA cm−2 | 43 | 22 h @ 217 mV | 1 M KOH |
| Fe-doped NiSe2 (ref. 171) | 231 mV @ 10 mA cm−2 | 83 | 20 h @ 15 mA cm−2 | 1 M KOH |
| Fe-doped Ni3Se4 (ref. 172) | 225 mV @ 10 mA cm−2 | 41 | 26 h @ 10 mA cm−2 | 1 M KOH |
| Fe-doped Ni3Se2 (ref. 173) | 225 mV @ 10 mA cm−2 | 35.3 | 12 h @ 20 mA cm−2 | 1 M KOH |
| Ni1.12Fe0.49Se2 (ref. 174) | 227 mV @ 10 mA cm−2 | 37.9 | 10 h @ 10 mA cm−2 | 1 M KOH |
| Ni0.04Fe0.16Co0.8Se2 (ref. 175) | 230 mV @ 10 mA cm−2 | 39 | 15 h @ 1.5 V vs. RHE | 1 M KOH |
| VSe–Ni0.70Fe0.30Se2 (ref. 176) | 210 mV @ 10 mA cm−2 | 61 | 20 h @ 10 mA cm−2 | 1 M KOH |
| Zn-doped CoSe2 (ref. 177) | 286 mV @ 10 mA cm−2 | 37 | 24 h @ 10, 20, and 50 mA cm−2 | 1 M KOH |
In addition to the Fe inclusion, doping other metals such as Ni,152,178 Co,148 Mn,150 Zn,160 Ce,147 and Al146 is also found to enhance the OER activity of metal sulfides. For instance, Lin et al. fabricated Ni-doped FeS nanosheets as an OER catalyst in alkaline electrolyte through a one-pot synthesis approach.152 The Ni-doped FeS nanosheets showed significant OER activity compared to commercial IrO2 with 228 mV overpotential at 10 mA cm−2 current density, by the consequent doping effect of exposing more active sites. Similarly, Kuila et al. synthesized binder-free self-standing Ni-doped FeS OER catalysts on Ni foam via an electrodeposition method.178 The Ni doping amount is controlled by varying the electrodeposition time, and the electrode fabricated by electrodeposition at 0.9 V for 30 min shows superior activity among them. The change in the electronic structure and charge carrier density via the incorporation of Ni into the lattice domain of FeS significantly improves its electrocatalytic activity. Likewise, Wen and coworkers incorporated Ce into the lattice of Ni3S2via a one-step electrodeposition method.179 Doping with Ce preserves the original structure and morphology. Still, it promotes the OER activity by regulating the active sites and chemical states on the surface of Ni3S2. Yang et al. significantly enhanced the electrocatalytic ability of WS2 by incorporating nearly 1% Co into its lattice.148 The overpotential was decreased from 492 to 303 mV after incorporation of Co, and the improved activity was ascribed to the presence of Co(II/III) species that improved the binding of reactants and facilitated the formation of the *OOH intermediate. Darr and coworkers optimized the OER activity of bimetallic cobalt–nickel sulfides by doping with various TMs, such as Ag, Fe, Mn, Cr, V and Ti.150 Among them, Mn-incorporated cobalt–nickel sulfide (Ni1.29Co1.49Mn0.22S4) shows enhanced activity through tuning of the electronic structure of the host by Mn(III) cations. Zn-doped Ni3S2 nanoarrays on nickel foam grown by Sun et al. have better OER activity than their pristine counterpart, with excellent stability for 20 h at 300 mV overpotential.160 The extended endurance and comparable activity were caused by modulation of the electronic structure of the Ni3S2 catalyst through Zn incorporation.
Recently, researchers reported incorporating non-metals, such as N, P, and F, into the lattice of metal sulfides to improve their electrocatalytic capability.149,151,153–159,180 N doping of a NiS2 catalyst using NH3·H2O as a precursor considerably increased its water oxidation ability by overcoming the intrinsic limits of reaction kinetics through optimizing d-states.153 3D N-doped Ni3S2 nanoflakes grown on Ni foam show superior durability for 20 h at 1.7 V with ∼93% retention of their initial activity.154 The experimental results indicate that N doping plays a key role in the quick discharging of OER intermediates and improved intrinsic activity.155 Dou et al. found that a Co9S8/graphene hybrid, doped with N using NH3 plasma etching, showed a significant improvement in its OER performance compared with a pristine sample.151 The synergetic effect of N-doping and surface etching considerably tuned the electronic structure of the Co9S8/graphene hybrid and thus enhanced the number of active sites on the surface. He et al. synthesized 3D self-supported and P-doped Ni3S2 on Ni foam by a one-step hydrothermal process by simultaneous phosphorization and sulfuration.157 The as-fabricated electrode exhibits enhanced OER activity with an overpotential of 256 mV at 10 mA cm−2. The inclusion of P anions optimizes the electronic structure and provides facile adsorption of reactants and thus enables a faster mass transfer. Similarly, P-doped bimetallic nickel–iron sulfide nanosheets on Ni foam were synthesized via a one-pot process.159 The incorporated P considerably enhances the electrochemical surface area and electrical conductivity by optimizing the adsorption of OER intermediates. F-doped CoS2 on Ni foam was synthesized using NH4F as a fluorine source by a hydrothermal process.180 The F dopant significantly enhances the wettability of the catalyst, and thus facilitates the maximum interaction with electrolyte in both alkaline and neutral media.
5.2 Metal selenides
Some doped metal selenides and their performances are listed in Table 3. The electrocatalytic behavior of metal selenides depends on the number of exposed edge sites.21 Edge sites are step-like apertures or defects generated at the basal plane because of doping.181 Besides the edge sites, the electrical conductivity and intrinsic activity of metal selenides are enhanced by doping; specifically, doping with TMs significantly improves the OER activity of metal selenides.169,171,173,174 For example, Zare et al. designed a self-supported electrocatalyst on Ni foam by doping Fe into the lattice domain of CoSe2, which only requires 256 mV overpotential to attain 100 mA cm−2 current density.161 Here, Fe improves the intrinsic electrical conductivity, and thus lowers the energy barrier for the OER. Zhao and coworkers improved the OER activity of CoSe2 nanobelts by simultaneous Fe doping and Co vacancy creation, as shown in Fig. 13a.182 LSV curves of CoSe2, CoSe2-DFe, CoSe2-DFe-VSe, and CoSe2-DFe-VCo catalysts, normalized by electrochemical double-layer capacitance, are shown in Fig. 13b, and suggest that CoSe2-DFe-VCo has superior OER activity among the catalysts. Fig. 13c displays the FeCoOOH with Co vacancies and the OER mechanism occurring on its surface, which involves three OER intermediates (*OH, O*, and *OOH) and four proton-coupled electron transfer steps. The Gibbs free energy profiles of this catalyst show different catalytic mechanisms for Co and Fe sites. The transformation of OH* to O* potentially limits the activity of Co active sites owing to the large Gibbs free energy difference (ΔG2). In contrast, the conversion of O* to *OOH is the RDS (ΔG3) in case of Fe sites, due to the high free energy difference. Theoretical simulations identified that the cobalt dimer (Co2) is the most active site for the OER, and is present adjacent to the Co vacancy near the surface Fe site. The combinatorial effect of Co vacancy and Fe doping significantly intensifies the OER performance by reducing the peak intensity near the Fermi level, so the binding of *OH with active sites is weakened. Zou et al. found that the minimal amount of Fe (7.4%) inclusion into the NiSe nanorod/nanosheet lattice significantly improved its OER activity.170 The Fe-doped NiSe only required 291 mV overpotential to achieve 1000 mA cm−2 current density. This superior activity is accredited to the increased electrical conductivity, active sites, and mass transfer. Xu et al. fabricated hierarchical Fe-doped Ni3Se4 on Ni foam using a hydrothermal method, and NiFe LDH was used as a precursor material.172 The doped sample shows superior OER activity than IrO2 and pristine catalysts, because of its higher electrical conductivity. Mohanty et al. reported an increase in OER activity of nickel selenide, owing to the synergetic effect of Fe doping along with the creation of Se vacancies by chemical reduction with NaBH4.176 Computational studies suggest an increase in DOS near the Fermi level because of Fe doping and Se vacancies, which helps in decreasing the energy barrier for OER intermediates.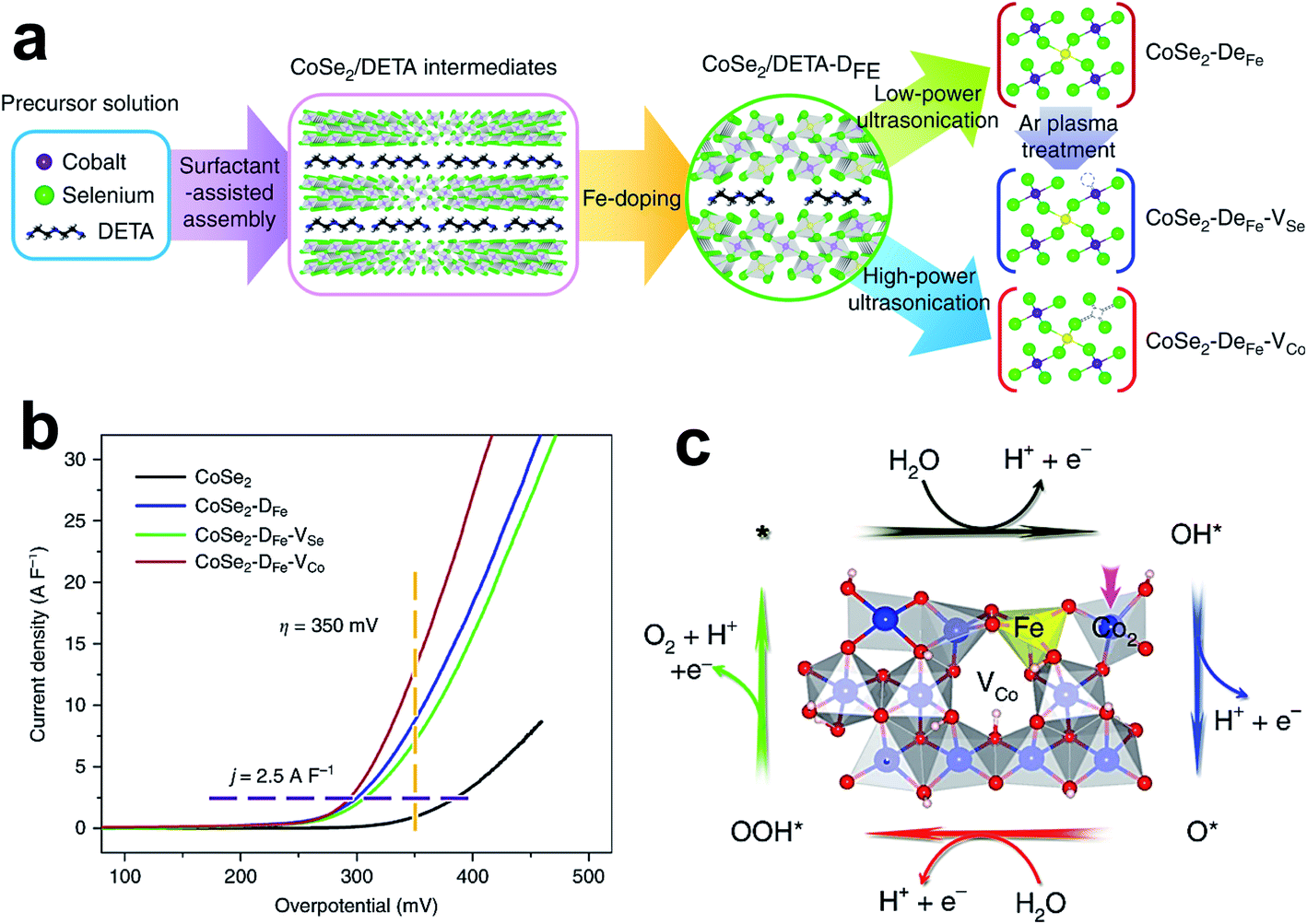 | ||
| Fig. 13 (a) Schematic illustration of the synthetic methods for different catalysts; (b) LSV curves normalized by electrochemical double-layer capacitance; and (c) crystal structure of FeCoOOH-VCo and the oxygen evolution reaction pathway on the (01−12) facets.182 Reproduced with open access. | ||
Besides Fe, Co was also used as a dopant to improve the OER performance of nickel selenides,165 and Co-doped NiSe nanoflowers grown on Ni foam exhibited considerable activity compared to their pristine counterpart. Here, Co doping generates more OER active sites via tuning the electronic structure of NiSe. Wang et al. showed MOF-derived Co-doped nickel selenide on Ni foam and analyzed its OER activity in alkaline electrolyte.164 The as-designed catalyst exhibits an overpotential of 275 mV at 30 mA cm−2, and the high activity is ascribed to an increase in electrical conductivity because of Co doping. Zhang et al. co-doped Ni and Fe into the matrix of CoSe2, and noticed that the catalyst with a stoichiometric formula of Ni0.04Fe0.16Co0.8Se2 has remarkable OER activity among the prepared catalysts.175 Ni doping does not alter the OER mechanism but reduces the energy barrier for the RDS and keeps Co-active sites intact, and Fe doping improves the charge transfer kinetics by transferring the active sites from Co to Fe. FeSe2 was doped with various transition elements (Co, Ni, Mn, and Cu) by Wei et al.168 Among these dopants, Cu is found to be more effective and shows enhanced OER activity. The theoretical and experimental results reveal that Cu doping optimizes the electronic structure of the FeSe2 lattice to enable faster electron transfer and thus boosts the reaction kinetics of the OER. In cobalt selenide, the Cu doping results in a phase transformation from orthorhombic CoSe2 to monoclinic Co3Se4, causing improved OER performance of cobalt selenide.167 The electronic structure change in cobalt selenide by Cu doping is evident from X-ray photoelectron spectroscopy and theoretical simulations. DFT calculations further reveal that because of structural transformation from orthorhombic CoSe2 to monoclinic Co3Se4, and the Gibbs free energy for the RDS was drastically decreased from 2.64 to 1.90 eV.
The minimal amount of Ag (1%) doping into the lattice domain of CoSe2 nanobelts significantly increases its electrical conductivity without much loss of active sites (Co).162 Ag-doped CoSe2 nanobelts exhibit high OER activity relative to pristine CoSe2 nanobelts, because of high electrical conductivity and active site retention. Incorporation of low-valence Zn into the CoSe2 lattice domain triggers subtle changes in the lattice and increases the number of active sites for the OER.177 Theoretical simulations suggest that Zn inclusion decreases the kinetic energy barrier for the adsorption of OER intermediates by encouraging adsorption on Co sites and neighboring Zn sites. Boron doping into the lattice of ternary pyrite selenide (Fe5Co4Ni20Se36) decreased its overpotential from 543 to 279.8 mV at 10 mA cm−2, with the Tafel slope lowered from 161 to 59.5 mV dec−1.163Fig. 14a and b show the schematic representation of NiSe2 and Fe5Co4Ni20Se36Bx crystal structures, and the doped B atoms are incorporated into the interstitial positions and some B atoms even bond with metals (Ni, Co, and Fe). The proposed OER mechanisms on pristine and doped pyrite are shown in Fig. 14c and d, and reveal that B doping facilitates an easy discharge of *OOH from the active sites, as a consequence of lower Gibbs free energy. The PDOS reveals that B doping significantly reduces the total d electrons around metals, enhancing the interaction between them and thus weakening the bond between *OOH and active sites. Recently, anion doping, i.e., doping on the Se site, was also found to be beneficial in enhancing the OER activity. For example, Xu et al. reported doping of sulfur on the Se site, enhancing the OER activity of bimetallic cobalt iron selenide Co0.75Fe0.25(S0.2Se0.8)2 as compared to non-doped Co0.75Fe0.25S2 and Co0.75Fe0.25Se2, since charge transfer and electrochemical surface area were increased by doping.166
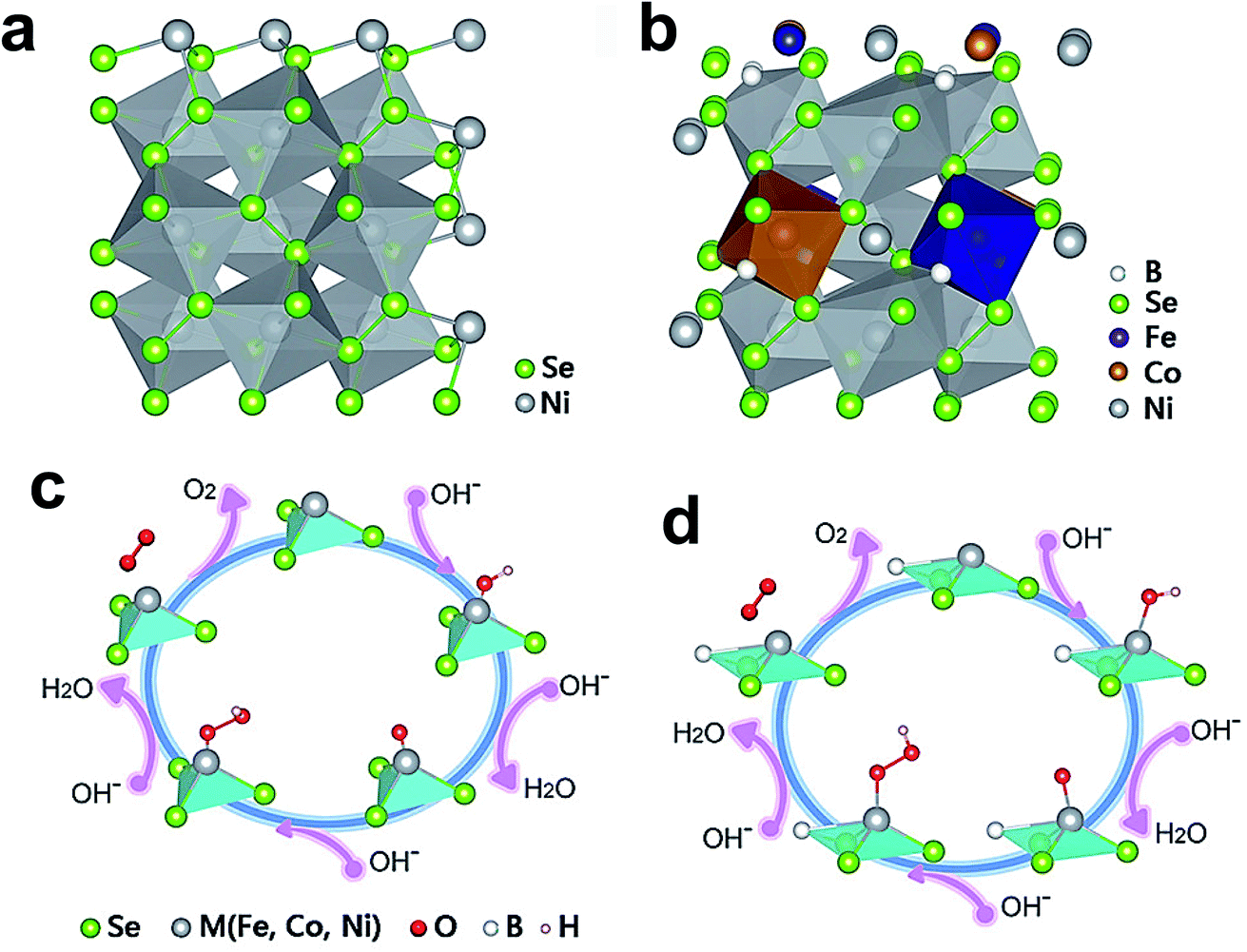 | ||
| Fig. 14 (a and b) Schematic illustration of NiSe2 and Fe5Co4Ni20Se36Bx crystal structures, and (c and d) proposed OER mechanisms for pyrite selenides before and after B dopant incorporation.163 Reproduced from ref. 163 with permission, © 2019 American Chemical Society. | ||
5.3 Metal phosphides
In the past five years, metal phosphides have emerged as one of the most promising types of catalyst for water oxidation, even though the first metal phosphide catalyst was reported as early as in 1990.183 Some doped metal phosphides and their performances are listed in Table 4. Incorporation of a second element into the matrix of a metal phosphide could enhance the OER performance, in terms of both activity and stability.184,185 For instance, Chen et al. doped Fe into the matrix of CoP ultrathin nanosheets grown on Ni foam, which exhibited a very low overpotential of 67 mV at 10 mA cm−2 in 1 M KOH.186 The excellent OER activity is attributed to the interference of Fe/Co/Ni in the low current density region. Post OER characterization revealed the complete absence of P because of the formation of CoOOH active species during the OER. Baek and coworkers synthesized Fe-doped porous CoP polyhedrons from MOFs by varying the Fe/Co ratio.187 Among them, Co0.68Fe0.32P had better OER activity over its pristine counterparts, i.e., CoP and FeP. Fe doping accelerated the discharge of OER intermediates by optimizing their adsorption energy and stabilized the P species during contact with air. Peng's group initially conducted theoretical simulations to predict the Fe influence on the water oxidation capability of Ni2P by constructing partially oxidized surfaces of Ni (111), Ni2P (111), and Fe-doped Ni2P (111) models.188 The Gibbs free energy studies reveal that the Fe-doped sample requires an energy 0.14 eV lower than that of Ni2P for the RDS (O* to *OOH). Experimental results were in accordance with the theoretical calculations, and they observed the formation of Fe, P co-doped γ-NiOOH during the electrochemical water oxidation reaction from the Fe-doped Ni2P. Fe, P co-doped γ-NiOOH is a good catalyst for improved OER activity, and Fe doping primarily enhances the active sites for the OER. Xiang et al. tuned the electronic structure of vanadium phosphide by including Fe into its lattice domain, which demonstrated remarkable OER activity.189 Among all the samples, the sample with a 1:3 ratio of Fe/V has the highest activity. XPS analysis identifies increased valency of V because of the incorporation of Fe, which optimizes the adsorption of *OH on active sites. Fe doping of bimetallic phosphides was also found to significantly contribute to improving electrochemical water oxidation.
| Catalyst | Overpotential at a specific current density | Tafel slope (mV dec−1) | Durability | Electrolyte |
|---|---|---|---|---|
| Metal phosphides | ||||
| Al-doped CoP193 | 360 mV @ 200 mA cm−2 | 38 | 150 h @ 10 mA cm−2 | 1 M KOH |
| Co0.68Fe0.32P187 | 289 mV @ 50 mA cm−2 | 64 | 3600 s @ 10 mA cm−2 | 1 M KOH |
| Fe0.27Co0.73P/NF184 | 251 mV @ 10 mA cm−2 | 59.1 | 40 h @ 40 mA cm−2 | 1 M KOH |
| Fe-doped CoP186 | 166 mV @ 50 mA cm−2 | 65.82 | 50 h @ 50 mA cm−2 | 1 M KOH |
| Fe-doped Ni2P192 | 330 mV @ 20 mA cm−2 | 39 | 10 h @ 20 mA cm−2 | 1 M KOH |
| Fe-doped Ni2P188 | 300 mV @ 1000 mA cm−2 | 52 | — | 1 M KOH |
| FexVy−xPC/NF189 | 201 mV @ 10 mA cm−2 | 75 | 24 h @ 10 mA cm−2 | 1 M KOH |
| Mn-doped CoP194 | 261 mV @ 10 mA cm−2 | 44.9 | 24 h @ 10 mA cm−2 | 1 M KOH |
| Mn-doped Ni2P195 | 330 mV @ 100 mA cm−2 | 116.7 | 18 h @ 320 mV | 1 M KOH |
| NiCoFeP/NF191 | 271 mV @ 200 mA cm−2 | 45 | 10 h @ 1.485 V vs. RHE | 1 M KOH |
| NiCoFexP/CC190 | 275 mV @ 50 mA cm−2 | 50 | 150 h @ 1.70 V vs. RHE | 1 M KOH |
| NCNP196 | 229 mV @ 10 mA cm−2 | 78.6 | 100 h @ 100 mA cm−2 | 1 M KOH |
| Ni1.85Fe0.15P NSAs/NF185 | 270 mV @ 20 mA cm−2 | 96 | 10 h @ 330 mV | 1 M KOH |
| Ni2P/(NiFe)2P(O) NAs197 | 530 mV @ 800 mA cm−2 | 60 | 100 h @ 500 mV | 1 M KOH |
| N-NiVFeP/NFF198 | 229 mV @ 10 mA cm−2 | 78.6 | 100 h @ 100 mA cm−2 | 1 M KOH |
| O-doped Co2P/CuO NWs/CF P199 | 270 mV @ 10 mA cm−2 | 74.4 | 30 h @ 10 mA cm−2 | 1 M KOH |
| S-doped CoP/NF200 | 300 mV @ 10 mA cm−2 | 82 | 20 h @ 350 mV | 1 M KOH |
|
||||
| Metal nitrides | ||||
| Co-doped MoNi nitride201 | 294 mV @ 10 mA cm−2 | 73 | 10 h @ 317 mV | 0.1 M KOH |
| Fe-doped cobalt nitride202 | 200 mV @ 10 mA cm−2 | 42.44 | 10 h @ 200 mV | 1 M KOH |
| Mn0.15 Co0.85N203 | 265 mV @ 10 mA cm−2 | 48 | 30 h @ 280 mV | 1 M KOH |
| Mn0.15 Co0.85N203 | 285 mV @ 10 mA cm−2 | 64 | 30 h @ 285 mV | 1 M phosphate-buffered saline |
| P,F-Ni1.5Co1.5N204 | 350 mV @ 100 mA cm−2 | 66.1 | — | 1 M KOH |
| P-NiMo4N5@Ni205 | 435 mV @ 100 mA cm−2 | 65 | 24 h @ 50 mV | 1 M KOH |
|
||||
| Metal carbides | ||||
| B, N:Mo2C@BCN206 | 360 mV @ 100 mA cm−2 | 61 | 20 h @ 350 mV | 1 M KOH |
| Co3C-NB207 | 354 mV @ 10 mA cm−2 | 90 | 24 h @ 50 mA cm−2 | 0.1 M KOH |
| Co-doped β-Mo2C208 | 262.2 mV @ 10 mA cm−2 | 28.8 | 16 h @ 1.49 V vs. RHE | 1 M KOH |
| Co-doped Ni3C/Ni@C209 | 325 mV @ 10 mA cm−2 | 67.76 | 100000 s @ 10 mA cm−2 |
1 M KOH |
| FCC@CNOs/NF210 | 271 mV @ 10 mA cm−2 | 48.9 | 40 h @ 100 mA cm−2 | 1 M KOH |
| Fe-doped Ni3C211 | 275 mV @ 10 mA cm−2 | 62 | 10 h @ 1.5 V vs. RHE | 1 M KOH |
| V0.28Co2.72C/CNFs212 | 210 mV @ 10 mA cm−2 | 147 | 10 h @ 1.4 V vs. RHE | 1 M KOH |
Like monometallic phosphides, bimetallic phosphides can also convert into their respective oxyhydroxides at high anodic potentials. Fe-doped NiCoP converts into Fe-doped NiOOH and CoOOH, and Fe doping improves the OER activity by accelerating the discharge of OER intermediates.190,191 Simultaneously, the conductive metallic phosphide support improves OER kinetics by promoting the proton transfer. Tsang and coworkers studied the comparative doping effects of TMs (Co, Fe, Mn, and Mo) on the OER activity of NiP.192 Among them, the Fe doped sample was found to have more prominent activity since Fe is able to tune the electronic structure of Ni to interact optimally with OER intermediates.
Mn doping of cobalt phosphide and nickel phosphide improves their catalytic activity for electrochemical water oxidation. In cobalt phosphide, Mn doping enhances the electrochemical surface area by increasing the number of exposed active sites.194 The optimal Mn/Ni ratio of 0.053 is found to provide superior activity, as the Ni2P electronic structure is optimized.195 Al doping was also used to enhance the electrochemical surface area of cobalt phosphide by dissolution during the OER. Al-doped cobalt phosphide shows a fourfold increase in electrochemical surface area compared to the pristine sample by the formation of Al/Co spinel oxide and partial leaching of Al.193
Along with doping of metals, doping of non-metals such as N, O, and S can also enhance the electrocatalytic activity of phosphide catalysts for the OER.196,198–200 N doping in the anionic site (P) of NiCoP improved its OER performance, with better activity compared to NiCoP and NiCoN. The high activity was induced by the formation of abundant oxygen vacancies, improving the charge transfer during oxygen generation.196 Likewise, oxygen and sulfur were also doped on the anionic sites to increase the OER activity of metallic phosphides. Oxygen doping of cobalt phosphide generates abundant defects and electroactive sites on the surface by optimizing the adsorption energy of reactants.199 In the case of bimetallic nickel–iron phosphide, the OER activity is enhanced by oxygen doping on account of an efficient decrease in the energy barrier of OER intermediates and products.197 Electronegative sulfur doping in the anionic site of cobalt phosphide significantly improved its OER activity, owing to the tuning of the electronic structure and increased number of active sites.200
5.4 Metal nitrides and carbides
Metal nitrides belong to an interstitial compound family, which contains nitrogen atoms on the interstitial sites of parent compounds. The presence of N atoms on the interstitial sites increases the distance between metal atoms; thus, it tunes the d band structure.20 Some doped metal nitrides and carbides and their performances are listed in Table 4. Recent studies on the electrochemical activity of metal nitrides suggest that they are highly active for the OER owing to their high electrical conductivity.204,205 However, the OER activity and stability were further improved by tuning their electronic structure by doping with TMs and non-metals. For instance, Mn-doped CoN nanowires grown on carbon cloth exhibit good OER activity in alkaline and neutral electrolytes.203 Among the doped catalysts, Mn0.15Co0.85N shows superior activity, and Mn doping considerably increases the charge transfer kinetics and facilitates the optimized discharge of OER intermediates. Zhou et al. investigated the doping effect of various metal atoms, such as Zn, Ni, Fe, and Mn, on the OER activity of cobalt nitride nanosheets.202 The Fe doped one has better OER activity, and XPS analysis suggests more Co4+ OER active species because of the doping effect of Lewis acid Fe3+.Cobalt doping into the lattice of bimetallic MoNi nitride nanowires shows a considerable increase in their OER activity by reduced charge transfer resistance and augmentation of the electrochemically active surface area.201 The dual modulation in the electronic structure of Mo and Ni was induced by cobalt doping that improved the electrical conductivity for highly active OER sites. Liang et al. theoretically studied the doping effect of first-row TMs (Sc to Zn) for improving the OER activity of Al/GaN.213 The inclusion of transition metals from Sc to Fe resulted in low OER activity because they bonded to OER intermediates too firmly and resulted in high overpotentials. Meanwhile, Cu and Zn also displayed no or low OER activity, as they attached to the intermediates very weakly. Only Ni doping had significant OER activity because it could switch from a high spin state to a low spin state and stabilized the OOH adsorbate. Tong and coworkers synthesized P and Co dual-doped vanadium nitride nanowires using a combined hydrothermal and annealing approach.214 The dual-doped vanadium nitrate catalyst showed better activity and durability for the OER, compared to the single-doped or pristine sample, or IrO2. Dual doping of Co and P synergistically tunes the number of active sites on the catalyst surface by moving the d states of vanadium closer to the Fermi level.
Zhang and coworkers demonstrated an increase in the OER performance of bi-metallic nitrides by co-doping with phosphorus and fluorine.204Fig. 15a shows the schematic representation of the fabrication process for P and F co-doped nickel–cobalt nitride (PF/Ni1.5Co1.5N) nanorods using an ionic liquid. The PF/Ni1.5Co1.5N prepared without using any ionic liquid results in a lamellar structure, as displayed in Fig. 15b. The SEM and elemental mapping analysis results of PF/Ni1.5Co1.5N nanorods are presented in Fig. 15c and d, which indicates the uniform distribution of N and F dopants all over the catalyst. Fig. 15e and f show the IR uncompensated LSV curves and comparison of η10 values (potentials required to reach 10 mA cm−2) for PF/CoxNi3−xN catalysts. Electrochemical studies reveal that the PF/Ni1.5Co1.5N catalyst shows augmented OER activity as well as facile reaction kinetics among the prepared catalysts. DFT calculations suggest that co-doping of P and F atoms into the lattice of the Ni1.5Co1.5N catalyst considerably decreases the free energy barrier for OER intermediates, thus helps in improving the electrocatalytic performance.
 | ||
| Fig. 15 (a) Schematic illustration of the fabrication processes for PF/Ni1.5Co1.5N nanorods; (b) SEM; (c) highly magnified SEM; (d) HAADF-STEM elemental mapping results of the heteroatom doped Ni–Co (1:1) precursor; (e) LSV curves of PF/CoxNi3−xN catalysts for the OER (400 mg of ionic liquid, without IR compensation); and (f) comparison of η10 values (potentials required to reach 10 mA cm−2) for various catalysts (Ni:Co = 1:1, 1:3, 1:0, 0:1, and 3:1).204 Reproduced from ref. 204 with permission, © 2017 John Wiley and Sons. | ||
Recently, researchers found that metal carbides, specifically transition metal carbides, can be applied as electrocatalysts for the OER owing to their high stability.215 Some doped metal carbides and their performances are listed in Table 4. The activity of TM carbides was improved by including one or more foreign dopants into their lattice domain. Yan et al. improved the OER activity of Ni3C by doping 2% atomic iron, which yields an overpotential of 275 mV at 10 mA cm−2.211 The optimized doping of Fe tuned the electronic structure of Ni and thus improved the number of active sites on the surface. Similarly, Jia et al. tuned the water oxidation ability of Ni3C via incorporation of a small amount of Co into it using a MOF-derived method.209 The as-fabricated catalyst had better activity than the IrO2 catalyst in the alkaline electrolyte, and the improved activity was associated with the active surface sites through optimization of electronic properties. In general, the Mo2C catalyst is inactive for the OER, but doping 10% cobalt into the Mo2C catalyst was found to increase its OER performance through significant enhancement in the density of active sites of the catalyst.208 Zhao et al. optimized the electronic structure of Co3C by doping with low electronegativity vanadium element.212 Specifically, V0.28Co2.72C shows superior activity by forming local negative Co centers through the intake of electrons from vanadium dopants. Xu et al. doped TMs such as Mn, Co, and Ni into the matrix of Fe3C and evaluated its OER performance.210 They noticed that Co-doped iron carbide had enhanced activity, and the Gibbs free energy was estimated in the different steps in the OER process. The free energy profiles of the Co and Fe sites were compared, and the Gibbs free energy required for the Co site (2.35 eV) was lower than that for the Fe site (2.75 eV), for the conversion of *O to *OOH (RDS). From the comparison, Fe has a stronger affinity towards *O compared to Co, hence doping of Co into the Fe3C facilitates the discharge of intermediates, thus lowering the potential.
Doping of metal carbides on the anionic site also improves their activity for electrocatalytic oxygen evolution. Generally, WC is a poor OER catalyst; however, when the carbon site is doped with nitrogen, it shows even better OER performance than IrO2 in an acid electrolyte.216 The N doping helps in accelerating the discharge of intermediates and thus improves activity, but its poor stability is still a concern. Dual doping of N and B on the anionic site of Co3C considerably increases its water oxidation ability compared to the pristine or single-doped sample, or IrO2. The synergetic effect of N and B dopants improves both activity and stability by boosting the charge transfer kinetics of intermediates.207 Likewise, Lee and coworkers synthesized dual-doped N and B into the matrix of Mo2C embedded in the B, N doped carbon network (B,N:Mo2C@BCN), which shows a considerable increase in OER activity compared to the mono-doped or pristine sample, or IrO2.206 The significant improvement in electrocatalytic performance is accredited to the formation of active B- and N-doped molybdenum oxide at high anodic potentials.
6. Tuning carbon-based catalysts
Although metal-based catalysts are employed in electrocatalytic OER applications widely because of their promising capabilities to deliver efficient catalytic activity by variable oxidation states,217 these catalysts suffer from relatively poor endurance, low selectivity, susceptibility towards potential gas poisoning, and hazardous environmental effects. In 2009, the first carbon-based catalyst was reported by Dai's group in fuel cells as an alternative to the platinum catalyst.218 Since then, carbon-based catalysts, particularly those doped with various heteroatoms or metals, have received extensive attention and tremendous progress has been made in electrocatalysis.219,220Table 5 provides a summary of some representative doped carbon-based electrocatalysts and their catalytic performances.| Catalyst | Overpotential at a specific current density | Tafel slope (mV dec−1) | Durability | Electrolyte |
|---|---|---|---|---|
| 20-NMWNT221 | 320 mV @ 10 mA cm−2 | 68 | — | 1 M NaOH |
| B, N-carbon222 | 340 mV @ 10 mA cm−2 | 84 | — | 1 M KOH |
| Co-C3N4/CNT223 | 380 mV @ 10 mA cm−2 | 68.4 | — | 1 M KOH |
| Co-N/GF224 | 313 mV @ 10 mA cm−2 | 84.1 | 30 h @ 1.6 V vs. RHE | 1 M KOH |
| Co-TA225 | 460 mV @ 10 mA cm−2 | 66 | — | 0.1 M KOH |
| GO-PANi31-FP226 | 300 mV @ 10 mA cm−2 | 136 | — | 0.1 M KOH |
| N/C227 | 380 mV @ 10 mA cm−2 | — | — | 0.1 M KOH |
| NCHCs228 | 290 mV onset potential | 90 | 12 h @ 1.6 V vs. RHE | 0.1 M KOH |
| NCNTFs229 | 370 mV @ 10 mA cm−2 | 93 | 10000 s @ 1.70 V vs. RHE |
1 M KOH |
| N-GRW230 | 360 mV @ 10 mA cm−2 | 47 | 24 h @ 10 mA cm−2 | 1 M KOH |
| N-HC@G-900 (ref. 231) | 350 mV @ 10 mA cm−2 | 88.1 | 25000 s @ 10 mA cm−2 |
1 M KOH |
| N-HC@G-900 (ref. 231) | 350 mV @ 10 mA cm−2 | 297.5 | 7500 s @ 1 mA cm−2 | 0.5 M H2SO4 |
| NiFe@g-C3N4/CNT232 | 326 mV @ 10 mA cm−2 | 67 | 16 h @ 10 mA cm−2 | 1 M KOH |
| Ni, Fe@PCN233 | 310 mV @ 10 mA cm−2 | 38 | 2 h @ 1.53 V vs. RHE | 1 M KOH |
| Ni, N doped graphene234 | 270 mV @ 10 mA cm−2 | 59 | 40 h @ 1.40 V vs. RHE | 1 M KOH |
| Ni-O-G SACs235 | 224 mV @ 10 mA cm−2 | 42 | 50 h @ 115 mA cm−2 | 1 M KOH |
| N, S-CNT236 | 360 mV @ 10 mA cm−2 | 56 | — | 1 M KOH |
| N, S doped graphene237 | 310 mV @ 10 mA cm−2 | 65 | 25 h @ 1.55 V vs. RHE | 0.1 M KOH |
| O-CNTs238 | 360 mV @ 10 mA cm−2 | 47.7 | 24 h @ 5 mA cm−2 | 1 M KOH |
| O Doped MWCNTs239 | 450 mV @ 10 mA cm−2 | 72 | 10000 s @ 5 mA cm−2 |
0.1 M KOH |
| P-doped C3N4 (ref. 240) | 400 mV @ 10 mA cm−2 | 61.6 | 15 h @ 10 mA cm−2 | 0.1 M KOH |
| P-doped graphene241 | 330 mV @ 10 mA cm−2 | 62 | — | 1 M KOH |
| S, N-Fe/N/C-CNT242 | 370 mV @ 10 mA cm−2 | 82 | 40000 s @ 10 mA cm−2 |
0.1 M KOH |
| S,S′-CNT243 | 350 mV @ 10 mA cm−2 | 95 | 75 h @ 10 mA cm−2 | 1 M KOH |
6.1 Graphene
Graphene is a thin layer of single atoms packed with sp2 hybridized carbon atoms in a hexagonal lattice. It is considered either a zero-bandgap semiconductor or conductor because of its symmetric band structure.244 Hence, pristine graphene shows low or no catalytic activity; however, the unique structural flexibility of graphene allows extensive functionalization via doping to improve catalytic activity. The dopants could interrupt the sp2 hybridized network and induce the formation of sp3 in the lattice, resulting in an electronic density change that imparts catalytic activity to it.244 For instance, doping of N into the lattice of graphene disrupts the charge distribution and spin density of neighboring carbon atoms and, consequently, imparts OER activity. Yang et al. conducted a study to gain mechanistic insights into N-doped graphene in the OER.230 They identified the formation of electron-withdrawing pyridinic N functionalities, which is the key reason for the superior OER performance of N-doped graphene. Xia's group studied the active sites in N-doped graphene nanoribbons combined with other dopants, such as B, Cl, P, and S, using DFT calculations.245 Gibbs free energy analysis for the OER on doped graphene nanoribbons suggests that the conversion of *O to *OOH is the RDS. The co-doping shows better activity than mono-doping, and the synergetic effect of co-doping is mostly related to the distance between them. In another report, N doping was done by using polyaniline and graphene oxide as precursors, which produced nanoporous layered and N-doped graphene that exhibits enhanced OER activity.246 Nearly 51% quaternary N plays a significant role in the considerable increase in OER activity. Wang et al. demonstrated a significant improvement in the OER performance of graphene by P doping.241 They found that the stronger oxygen adsorbing ability of P, which was present at the edges of graphene, played a crucial part in stabilizing OER intermediates.Besides single atom doping of graphene, dual- and multi-atom doping were also found to deliver significant results in improving OER activity by their synergistic effects. Zhao and Xia studied several combinations of dual doping such as N–N, N–P, N–S, N–B, and N–Cl in optimizing the OER activity of graphene using first-principles modeling.247 They suggested that optimizing the distance between dopants, selecting the right combination of dopants, and efficient utilization of edge effect are the vital factors in designing a highly efficient electrocatalyst. For example, the co-doping of P and N into the graphene lattice results in graphene formation with P, N population at the edges, as a consequence of the large atomic size of P that always moves away from the planar surface. This co-doped catalyst exhibits a significant improvement in OER performance, and the elevated activity is attributed to the synergetic effect of both dopants, the high oxidation ability of the P-site that initially adsorbs O2, and the N-site that stabilizes the OER intermediates and promotes desorption.248
Qiao's group synthesized N and S dual-doped graphene microwires using an ionic liquid assisted method, which showed an overpotential of 310 mV at 10 mA cm−2, stability for 25 h and 95% faradaic efficiency.237 Moreover, this electrode was employed as an efficient cathode for Zn–air batteries, and this superior OER activity is attributed to the numerous active sites created by C–N–S spin density. Furthermore, Zhang and Dai demonstrated tri-doping of graphene with N, P and F atoms, yielding a multifunctional electrocatalyst.226 The N and P doping roles in improving OER performance were elaborated. Still, it is inconclusive how F inclusion into the graphene lattice contributes to the electrocatalytic activity. Li et al. demonstrated a significant improvement of the OER performance of graphene by including single nickel atoms.235 The single-atom doped graphene displayed 50 h of long-term stability at a very high current density of 115 mA cm−2. This unusual activity is accredited to the bonding between single nickel atoms and oxygen sites of graphene. Chen et al. doped Ni single atoms on N-doped nano-porous graphene via a chemical vapor deposition (CVD) method, followed by chemical etching.234 The dual-doped catalyst showed superior OER activity to other catalysts, and even outperformed the IrO2 catalyst. The DFT calculations suggest that Ni, N co-doping significantly decreased the stability difference between O* and *OH intermediates by 0.3 eV.
6.2 Carbon nanotubes (CNTs)
CNTs are 1D structural allotropes of carbon that consist of hexagons of carbons arranged in a concentric fashion. CNTs possess good electrical–thermal properties due to their unique hollow geometry and conjugated long-range π electron structure.249 However, CNTs in their pristine form exhibit poor electrocatalytic properties. To improve the electrocatalytic properties, CNTs need to be functionalized with several dopants.250 This functionalization can be done through two methodologies, i.e., covalent and non-covalent methods. The covalent functionalization proceeds via forming a covalent bond between the functional group and CNTs, which could disrupt the unique structure of CNTs. In the non-covalent method, the functionalization happens through physical processes such as electrostatic, hydrogen bonding, π stacking and van der Waals interactions, which leaves the CNT structure undisturbed.249 For example, Tian et al. doped nitrogen into CNTs and synthesized highly active CNT@NCNT for the OER as a catalyst.251 The superior OER activity is ascribed to the highly exposed integrated N atoms present on the surface with the combined mechanical stability of the undisrupted inner walls of CNTs. Likewise, the Kallio group doped nitrogen into multiwalled CNTs (MWCNTs) and observed a considerable increase in OER performance after doping, compared with pristine MWCNTs.221 The significant OER activity of N-doped MWCNTs is induced by pyridinic N functionalities that are electron-withdrawing. Wang's group designed a generalized approach to make highly efficient hollow N-doped CNTs as a catalyst for the OER from a MOF. The remarkable electrochemical activity is attributed to the synergistic effects from interconnected N-doped CNT structures and chemical compositions with high mechanical stability.229Qiao's group synthesized dual-doped CNTs with nitrogen and sulfur inspired by the features of polydopamine (PDA), as depicted in Fig. 16a.236 N and S doped CNTs were prepared through a two-step “graft-and-pyrolyze” route using PDA. Initially, PDA was deposited on the surface of multiwalled CNTs and sulfur was incorporated at room temperature via chemical grafting, and subsequent pyrolysis at 700 °C resulted in a N and S uniformly co-doped carbon framework with enhanced S-doping efficiency. Even after doping and pyrolysis, the nanotube morphology of CNTs was preserved, as shown in Fig. 16b. The OER performance of pristine and doped CNT catalysts is displayed in Fig. 16c, which shows that the N, S-CNT catalyst exhibited superior OER activity among them with an overpotential of 360 mV and a Tafel slope of 56 mV dec−1 (Fig. 16d). Theoretical studies show a considerable increase in spin-charge density difference among carbon atoms with doping; these carbon atoms act as electrocatalytically active sites. As a consequence, the apparent OER activity improved with dual doping compared to single doping. Liu's group has extensively studied the doping effect of oxygen on the OER performance of CNTs by synthesis via two different methods, i.e., oxygen plasma oxidation (O-CNTs) and thermal reduction (R-CNTs).238 O-CNTs exhibit enhanced OER activity over R-CNTs and pristine CNTs, and DFT calculations reveal that both O-CNTs and R-CNTs have the same active sites, i.e., carbon near CO. The high-water oxidation ability of O-CNTs was thought to be possible due to the presence of a high density of active sites, with high Ea,app at the specific reaction temperature.
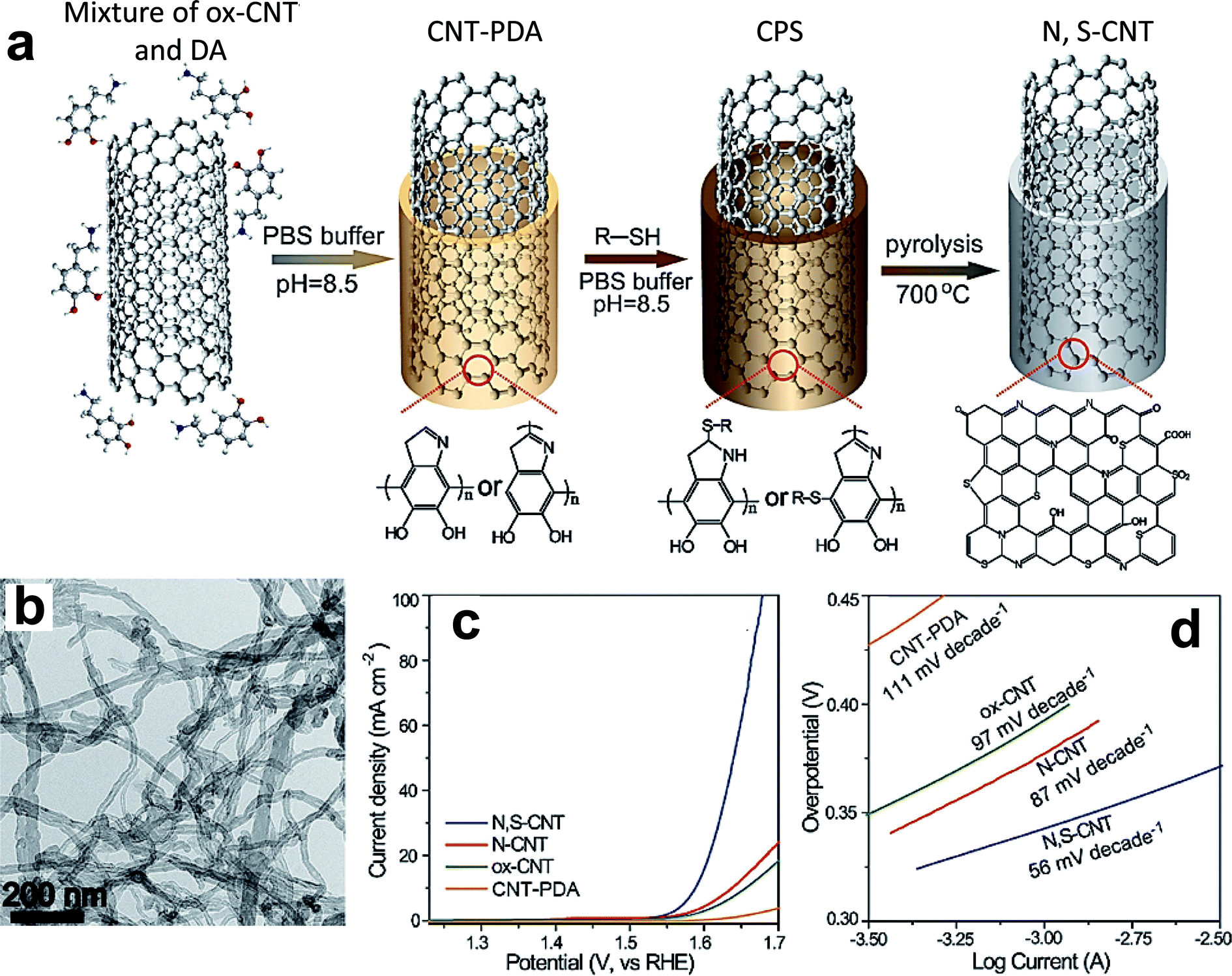 | ||
| Fig. 16 (a) Fabrication of the N, S-CNT following a two-step “graft-and-pyrolyze” route. The oxidized CNT (ox-CNT) was mixed with dopamine (DA) in PBS buffer to obtain a CNT–PDA hybrid. After the addition of R-SH, CNT–PDA modified with thiol (CPS) was synthesized, which produced N, S-CNT after pyrolysis. R-SH = 2-mercaptoethanol; (b) TEM image of the N, S-CNT; (c) OER polarization curves; and (d) corresponding Tafel plots of CNT–PDA, ox-CNT, N-CNT, and N, S-CNT in 1 M KOH solution.236 Reproduced from ref. 236 with permission, © 2017 John Wiley and Sons. | ||
Lu et al. oxidized the surface of MWCNTs through hydrothermal annealing and electrochemical activation, which results in a highly active OER catalyst.239 The superior OER activity originated from the formation of carbonyl functionalities on the outer wall, which altered the electronic structure of neighboring carbon atoms and facilitated adsorption of OER intermediates. Suib et al. fabricated a highly stable metal-free catalyst at high anodic potentials by successive two-step doping of sulfur into the MWCNTs, displaying two orders of magnitude higher turnover frequency (TOF) compared to the Ir/C catalyst.243 The enhanced OER activity was caused by the inclusion of heterocyclic sulfur into the hexagonal rings of CNTs. Wu and co-workers developed a single-atom doped catalyst for the water oxidation reaction by coating CNTs with Fe salt and 2,2 bipyridine, followed by pyrolysis and acid leaching, yielding FeNx species on the S and N co-doped CNT catalyst.242 This catalyst exhibits superior OER activity in alkaline medium, as a result of high exposure of Fe atoms and synergetic effects of S and N doping.
6.3 Other carbon-based catalysts
In addition to graphene and CNTs, other forms of carbon-based catalysts were also reported for OER activity. Graphitic carbon nitride (g-C3N4) has a C–N structure with graphite like sp2 bonding, which exhibits semiconducting behavior due to π-conjugation.252 Like graphene and CNTs, g-C3N4 is also inactive in its pristine form, and tuning its electronic structure by doping non-metal or metal elements significantly improves its water oxidation ability.233,240 For instance, Huang et al. dual-doped N and B into 3D porous g-C3N4 and observed superior OER activity in alkaline solution, compared to N-doped or N and P co-doped 3D g-C3N4.253 It was found that the N and B co-doped g-C3N4 has a high population of pyrrolic and pyridinic nitrogen for enhanced OER.222 Based on DFT calculations, Jiang and coworkers proposed doping single transition elements such as Pt, Pd, Co, Ni, and Cu into carbon nitride (CN) that could lead to a cost-efficient, highly efficient OER catalyst.254 Specifically, the electrocatalysts of non-noble metal Co/Ni@CN and Co–O@CN exhibit excellent activity and function with extremely low overpotential. Qiao's group conducted the theoretical study and experimental validation on cobalt inclusion on g-C3N4 for OER activity in alkaline medium.223 The combinative evaluation of theoretical and experimental methods reveals that metal–nitrogen M–N2 coordination in the matrix of g-C3N4 is the origin of high electrocatalytic activity compared to the pristine sample. Song et al. studied the effect of dual metal doping in enhancing the OER activity, and doped Ni and Fe metals in tri-s-triazine units of g-C3N4 enveloped by CNTs.232 Synchrotron-based X-ray absorption with combination of atomic microscopy indicated that the electronic configuration of both Fe and Ni was reconfigured, and the valence state of Ni was increased, and thus more OER active sites were created compared to single doping. Sun and coworkers dual-doped Ni and Fe into the framework of polymeric carbon nitride (Ni, Fe@PCN), in which superior activity was found.233 Computational studies suggest that the enhanced OER activity of NiFeOH@PCN is a consequence of the vastly reduced energy barrier (0.10 or 0.22 eV for U ¼ 1.58 V) through synergetic contribution of adjacent Ni and Fe atoms.In 2013, Zhao et al. reported N-doped carbon nanomaterials as metal-free OER catalysts other than graphene and CNTs for the first time.227 In alkaline electrolyte, the catalyst exhibits 380 mV overpotential at a current density of 10 mA cm−2, which is comparable to iridium and cobalt oxide catalysts. The pyridinic and quaternary nitrogen species shorten the diffusion path of OER intermediates. The N-doped carbon hollow cubes (NCHCs) for the OER were synthesized from biomass lysine using a NaCl-template method by Yang and coworkers.228 The NCHCs show comparable OER activity and high endurance compared with the RuO2/C catalyst in alkaline medium. Synergetic effects of N-doping and high specific surface area were found. Ozkan et al. demonstrated the OER activity of N-doped carbon nanostructures (CNx) in an acidic medium, which showed significantly higher OER performance than the Ir/C catalyst.255 XPS results suggested the presence of pyridinic N species for the enhanced OER activity of the CNx catalyst. Zhao's group demonstrated a novel strategy to fabricate a highly efficient OER catalyst by synthesizing an ultra-thin N-doped holey carbon layer (HCL) on graphene through a two-step process as displayed in Fig. 17a.231Fig. 17b presents the HR-TEM image of Zn@G-hydro annealed at 900 °C, which exhibits nanoholes with an approximate diameter of 2 nm, as a result of Zn evaporation (N-HC@G-900). The nano-holes are not observed when zinc gluconate is not used in the synthesis (N-C@G-900). N-HC@G-900 displayed enhanced OER activity, as observed in Fig. 17c, because of pyridinic N moieties at the edges of the HCL, and the sandwiched graphene sheet provided mechanical support.
 | ||
| Fig. 17 (a) Schematic representation of the N-HC@G@x synthesis procedure; (b) HRTEM image of N-HC@G-900 (nanoholes marked in red); (c) LSV curves of N–C@G-900, N-HC@G-900 and Ir/C in 1 M KOH.231 Reproduced from ref. 231 with permission, © 2018 John Wiley and Sons. | ||
The combination of N and transition metal doping into the carbon materials effectively improved the OER performance and stability by creating more active sites.224,225,256 For instance, Mu et al. co-doped Co and N into the matrix of graphene-like porous carbon nanosheets (Co@N-PGCS), which possess a high specific surface area of 1716 cm2 g−1.256 The Co@N-PGCS catalyst showed comparable activity over the state-of-the-art catalyst IrO2 by a synergetic effect of Co and N co-doping, and the porous surfaces open more diffusion channels. In another report, Tong and coworkers designed an efficient self-standing OER catalyst by dual doping Co and N into a graphite foam matrix (Co–N/GF).224 The Co–N/GF catalyst prepared at 700 °C could deliver significant OER activity, compared to the catalyst prepared at other temperatures in alkaline electrolyte. Initially, Co–N species were partly oxidized to CoOOH/Co–N. At higher anodic potentials, CoOOH was further oxidized to the most active CoO2, which considerably promoted the oxidation of OH− species. Wang's group, for the first time, fabricated a Co/N co-doped carbon composite for water oxidation from metal–polyphenol (Tanin) crystals, which are highly abundant in nature.225 Co/N co-doping in a carbon composite was found to produce a synergistic effect with a high specific surface area (180 m2 g−1), producing excellent OER activity in the alkaline electrolyte. Lee et al. demonstrated the synthesis of a tri-doped carbon catalyst for acidic OER.257 Here, they doped N, O, and TMs, such as Fe, Co, and Ni, onto the carbon, and they noticed that N/Fe/O and N/Ni/O doped carbon catalysts showed high OER activity, and N/Co/O displayed better stability.
7. Summary and concluding remarks
Searching for eco-friendly renewable energy that can replace carbon-based fossil fuels for future fuel is a prime goal of the research community. Electrochemical oxygen evolution plays a crucial role in energy storage by converting renewable energies, such as solar, wind, and tidal forms, into storable chemical fuel of hydrogen. Hence, the fundamental priority is to fabricate efficient OER catalysts that can be applied in the real world. This review summarized the research on the tuning of OER catalyst properties by doping. Doping of foreign elements can significantly alter the electrical conductivity and binding ability of OER intermediates by optimizing the host electronic structure. The considerable changes in the OER activity of metal oxides, non-metal oxides, and carbon-based catalysts via doping were reviewed. Experimental and theoretical studies were discussed to explain the OER mechanisms. Among the catalysts discussed above, doping in hydroxides, sulfides, selenides, and phosphides has been shown to be relatively more effective compared to the others discussed in this paper. Overall, this review outlines the systematic understanding of experimental and theoretical effects of doping on OER catalysts, which is associated with several challenges involving energy processes such as carbon dioxide reduction, nitrogen reduction, and production of hydrogen peroxide.Considering the studies reported in this paper, an efficient OER catalyst should have optimum adsorption ability for OER intermediates, neither too weak nor too strong, and possess a good electrical conductivity. The optimized eg filling value (∼1.2) can be obtained by tuning the electronic structure of TM ions present in metal oxides, and optimizing the covalency of metal and oxygen orbitals, including other metal ions along with TM ions. An optimized surface area can be achieved by preparing nanoforms. Furthermore, the catalyst should possess excellent resistance under extreme OER conditions with high anodic potentials and a good redox center that can lose or gain electrons.
In future, the development of advanced characterization techniques, such as in situ XPS, Fourier-transform infrared spectroscopy (FT-IR), X-ray absorption spectroscopy, XRD, etc., will provide more insights into OER intermediates on the surface of a catalyst. To design a highly stable catalyst for industrial-scale applications, it is essential to have the metric of long-term stability without dropping its initial activity evidently. Several catalysts display high activity, but a few of them can reach long-term endurance. Besides, large surface area and porosity are the other vital parameters in producing catalysts for large-scale use, which could provide high activity with a small catalyst. Most of the catalysts described in this paper were synthesized on a lab scale, while mass production of the same type of catalyst will be challenging in terms of cost effectiveness to fulfill the practical needs. Interdisciplinary collaborative research efforts are needed in science and engineering towards widespread applications of fuel cells.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation (DMR-1827731).References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- J. Yin, A. Molini and A. Porporato, Nat. Commun., 2020, 11, 4781 CrossRef CAS.
- J. Jurasz, F. A. Canales, A. Kies, M. Guezgouz and A. Beluco, Sol. Energy, 2020, 195, 703–724 CrossRef.
- Z. Yan, J. L. Hitt, J. A. Turner and T. E. Mallouk, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 12558–12563 CrossRef CAS.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- K. Scott, in Electrochemical Methods for Hydrogen Production, ed. K. Scott, Royal Society of Chemistry, London, UK, 2019, ch. 1, pp. 1–27 Search PubMed.
- T. Schuler, T. Kimura, T. J. Schmidt and F. N. Büchi, Energy Environ. Sci., 2020, 13, 2153–2166 RSC.
- K. S. Exner, I. Sohrabnejad-Eskan and H. Over, ACS Catal., 2018, 8, 1864–1879 CrossRef CAS.
- W. Yang, Z. Wang, W. Zhang and S. Guo, Trends Chem., 2019, 1, 259–271 CrossRef CAS.
- E. Fabbri and T. J. Schmidt, ACS Catal., 2018, 8, 9765–9774 CrossRef CAS.
- K. Kannimuthu, K. Sangeetha, S. S. Sankar, A. Karmakar, R. Madhu and S. Kundu, Inorg. Chem. Front., 2021, 8, 234–272 RSC.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS.
- Z. Chen, X. Duan, W. Wei, S. Wang and B.-J. Ni, Nano Energy, 2020, 78, 105392 CrossRef CAS.
- J. Huang, Y. Jiang, T. An and M. Cao, J. Mater. Chem. A, 2020, 8, 25465–25498 RSC.
- F. Lu, M. Zhou, Y. Zhou and X. Zeng, Small, 2017, 13, 1701931 CrossRef PubMed.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 1–13 CrossRef.
- F. Dionigi and P. Strasser, Adv. Energy Mater., 2016, 6, 1600621 CrossRef.
- W.-J. Yin, B. Weng, J. Ge, Q. Sun, Z. Li and Y. Yan, Energy Environ. Sci., 2019, 12, 442–462 RSC.
- A. Dutta and N. Pradhan, J. Phys. Chem. Lett., 2017, 8, 144–152 CrossRef CAS.
- N. Han, P. Liu, J. Jiang, L. Ai, Z. Shao and S. Liu, J. Mater. Chem. A, 2018, 6, 19912–19933 RSC.
- X. Peng, Y. Yan, X. Jin, C. Huang, W. Jin, B. Gao and P. K. Chu, Nano Energy, 2020, 78, 105234 CrossRef CAS.
- Q. Zhao, Z. Yan, C. Chen and J. Chen, Chem. Rev., 2017, 117, 10121–10211 CrossRef CAS.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- M. J. Craig, G. Coulter, E. Dolan, J. Soriano-López, E. Mates-Torres, W. Schmitt and M. García-Melchor, Nat. Commun., 2019, 10, 4993 CrossRef.
- Z.-P. Wu, X. F. Lu, S.-Q. Zang and X. W. (David) Lou, Adv. Funct. Mater., 2020, 30, 1910274 CrossRef CAS.
- J. O. Bockris, J. Chem. Phys., 1956, 24, 817–827 CrossRef CAS.
- Y. Matsumoto and E. Sato, Mater. Chem. Phys., 1986, 14, 397–426 CrossRef CAS.
- W. H. Wade and N. Hackerman, Trans. Faraday Soc., 1957, 53, 1636–1647 RSC.
- M. R. Tarasevich, A. Sadkowski and E. Yeager, in Comprehensive Treatise of Electrochemistry: Kinetics and Mechanisms of Electrode Processes, ed. B. E. Conway, J. O. Bockris, E. Yeager, S. U. M. Khan and R. E. White, Springer US, Boston, MA, 1983, vol. 7, pp. 301–398 Search PubMed.
- J. O. Bockris and T. Otagawa, J. Electrochem. Soc., 1984, 131, 290 CrossRef CAS.
- M. Kim, J. Park, M. Kang, J. Y. Kim and S. W. Lee, ACS Cent. Sci., 2020, 6, 880–891 CrossRef CAS.
- S. T. Dix, S. Lu and S. Linic, ACS Catal., 2020, 10, 10735–10741 CrossRef CAS.
- Y. Liu, Y. Ying, L. Fei, Y. Liu, Q. Hu, G. Zhang, S. Y. Pang, W. Lu, C. L. Mak, X. Luo, L. Zhou, M. Wei and H. Huang, J. Am. Chem. Soc., 2019, 141, 8136–8145 CrossRef CAS.
- A. Zunger and O. I. Malyi, Chem. Rev., 2021, 121, 3031–3060 CrossRef CAS PubMed.
- A. Goryachev, M. Etzi Coller Pascuzzi, F. Carlà, T. Weber, H. Over, E. J. M. Hensen and J. P. Hofmann, Electrochim. Acta, 2020, 336, 135713 CrossRef CAS.
- O. Kasian, J.-P. Grote, S. Geiger, S. Cherevko and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2018, 57, 2488–2491 CrossRef CAS PubMed.
- T. D. Nguyen, H. H. Nguyen, C. Dai, J. Wang and G. G. Scherer, Int. J. Hydrogen Energy, 2020, 45, 46–55 CrossRef CAS.
- J. Su, R. Ge, K. Jiang, Y. Dong, F. Hao, Z. Tian, G. Chen and L. Chen, Adv. Mater., 2018, 30, 1801351 CrossRef PubMed.
- W. Sun, Y. Song, X.-Q. Gong, L. Cao and J. Yang, Chem. Sci., 2015, 6, 4993–4999 RSC.
- J. Feng, F. Lv, W. Zhang, P. Li, K. Wang, C. Yang, B. Wang, Y. Yang, J. Zhou, F. Lin, G.-C. Wang and S. Guo, Adv. Mater., 2017, 29, 1703798 CrossRef.
- Y. Tian, S. Wang, E. Velasco, Y. Yang, L. Cao, L. Zhang, X. Li, Y. Lin, Q. Zhang and L. Chen, iScience, 2020, 23, 100756 CrossRef CAS PubMed.
- S. Chen, H. Huang, P. Jiang, K. Yang, J. Diao, S. Gong, S. Liu, M. Huang, H. Wang and Q. Chen, ACS Catal., 2020, 10, 1152–1160 CrossRef CAS.
- W. Gou, M. Zhang, Y. Zou, X. Zhou and Y. Qu, ChemCatChem, 2019, 11, 6008–6014 CrossRef CAS.
- Y. Lin, Z. Tian, L. Zhang, J. Ma, Z. Jiang, B. J. Deibert, R. Ge and L. Chen, Nat. Commun., 2019, 10, 162 CrossRef.
- Y. Wang, S. Hao, X. Liu, Q. Wang, Z. Su, L. Lei and X. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 37006–37012 CrossRef CAS.
- F. Lv, J. Feng, K. Wang, Z. Dou, W. Zhang, J. Zhou, C. Yang, M. Luo, Y. Yang, Y. Li, P. Gao and S. Guo, ACS Cent. Sci., 2018, 4, 1244–1252 CrossRef CAS.
- Z. Li, S. Wang, Y. Tian, B. Li, H. jun Yan, S. Zhang, Z. Liu, Q. Zhang, Y. Lin and L. Chen, Chem. Commun., 2020, 56, 1749–1752 RSC.
- R. Ge, L. Li, J. Su, Y. Lin, Z. Tian and L. Chen, Adv. Energy Mater., 2019, 9, 1901313 CrossRef.
- H. Sun, G. Chen, J. Sunarso, J. Dai, W. Zhou and Z. Shao, ACS Appl. Mater. Interfaces, 2018, 10, 16939–16942 CrossRef CAS PubMed.
- G. Chen, W. Zhou, D. Guan, J. Sunarso, Y. Zhu, X. Hu, W. Zhang and Z. Shao, Sci. Adv., 2017, 3, e1603206 CrossRef PubMed.
- Y.-Q. Lyu and F. Ciucci, ACS Appl. Mater. Interfaces, 2017, 9, 35829–35836 CrossRef CAS PubMed.
- L. Yang, H. Chen, L. Shi, X. Li, X. Chu, W. Chen, N. Li and X. Zou, ACS Appl. Mater. Interfaces, 2019, 11, 42006–42013 CrossRef CAS PubMed.
- A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 1–7 Search PubMed.
- B. Zhao, L. Zhang, D. Zhen, S. Yoo, Y. Ding, D. Chen, Y. Chen, Q. Zhang, B. Doyle, X. Xiong and M. Liu, Nat. Commun., 2017, 8, 14586 CrossRef CAS.
- Z. Li, L. Lv, J. Wang, X. Ao, Y. Ruan, D. Zha, G. Hong, Q. Wu, Y. Lan, C. Wang, J. Jiang and M. Liu, Nano Energy, 2018, 47, 199–209 CrossRef CAS.
- G. Chen, Z. Hu, Y. Zhu, Z.-G. Chen, Y. Zhong, H.-J. Lin, C.-T. Chen, L. H. Tjeng, W. Zhou and Z. Shao, J. Mater. Chem. A, 2018, 6, 9854–9859 RSC.
- S. R. Ede, C. N. Collins, C. D. Posada, G. George, H. Wu, W. D. Ratcliff, Y. Lin, J. Wen, S. Han and Z. Luo, ACS Catal., 2021, 4327–4337 CrossRef CAS.
- Y. Zhu, W. Zhou, J. Sunarso, Y. Zhong and Z. Shao, Adv. Funct. Mater., 2016, 26, 5862–5872 CrossRef CAS.
- Z. Wu, L.-P. Sun, T. Xia, L.-H. Huo, H. Zhao, A. Rougier and J.-C. Grenier, J. Power Sources, 2016, 334, 86–93 CrossRef CAS.
- C. Su, W. Wang, Y. Chen, G. Yang, X. Xu, M. O. Tadé and Z. Shao, ACS Appl. Mater. Interfaces, 2015, 7, 17663–17670 CrossRef CAS.
- Y. Guo, Y. Tong, P. Chen, K. Xu, J. Zhao, Y. Lin, W. Chu, Z. Peng, C. Wu and Y. Xie, Adv. Mater., 2015, 27, 5989–5994 CrossRef CAS.
- H. Jin, Y. Hong, J. Yoon, A. Oh, N. K. Chaudhari, H. Baik, S. H. Joo and K. Lee, Nano Energy, 2017, 42, 17–25 CrossRef CAS.
- J. Xu, C. Chen, Z. Han, Y. Yang, J. Li and Q. Deng, Nanomaterials, 2019, 9, 1161 CrossRef CAS PubMed.
- G. George, S. R. Ede and Z. Luo, Fundamentals of Perovskite Oxides: Synthesis, Structure, Properties and Applications, CRC Press, Boca Raton, Florida, 2020 Search PubMed.
- J. Suntivich, W. T. Hong, Y.-L. Lee, J. M. Rondinelli, W. Yang, J. B. Goodenough, B. Dabrowski, J. W. Freeland and Y. Shao-Horn, J. Phys. Chem. C, 2014, 118, 1856–1863 CrossRef CAS.
- G. George, S. L. Jackson, C. Q. Luo, D. Fang, D. Luo, D. Hu, J. Wen and Z. Luo, Ceram. Int., 2018, 44, 21982–21992 CrossRef CAS.
- M. Retuerto, L. Pascual, F. Calle-Vallejo, P. Ferrer, D. Gianolio, A. G. Pereira, Á. García, J. Torrero, M. T. Fernández-Díaz, P. Bencok, M. A. Peña, J. L. G. Fierro and S. Rojas, Nat. Commun., 2019, 10, 2041 CrossRef PubMed.
- Y. Zhao, L. Xu, L. Mai, C. Han, Q. An, X. Xu, X. Liu and Q. Zhang, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 19569–19574 CrossRef CAS.
- B. Hua, Y.-F. Sun, M. Li, N. Yan, J. Chen, Y.-Q. Zhang, Y. Zeng, B. Shalchi Amirkhiz and J.-L. Luo, Chem. Mater., 2017, 29, 6228–6237 CrossRef CAS.
- X. Cheng, E. Fabbri, M. Nachtegaal, I. E. Castelli, M. El Kazzi, R. Haumont, N. Marzari and T. J. Schmidt, Chem. Mater., 2015, 27, 7662–7672 CrossRef CAS.
- Y. Zhou, S. Sun, C. Wei, Y. Sun, P. Xi, Z. Feng and Z. J. Xu, Adv. Mater., 2019, 31, 1902509 CrossRef CAS PubMed.
- J. Li, D. Chu, H. Dong, D. R. Baker and R. Jiang, J. Am. Chem. Soc., 2020, 142, 50–54 CrossRef CAS PubMed.
- K.-L. Yan, J.-Q. Chi, J.-Y. Xie, B. Dong, Z.-Z. Liu, W.-K. Gao, J.-H. Lin, Y.-M. Chai and C.-G. Liu, Renewable Energy, 2018, 119, 54–61 CrossRef CAS.
- C.-C. Lin and C. C. L. McCrory, ACS Catal., 2017, 7, 443–451 CrossRef CAS.
- Y. Tian, L. Cao and P. Qin, ChemCatChem, 2019, 11, 4420–4426 CrossRef CAS.
- S. L. Zhang, B. Y. Guan, X. F. Lu, S. Xi, Y. Du and X. W. (David) Lou, Adv. Mater., 2020, 32, 2002235 CrossRef CAS PubMed.
- J. Swaminathan, A. B. Puthirath, M. R. Sahoo, S. K. Nayak, G. Costin, R. Vajtai, T. Sharifi and P. M. Ajayan, ACS Appl. Mater. Interfaces, 2019, 11, 39706–39714 CrossRef CAS.
- C. W. Cady, G. Gardner, Z. O. Maron, M. Retuerto, Y. B. Go, S. Segan, M. Greenblatt and G. C. Dismukes, ACS Catal., 2015, 5, 3403–3410 CrossRef CAS.
- K. Lankauf, K. Cysewska, J. Karczewski, A. Mielewczyk-Gryń, K. Górnicka, G. Cempura, M. Chen, P. Jasiński and S. Molin, Int. J. Hydrogen Energy, 2020, 45, 14867–14879 CrossRef CAS.
- J. S. Hong, H. Seo, Y. H. Lee, K. H. Cho, C. Ko, S. Park and K. T. Nam, Small Methods, 2020, 4, 1900733 CrossRef CAS.
- X. Liu, W. Xi, C. Li, X. Li, J. Shi, Y. Shen, J. He, L. Zhang, L. Xie, X. Sun, P. Wang, J. Luo, L.-M. Liu and Y. Ding, Nano Energy, 2018, 44, 371–377 CrossRef CAS.
- Z. Wang, H. Liu, R. Ge, X. Ren, J. Ren, D. Yang, L. Zhang and X. Sun, ACS Catal., 2018, 8, 2236–2241 CrossRef CAS.
- Z. Xiao, Y. Wang, Y.-C. Huang, Z. Wei, C.-L. Dong, J. Ma, S. Shen, Y. Li and S. Wang, Energy Environ. Sci., 2017, 10, 2563–2569 RSC.
- R. Li, Y. Guo, H. Chen, K. Wang, R. Tan, B. Long, Y. Tong, P. Tsiakaras, S. Song and Y. Wang, ACS Sustainable Chem. Eng., 2019, 7, 11901–11910 CrossRef CAS.
- R. Wei, X. Bu, W. Gao, R. A. B. Villaos, G. Macam, Z.-Q. Huang, C. Lan, F.-C. Chuang, Y. Qu and J. C. Ho, ACS Appl. Mater. Interfaces, 2019, 11, 33012–33021 CrossRef CAS.
- H. Xu, B. Wang, C. Shan, P. Xi, W. Liu and Y. Tang, ACS Appl. Mater. Interfaces, 2018, 10, 6336–6345 CrossRef CAS.
- X. Zhang, L. An, J. Yin, P. Xi, Z. Zheng and Y. Du, Sci. Rep., 2017, 7, 43590 CrossRef.
- L. Chen, H. Zhang, L. Chen, X. Wei, J. Shi and M. He, J. Mater. Chem. A, 2017, 5, 22568–22575 RSC.
- B. Cao, C. Luo, J. Lao, H. Chen, R. Qi, H. Lin and H. Peng, ACS Omega, 2019, 4, 16612–16618 CrossRef CAS PubMed.
- Y. Dou, C.-T. He, L. Zhang, M. Al-Mamun, H. Guo, W. Zhang, Q. Xia, J. Xu, L. Jiang, Y. Wang, P. Liu, X.-M. Chen, H. Yin and H. Zhao, Cell Rep. Phys. Sci., 2020, 1, 100077 CrossRef.
- J. Lv, X. Yang, K. Li, X. Chen, S. Sun, H.-Y. Zang, Y.-F. Chang, Y.-H. Wang and Y.-G. Li, Nanoscale Adv., 2019, 1, 4099–4108 RSC.
- Z. Chen, C. X. Kronawitter, Y.-W. Yeh, X. Yang, P. Zhao, N. Yao and B. E. Koel, J. Mater. Chem. A, 2017, 5, 842–850 RSC.
- V. P. Tolstoy, L. I. Kuklo and L. B. Gulina, J. Alloys Compd., 2019, 786, 198–204 CrossRef CAS.
- Q. Zhou, Y. Chen, G. Zhao, Y. Lin, Z. Yu, X. Xu, X. Wang, H. K. Liu, W. Sun and S. X. Dou, ACS Catal., 2018, 8, 5382–5390 CrossRef CAS.
- S. Niu, W.-J. Jiang, Z. Wei, T. Tang, J. Ma, J.-S. Hu and L.-J. Wan, J. Am. Chem. Soc., 2019, 141, 7005–7013 CrossRef CAS.
- J. Yan, L. Kong, Y. Ji, J. White, Y. Li, J. Zhang, P. An, S. Liu, S.-T. Lee and T. Ma, Nat. Commun., 2019, 10, 2149 CrossRef PubMed.
- J. W. D. Ng, M. García-Melchor, M. Bajdich, P. Chakthranont, C. Kirk, A. Vojvodic and T. F. Jaramillo, Nat. Energy, 2016, 1, 1–8 Search PubMed.
- X. Xiong, C. You, Z. Liu, A. M. Asiri and X. Sun, ACS Sustainable Chem. Eng., 2018, 6, 2883–2887 CrossRef CAS.
- Y. Zhao, J. Zhang, W. Wu, X. Guo, P. Xiong, H. Liu and G. Wang, Nano Energy, 2018, 54, 129–137 CrossRef CAS.
- C. Meng, Y.-F. Gao, X.-M. Chen, Y.-X. Li, M.-C. Lin and Y. Zhou, ACS Sustainable Chem. Eng., 2019, 7, 18055–18060 CrossRef CAS.
- W. Li, M. Li, C. Wang, Y. Wei and X. Lu, Appl. Surf. Sci., 2020, 506, 144680 CrossRef CAS.
- R. Mondal, H. Ratnawat, S. Kumar, A. Kumar and P. Singh, RSC Adv., 2020, 10, 17845–17853 RSC.
- Y. Yang, X. Su, L. Zhang, P. Kerns, L. Achola, V. Hayes, R. Quardokus, S. L. Suib and J. He, ChemCatChem, 2019, 11, 1689–1700 CrossRef CAS.
- Z. Ye, T. Li, G. Ma, Y. Dong and X. Zhou, Adv. Funct. Mater., 2017, 27, 1704083 CrossRef.
- Q. Feng, J. Zou, Y. Wang, Z. Zhao, M. C. Williams, H. Li and H. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 4520–4530 CrossRef CAS.
- M. Huo, Z. Yang, C. Yang, Z. Gao, J. Qi, Z. Liang, K. Liu, H. Chen, H. Zheng and R. Cao, ChemCatChem, 2019, 11, 1480–1486 CrossRef CAS.
- F. Cheng, J. Shen, B. Peng, Y. Pan, Z. Tao and J. Chen, Nat. Chem., 2011, 3, 79–84 CrossRef CAS PubMed.
- T. Maiyalagan, K. R. Chemelewski and A. Manthiram, ACS Catal., 2014, 4, 421–425 CrossRef CAS.
- M. H. Mendonça, M. I. Godinho, M. A. Catarino, M. I. da Silva Pereira and F. M. Costa, Solid State Sci., 2002, 4, 175–182 CrossRef.
- A. S. Anindita and R. N. Singh, Int. J. Hydrogen Energy, 2010, 35, 3243–3248 CrossRef CAS.
- R. N. Singh, J. P. Singh, B. Lal, M. J. K. Thomas and S. Bera, Electrochim. Acta, 2006, 51, 5515–5523 CrossRef CAS.
- M. Cui, X. Ding, X. Huang, Z. Shen, T.-L. Lee, F. E. Oropeza, J. P. Hofmann, E. J. M. Hensen and K. H. L. Zhang, Chem. Mater., 2019, 31, 7618–7625 CrossRef CAS.
- I. Nikolov, R. Darkaoui, E. Zhecheva, R. Stoyanova, N. Dimitrov and T. Vitanov, J. Electroanal. Chem., 1997, 429, 157–168 CrossRef CAS.
- D. Chanda, J. Hnát, M. Paidar and K. Bouzek, Int. J. Hydrogen Energy, 2014, 39, 5713–5722 CrossRef CAS.
- B. Chi, H. Lin and J. Li, Int. J. Hydrogen Energy, 2008, 33, 4763–4768 CrossRef CAS.
- T. Sun, P. Liu, Y. Zhang, Z. Chen, C. Zhang, X. Guo, C. Ma, Y. Gao and S. Zhang, Chem. Eng. J., 2020, 390, 124591 CrossRef CAS.
- Z. Cai, X. Bu, P. Wang, J. C. Ho, J. Yang and X. Wang, J. Mater. Chem. A, 2019, 7, 5069–5089 RSC.
- A. Karmakar, K. Karthick, S. S. Sankar, S. Kumaravel, R. Madhu and S. Kundu, J. Mater. Chem. A, 2021, 9, 1314–1352 RSC.
- S. Anantharaj, S. Kundu and S. Noda, Nano Energy, 2021, 80, 105514 CrossRef CAS.
- M. Gong and H. Dai, Nano Res., 2015, 8, 23–39 CrossRef CAS.
- Y.-F. Li and A. Selloni, ACS Catal., 2014, 4, 1148–1153 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- J. M. P. Martirez and E. A. Carter, J. Am. Chem. Soc., 2019, 141, 693–705 CrossRef CAS.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS.
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549–7558 CrossRef CAS.
- T. A. Edison, US Pat., US876445A, 1908 Search PubMed.
- R. L. Tichenor, Ind. Eng. Chem., 1952, 44, 973–977 CrossRef CAS.
- G. Młynarek, M. Paszkiewicz and A. Radniecka, J. Appl. Electrochem., 1984, 14, 145–149 CrossRef.
- D. A. Corrigan, J. Electrochem. Soc., 1987, 134, 377 CrossRef CAS.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- J. Landon, E. Demeter, N. İnoğlu, C. Keturakis, I. E. Wachs, R. Vasić, A. I. Frenkel and J. R. Kitchin, ACS Catal., 2012, 2, 1793–1801 CrossRef CAS.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- G. Fu, X. Wen, S. Xi, Z. Chen, W. Li, J.-Y. Zhang, A. Tadich, R. Wu, D.-C. Qi, Y. Du, J. Cheng and K. H. L. Zhang, Chem. Mater., 2019, 31, 419–428 CrossRef CAS.
- D. A. Corrigan and R. M. Bendert, J. Electrochem. Soc., 1989, 136, 723 CrossRef CAS.
- N. Li, D. K. Bediako, R. G. Hadt, D. Hayes, T. J. Kempa, F. von Cube, D. C. Bell, L. X. Chen and D. G. Nocera, Proc. Natl. Acad. Sci. U.S.A., 2017, 114, 1486–1491 CrossRef CAS PubMed.
- C. A. Kent, J. J. Concepcion, C. J. Dares, D. A. Torelli, A. J. Rieth, A. S. Miller, P. G. Hoertz and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 8432–8435 CrossRef CAS PubMed.
- Y. Sun, T. Zhang, C. Li, K. Xu and Y. Li, J. Mater. Chem. A, 2020, 8, 13415–13436 RSC.
- G. Zhang, Y.-S. Feng, W.-T. Lu, D. He, C.-Y. Wang, Y.-K. Li, X.-Y. Wang and F.-F. Cao, ACS Catal., 2018, 8, 5431–5441 CrossRef CAS.
- L. Wang, Y. Li, Q. Sun, Q. Qiang, Y. Shen, Y. Ma, Z. Wang and C. Zhao, ChemCatChem, 2019, 11, 2011–2016 CrossRef CAS.
- Y. Zhou, M. Luo, Z. Zhang, W. Li, X. Shen, W. Xia, M. Zhou and X. Zeng, Appl. Surf. Sci., 2018, 448, 9–15 CrossRef CAS.
- W.-K. Gao, J.-F. Qin, K. Wang, K.-L. Yan, Z.-Z. Liu, J.-H. Lin, Y.-M. Chai, C.-G. Liu and B. Dong, Appl. Surf. Sci., 2018, 454, 46–53 CrossRef CAS.
- N. Yang, C. Tang, K. Wang, G. Du, A. M. Asiri and X. Sun, Nano Res., 2016, 9, 3346–3354 CrossRef CAS.
- X. Wang, W. Zhang, J. Zhang and Z. Wu, ChemElectroChem, 2019, 6, 4550–4559 CrossRef CAS.
- Y. Guo, X. Zhou, J. Tang, S. Tanaka, Y. V. Kaneti, J. Na, B. Jiang, Y. Yamauchi, Y. Bando and Y. Sugahara, Nano Energy, 2020, 75, 104913 CrossRef CAS.
- W. He, F. Wang, D. Jia, Y. Li, L. Liang, J. Zhang, Q. Hao, C. Liu, H. Liu and J. Zhao, Nanoscale, 2020, 12, 24244–24250 RSC.
- W. Gao, F. Ma, C. Wang and D. Wen, J. Power Sources, 2020, 450, 227654 CrossRef CAS.
- R. Xu, Z. Xu, X. Zhang, Y. Ling, M. Li and Z. Yang, ChemElectroChem, 2020, 7, 148–154 CrossRef CAS.
- L. Huang, H. Wu, H. Liu and Y. Zhang, Electrochim. Acta, 2019, 318, 892–900 CrossRef CAS.
- Y. Xu, A. Sumboja, A. Groves, T. Ashton, Y. Zong and J. A. Darr, RSC Adv., 2020, 10, 41871–41882 RSC.
- S. Dou, L. Tao, J. Huo, S. Wang and L. Dai, Energy Environ. Sci., 2016, 9, 1320–1326 RSC.
- Z. Jing, Q. Zhao, D. Zheng, L. Sun, J. Geng, Q. Zhou and J. Lin, J. Mater. Chem. A, 2020, 8, 20323–20330 RSC.
- J. Hao, W. Yang, J. Hou, B. Mao, Z. Huang and W. Shi, J. Mater. Chem. A, 2017, 5, 17811–17816 RSC.
- Q. Hao, S. Li, H. Liu, J. Mao, Y. Li, C. Liu, J. Zhang and C. Tang, Catal. Sci. Technol., 2019, 9, 3099–3108 RSC.
- H. Liu, Z. Liu, F. Wang and L. Feng, Chem. Eng. J., 2020, 397, 125507 CrossRef CAS.
- F. Zhang, Y. Ge, H. Chu, P. Dong, R. Baines, Y. Pei, M. Ye and J. Shen, ACS Appl. Mater. Interfaces, 2018, 10, 7087–7095 CrossRef CAS.
- W. He, D. Jia, J. Cheng, F. Wang, L. Zhang, Y. Li, C. Liu, Q. Hao and J. Zhao, Catal. Sci. Technol., 2020, 10, 7581–7590 RSC.
- Y. Ding, H. Li and Y. Hou, Int. J. Hydrogen Energy, 2018, 43, 19002–19009 CrossRef CAS.
- C. Liu, D. Jia, Q. Hao, X. Zheng, Y. Li, C. Tang, H. Liu, J. Zhang and X. Zheng, ACS Appl. Mater. Interfaces, 2019, 11, 27667–27676 CrossRef CAS PubMed.
- Q. Liu, L. Xie, Z. Liu, G. Du, A. M. Asiri and X. Sun, Chem. Commun., 2017, 53, 12446–12449 RSC.
- A. Zare, A. Bayat, E. Saievar-Iranizad and H. Naffakh-Moosavy, J. Electroanal. Chem., 2020, 878, 114595 CrossRef CAS.
- X. Zhao, H. Zhang, Y. Yan, J. Cao, X. Li, S. Zhou, Z. Peng and J. Zeng, Angew. Chem., 2017, 129, 334–338 CrossRef.
- Y. Zuo, D. Rao, S. Ma, T. Li, Y. H. Tsang, S. Kment and Y. Chai, ACS Nano, 2019, 13, 11469–11476 CrossRef CAS.
- D. Liang, J. Mao, P. Liu, J. Li, J. Yan and W. Song, Int. J. Hydrogen Energy, 2020, 45, 27047–27055 CrossRef CAS.
- F. Ming, H. Liang, H. Shi, X. Xu, G. Mei and Z. Wang, J. Mater. Chem. A, 2016, 4, 15148–15155 RSC.
- H. Kang, H. Li, X. Zhao, L. Yang and S. Xu, Ceram. Int., 2020, 46, 2792–2797 CrossRef CAS.
- J. Dai, D. Zhao, W. Sun, X. Zhu, L.-J. Ma, Z. Wu, C. Yang, Z. Cui, L. Li and S. Chen, ACS Catal., 2019, 9, 10761–10772 CrossRef CAS.
- X. Wei, Y. Xie, M. Liu, J. Zhou, A. Zhou, P. He, Y. Dou and J.-R. Li, APL Mater., 2019, 7, 101106 CrossRef.
- J. Li, G. Liu, B. Liu, Z. Min, D. Qian, J. Jiang and J. Li, Electrochim. Acta, 2018, 265, 577–585 CrossRef CAS.
- Z. Zou, X. Wang, J. Huang, Z. Wu and F. Gao, J. Mater. Chem. A, 2019, 7, 2233–2241 RSC.
- L. Lin, M. Chen and L. Wu, ACS Appl. Energy Mater., 2019, 2, 4737–4744 CrossRef CAS.
- J. Du, Z. Zou, C. Liu and C. Xu, Nanoscale, 2018, 10, 5163–5170 RSC.
- M. Ghaemmaghami, Y. Yamini, E. Saievar-Iranizad and A. Bayat, Sustainable Energy Fuels, 2020, 4, 1150–1156 RSC.
- Y. Du, G. Cheng and W. Luo, Nanoscale, 2017, 9, 6821–6825 RSC.
- Y. Tuo, X. Wang, C. Chen, X. Feng, Z. Liu, Y. Zhou and J. Zhang, Electrochim. Acta, 2020, 335, 135682 CrossRef CAS.
- B. Mohanty, B. K. Jena, M. Kandasamy, N. Dalai, R. K. Sahu, R. M. Kadam, B. Chakraborty and B. Jena, Sustainable Energy Fuels, 2020, 4, 3058–3065 RSC.
- K. Zhang, G. Zhang, J. Qu and H. Liu, Small, 2020, 16, 1907001 CrossRef CAS PubMed.
- S. Shit, S. Bolar, N. C. Murmu and T. Kuila, ACS Sustainable Chem. Eng., 2019, 7, 18015–18026 CrossRef CAS.
- W. Gao, F. Ma, C. Wang and D. Wen, J. Power Sources, 2020, 450, 227654 CrossRef CAS.
- L. Lei, D. Huang, C. Zhang, R. Deng, S. Chen and Z. Li, J. Catal., 2020, 385, 129–139 CrossRef CAS.
- B. Garlyyev, J. Fichtner, O. Piqué, O. Schneider, A. S. Bandarenka and F. Calle-Vallejo, Chem. Sci., 2019, 10, 8060–8075 RSC.
- Y. Dou, C.-T. He, L. Zhang, H. Yin, M. Al-Mamun, J. Ma and H. Zhao, Nat. Commun., 2020, 11, 1664 CrossRef CAS.
- J. Kupka and A. Budniok, J. Appl. Electrochem., 1990, 20, 1015–1020 CrossRef CAS.
- C. Lin, P. Wang, H. Jin, J. Zhao, D. Chen, S. Liu, C. Zhang and S. Mu, Dalton Trans., 2019, 48, 16555–16561 RSC.
- P. Wang, Z. Pu, Y. Li, L. Wu, Z. Tu, M. Jiang, Z. Kou, I. S. Amiinu and S. Mu, ACS Appl. Mater. Interfaces, 2017, 9, 26001–26007 CrossRef CAS.
- Y. Li, F. Li, Y. Zhao, S.-N. Li, J.-H. Zeng, H.-C. Yao and Y. Chen, J. Mater. Chem. A, 2019, 7, 20658–20666 RSC.
- F. Li, Y. Bu, Z. Lv, J. Mahmood, G.-F. Han, I. Ahmad, G. Kim, Q. Zhong and J.-B. Baek, Small, 2017, 13, 1701167 CrossRef PubMed.
- H. Sun, Y. Min, W. Yang, Y. Lian, L. Lin, K. Feng, Z. Deng, M. Chen, J. Zhong, L. Xu and Y. Peng, ACS Catal., 2019, 9, 8882–8892 CrossRef CAS.
- N. Suo, X. Han, C. Chen, X. He, Z. Dou, Z. Lin, L. Cui and J. Xiang, Electrochim. Acta, 2020, 333, 135531 CrossRef CAS.
- C. Ray, S. C. Lee, B. Jin, A. Kundu, J. H. Park and S. C. Jun, ACS Sustainable Chem. Eng., 2018, 6, 6146–6156 CrossRef CAS.
- Q. Zhang, D. Yan, Z. Nie, X. Qiu, S. Wang, J. Yuan, D. Su, G. Wang and Z. Wu, ACS Appl. Energy Mater., 2018, 1, 571–579 CrossRef CAS.
- H.-W. Man, C.-S. Tsang, M. M.-J. Li, J. Mo, B. Huang, L. Y. S. Lee, Y. Leung, K.-Y. Wong and S. C. E. Tsang, Chem. Commun., 2018, 54, 8630–8633 RSC.
- T. E. Rosser, J. P. S. Sousa, Y. Ziouani, O. Bondarchuk, D. Y. Petrovykh, X.-K. Wei, J. J. L. Humphrey, M. Heggen, Y. V. Kolen’ko and A. J. Wain, Catal. Sci. Technol., 2020, 10, 2398–2406 RSC.
- M. Wang, W. Fu, L. Du, Y. Wei, P. Rao, L. Wei, X. Zhao, Y. Wang and S. Sun, Appl. Surf. Sci., 2020, 515, 146059 CrossRef CAS.
- P. Xu, L. Qiu, L. Wei, Y. Liu, D. Yuan, Y. Wang and P. Tsiakaras, Catal. Today, 2020, 355, 815–821 CrossRef CAS.
- Z. Shao, J. Sun, N. Guo, F. He, K. Huang, F. Tian and Q. Wang, J. Power Sources, 2019, 422, 33–41 CrossRef CAS.
- W. Xi, G. Yan, Z. Lang, Y. Ma, H. Tan, H. Zhu, Y. Wang and Y. Li, Small, 2018, 14, 1802204 CrossRef.
- H. Fan, W. Chen, G. Chen, J. Huang, C. Song, Y. Du, C. Li and K. (Ken) Ostrikov, Appl. Catal., B, 2020, 268, 118440 CrossRef CAS.
- T. L. Luyen Doan, D. T. Tran, D. C. Nguyen, H. Tuan Le, N. H. Kim and J. H. Lee, Appl. Catal., B, 2020, 261, 118268 CrossRef.
- M. A. R. Anjum, M. D. Bhatt, M. H. Lee and J. S. Lee, Chem. Mater., 2018, 30, 8861–8870 CrossRef CAS.
- Z. Yin, Y. Sun, Y. Jiang, F. Yan, C. Zhu and Y. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 27751–27759 CrossRef CAS.
- T. Liu, M. Li, X. Bo and M. Zhou, ACS Sustainable Chem. Eng., 2018, 6, 11457–11465 CrossRef CAS.
- Y. Sun, T. Zhang, X. Li, D. Liu, G. Liu, X. Zhang, X. Lyu, W. Cai and Y. Li, Chem. Commun., 2017, 53, 13237–13240 RSC.
- X. Bai, Q. Wang, G. Xu, Y. Ning, K. Huang, F. He, Z. Wu and J. Zhang, Chem.–Eur. J., 2017, 23, 16862–16870 CrossRef CAS PubMed.
- F.-C. Shen, S.-N. Sun, Z.-F. Xin, S.-L. Li, L.-Z. Dong, Q. Huang, Y.-R. Wang, J. Liu and Y.-Q. Lan, Appl. Catal., B, 2019, 243, 470–480 CrossRef CAS.
- M. A. R. Anjum, M. H. Lee and J. S. Lee, ACS Catal., 2018, 8, 8296–8305 CrossRef CAS.
- X. Ma, K. Li, X. Zhang, B. Wei, H. Yang, L. Liu, M. Zhang, X. Zhang and Y. Chen, J. Mater. Chem. A, 2019, 7, 14904–14915 RSC.
- X. Zhu, X. Zhang, L. Huang, Y. Liu, H. Zhang and S. Dong, Chem. Commun., 2019, 55, 9995–9998 RSC.
- X. Jia, M. Wang, G. Liu, Y. Wang, J. Yang and J. Li, Int. J. Hydrogen Energy, 2019, 44, 24572–24579 CrossRef CAS.
- S. Xu, M. Wang, G. Saranya, N. Chen, L. Zhang, Y. He, L. Wu, Y. Gong, Z. Yao, G. Wang, Z. Wang, S. Zhao, H. Tang, M. Chen and H. Gou, Appl. Catal., B, 2020, 268, 118385 CrossRef CAS.
- H. Fan, H. Yu, Y. Zhang, Y. Zheng, Y. Luo, Z. Dai, B. Li, Y. Zong and Q. Yan, Angew. Chem., Int. Ed., 2017, 56, 12566–12570 CrossRef CAS PubMed.
- S. Zhang, G. Gao, J. Hao, M. Wang, H. Zhu, S. Lu, F. Duan, W. Dong, M. Du and Y. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 43261–43269 CrossRef CAS.
- Q. Liang, G. Brocks, X. Zhang and A. Bieberle-Hütter, J. Phys. Chem. C, 2019, 123, 26289–26298 CrossRef CAS.
- H. Yang, Y. Hu, D. Huang, T. Xiong, M. Li, M.-S. Balogun and Y. Tong, Mater. Today Chem., 2019, 11, 1–7 CrossRef CAS.
- Y. Wang, Q. Wu, B. Zhang, L. Tian, K. Li and X. Zhang, Catalysts, 2020, 10, 1164 CrossRef CAS.
- N. Han, K. R. Yang, Z. Lu, Y. Li, W. Xu, T. Gao, Z. Cai, Y. Zhang, V. S. Batista, W. Liu and X. Sun, Nat. Commun., 2018, 9, 924 CrossRef.
- Y. Xu, M. Kraft and R. Xu, Chem. Soc. Rev., 2016, 45, 3039–3052 RSC.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- J. Wang, H. Kong, J. Zhang, Y. Hao, Z. Shao and F. Ciucci, Prog. Mater. Sci., 2021, 116, 100717 CrossRef CAS.
- X. Liu and L. Dai, Nat. Rev. Mater., 2016, 1, 1–12 CrossRef.
- F. Davodi, M. Tavakkoli, J. Lahtinen and T. Kallio, J. Catal., 2017, 353, 19–27 CrossRef CAS.
- T. Sun, J. Wang, C. Qiu, X. Ling, B. Tian, W. Chen and C. Su, Adv. Sci., 2018, 5, 1800036 CrossRef.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336–3339 CrossRef CAS PubMed.
- Y. Tong, X. Yu, H. Wang, B. Yao, C. Li and G. Shi, ACS Catal., 2018, 8, 4637–4644 CrossRef CAS.
- J. Wei, Y. Liang, Y. Hu, B. Kong, J. Zhang, Q. Gu, Y. Tong, X. Wang, S. P. Jiang and H. Wang, Angew. Chem., Int. Ed., 2016, 55, 12470–12474 CrossRef CAS.
- J. Zhang and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 13296–13300 CrossRef CAS.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 CrossRef PubMed.
- X. Zheng, X. Cao, X. Li, J. Tian, C. Jin and R. Yang, Nanoscale, 2017, 9, 1059–1067 RSC.
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. (David) Lou and X. Wang, Nat. Energy, 2016, 1, 1–8 Search PubMed.
- H. B. Yang, J. Miao, S.-F. Hung, J. Chen, H. B. Tao, X. Wang, L. Zhang, R. Chen, J. Gao, H. M. Chen, L. Dai and B. Liu, Sci. Adv., 2016, 2, e1501122 CrossRef PubMed.
- J. Sun, S. E. Lowe, L. Zhang, Y. Wang, K. Pang, Y. Wang, Y. Zhong, P. Liu, K. Zhao, Z. Tang and H. Zhao, Angew. Chem., Int. Ed., 2018, 57, 16511–16515 CrossRef CAS.
- D. Liu, S. Ding, C. Wu, W. Gan, C. Wang, D. Cao, Z. ur Rehman, Y. Sang, S. Chen, X. Zheng, Y. Wang, B. Ge and L. Song, J. Mater. Chem. A, 2018, 6, 6840–6846 RSC.
- C. Wu, X. Zhang, Z. Xia, M. Shu, H. Li, X. Xu, R. Si, A. I. Rykov, J. Wang, S. Yu, S. Wang and G. Sun, J. Mater. Chem. A, 2019, 7, 14001–14010 RSC.
- H.-J. Qiu, P. Du, K. Hu, J. Gao, H. Li, P. Liu, T. Ina, K. Ohara, Y. Ito and M. Chen, Adv. Mater., 2019, 31, 1900843 CrossRef.
- Y. Li, Z.-S. Wu, P. Lu, X. Wang, W. Liu, Z. Liu, J. Ma, W. Ren, Z. Jiang and X. Bao, Adv. Sci., 2020, 7, 1903089 CrossRef CAS PubMed.
- K. Qu, Y. Zheng, Y. Jiao, X. Zhang, S. Dai and S.-Z. Qiao, Adv. Energy Mater., 2017, 7, 1602068 CrossRef.
- S. Chen, J. Duan, Y. Zheng, X. Chen, X. W. Du, M. Jaroniec and S.-Z. Qiao, Energy Storage Mater., 2015, 1, 17–24 CrossRef.
- L. Li, H. Yang, J. Miao, L. Zhang, H.-Y. Wang, Z. Zeng, W. Huang, X. Dong and B. Liu, ACS Energy Lett., 2017, 2, 294–300 CrossRef CAS.
- X. Lu, W.-L. Yim, B. H. R. Suryanto and C. Zhao, J. Am. Chem. Soc., 2015, 137, 2901–2907 CrossRef CAS.
- T. Y. Ma, J. Ran, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., 2015, 127, 4729–4733 CrossRef.
- Z. Xiao, X. Huang, L. Xu, D. Yan, J. Huo and S. Wang, Chem. Commun., 2016, 52, 13008–13011 RSC.
- P. Chen, T. Zhou, L. Xing, K. Xu, Y. Tong, H. Xie, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 610–614 CrossRef CAS.
- A. M. El-Sawy, I. M. Mosa, D. Su, C. J. Guild, S. Khalid, R. Joesten, J. F. Rusling and S. L. Suib, Adv. Energy Mater., 2016, 6, 1501966 CrossRef.
- D. G. Papageorgiou, I. A. Kinloch and R. J. Young, Prog. Mater. Sci., 2017, 90, 75–127 CrossRef CAS.
- Z. Zhao, M. Li, L. Zhang, L. Dai and Z. Xia, Adv. Mater., 2015, 27, 6834–6840 CrossRef CAS.
- Z. Lin, G. H. Waller, Y. Liu, M. Liu and C. Wong, Carbon, 2013, 53, 130–136 CrossRef CAS.
- Z. Zhao and Z. Xia, ACS Catal., 2016, 6, 1553–1558 CrossRef CAS.
- G.-L. Chai, K. Qiu, M. Qiao, M.-M. Titirici, C. Shang and Z. Guo, Energy Environ. Sci., 2017, 10, 1186–1195 RSC.
- V. N. Popov, Mater. Sci. Eng., R, 2004, 43, 61–102 CrossRef.
- Z. Luo, A. Oki, L. Carson, L. Adams, G. Neelgund, N. Soboyejo, G. Regisford, M. Stewart, K. Hibbert, G. Beharie, C. Kelly-Brown and P. Traisawatwong, Chem. Phys. Lett., 2011, 513, 88–93 CrossRef CAS PubMed.
- G.-L. Tian, Q. Zhang, B. Zhang, Y.-G. Jin, J.-Q. Huang, D. S. Su and F. Wei, Adv. Funct. Mater., 2014, 24, 5956–5961 CrossRef CAS.
- M. Inagaki, T. Tsumura, T. Kinumoto and M. Toyoda, Carbon, 2019, 141, 580–607 CrossRef CAS.
- X. Huang, Q. Wang, D. Jiang and Y. Huang, Catal. Commun., 2017, 100, 89–92 CrossRef CAS.
- X. Li, P. Cui, W. Zhong, J. Li, X. Wang, Z. Wang and J. Jiang, Chem. Commun., 2016, 52, 13233–13236 RSC.
- K. Mamtani, D. Jain, D. Dogu, V. Gustin, S. Gunduz, A. C. Co and U. S. Ozkan, Appl. Catal., B, 2018, 220, 88–97 CrossRef CAS.
- X. Liu, I. S. Amiinu, S. Liu, K. Cheng and S. Mu, Nanoscale, 2016, 8, 13311–13320 RSC.
- S. Yang, T. Zhang, G. Li, L. Yang and J. Y. Lee, Energy Storage Mater., 2017, 6, 140–148 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |