
Controlling nanostructure and mechanical properties in triblock copolymer/monomer blends via reaction-induced phase transitions†
Vincent M.
Torres
 a,
Jacob A.
LaNasa
b,
Bryan D.
Vogt
c and
Robert J.
Hickey
*bd
a,
Jacob A.
LaNasa
b,
Bryan D.
Vogt
c and
Robert J.
Hickey
*bd
aDepartment of Chemistry, The Pennsylvania State University, University Park, Pennsylvania 16801, USA. E-mail: rjh64@psu.edu
bDepartment of Materials Science and Engineering, The Pennsylvania State University, University Park, Pennsylvania 16801, USA
cDepartment of Chemical Engineering, The Pennsylvania State University, University Park, Pennsylvania 16801, USA
dMaterials Research Institute, The Pennsylvania State University, University Park, Pennsylvania 16801, USA
First published on 15th December 2020
Abstract
Thermoplastic elastomers based on ABA triblock copolymers are typically limited in modulus and strength due to crack propagation within the brittle regions when the hard end-block composition favors morphologies that exhibit connected domains. Increasing the threshold end-block composition to achieve enhanced mechanical performance is possible by increasing the number of junctions or bridging points per chain, but these copolymer characteristics also tend to increase the complexity of the synthesis. Here, we report an in situ polymerization method to successfully increase the number of effective junctions per chain through grafting of poly(styrene) (PS) to a commercial thermoplastic elastomer, poly(styrene)–poly(butadiene)–poly(styrene) (SBS). The strategy described here transforms a linear SBS triblock copolymer–styrene mixture into a linear-comb-linear architecture in which poly(styrene) (PS) grafts from the mid-poly(butadiene) (PBD) block during the polymerization of styrene. Through systematic variation in the initial SBS/styrene content, nanostructural transitions from disordered spheres to lamellar through reaction-induced phase transitions (RIPT) were identified as the styrene content increased. Surprisingly, maximum mechanical performance (Young's modulus, tensile strength, and elongation at break) was obtained with samples exhibiting lamellar nanostructures, corresponding to overall PS contents of 61–77 wt% PS (including the original PS in SBS). The PS grafting from the PBD block increases the modulus and the strength of the thermoplastic elastomer while preventing brittle fracture due to the greater number of junctions afforded by the PS grafts. The work presented here demonstrates the use of RIPT to transform standard SBS materials into polymer systems with enhanced mechanical properties.
Introduction
Thermoplastic elastomers (TPEs) are a class of block polymers in which the microphase separated domains of low glass transition temperature (Tg) regions (“soft” domains) are reinforced with high-Tg or crystalline domains (“hard” domains) that act as physical crosslinks.1,2 The strength and resilience of TPEs has enabled their commercial use in a variety of applications ranging from footwear to automotive products.3–5 Unlike chemically crosslinked thermoset elastomers, the performance of TPEs is directly tied to the microstructure in which the hard domains create physical crosslinks and the rubbery mid-blocks act as bridges between the hard domains.4,5 While many advancements in TPEs, led by supramolecular design,4 macromolecular architecture,6 and polymerization catalysts,7 have enabled self-healing properties and tunable mechanical responses, these strategies are focused on designing the static polymer structure resulting in hard segments embedded within a soft matrix. Although synthetic improvements for controlling polymer topology and chemical composition have led to TPE advances, there are a wealth of opportunities in utilizing in situ reaction and processing modalities to tune macromolecular structures and nanoscale phases not easily accessible via traditional methods.8–11The molecular architecture of TPEs is based on a block polymer framework in which covalent bonds chemically link distinct repeat segments or “blocks” (e.g., A or B blocks in an ABA triblock copolymer) to form a single macromolecule.12 Block polymers will microphase separate into distinct domains as a result of the incompatibility between the polymer blocks.12 In TPEs, the simplest ABA triblock copolymer architecture in which the end A-blocks are composed of hard glassy domains enables bridging of the mid-block chains between two different A-block domains, reinforcing the polymer material and preventing macrophase separation. TPEs are designed to favor isolated hard domains to prevent crack propagation during fracture by controlling the volume fraction of the different polymer blocks. Therefore, isolated ordered phases (e.g., body-centered cubic spheres and hexagonally-packed cylinders) are preferred due to the ability to prevent crack propagation during deformation. When designing TPEs with desirable nanoscale morphologies, one must consider the block volume fraction (φA, for the A-block of an ABA triblock copolymer), degree of polymerization (N), Flory–Huggins interaction parameter (χ), and macromolecular architecture and block sequence (e.g., linear and brush, or AB, ABA and ABC).12,13 The self-assembled nanoscale phases that exhibit either periodic or non-periodic ordering of hard and soft regions in TPEs increases the hard–soft component interface compared to traditional blends, and effectively increases the number of physical crosslinks per unit volume.14 However, an inherent limitation to increasing the modulus of TPEs with linear macromolecular architectures via increases in hard-block composition is that when hard domains form connected phases such as gyroid or lamellar, materials tend to become brittle and prone to fracture. To circumvent the impact of morphology on the TPE properties, previously reported results show that multigraft copolymers lead to enhancement in tensile strength and elongation at break as a result of an individual copolymer chain bridging domains.15,16 Although complex macromolecular architectures have shown to be a viable method for enhancing or tuning the properties of TPEs,17 the synthetic rigor required to produce such is a potential issue. Therefore, facile synthetic methods to generate graft copolymers that increase the number of junctions per chain are highly desirable for widespread implementation of complex macromolecular architectures in commercial applications.
In situ chemical methods to transform the state of a material (i.e., reaction-induced phase transitions (RIPT)) have been recently utilized to control the nanostructure of polymer materials in solution and in the bulk.8 Although polymerization processes to drive nanoscale morphology transitions are being actively explored, the concept of RIPT has been broadly used to create high-impact poly(styrene) (HIPS),18 polymer monoliths for separations,19 siloxane hydrogels for contact lenses,20 and polyurethanes.21–23 In many of the RIPT examples, the underlying driver of the phase transition is polymer incompatibility between either a different polymer or solvent during the polymerization of monomer.24In situ polymerization methods resulting in a variety of block polymer topologies (linear or grafted) have demonstrated the usefulness of RIPT as a means to control nanostructure during bulk polymerizations as opposed to simply blending block polymers and homopolymers.25–27 The synthesis of HIPS is an excellent example of the in situ process of creating graft copolymers during the polymerization of styrene in the presence of poly(butadiene) (PBD). The formation of graft copolymers is a result of using a radical generator that creates allylic radicals along the PBD backbone, leading to the formation of poly(styrene) (PS) grafted from PBD. Homopolymer PS also forms during the polymerization due to the presence of free radicals in the mixture. The grafting of PS from PBD prevents macrophase separation of PBD and PS during the polymerization, enhances interfacial adhesion, and leads to intricate PBD droplet morphologies embedded within a PS matrix, which are critical for the desirable properties of HIPS.28–30 While HIPS is an excellent example of how in situ polymerizations drive phase transitions, there are unlimited possibilities for harnessing in situ polymerization methods that result in complex macromolecular architectures favoring unconventional nanostructural transitions, opening new directions for creating nanoscale morphologies with enhanced mechanical properties that are not easily accessible using traditional self-assembly methods.
Here, we report on controlling nanoscale morphologies using in situ polymer grafting chemistry, similar to the synthesis of HIPS, and correlate the enhanced mechanical properties to PS content and chain architecture after polymerization. The in situ polymer grafting strategy described here transforms a linear poly(styrene)–poly(butadiene)–poly(styrene) (SBS) triblock copolymer to a linear-comb-linear architecture in which PS grafts from the mid-PBD block during the polymerization of styrene (Fig. 1). Our strategy follows previously published work in which PS is grafted from the PBD backbone of a PS-PBD diblock copolymer via the generation of an allylic radical.9,10 The in situ grafting during the polymerization of styrene resulted in both order–order and disorder–order nanostructural transitions,9,10 but the impact of these changes on properties was not previously investigated. The polymer grafting chemistry has been shown to be generalizable to other unsaturated polymer motifs (hybrid inorganic nanoparticle/polymer materials) and grafting polymers (PS and poly(methyl methacrylate)).31 Here combining in situ grafting chemistry with the SBS TPE leads to nanoscale morphology transitions from an originally microphase separated but disordered sphere morphology (DIS Sphere) for the neat SBS to lamellar (LAM) morphologies or co-existing morphologies in which PS is the majority phase with increasing styrene content (Fig. 1). Despite the transition to an unfavorable morphology (LAM) for high performance TPEs, the increases in Young's modulus (E), tensile strength (TS), and elongation at break (εb) relative to the original SBS occur at intermediate PS wt%, while the mechanical performance degrades at high styrene content. The maximum values in E, TS, and εb occur at an overall PS content of around 77 wt%, which highlights the potential to eschew common design limitations for TPEs through in situ grafting chemistry.
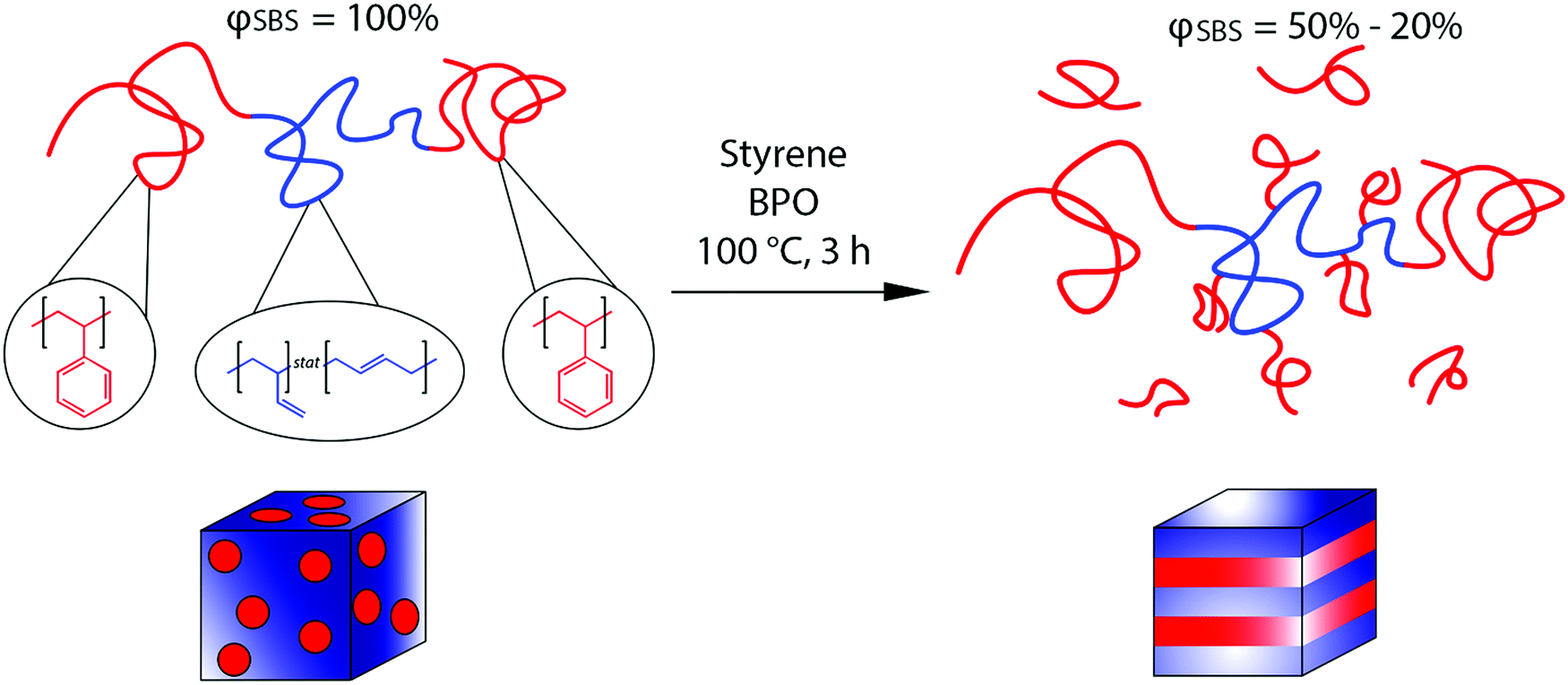 | ||
| Fig. 1 In situ polymerization and polymer grafting scheme to create linear-comb-linear grafted block polymers. Initially, a SBS triblock copolymer (φSBS = 100%), exhibiting a microphase separated but disordered sphere morphology, is blended with styrene and benzoyl peroxide (BPO). The blend is heated to 100 °C and reacted for 3 h. The reaction produces linear-comb-linear grafted block polymers and a small fraction of PS homopolymer, and results in a morphology transition to either lamellar morphologies (samples in the φSBS = 50–20% range) or co-existing morphologies (φSBS = 10%) in which PS is the majority phase. | ||
Experimental
Materials
Poly(styrene)–poly(butadiene)–poly(styrene) (SBS) triblock copolymer (styrene 30 wt%), benzoyl peroxide (BPO), styrene, and deuterated chloroform (CDCl3) were purchased from Sigma Aldrich (St. Louis, USA). Tetrahydrofuran (THF) was purchased from ThermoFisher Scientific (Waltham, USA). Methanol was purchased from VWR (Radnor, USA). Alumina was purchased from Honeywell (Charlotte, USA). The tin-cure silicone rubber mold mixture was purchased from Smooth-On (Macungie, USA).Mold preparation
The silicone molds used to polymerize bulk dog bone samples were based on ASTM standard D638 type IV dog bones for tensile measurements. 3D printed dog bones following the ASTM specifications (3D Hubs, 115 × 19 × 4 mm) were laid across packing tape lining the bottom of a disposable aluminum baking pan, and the silicone mold mix was poured over the dog bones. The resulting mold was cured for 6 hours at room temperature then placed in an oven for 4 h at 65 °C.SBS dog bone preparation
Pre-polymerized blends containing SBS, styrene, and BPO were prepared with desired SBS/styrene volume fractions, mixed until homogeneous, injected into the silicone dog bone mold, and placed into an oven that had been preheated to 100 °C. The polymerization was run for 3 h, and the resulting dog bones were removed from the mold and placed under vacuum overnight to remove any unreacted styrene, giving an overall yield of around 80%. All initial pre-polymerized blends used freshly purified styrene that was passed over basic alumina, and SBS that had been reprecipitated in methanol to remove inhibitor. A molar ratio of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of styrene to BPO was used for all samples.
:1 of styrene to BPO was used for all samples.
Neat SBS dog bones were fabricated by injecting a SBS/THF solution (0.6 g mL−1) into the silicone dog bone mold and removing the THF via initial ambient evaporation in the hood and then placed under vacuum to remove any remaining THF.
Nuclear magnetic resonance (NMR)
1H NMR spectroscopy was used to determine the wt% of PS in the neat SBS as well as the relative amount of 1,2- and 1,4-PBD using an AVANCE III HD 500 NMR (Bruker) instrument fitted with a 5 mm Prodigy BBO cryoprobe (Bruker) at 25 °C. Samples were prepared by dissolving 10–15 mg in 1 mL of deuterated chloroform, then transferred to standard NMR tubes. The SBS PS wt% was determined to be 35% and was found to be 89% 1,4-PBD (Fig. S1, ESI†).Size exclusion chromatography (SEC)
The molecular weight of the neat SBS and the grafted SBS polymers were determined using a Tosoh EcoSEC (Tosoh Co.) equipped with a Wyatt Dawn Heleos-II eight angle light scattering detector (Wyatt Technology Corp.) with a THF mobile phase at 40 °C. Samples with a concentration of 2.5 mg mL−1 were filtered with 0.2 μm PTFE filter prior to injection. The molecular weight of the neat SBS was determined from SEC using dn/dc = 0.1495 for the absolute calculation.Small-angle X-ray scattering (SAXS)
SAXS analysis was conducted at the National Synchrotron Light Source II (NSLS-II) at Brookhaven National Laboratory using the Complex Materials Scattering (CMS/11-BM) beamline. Samples were mounted with Kapton tape and exposed for 10 s using a 13.50 keV beam, where the wavelength was 0.9184 Å. A sample to detector distance of 2 m was used to probe the q range 0.008–0.444 Å−1. The scattering images were captured with a Dectris Pilatus 2M detector (pixel size 172 μm × 172 μm). These 2D scattering data were corrected for background using an empty glass capillary as the reference. The raw 2D SAXS data were converted to 1D by circular averaging using the software SciAnalysis.Transmission electron microscopy (TEM)
Micrographs of the polymer systems were obtained using a FEI Tecnai G2 Spirit BioTwin TEM. Polymer samples were prepared by first microtoming polymerized dog bone samples (approximately 70–90 nm thick sections using a Leica UC6 ultramicrotome), placing the microtomed samples onto TEM grids (Electron Microscopy Sciences, Formvar/Carbon 200 Mesh, Copper), and staining the samples with osmium tetroxide to differentiate between the PS and PBD domains. The OsO4 stain enables the differentiation between the PS and PBD domains by selectively staining the vinyl groups of the PBD mid-block.32Tensile measurements
Uniaxial extension measurements were conducted using an MTS Exceed load frame with a 10 kN transducer. The gauge length was measured with calipers once the samples were mounted, and the width and thickness were measured at both grips and averaged. The applied rate of strain was 5 mm min−1. The Young's modulus (E), yield strength (YS), yield strain (εy), tensile strength (TS), and strain at break (εb) were determined from the measured stress versus strain plots that were generated from the tensile measurements. For each composition, at least five dog bone samples were prepared and measured. The reported mechanical properties are the average obtained from five (or more) individual stress–strain curves at the respective composition.Results and discussion
Nanostructure morphologies and mechanical properties were investigated for a series of PS-grafted SBS samples after conducting the RIPT process. Dog bone samples (ASTM standard D638 type IV) with varying PS content were synthesized by conducting in situ polymerizations on mixtures of SBS, styrene, and BPO using silicone dog bone molds (Fig. 2). Sample nomenclature is based on the initial SBS volume fraction (φSBS) with respect to styrene before polymerization (i.e., φSBS = 50% initially had a SBS volume fraction of 50% before polymerization). The number-average molecular weight (Mn), dispersity (Đ), weight percent of PS (wt%), and 1,4 versus 1,2 microstructural content of the PBD block for the neat SBS triblock copolymer used in the work were 62 kg mol−1, 1.11, 35%, and 89%, respectively (see ESI†). Polymerizations were run at 100 °C for 3 h. After polymerization, dog bone samples were removed from the silicon mold and vacuum dried to remove unreacted styrene. The yield from the polymerization was approximately 80% for all samples synthesized in this work. | ||
| Fig. 2 Digital photograph of the dog bone samples after the RIPT process and drying under vacuum. | ||
The proposed PS-grafting mechanism from PBD during the in situ polymerization is predicted to occur via an allylic radical that forms on the PBD backbone when BPO abstracts a hydrogen from a carbon adjacent to the vinyl groups.33,34 In addition to forming grafted PS, which is initiated from the allylic radical, PS homopolymer is also produced due to the presence of free radicals. PS grafting was verified for the polymerization process described here by conducting the styrene polymerization at low SBS volume fractions (φSBS = 2.5%). The size-exclusion chromatogram indicates a shift in the elution peak to shorter retention times, corresponding to an increase in the molecular weight for the PS-grafted SBS compared to the neat SBS (Fig. S2, ESI†). At higher φSBS (φSBS ≥ 5%), the samples became crosslinked and thus could not be analyzed via SEC or solution 1H NMR. Therefore, the PS content was determined gravimetrically by measuring the change in mass after polymerization and vacuum drying in comparison to the styrene and polystyrene in the SBS in the original solution. The amount of uncrosslinked homopolymer in each sample after polymerization was determined by conducting swelling experiments and measuring the weight of the sample before and after swelling. Specifically, dog bone samples (approximately 0.5 g) were swollen in THF for 1 h. During swelling, uncrosslinked homopolymer (sol) was extracted from the crosslinked network (gel). The swollen crosslinked samples were retrieved from the THF (20 mL), dried, and weighed. The difference in mass before and after swelling for samples φSBS = 50–20% was between 3 and 7 wt%, suggesting that the majority of PS formed during polymerization was grafted onto the PBD mid-block (Table 1). For the φSBS = 10%, the mass loss was greater (37%), indicating that at sufficiently high styrene concentration that a fair amount of PS homopolymer forms during the RIPT process.
| Samplea | PSb wt% | H-PSc wt% | T g,PBD (°C) | T g,PS (°C) | Morphologyd |
|---|---|---|---|---|---|
| a Sample name corresponds to the initial SBS volume fraction in the pre-polymerized SBS/styrene mixture. b PS wt% referrers to the total PS content in the samples after RIPT and was gravimetrically determined. The value includes PS end blocks in SBS, PS grafted onto the PBD midblock, and uncrosslinked homopolymer PS. c PS homopolymer wt% was determined by conducting swelling experiments. Swelling experiments were not conducted for the φSBS = 100% sample. d The morphology of the dog bone samples after RIPT was determined using a combination of TEM and SAXS. | |||||
| φ SBS = 100% | 35 | — | −91 | 89 | DIS Spheres |
| φ SBS = 50% | 61 | 3 | −85 | 95 | LAM |
| φ SBS = 40% | 69 | 5 | −92 | 91 | LAM |
| φ SBS = 30% | 77 | 4 | −91 | 90 | LAM |
| φ SBS = 20% | 84 | 7 | −90 | 82 | LAM |
| φ SBS = 10% | 92 | 37 | −91 | 81 | Co-existing |
There is a potential for oligomer styrene (either homopolymer or grafted) to form during the free radical polymerization and that is miscible in the PBD domain. However, the glass transitions for the PBD (Tg,PBD) and PS (Tg,PS) domains remain relatively constant across all samples (Table 1). The Tg,PS varies from 89 °C for φSBS = 100% to 81 °C for φSBS = 10% (Table 1). The decrease by approximately 10 °C is attributed to plasticization by low molecular weight PS chain formed during polymerization (Fig. S3, ESI†). Similar reductions in Tg,PS have been reported using related PS polymerization and processing procedures.35 An estimate of the segregation of the PS and PBD domain was estimated using the Fox equation to determine the weight fraction of either PBD or PS in the PS or PBD domains, respectively (Table S1, ESI†). These results infer that the domains are relatively pure with limited difference in the Tg of either phase relative to the original ABA triblock copolymer. Table 1 summarizes some of the characteristics of the samples produced by RIPT including the total PS wt% (SBS, grafted PS, and uncrosslinked PS), PS homopolymer wt% (H-PS wt%), glass transition temperatures of the PS and PBD phases, and resulting morphologies explored in this study.
The morphologies of the dog bone samples after the RIPT process were determined using a combination of SAXS and TEM. TEM micrographs clearly illustrate the change in the morphologies of the samples due to the RIPT process (Fig. 3). The TEM images confirm that all samples are microphase separated with the PS and PBD domains easily resolved after staining with OsO4. The original ABA triblock copolymer (φSBS = 100%) forms a disordered sphere (DIS Sphere) in which isolated spherical PS domains are dispersed within a PBD matrix (Fig. 3a). After the RIPT process for the φSBS = 50% sample, the TEM micrograph indicates that the phase is transformed to what can be interpreted as either a hexagonally-packed cylinder or a LAM morphology (Fig. 3b) with no long-range order. As φSBS decreases, the TEM micrographs illustrate the typical striped pattern associated with LAM for samples φSBS = 40%, 30%, and 20% (Fig. 3c–e). At φSBS = 10%, there is a shift in the morphology with some undulations on the lamellae and apparent cylindrical or spherical phase co-existence. The TEM is interpreted that the φSBS = 10% consists of co-existing multiple phases that include disordered microphase separated regions (PBD is the minority phase) and ordered regions of LAM and/or hexagonally-packed cylinders (Fig. 3f).
 | ||
| Fig. 3 TEM images for microtomed and stained RIPT dog bone samples. (a) φSBS = 100%, (b) φSBS = 50%, (c) φSBS = 40%, (d) φSBS = 30%, (e) φSBS = 20%, and (f) φSBS = 10%. Samples were stained with OsO4. | ||
To confirm the phase assignment based on TEM images, 1D SAXS patterns are shown in Fig. 4. The results from the SAXS measurements are consistent with the finding that the φSBS = 100% sample transitions from a DIS Sphere to a LAM morphology with increasing PS content (φSBS = 50%, 40%, 30%, and 20%) after the polymerization process based on higher order reflections present in the scattering profiles. The 1D SAXS pattern for the φSBS = 100% sample exhibits a broad primary scattering peak, which is due to the distribution in distances between spherical domains associated with DIS Sphere. Samples φSBS = 50% and 40% are indexed to a LAM morphology (Bragg reflections, q/q* = √1, √4, √9, and √16, where q* is the principle scattering peak). Additionally, the primary Bragg reflections become sharper than the broad scattering peak of the φSBS = 100% sample, indicating that the system becomes ordered as a result of the polymerization process. Analysis of the 1D SAXS patterns display a nanostructural transition from a DIS Sphere (still microphase separated) to LAM morphology during the in situ polymerization process in which PS grafts from SBS along with some formation of PS homopolymer. The scattering for φSBS = 30% is suggestive of an approach to a transition point due to the reduction in the higher-order Bragg reflections. At φSBS = 20%, there is both a decrease in the scattering intensity and an increase in the peak width for the primary scattering peak, signifying a continual disordering process. Finally, at φSBS = 10%, there is a change in the scattering reflections, but these peaks are broad, indicating that the sample transitions from a LAM phase to a different phase. Based on the TEM and the asymmetry of the peak at q ≈ 0.035 Å−1, the sample likely consists of a co-existing microphase separated morphology in which the PBD domains become the isolated minority phase within a PS matrix.
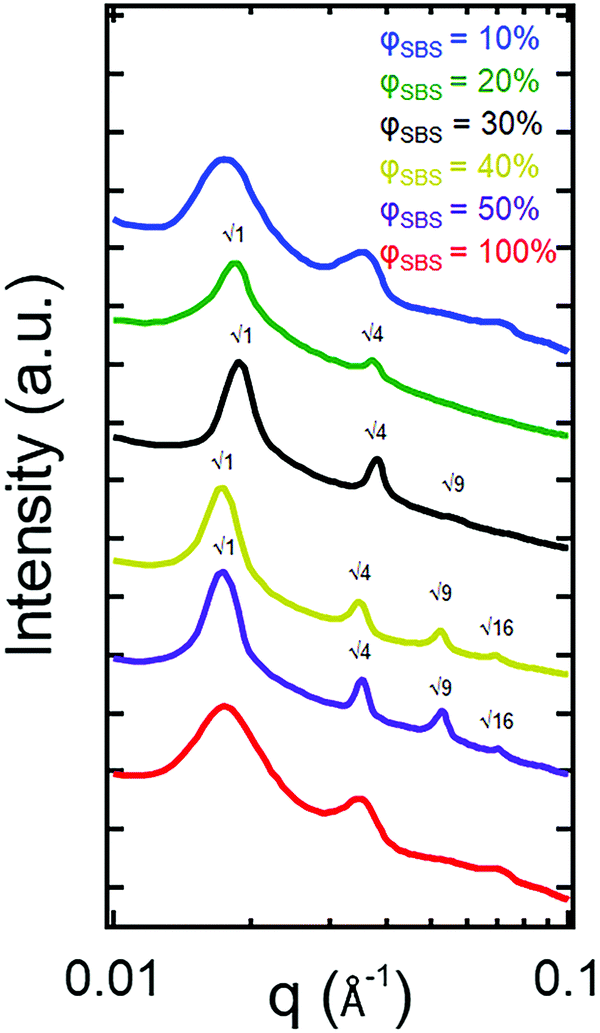 | ||
| Fig. 4 1D SAXS patterns for the dog bone samples after polymerization and vacuum drying. A transition to LAM is seen with samples φSBS = 50%, 40%, 30%, and 20% from the higher-order reflections indexed to the primary scattering peak as indicated on the scattering curves. Scattering plots have been vertically shifted. | ||
The TEM and SAXS results are consistent in concluding that swelling the PS–PBD–PS triblock copolymer with styrene monomer and conducting in situ polymerizations leads to nanostructural transitions. Comparing the TEM images, it is evident that the morphology of the φSBS = 100% sample consists of disordered spheres in which the PS minority phase is embedded within a PBD matrix transitions to a lamellar phase after polymerization. Although there are similarities between the 1D SAXS plots for the different samples, the examination of the peak positions can eliminate the possibility of a pure LAM phase for φSBS = 100% and 10%. The lack of what could be indexed as q/q* = √9 peak for the φSBS = 100% sample is not a possible systematic absence for a lamellar morphology if the q/q* = √1, √4, and √16 are present, further supporting the SAXS identification that the φSBS = 100% sample is consistent with a disordered sphere phase. Additionally, the TEM images show that the φSBS = 100% sample is not lamellar.
Interestingly, all φSBS compositions exhibit only minor variations in the domain spacing (d), despite the large differences in the PS content. In linear diblock copolymers, d increases with increasing molecular weight. Here, two different processes are occurring: (1) the increase in the SBS molecular weight is a result of PS grafting from the PBD mid-block, and (2) PS homopolymer is forming. Therefore, the overall domain spacing will be affected by both the molecular architecture and the swelling of the PS domains with PS homopolymer. Here, the polymerization process creates grafted block polymers, which architecturally resemble miktoarm block polymers. Miktoarm block polymers have been shown to result in smaller domain sizes as compared to the linear counterparts.36 The reason for the minimal change in d for all φSBS compositions is being currently explored, but we posit that the PS that grafts from the PBD mid-block undergoes microphase separation to reside in the PS domains formed by the PS end-blocks, potentially reducing increases in d with increasing PS content.
Uniaxial extension measurements were conducted to establish the influence of PS content on the mechanical properties of the dog bone samples using the described RIPT procedure. At least five dog bone samples for each composition (φSBS = 100–10%) were tested using tensile measurements to validate the stress–strain response (see ESI,† Fig. S4–S9). Fig. 5 shows representative stress–strain curves for the φSBS samples. The Young's modulus (E), yield strength (YS), elongation at yield (εy), tensile strength (TS), and the elongation at break (εb) were determined from the stress–stain curves and are summarized in Table 2. The mold preparation method used in the current study was chosen due to the ease of processing the dog bone geometry. However, solvent casting is known to lead to the formation of air bubbles, which is the reason for the variance in the stress–strain responses.37
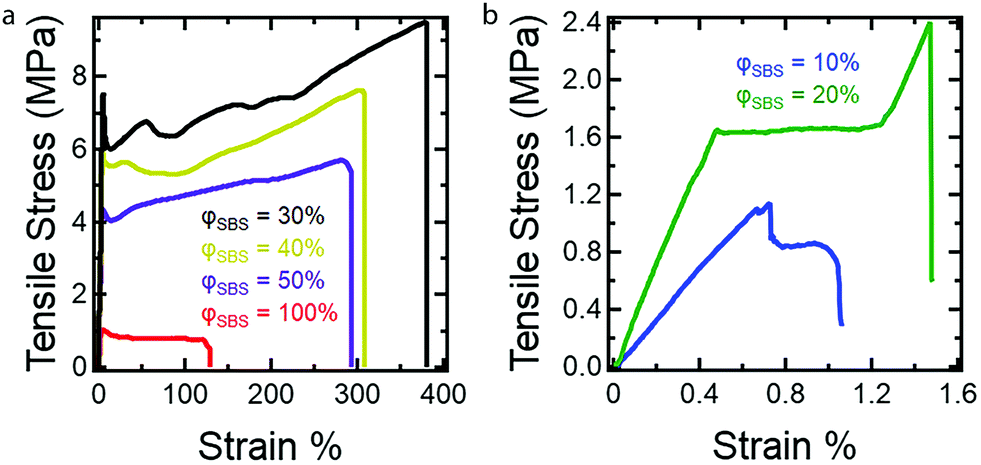 | ||
| Fig. 5 Representative stress–strain curves for dog bone samples investigated in the current study. All samples were run until fracture at a strain rate of 5 mm min−1. All mechanical property values determined from the stress–stain curves are found in Table 2. Stress–strain curves for (a) φSBS = 100%, 50%, 40%, and 30%, and (b) φSBS = 20% and 10%. | ||
| Sample | E (MPa) | YS (MPa) | ε y (%) | TS (MPa) | ε b (%) |
|---|---|---|---|---|---|
| φ SBS = 100% | 0.4 ± 0.1 | 0.8 ± 0.1 | 4.5 ± 0.9 | 0.9 ± 0.1 | 128.7 ± 21.3 |
| φ SBS = 50% | 1.9 ± 0.1 | 4.0 ± 0.3 | 3.7 ± 0.2 | 5.9 ± 0.5 | 342.0 ± 36.4 |
| φ SBS = 40% | 2.0 ± 0.1 | 4.8 ± 0.4 | 4.1 ± 0.3 | 7.1 ± 0.5 | 301.1 ± 63.1 |
| φ SBS = 30% | 2.5 ± 0.2 | 7.6 ± 0.3 | 4.6 ± 0.1 | 8.7 ± 0.4 | 267.9 ± 63.6 |
| φ SBS = 20% | 3.6 ± 0.3 | 2.6 ± 0.3 | 1.6 ± 0.2 | 2.6 ± 0.3 | 1.8 ± 0.1 |
| φ SBS = 10% | 1.5 ± 0.3 | 1.1 ± 0.2 | 0.8 ± 0.1 | 1.2 ± 0.2 | 1.5 ± 0.4 |
Overall, the dog bone samples prepared using RIPT spanned a range of mechanical properties, which correspond well with the final styrene content (PS wt% in Table 1). With increasing styrene content, from 35 PS wt% to 61 PS wt%, which correspond to samples φSBS = 100% and φSBS = 50%, respectively, E, YS, TS, and εb increase, while εy is invariant within the experimental uncertainty (Table 2). There are three possible reasons for the increases in E, YS, and TS for sample φSBS = 50% when compared to φSBS = 100%: (1) increase in the PS wt%, (2) morphology transition to LAM, or (3) greater number of branch points per molecule due to PS grafting. At this point, additional experiments need to be conducted to identify the various factors that account for the change in mechanical properties, but previously published results indicate increasing the number of branch points plays a significant role.15,16 Comparing the mechanical properties of samples φSBS = 50%, 40%, and 30%, all of which exhibit LAM morphologies, shows an increase in E, YS, and TS as PS content increases. Therefore, the higher PS content is a major contributor to the mechanical properties, as expected from the known mechanical performance of styrenic TPEs.38 Both YS and TS are maximized at 77 wt% PS, while E reaches a maximum at 84 wt% PS. Further increasing the PS wt% to 84 wt% leads to a significant drop in YS and TS, while increasing in E, which is expected due to the increased brittleness of the materials imparted by the glassy PS domains. Remarkably, samples φSBS = 50–30% are extendable beyond 250%, which corresponds to 61–77 wt% PS. The high PS contents and large εb values highlight how in situ polymer grafting during RIPT leads to new nanostructured materials not easily accessible using current polymerization and self-assembly methods. However, the mechanical properties are significantly reduced above 84 wt% PS. Fig. 6 clearly displays the variation in E and TS with respect to PS wt%. Overall, the work presented here indicates that the RIPT process leads to an enhancement in the mechanical properties (Young's modulus, yield stress and strain to break) despite the resulting materials exhibiting a LAM morphology, which is generally not optimal for mechanical properties. Increasing the PS content will intrinsically increase E from a simple rule of mixtures based on the modulus of PBD and PS. The increased glassy PS content will also increase the yield stress. We hypothesize that the PS grafting from the PBD block results in increasing polymer chain bridging between brittle domains that provide a mechanism to prevent fracture propagation, ultimately increasing εb.
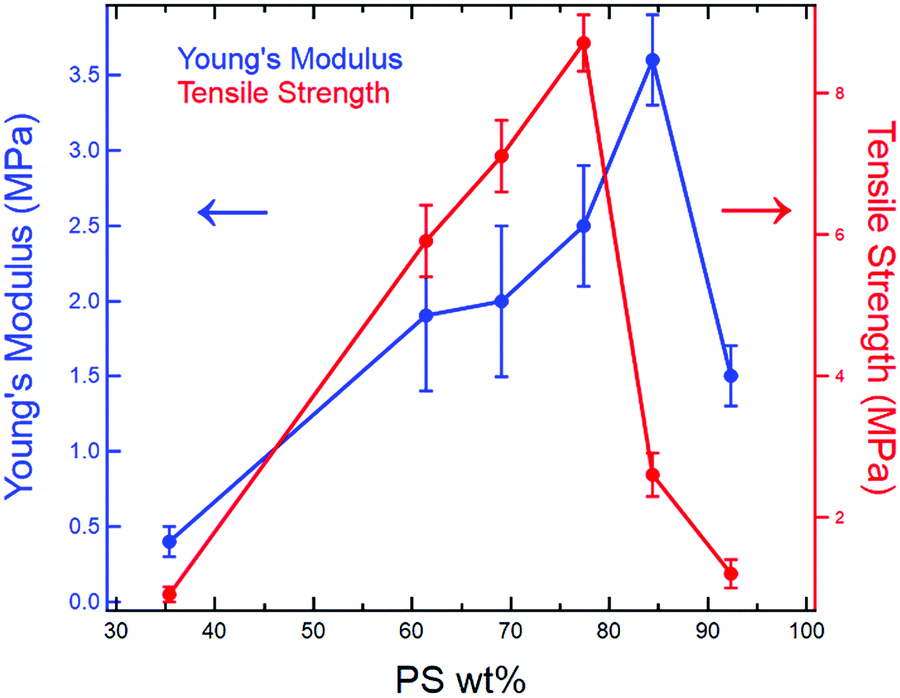 | ||
| Fig. 6 E (left, blue) and TS (right, red) with respect to PS wt%. As PS wt% increases, the modulus and the tensile strength increase. However, a significant reduction in mechanical properties occurs when the PS content increases beyond a specific amount. | ||
As the PS content of the samples is increased, there is a discernible change in the fracture mechanism. For φSBS = 100%, defects caused by the escaping solvent caused the samples to form sparse tendrils connecting the sample at the fracture point. For samples in the φSBS = 50–30% range, the samples stress whiten, indicating that crazing or microvoids are produced during deformation. Crazing in the samples leads to greater εb, as both crazing and microvoids help to inhibit crack growth by acting as energy sinks.39 Furthermore, crazing should suppress the nucleation of cavities around the fracture point by allowing continual extension of the fibers of the material until fracture occurs.14 The φSBS = 20% sample exhibits an upper yield point during the onset of plastic deformation and deforms at a slightly lower and constant stress before resuming plastic deformation. The reported deformation behavior of the φSBS = 20% sample is consistent with the formation of Lüder's bands during uniaxial extension, similar to what has been seen in a variety of materials including poly(carbonate). Lüder's bands are caused by heterogeneity in the plastic deformation in the sample due to defects.40 For φSBS = 10%, the high PS content caused these samples to fracture almost immediately at the yield point with no observable change in the samples. Without crazing or microvoids, the nucleation of cavities caused by the fracture cannot be suppressed, leading to the sudden failure. Overall, the work presented here indicates that the RIPT process leads to an enhancement in the mechanical properties of the materials by increasing the PS content. This is even despite the resulting materials exhibiting a LAM morphology, which is expected to perform poorly due to the interconnected brittle domains, thus providing a fracture propagation mechanism leading to the early onset of fracture.
Conclusion
The RIPT process presented here is a facile synthetic method for transforming linear block copolymers into graft copolymers to control the nanostructure and, as a result, the mechanical properties. Starting with a SBS triblock copolymer with a DIS Sphere morphology, an in situ polymerization was performed, leading to PS grafting on the PBD mid-block as well as forming homopolymer PS. Increasing the PS wt% from 35% to 61% caused a nanostructural transition to LAM. The DIS Sphere-to-LAM transition and increases in PS content led to significant improvements to the mechanical properties, which was unexpected as connected phases such as LAM are known to be brittle. At compositions greater than 84 wt% PS, the morphology of the system begins transitioning to a morphology in which multiple phases are co-existing. Furthermore, the mechanical properties maximize in the 77 PS wt%, and then significantly reduce due to increased brittleness caused by the abundant PS. The reported work highlights that the in situ polymer grafting during RIPT results in nanostructured materials with high PS content and large εb values, which is surprising with materials that have large PS content. The results presented here indicate that the PS content plays a major role in the mechanical properties of the materials, however it is also suspected that the increased number of branch points per molecule is a significant factor in the mechanical properties of the materials.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Science Foundation, Division of Materials Research Polymers Program (CAREER Proposal: DMR-1942508) and start-up funds from the Penn State University. This research used the Complex Materials Scattering beamline of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. We thank Masafumi Fukuto and Ruipeng Li for their help in obtaining the SAXS data. TEM measurements were taken at the Materials Characterization Lab (MCL) in the Materials Research Institute (MRI) at the Penn State University. We are grateful to Missy Hazen for her help with microtoming and taking the polymer samples for TEM.References
- R. J. Spontak and N. P. Pate, Curr. Opin. Colloid Interface Sci., 2000, 5, 334–341 CrossRef CAS.
- C. R. Kumar, I. Fuhrmann and J. Karger-Kocsis, Polym. Degrad. Stab., 2002, 76, 137–144 CrossRef CAS.
- A. Watts and M. A. Hillmyer, Biomacromolecules, 2019, 20, 2598–2609 CrossRef CAS.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467–472 CrossRef CAS.
- O. Colombani, C. Barioz, L. Bouteiller and C. Chane, Macromolecules, 2005, 38, 1752–1759 CrossRef CAS.
- M. Vatankhah-Varnosfaderani, W. F. M. Daniel, M. H. Everhart, A. A. Pandya, H. Liang, K. Matyjaszewski, A. V. Dobrynin and S. S. Sheiko, Nature, 2017, 549, 497–501 CrossRef.
- D. J. Arriola, E. M. Carnahan, P. D. Hustad, R. L. Kuhlman and T. T. Wenzel, Science, 2006, 312, 714–719 CrossRef CAS.
- J. Lequieu and A. J. D. Magenau, Polym. Chem., 2021 10.1039/D0PY00722F.
- E. S. Zofchak, J. A. Lanasa, W. Mei and R. J. Hickey, ACS Macro Lett., 2018, 7, 822–827 CrossRef CAS.
- E. S. Zofchak, J. A. Lanasa, V. M. Torres and R. J. Hickey, Macromolecules, 2020, 53, 835–843 CrossRef CAS.
- C. Lang, J. A. LaNasa, N. Utomo, Y. Xu, M. J. Nelson, W. Song, M. A. Hickner, R. H. Colby, M. Kumar and R. J. Hickey, Nat. Commun., 2019, 10, 1–10 CrossRef CAS.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434–441 CrossRef CAS.
- C. Creton, G. Hu, F. Deplace, L. Morgret and K. R. Shull, Macromolecules, 2009, 42, 7605–7615 CrossRef CAS.
- R. Weidisch, S. P. Gido, D. Uhrig, H. Iatrou, J. Mays and N. Hadjichristidis, Macromolecules, 2001, 34, 6333–6337 CrossRef CAS.
- Y. Zhu, R. Weidisch, S. P. Gido, G. Velis and N. Hadjichristidis, Macromolecules, 2002, 35, 5903–5909 CrossRef CAS.
- T. R. Panthani and F. S. Bates, Macromolecules, 2015, 48, 4529–4540 CrossRef CAS.
- M. Fischer and G. P. Hellmann, Macromolecules, 1996, 29, 2498–2509 CrossRef CAS.
- F. Svec, J. Sep. Sci., 2004, 27, 747–766 CrossRef CAS.
- P. C. Nicolson and J. Vogt, Biomaterials, 2001, 22, 3273–3283 CrossRef CAS.
- W. Li, A. J. Ryan and I. K. Meier, Macromolecules, 2002, 35, 5034–5042 CrossRef CAS.
- A. J. Ryan, Polymer, 1990, 31, 707–712 CrossRef CAS.
- W. P. Yang and C. W. Macosko, Makromol. Chem., Macromol. Symp., 1989, 25, 23–44 CrossRef CAS.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS.
- R. J. Hickey, T. M. Gillard, M. T. Irwin, D. C. Morse, T. P. Lodge and F. S. Bates, Macromolecules, 2016, 49, 7928–7944 CrossRef CAS.
- A. N. Semenov, Macromolecules, 1993, 26, 2273–2281 CrossRef CAS.
- M. W. Matsen, Phys. Rev. Lett., 1995, 74, 4225–4228 CrossRef CAS.
- K. Sardelis and G. Allen, Polymer, 1987, 28, 244–250 CrossRef CAS.
- M. Asua and G. P. Leal, Polymer, 2009, 50, 68–76 CrossRef.
- G. R. Meira, C. V. Luciani and D. A. Estenoz, Macromol. React. Eng., 2007, 1, 25–39 CrossRef CAS.
- J. A. Lanasa, V. M. Torres and R. J. Hickey, J. Appl. Phys., 2020, 127, 134701 CrossRef CAS.
- R. L. Kruse, J. Microsc., 1981, 123, 323–327 CrossRef CAS.
- N.-J. Huang and D. C. Sundberg, J. Polym. Sci. Part A Polym. Chem., 1995, 33, 2571–2586 CrossRef CAS.
- N.-J. Huang and D. C. Sundberg, J. Polym. Sci. Part A Polym. Chem., 1995, 33, 2587–2603 CrossRef CAS.
- D. Hou, J. E. Bostwick, J. R. Shallenberger, E. S. Zofchak, R. H. Colby, Q. Liu and R. J. Hickey, ACS Appl. Nano Mater., 2020, 3, 962–968 CrossRef CAS.
- W. Shi, Y. Tateishi, W. Li, C. J. Hawker, G. H. Fredrickson and E. J. Kramer, ACS Macro Lett., 2015, 4, 1287–1292 CrossRef CAS.
- H.-M. Tong and A. C. Ouano, Polym. Eng. Sci., 1985, 25, 75–82 CrossRef CAS.
- M. Morton, J. E. McGrath and P. C. Juliano, J. Polym. Sci., Part C: Polym. Symp., 1969, 26, 99–115 CrossRef.
- J. G. Williams, Fracture Mechanics of Polymers, Ellis Horwood Limited, Chichester, 1st edn, 1984, pp. 143–147 Search PubMed.
- H. F. Brinson and L. C. Brinson, Polymer Engineering Science and Viscoelasticity: An Introduction, Springer, US, Boston, MA, 2015, pp. 57–100 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sm01661f |
| This journal is © The Royal Society of Chemistry 2021 |