
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective propargylic C(sp3)–H activation of methyl-substituted alkynes versus [2 + 2] cycloaddition at a titanium imido template†
Malte
Fischer‡
 *ac,
Manfred
Manßen‡
*bc,
Marc
Schmidtmann
c,
Thorsten
Klüner
*c and
Rüdiger
Beckhaus
*c
*ac,
Manfred
Manßen‡
*bc,
Marc
Schmidtmann
c,
Thorsten
Klüner
*c and
Rüdiger
Beckhaus
*c
aInorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK. E-mail: malte.fischer@chem.ox.ac.uk
bAnorganische Chemie Universität Tübingen, Auf der Morgenstelle 18, D-72076 Tübingen, Germany. E-mail: manfred.manssen@anorg.uni-tuebingen.de;
cInstitut für Chemie, Fakultät für Mathematik und Naturwissenschaften, Carl von Ossietzky Universität Oldenburg, Postfach 2503, D-26111 Oldenburg, Germany. E-mail: ruediger.beckhaus@uni-oldenburg.de
First published on 28th September 2021
Abstract
The reaction of the titanium imido complex 1b with 2-butyne leads to the formation of the titanium azadiene complex 2a at ambient temperature instead of yielding the archetypical [2 + 2] cycloaddition product (titanaazacyclobutene) which is usually obtained by combining titanium imido complexes and internal alkynes. The formation of 2a is presumably caused by an initial propargylic C(sp3)–H activation step and quantum chemical calculations suggest that the outcome of this unexpected reactivity is thermodynamically favored. The previously reported titanaazacyclobutene I (which is obtained by reacting 1b with 1-phenyl-1-propyne) undergoes a rearrangement reaction at elevated temperature to give the corresponding five-membered titanium azadiene complex 2b.
Introduction
The formation of multiple bonds between d- and p-block elements and their resulting reactivities have always excited chemists, and have lead (and presumably will always lead) to novel groundbreaking discoveries which ask fundamental questions in the analyses of structure and bonding.1 Among these multiply bonded compounds, early transition metal imido complexes bearing [RN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ]2− ligands (formally the dianion of a primary amine) are arguably one of the most important and well-understood.2 In this context, solely for the chemistry of titanium imido complexes diverse reactivities have been reported ranging from metathesis reactions, insertions, and cycloadditions to C–H bond activations.2 With regard to catalysis, imido complexes have been employed as robust supporting ligands in Ziegler–Natta type olefin polymerization, ring-opening metathesis, and are used in the industrial production of acrylonitrile.2e,3,4 Furthermore, titanium imido complexes continue to gain momentum in stoichiometric and catalytic hydroamination, nitrene transfer, and oxidative amination reactions.5,6 The fundamental initial reaction step of these last mentioned transformations is the [2 + 2] cycloaddition of a multiple bond substrate (e.g. an alkyne) to the titanium imido fragment to give four-membered systems which can then be further functionalized (e.g. by subsequent insertion of another substrate). These [2 + 2] cycloaddition products of alkynes and terminal imido complexes were observed, isolated, and characterized on several occasions, and undoubtedly the early work by Bergman and co-workers laid the foundation for the later developed catalytic applications.7 Although the [2 + 2] cycloaddition reaction between titanium imido complexes and alkynes is by far the most dominantly observed pathway for early transition metals (selected example Scheme 1, top), two unusual reaction pathways between these compounds were unveiled in recent years. These exceptions might be particularly interesting in the future because they might lead to novel catalytic transformations, give additional mechanistic insight, and allow the isolation of heterocycles which are not accessible by the [2 + 2] cycloaddition route. For terminal titanium imido complexes, Mountford et al. reported the formation of a titanaazetidine (e.g.III) with an exocyclic carbon carbon double bond by reacting titanium imido complexes (II) bearing tridentate N,N,N-supporting ligands with methyl-substituted alkynes (Scheme 1, second from top).8 Mashima, Tsurugi, and co-workers reported the activation of ortho C(sp2)–H aryl bonds at the N-aryl-substituent in imido bridged titanium complexes IV when reacted with 1-(trimethylsilyl)propyne to give the six-membered dinuclear titanium complex V (Scheme 1, third from top).9 Herein, we report on another exception to the perceived well-understood chemistry of early transition metal imido complexes. The reactions of the bis(cyclopentadienyl)-based titanium imido complex 1b with methyl-substituted alkynes lead to the formation of five-membered titanium azadiene complexes 2a,b by initial C–H activation of an internal alkyne (Scheme 1, bottom).
]2− ligands (formally the dianion of a primary amine) are arguably one of the most important and well-understood.2 In this context, solely for the chemistry of titanium imido complexes diverse reactivities have been reported ranging from metathesis reactions, insertions, and cycloadditions to C–H bond activations.2 With regard to catalysis, imido complexes have been employed as robust supporting ligands in Ziegler–Natta type olefin polymerization, ring-opening metathesis, and are used in the industrial production of acrylonitrile.2e,3,4 Furthermore, titanium imido complexes continue to gain momentum in stoichiometric and catalytic hydroamination, nitrene transfer, and oxidative amination reactions.5,6 The fundamental initial reaction step of these last mentioned transformations is the [2 + 2] cycloaddition of a multiple bond substrate (e.g. an alkyne) to the titanium imido fragment to give four-membered systems which can then be further functionalized (e.g. by subsequent insertion of another substrate). These [2 + 2] cycloaddition products of alkynes and terminal imido complexes were observed, isolated, and characterized on several occasions, and undoubtedly the early work by Bergman and co-workers laid the foundation for the later developed catalytic applications.7 Although the [2 + 2] cycloaddition reaction between titanium imido complexes and alkynes is by far the most dominantly observed pathway for early transition metals (selected example Scheme 1, top), two unusual reaction pathways between these compounds were unveiled in recent years. These exceptions might be particularly interesting in the future because they might lead to novel catalytic transformations, give additional mechanistic insight, and allow the isolation of heterocycles which are not accessible by the [2 + 2] cycloaddition route. For terminal titanium imido complexes, Mountford et al. reported the formation of a titanaazetidine (e.g.III) with an exocyclic carbon carbon double bond by reacting titanium imido complexes (II) bearing tridentate N,N,N-supporting ligands with methyl-substituted alkynes (Scheme 1, second from top).8 Mashima, Tsurugi, and co-workers reported the activation of ortho C(sp2)–H aryl bonds at the N-aryl-substituent in imido bridged titanium complexes IV when reacted with 1-(trimethylsilyl)propyne to give the six-membered dinuclear titanium complex V (Scheme 1, third from top).9 Herein, we report on another exception to the perceived well-understood chemistry of early transition metal imido complexes. The reactions of the bis(cyclopentadienyl)-based titanium imido complex 1b with methyl-substituted alkynes lead to the formation of five-membered titanium azadiene complexes 2a,b by initial C–H activation of an internal alkyne (Scheme 1, bottom).
 | ||
| Scheme 1 Overview of the reactivity of titanium imido complexes with internal alkynes. | ||
These results are supported by multinuclear NMR experiments, single crystal X-ray diffraction and quantum chemical calculations.
Results and discussion
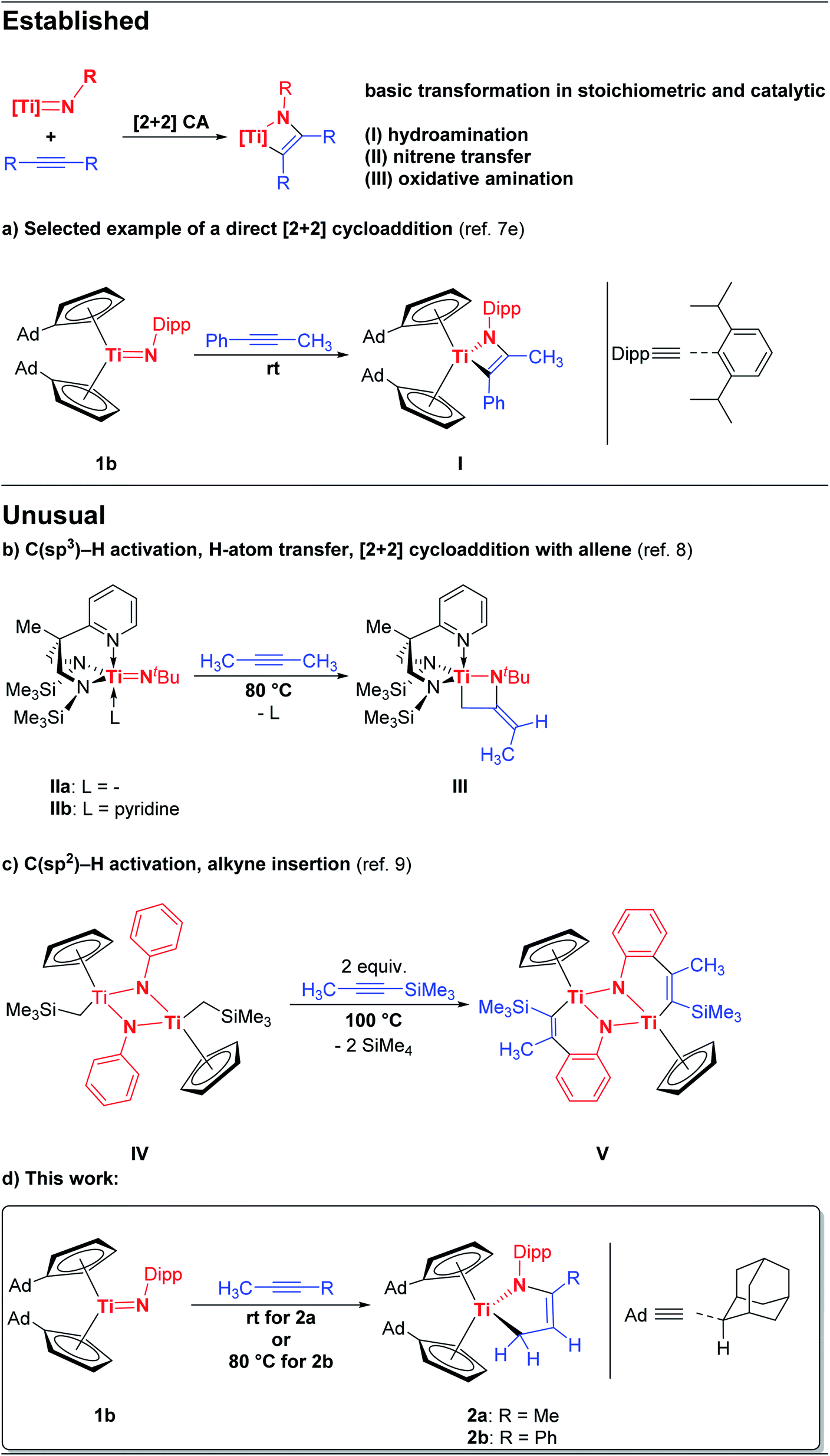
In a recent work we reported the preparation of the chemical equilibrium between the titanium monoamide 1a and the titanium imido complex 1b.7e Enclosed reactivity studies of 1a/1b with multiple bond substrates furnished a broad range of [2 + 2] cycloaddition products.7e,10 As a representative example, the reaction of 1a/1b with 1-phenyl-1-propyne regioselectively yielded the corresponding titatanaazacyclobutene I, in which the phenyl substituent is localized in position α to the titanium centre (Scheme 1, top). Interestingly, the reaction of 1a/1b with 2-butyne under the same reaction conditions as the reaction of 1a/1b with 1-phenyl-1-propyne (n-hexane, 20 h, rt) was already being probed at the same time, but quite complex reaction mixtures were obtained and the expected [2 + 2] cycloaddition product was not observed, which naturally inspired us to take a closer look at this outcome.Accordingly, the equilibrium of 1a/1b was reacted with 2-butyne in n-hexane over a significantly increased time period of five days at room temperature which was accompanied by an observable color change of the reaction mixture from red to maroon. Gratifyingly, removal of all volatile components and subsequent 1H NMR analysis revealed, that the starting material 1a/1b was completely consumed and a new complex was formed. Unexpectedly, 3 was unequivocally identified as the titanium azadiene complex 2a (Scheme 2), thus the reaction of 1a/1b with 2-butyne neither yielded the expected [2 + 2] cycloaddition product comparable to I nor a titanaazetidine (cf.III) which was previously observed by Mountford et al. (Scheme 1).
 | ||
| Scheme 2 Reaction of 1a/1b with 2-butyne to yield the titanium azadiene complex 2a. | ||
Noteworthy, titanium azadiene complexes are typically prepared by reductive complexation of azadiene ligand precursors and, due to the unavailability of CH2 terminated azadienes, only α-C-substituted titanium azadiene complexes are synthetically available by this route.11,12 The hitherto only other reported procedure of preparing TiCH2 substituted titanium azadiene complexes employs allylamines and (FvAd)2Ti.13 Complex 2a is obtained in a good isolated yield of 75% and shows good solubilities in aliphatic and aromatic hydrocarbons such as n-hexane, toluene, and tetrahydrofuran. Multinuclear NMR spectroscopy is already indicative for the formation of the five-membered heterocycle 2a (Fig. S1–S7†). Highly characteristic is the 13C{1H} NMR signal at δ13C{1H} = 64.9 ppm with a negative phase in the 13C{1H} DEPT 135 NMR spectrum, being in the typical region for methylene groups in position α to titanium.13 The signals of the respective chemically non-equivalent Ti–CH2 hydrogen atoms are localized at δ1H = 0.82 and 2.37 ppm. Both signals couple in the 1H/1H COSY spectrum to a multiplet signal at δ1H = 5.10 ppm with the corresponding 13C{1H} NMR signal at δ13C{1H} = 111.6 ppm. The remaining signals of the methyl group and the belonging quaternary carbon atom at δ1H = 1.69 ppm (δ13C{1H} = 20.6 ppm) and δ13C{1H} = 140.6 ppm, together with the expected cross peaks in the 2D NMR spectra already allow for the assignment of connectivity in 2a. Additionally, both the 1H NMR signals of the methyl group and the olefinic hydrogen atoms show coupling to the nitrogen atom in the 1H/15N HMBC spectrum (δ15N = 266.3 ppm). All these chemical shifts are in good agreement to other structurally characterized titanium azadienes,11,13e.g. the recently reported complex VI13 (Table 1).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)

The presence of eight signals in the 1H NMR spectrum (δ1H = 4.93–5.93 ppm) for the cyclopentadienyl hydrogen atoms and distinct signals for the Dipp (2,6-diisopropylphenyl) moiety are caused by the different chemical environments above and below the central five-membered ring system. In this context, the inhibition of the characteristic envelope rearrangement of titanium azadiene complexes11,13 is not observed in the 1H NMR spectrum of 2a in the temperature range from 213 K to 353 K (excerpt of the VT 1H NMR experiment: Fig. S8†).
Crystals of 2a suitable for single crystal X-ray diffraction were obtained by slow evaporation of an n-hexane solution of 2a. The molecular structure is shown in Fig. 1 and confirms the information obtained from the solution NMR analyses.
 | ||
| Fig. 1 Molecular structure of 2a. Hydrogen atoms (except H6, H21, and those of the central five-membered ring system) and fragments of the adamantyl groups are omitted for clarity. Thermal ellipsoids are shown at the 50% probability level. Selected bond lengths (Å) and angles (deg): Ti1–N1 2.0201(7), Ti1–C43 2.1840(9), N1–C45 1.3851(11), C43–C44 1.4561(14), C44–C45 1.3807(13), N1–Ti1–C43 83.89(3), Ct1–Ti1–Ct2 128.3, Σ∠N1 359.9, Σ∠C44 359.4, Σ∠C45 359.5, Ti1–C43–H43A/H43A–C43–C44 70.4, Ti1–C43–H43B/H43B–C43–C44 86.5 (Ct1 = centroid of C1–C5; Ct2 = centroid of C16–C20). | ||
The central titanium atom is in a trigonal pyramidal coordination environment with respect to the τ4/τδ values of τ4 = 0.83 and τδ = 0.69, respectively (the centroids of the Cp ligands have been used for the calculation).14 The above mentioned envelope structure of this diene type complex is demonstrated by a fold angle of the central five-membered ring system of 49.3° which is in good agreement to other structurally characterized titanium azadiene complexes (e.g. 44.4° for VI).11,13 The Ti1–N1 bond length of 2.0201(7) Å and the Ti1–C43 bond length of 2.1840(9) Å are in reasonable accordance to the respective sums of covalent radii (∑cov(Ti–N) = 2.02 Å, ∑cov(Ti–C) = 2.11 Å (ref. 15)). The C43–C44 bond length of 1.4561(14) Å and the N1–C45 bond length of 1.3851(11) constitute a slightly shortened C(sp3)–C(sp2) single bond and a N–C(sp2) single bond (C(sp3)–C(sp2) = 1.51 Å, N–C(sp2) = 1.38 Å (ref. 16)), respectively. The C44–C45 bond length of 1.3807(13) Å is elongated compared to a typical double bond between two sp2-hybridized carbon atoms (C(sp2)C(sp2) = 1.32 Å (ref. 16)).
To gain further insight into the significantly different reactivity of 1a/1b toward 2-butyne compared to the reactions of internal alkynes with other titanium imido complexes, quantum chemical calculations were performed.17
The Gibbs free enthalpy difference ΔrG for the formation of 2a was found to be ΔrG = −0.63 eV which is significantly more exergonic as compared to the corresponding [2 + 2] cycloaddition product (ΔrG = −0.16 eV). The formation of a titanaazetidine (cf.III) could further be excluded being only slightly exergonic (ΔrG = −0.10 eV). Thus, the formation of 2a is thermodynamically favored.
With regard to titanium catalyzed hydroamination reactions, this unexpected titanium azadiene formation could add another discussable part to the selectivity of this reaction type since both a typical [2 + 2] cycloaddition product VII and the corresponding titanium azadiene complex VIII would (in principle) give the same enamine IX after formal twofold protonation (Scheme 3).
 | ||
| Scheme 3 Formal twofold protonation of both a typical [2 + 2] cycloaddition product VII and of a corresponding titanium azadiene VIII to yield the same enamine IX. | ||
Intriguingly, the quantum chemical calculations showed that even in the previously reported reaction of 1a/1b with 1-phenyl-1-propyne, the corresponding titanium azadiene complex should be the thermodynamically favored product of this reaction and not the [2 + 2] cycloaddition product I. Here, a Gibbs free energy difference of ΔrG = −0.52 eV for the formation of the titanium azadiene complex, and ΔrG = −0.26 eV for the formation of I was found.
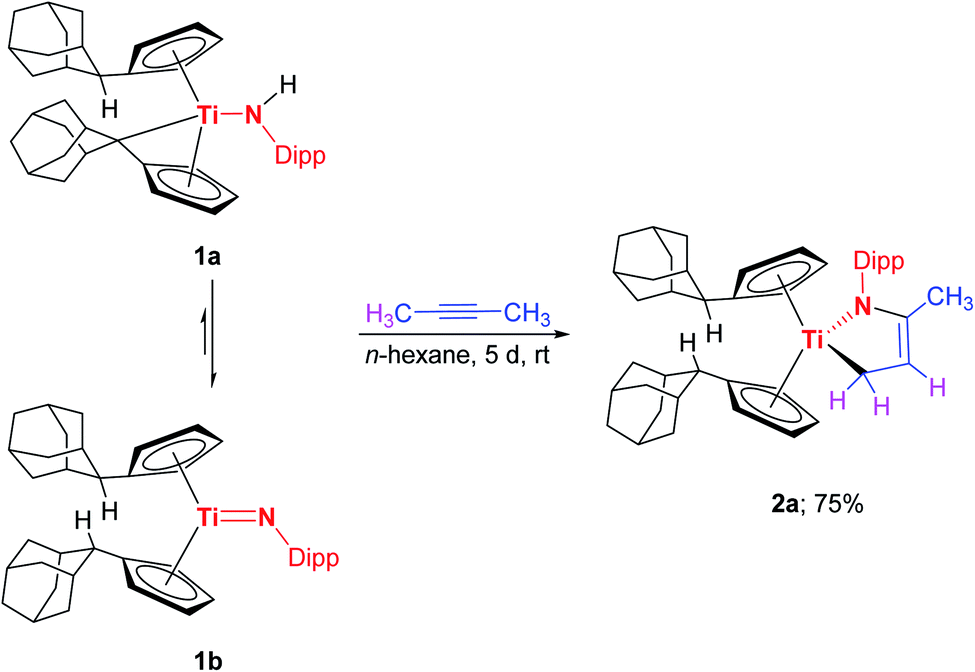
To verify this quantum chemical results, I was synthesized according to the literature procedure (Scheme 4).7e
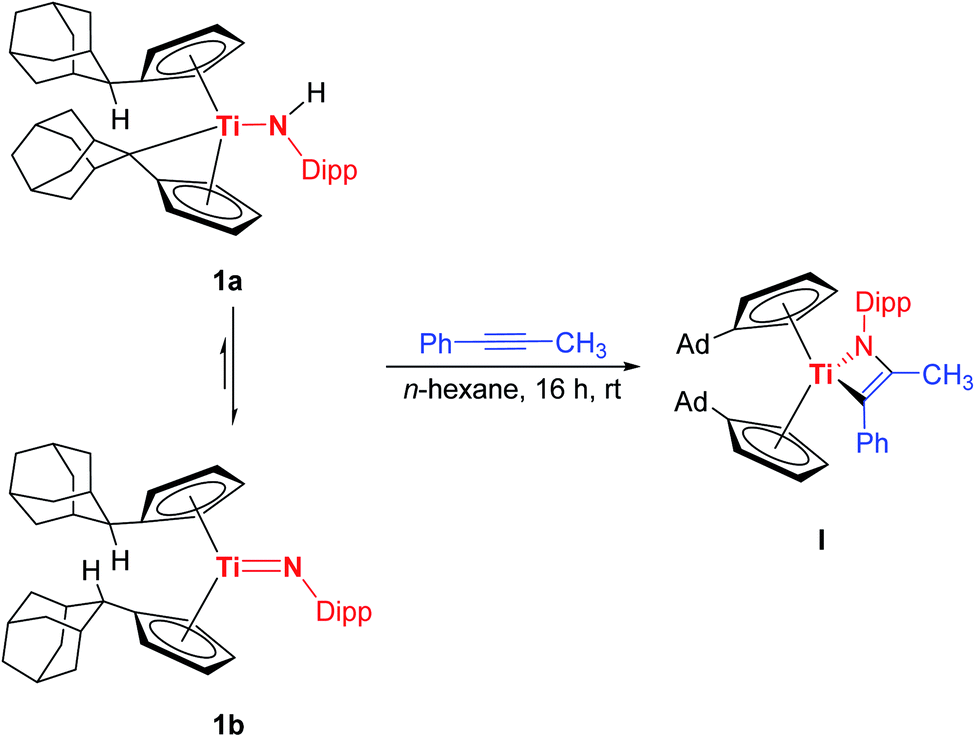 | ||
| Scheme 4 Reaction of 1a/1b with 1-phenyl-1-propyne to yield the titanaazacyclobutene I. | ||
I has been comprehensively characterized before.7e In the course of this work crystals of I suitable for single crystal X-ray diffraction could be obtained with the molecular structure being shown in Fig. 2, unambiguously verifying that indeed the formal [2 + 2] cycloaddition product is formed with the phenyl substituent being in position α to the titanium centre. It should be mentioned here, although we consider it unlikely, that in principle a formal σ-bond insertion starting from the titanium amide 1a could also lead to the formation of I.18
 | ||
| Fig. 2 Molecular structure of I. Hydrogen atoms (except H6 and H21) and fragments of the adamantyl groups are omitted for clarity. Thermal ellipsoids are shown at the 50% probability level. Selected bond lengths (Å) and angles (deg): Ti1–N1 2.034(2), Ti1–C45 2.107(3), N1–C44 1.413(3), C44–C45 1.364(4), N1–Ti1–C45 67.47(10), Ct1–Ti1–Ct2 130.9, Σ∠N1 355.1, Σ∠C44 359.1, Σ∠C45 356.0 (Ct1 = centroid of C1–C5; Ct2 = centroid of C16–C20). | ||
According to the τ4/τδ values of τ4 = 0.78 (τd = 0.71), the coordination environment at the titanium centre is best described as trigonal pyramidal and the central four-membered ring system forms a nonplanar kite shape.14 The bond lengths within the four-membered ring are as one would expect. The Ti1–N1 and Ti1–C45 bond lengths of 2.034(2) Å and 2.107(3) Å are in good accordance to the respective single bond covalent radii (∑cov(Ti–N) = 2.07 Å, ∑cov(Ti–C) = 2.11 Å (ref. 15)), the C44–C45 bond length of 1.364(4) Å is in the range of the respective double bond (C(sp2)C(sp2) = 1.32 Å (ref. 16)), and the N1–C44 bond length of 1.413(3) Å constitutes an elongated N–C(sp2) single bond (N–C(sp2) = 1.38 Å (ref. 8)), indicative for electron donation from the nitrogen lone pair to the electropositive metal centre.
Given the results of the quantum chemical calculations, and the energy difference of 0.26 eV between I and the to 2a corresponding titanium azadiene complex 2b, a solution of I in deuterated benzene was first heated to 50 °C for several hours, and subsequently to 80 °C due to the slow reaction process at 50 °C (Fig. S17†). This variable temperature 1H NMR experiment showed a quite clean and irreversible conversion of I to the corresponding titanium azadiene complex 2b (Scheme 5).
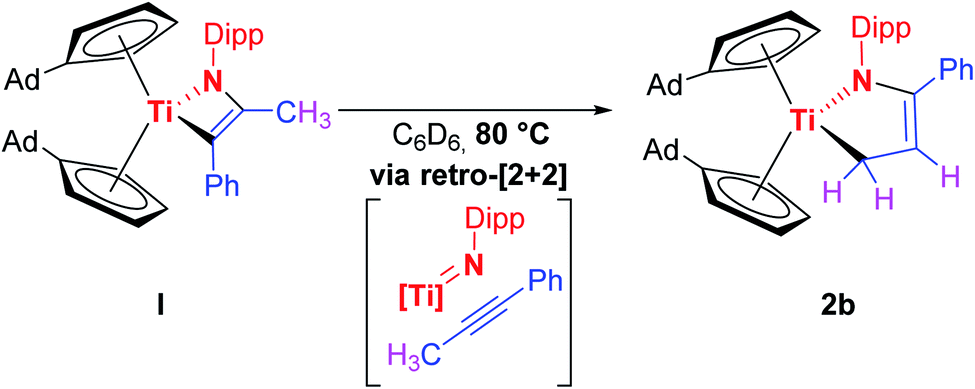 | ||
| Scheme 5 Thermally induced formation of 2b from I. | ||
Multinuclear NMR experiments (Fig. S10–S16†) show excellent agreement of the significant signals to those observed for 2a. Furthermore, crystals suitable for single crystal X-ray diffraction were obtained by slow evaporation of a C6D6 solution of 2b at room temperature, unambiguously verifying this thermally induced rearrangement (Fig. 3).
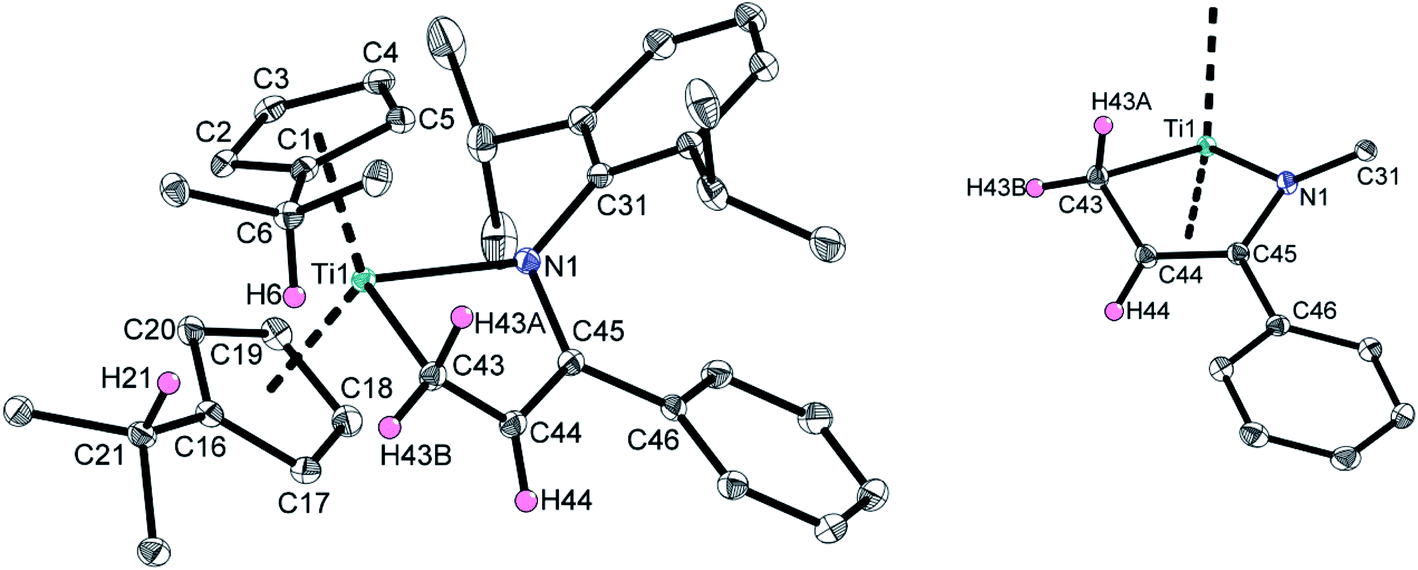 | ||
| Fig. 3 Molecular structure of 2b. Hydrogen atoms (except H6, H21, and except those of the central five-membered ring system) and fragments of the adamantyl groups are omitted for clarity. Thermal ellipsoids are shown at the 50% probability level. Selected bond lengths (Å) and angles (deg): Ti1–N1 2.0761(10), Ti1–C43 2.1724(12), N1–C45 1.3861(14), C43–C44 1.4535(16), C44–C45 1.3851(16), N1–Ti1–C43 82.50(4), Ct1–Ti1–Ct2 128.6, Σ∠N1 359.6, Σ∠C44 360.0, Σ∠C45 360.0 (Ct1 = centroid of C1–C5; Ct2 = centroid of C16–C20). | ||
Since the structural parameters and solution NMR data of 2a and 2b are in very good agreement, and have been comprehensively discussed for 2a, no detailed discussion for 2b is given here.
Mechanistically, we think that the initial step of the reaction is the C–H activation of the methyl group of the alkyne by the titanium imido complex to give the corresponding titanapropargyl XI. A precoordination of the alkyne to the titanium center to give an alkyne-titanium π-complex (X) is suggested to lower the pKa of the propargylic hydrogen atom(s) in the first place.19 Even though this type of reactivity is very rare for titanium imido complexes, it was observed for reversible C(sp3)–H activation processes in the case of alkanes.2b,20 Just recently, Tonks et al. also observed propargylic C–H activation of 3-hexyne by a titanium bisamido complex.21 Subsequently, the carbon–carbon triple bond inserts into the Ti–N bond which leads to the formation of the titanacyclopropane derivative XII with an enamine functionality in the backbone. This insertion step was already discussed by the Doye group as a possible alternative reaction pathway for titanium catalyzed hydroamination reactions,22 and is common for group 3 metals.5c,23 Enamine–imine tautomerization and ring expansion, similar to the chemistry of group 4 cumulene complexes,24 finally yields the titanium azadiene complex 2a. The thermally induced rearrangement of I to give 2b suggests that in this case a [2 + 2] retro-cycloaddition is the first step (Scheme 5). Alternatively, a loose coordination of the nitrogen lone pair to the metal centre in XII could trigger a 1,3-H-shift to directly give 2a,b. Noteworthy, the general possibility for a [2 + 2] cycloaddition pathway followed by multiple H-shifts can not be ruled out completely at this point. The proposed mechanism is shown in Scheme 6.
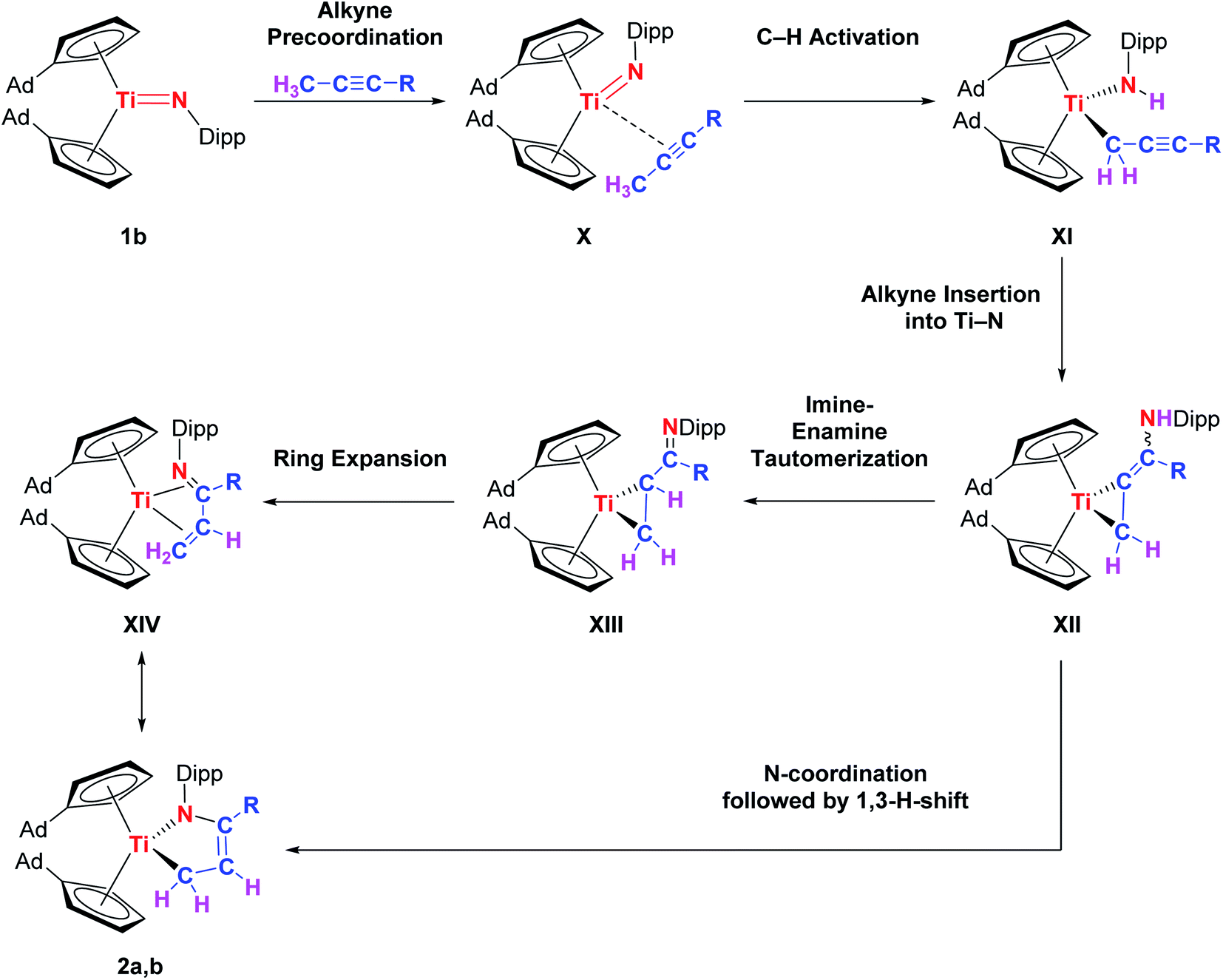 | ||
| Scheme 6 Proposed mechanism for the formation of 2a,b. | ||
Conclusion
In this contribution an exception from the so far observed reactivity between titanium imido complexes and internal alkynes is reported. By employing the bis(cyclopentadienyl) titanium imido complex 1b, the formation of the titanium azadienes 2a,b can be achieved in reactions with the methyl-substituted alkynes 2-butyne and 1-phenyl-1-propyne, respectively. Whereas, in the case of 2-butyne the reaction proceeds at room temperature, the formation of the titanium azadiene complex 2b is realized by heating a solution of the known [2 + 2] cycloaddition product I for a prolonged time. The titanium azadiene complexes were comprehensively characterized in solution by multinuclear NMR spectroscopy and in the solid state by single crystal X-ray diffraction. These unusual reactions between an early transition metal imido complex and internal alkynes via an initial C(sp3)–H activation step might induce new possibilities in hydroamination and nitrene transfer chemistry in the near future. Currently, extensive experimental and theoretical investigations on the proposed mechanism are being investigated in our laboratories.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
M. F.: conceptualization, experimentation, data analysis, drafting the manuscript and writing the final version with contributions from all authors; M. M.: conceptualization, experimentation, data analysis; M. S.: single crystal X-ray diffraction; T. K.: computational analysis; R. B.: conceptualization, project coordination.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. F. and M. M. are thankful to the Alexander von Humboldt Foundation for funding and support. The simulations were performed at the HPC Cluster CARL, located at the University of Oldenburg (Germany) and funded by the DFG through its Major Research Instrumentation Programme (INST 184/157-1 FUGG) and the Ministry of Science and Culture (MWK) of the State of Lower Saxony. M. F. would like to thank his friend Vladyslav Kukulevskyy for the drawing of the table of content.References
- (a) W. A. Herrmann, Angew. Chem. Int. Ed., 1986, 25, 56–76 ( Angew. Chem. , 1986 , 98 , 57–77 ) CrossRef; (b) E. Rivard and P. P. Power, Inorg. Chem., 2007, 46, 10047–10064 CrossRef CAS PubMed.
- (a) P. D. Bolton and P. Mountford, Adv. Synth. Catal., 2005, 347, 355–366 CrossRef CAS; (b) N. Hazari and P. Mountford, Acc. Chem. Res., 2005, 38, 839–849 CrossRef CAS PubMed; (c) D. N. Zarubin and N. A. Ustynyuk, Russ. Chem. Rev., 2006, 75, 671–707 CrossRef CAS; (d) A. R. Fout, U. J. Kilgore and D. J. Mindiola, Chem.–Eur. J., 2007, 13, 9428–9440 CrossRef CAS PubMed; (e) R. R. Schrock, Chem. Rev., 2009, 109, 3211–3226 CrossRef CAS PubMed; (f) T. L. Gianetti, H. S. La Pierre and J. Arnold, Eur. J. Inorg. Chem., 2013, 2013, 3771–3783 CrossRef CAS; (g) K. Kawakita, B. F. Parker, Y. Kakiuchi, H. Tsurugi, K. Mashima, J. Arnold and I. A. Tonks, Coord. Chem. Rev., 2020, 407, 213118 CrossRef CAS PubMed.
- (a) C. T. Owen, P. D. Bolton, A. R. Cowley and P. Mountford, Organometallics, 2007, 26, 83–92 CrossRef CAS; (b) R. Arteaga-Müller, J. Sánchez-Nieves, J. Ramos, P. Royo and M. E. G. Mosquera, Organometallics, 2008, 27, 1417–1426 CrossRef.
- T. Hanna, Coord. Chem. Rev., 2004, 248, 429–440 CrossRef CAS.
- Selected reviews: (a) T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675–703 CrossRef PubMed; (b) R. Severin and S. Doye, Chem. Soc. Rev., 2007, 36, 1407–1420 RSC; (c) T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed; (d) A. L. Odom and T. J. McDaniel, Acc. Chem. Res., 2015, 48, 2822–2833 CrossRef CAS PubMed; (e) M. Manßen and L. L. Schafer, Chem. Soc. Rev., 2020, 49, 6947–6994 RSC.
- Selected examples for nitrene transfer reactions with group 4 complexes: (a) A. I. Nguyen, R. A. Zarkesh, D. C. Lacy, M. K. Thorson and A. F. Heyduk, Chem. Sci., 2011, 2, 166–169 RSC; (b) A. A. Dissanayake and A. L. Odom, Chem. Commun., 2012, 48, 440–442 RSC; (c) C. M. Pasko, A. A. Dissanayake, B. S. Billow and A. L. Odom, Tetrahedron, 2016, 72, 1168–1176 CrossRef CAS; (d) Z. W. Gilbert, R. J. Hue and I. A. Tonks, Nat. Chem., 2016, 8, 63–68 CrossRef CAS PubMed; (e) S. P. Heins, P. T. Wolczanski, T. R. Cundari and S. N. MacMillan, Chem. Sci., 2017, 8, 3410–3418 RSC; (f) Z. W. Davis-Gilbert, L. J. Yao and I. A. Tonks, J. Am. Chem. Soc., 2016, 138, 14570–14573 CrossRef CAS PubMed; (g) A. J. Pearce, X. Y. See and I. A. Tonks, Chem. Commun., 2018, 54, 6891–6894 RSC; (h) H.-C. Chiu and I. A. Tonks, Angew. Chem. Int. Ed., 2018, 57, 6090–6094 ( Angew. Chem. , 2018 , 130 , 6198–6202 ) CrossRef CAS PubMed; (i) Z. W. Davis-Gilbert, X. Wen, G. D. Goodpaster and I. A. Tonks, J. Am. Chem. Soc., 2018, 140, 7267–7281 CrossRef CAS PubMed; (j) Z. W. Davis-Gilbert, K. Kawakita, D. R. Blechschmidt, H. Tsurugi, K. Mashima and I. A. Tonks, Organometallics, 2018, 37, 4439–4445 CrossRef CAS PubMed; (k) E. P. Beaumier, M. E. McGreal, A. R. Pancoast, R. H. Wilson, K. T. Moore, B. J. Graziano, J. D. Goodpaster and I. A. Tonks, ACS Catal., 2019, 9, 11753–11762 CrossRef CAS PubMed; (l) A. J. Pearce, R. P. Harkins, B. R. Reiner, A. C. Wotal, R. J. Dunscomb and I. A. Tonks, J. Am. Chem. Soc., 2020, 142, 4390–4399 CrossRef CAS PubMed; (m) E. P. Beaumier, A. A. Ott, X. Wen, Z. W. Davis-Gilbert, T. A. Wheeler, J. J. Topczewski, J. D. Goodpaster and I. A. Tonks, Chem. Sci., 2020, 11, 7204–7209 RSC.
- Further selected examples in which group 4 azacyclobutenes were isolated: (a) P. J. Walsh, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1988, 110, 8729–8731 CrossRef CAS; (b) P. J. Walsh, A. M. Baranger and R. G. Bergman, J. Am. Chem. Soc., 1992, 114, 1708–1719 CrossRef CAS; (c) N. Vujikovic, B. D. Ward, A. Maisse-Francois, H. Wadepohl, P. Mountfourd and L. Gade, Organometallics, 2007, 26, 5522–5534 CrossRef; (d) P. D. Bolton, M. Feliz, A. R. Cowley, E. Clot and P. Mountford, Organometallics, 2008, 27, 6096–6110 CrossRef CAS; (e) M. Manßen, S. de Graaff, M. F. Meyer, M. Schmidtmann and R. Beckhaus, Organometallics, 2018, 37, 4506–4514 CrossRef; (f) M. Fischer, M. C. Wolff, E. del Horno, M. Schmidtmann and R. Beckhaus, Organometallics, 2020, 39, 3232–3239 CrossRef CAS.
- (a) A. Bashall, M. McParlin, P. E. Collier, P. Mountford, L. H. Gade and D. J. M. Trösch, Chem. Commun., 1998, 2555–2556 RSC; (b) D. J. M. Trösch, P. E. Collier, A. Bashall, L. H. Gade, M. McPartlin, P. Mountford and S. Radojevic, Organometallics, 2001, 20, 3308–3313 CrossRef.
- H. Nagae, W. Hato, K. Kawakita, H. Tsurugi and K. Mashima, Chem.–Eur. J., 2017, 23, 586–596 CrossRef CAS PubMed.
- The corresponding titanium imido pyridine adduct of 1a/1b proved to be completely inert in reactions with multiple bond substrates.7e.
- (a) M. Fischer, M. Schmidtmann and R. Beckhaus, Organometallics, 2017, 36, 4779–4793 CrossRef CAS; (b) F. Loose, I. Plettenberg, D. Haase, W. Saak, M. Schmidtmann, A. Schäfer, T. Müller and R. Beckhaus, Organometallics, 2014, 33, 6785–6795 CrossRef CAS; (c) S. Kahlert, H. Görls and J. Scholz, Angew. Chem. Int. Ed., 1998, 37, 1857–1861 ( Angew. Chem. , 1998 , 37 , 1857–1861 ) CrossRef CAS; (d) J. Scholz, S. Kahlert and H. Görls, Organometallics, 1998, 17, 2876–2884 CrossRef CAS; (e) J. Scholz, M. Nolte and C. Krüger, Chem. Ber., 1993, 126, 803–809 CrossRef CAS; (f) J. M. Davis, R. J. Whitby and A. Jaxa-Chamiec, J. Chem. Soc., Chem. Commun., 1991, 1743–1745 RSC.
- H. K. Hall, Makromol. Chem., Macromol. Symp., 1992, 54–55, 73–81 CrossRef.
- (a) M. Manßen, I. Töben, C. Kahrs, J.-H. Bölte, M. Schmidtmann and R. Beckhaus, Organometallics, 2017, 36, 2973–2981 CrossRef; (b) M. Manßen, C. Kahrs, I. Töben, J.-H. Bölte, M. Schmidtmann and R. Beckhaus, Chem.–Eur. J., 2017, 23, 15827–15833 CrossRef PubMed.
- (a) M. H. Reineke, M. D. Sampson, A. L. Rheingold and C. P. Kubiak, Inorg. Chem., 2015, 54, 3211–3217 CrossRef CAS PubMed; (b) L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- (a) P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed; (b) P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- M. B. Smith and J. March, Advanced Organic Chemistry, 6th edn, Wiley, New York, 2007 Search PubMed.
- All calculations were performed using version ES64L-G16RevA.03 of Gaussian 16. Using a cc-pVTZ basis set, the geometry of all species were optimized within density functional theory using the B97D3 functional for exchange and correlation. Subsequent frequency calculations were performed to exclude imaginary frequencies of all obtained structures and to obtain thermodynamic properties (T = 298, 15 K, p = 1 atm). M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas,J. B. Foresman, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- Selected examples for this reactivity: (a) D. C. Leitch, C. S. Turner and L. L. Schafer, Angew. Chem. Int. Ed., 2010, 49, 6382–6386 ( Angew. Chem. , 2010 , 122 , 6526–6530 ) CrossRef CAS PubMed; (b) D. V. Gribkov and K. C. Hultzsch, Angew. Chem. Int. Ed., 2004, 43, 5542–5546 ( Angew. Chem. , 2004 , 116 , 5659–5663 ) CrossRef CAS PubMed.
- Y. Wang, J. Zhu, A. C. Durham, H. Lindberg and Y.-M. Wang, J. Am. Chem. Soc., 2019, 141, 19594–19599 CrossRef CAS PubMed.
- J. L. Bennett and P. T. Wolczanski, J. Am. Chem. Soc., 1997, 119, 10696–10719 CrossRef CAS.
- A. J. Pearce, Y. Cheng, R. J. Dunscomb and I. Tonks, Organometallics, 2020, 39, 3771–3774 CrossRef CAS PubMed.
- C. Müller, R. Koch and S. Doye, Chem.–Eur. J., 2008, 14, 10430–10436 CrossRef PubMed.
- (a) F. Lauterwasser, P. G. Hayer, S. Bräse, W. E. Piers and L. L. Schafer, Organometallics, 2004, 23, 2234–2237 CrossRef CAS; (b) L. J. E. Stanlake and L. L. Schafer, Organometallics, 2009, 28, 3990–3998 CrossRef CAS.
- U. Rosenthal, V. V. Burlakov, M. A. Bach and T. Beweries, Chem. Soc. Rev., 2007, 36, 719–728 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2070445–2070447. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04334j |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |