
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulation of amyloid-β aggregation by metal complexes with a dual binding mode and their delivery across the blood–brain barrier using focused ultrasound†
Tiffany G.
Chan
 abc,
Carmen L.
Ruehl
a,
Sophie V.
Morse
b,
Michelle
Simon
a,
Viktoria
Rakers
a,
Helena
Watts
bd,
Francesco A.
Aprile
a,
James J.
Choi
*b and
Ramon
Vilar
*a
abc,
Carmen L.
Ruehl
a,
Sophie V.
Morse
b,
Michelle
Simon
a,
Viktoria
Rakers
a,
Helena
Watts
bd,
Francesco A.
Aprile
a,
James J.
Choi
*b and
Ramon
Vilar
*a
aDepartment of Chemistry, Imperial College London, White City Campus, 82 Wood Lane, London, W12 0BZ, UK. E-mail: r.vilar@imperial.ac.uk
bDepartment of Bioengineering, Imperial College London, London, SW7 2AZ, UK. E-mail: j.choi@imperial.ac.uk
cCentre of Excellence in Neurotechnology, Imperial College London, London, SW7 2AZ, UK
dDepartment of Brain Sciences, Imperial College London, London, W12 0NN, UK
First published on 15th June 2021
Abstract
One of the key hallmarks of Alzheimer's disease is the aggregation of the amyloid-β peptide to form fibrils. Consequently, there has been great interest in studying molecules that can disrupt amyloid-β aggregation. While a handful of molecules have been shown to inhibit amyloid-β aggregation in vitro, there remains a lack of in vivo data reported due to their inability to cross the blood–brain barrier. Here, we investigate a series of new metal complexes for their ability to inhibit amyloid-β aggregation in vitro. We demonstrate that octahedral cobalt complexes with polyaromatic ligands have high inhibitory activity thanks to their dual binding mode involving π–π stacking and metal coordination to amyloid-β (confirmed via a range of spectroscopic and biophysical techniques). In addition to their high activity, these complexes are not cytotoxic to human neuroblastoma cells. Finally, we report for the first time that these metal complexes can be safely delivered across the blood–brain barrier to specific locations in the brains of mice using focused ultrasound.
Introduction
Alzheimer's disease is the most common form of dementia, affecting approximately 50 million people worldwide. One of the key hallmarks of Alzheimer's disease is the accumulation of the amyloid-β (Aβ) peptide in the brain, with post-mortem analyses showing that the total amount of Aβ in Alzheimer's disease brains is significantly higher than in healthy brains. For example, in a patient cohort with an average age of 78 years, previous studies have determined that there was an approximately 3-fold increase in total Aβ in the brains of Alzheimer's disease patients compared to in control patients.1–3 Aβ is produced via the sequential cleavage of the transmembrane protein amyloid precursor protein (APP) by the enzymes β- and γ-secretases.2,4 The resulting peptides are typically 40 (Aβ1-40) or 42 (Aβ1-42) amino acids in length, and they aggregate over time to form soluble oligomers and insoluble plaques in the extracellular space.2,4 The presence of these aggregates in the brain ultimately leads to impaired cell signalling, neuronal death and brain atrophy.5,6Cerebral metal ion levels are known to be altered in Alzheimer's disease and it has been proposed that they could play an important role in the pathology of the disease.7–11 Structural studies have identified a high affinity metal ion binding site at the N-terminal of Aβ (residues 1–16) which can modulate aggregation and the generation of reactive oxygen species upon the binding of endogenous metal ions such as Cu2+, Fe3+ and Zn2+.12–17 Copper(I) is believed to bind to Aβ in a linear two-coordinate fashion through interactions with the histidine residues (His6, His13 and His14), whilst copper(II) is proposed to form square planar complexes with Aβ involving two histidine residues and additional interactions from Asp1 or Ala2.18–22 Binding to His6, His13 and His14 have also been implicated in the binding of iron(II) and zinc(II) ions to Aβ, but the precise mode of binding remains unclear.23,24 Numerous studies have shown that the binding of metal ions to Aβ can promote aggregation, resulting in increased neurotoxic effects.13,14,25,26
It has been suggested that metal complexes that can preferentially bind to the N-terminal metal ion binding site of Aβ could prove to be therapeutically useful.27–30 In 2008, Barnham et al. reported a series of platinum-based complexes containing 1,10-phenanthroline ligands, which were able to interact with this site and inhibit Aβ aggregation in vitro, as well as rescue Aβ-induced neurotoxicity in mouse hippocampal slices ex vivo.31 Subsequent experimental and theoretical studies identified the binding mode of these platinum complexes to be via π–π stacking with the aromatic residues in the pocket (e.g. Tyr10) and coordination to the histidine residues (His6, His13 and His14).32,33 Since this pioneering study, a handful of other metal complexes have been developed for this application, including cobalt Schiff base complexes reported by Meade et al. and iridium benzimidazole complexes reported by Ruiz et al., with promising results being obtained in vitro.34–37 However, as of now, there is just one example of this therapeutic approach being tested in vivo as many of these metal complexes are presumed to be either highly toxic or unable to cross the blood–brain barrier (BBB).36,38
The presence of the BBB remains one of the biggest obstacles to the successful treatment of brain diseases. To date, the only reported method to open the BBB in a manner that is non-invasive, targeted and transient is the use of focused ultrasound and microbubbles.39 In this approach, microbubbles followed by the compound of choice are injected intravenously into the bloodstream and the target region is exposed to ultrasound (Fig. S1†). Microbubbles in an ultrasound field radially oscillate, exerting mechanical stress to their surroundings, delivering the compound across the BBB. Focused ultrasound-mediated BBB opening has been used to successfully deliver a wide range of small molecules, antibodies and nanoparticles into the brains of rodents and non-human primates, and the first human clinical trials are currently underway.40–47
In this work, we report a series of novel octahedral cobalt(III) salphen and salnaph complexes (Fig. 1) as inhibitors of Aβ aggregation. These complexes were designed to interact with Aβ via a dual binding mode involving π–π stacking interactions from the salphen/salnaph ligands and direct coordination via the two axial sites on the cobalt. Furthermore, trimethylammonium substituents were added to the ligands to increase water solubility as well as prevent cellular uptake due to the higher hydrophilicity/charge of the resulting complexes. Reduction of cellular permeability would in turn reduce unwanted cytotoxicity. This design was aimed to address some of the limitations of previously reported metal complexes such as high cytotoxicity and low specificity. Through a series of spectroscopic and biophysical experiments, we have shown that these cobalt complexes have high activity as inhibitors of Aβ aggregation through interactions with its N-terminal. Furthermore, the complexes show limited cytotoxicity against human neuroblastoma cells. Following their excellent in vitro activity, the most promising cobalt(III)–salnaph complex was successfully delivered across the BBB in both wild-type C57BL/6J and transgenic Alzheimer's disease mice using focused ultrasound-mediated BBB opening methods. Preliminary in vivo results indicate that the cobalt(III)–salnaph complex under study is well-tolerated in mice over several weeks with no signs of toxicity observed.
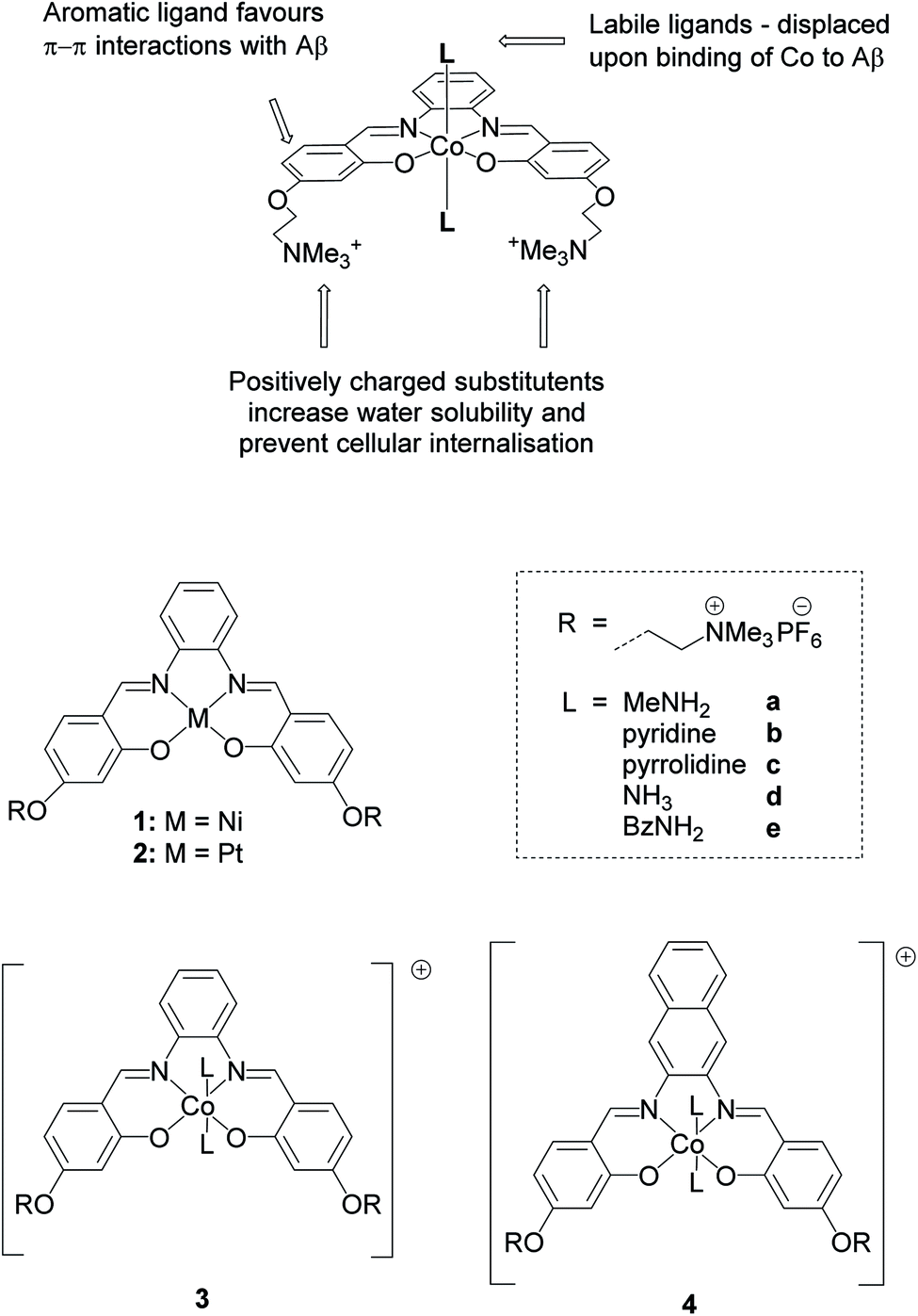 | ||
| Fig. 1 Structures of metal salphens (1, 2 and 3a–3e) and salnaphs (4a–4e) investigated. | ||
Results and discussion
As indicated above, metal complexes that are able to interact with the N-terminal metal binding site of Aβ via π–π stacking and/or histidine coordination have been shown to prevent Aβ aggregation in vitro.31,32,34 Inspired by these previously reported examples, we designed and synthesised a series of metal salphens and salnaphs (Fig. 1), and investigated their potential as inhibitors of Aβ aggregation. Our complexes include square planar nickel(II)- and platinum(II) salphen complexes (1 and 2) that would only be expected to π–π stack with Aβ, as well as a series of octahedral cobalt complexes (3a–3e and 4a–4e) that, in addition to displaying π–π stacking interactions, were designed to coordinate to the histidine residues of Aβ upon release of their axial ligands. Comparison between the square planar and octahedral salphen complexes (1 and 2vs.3a–3e) allows the effects of coordination to be investigated, whilst comparison between the octahedral salphen and salnaph complexes (3a–3evs.4a–4e) allows the probing of non-covalent interactions – in particular, π–π stacking. The octahedral complexes were synthesised with a variety of axial ligands to probe any effects related to the different dissociation kinetics of these ligands from the complexes.Synthesis of metal complexes
Nickel(II) salphen 1 and cobalt(III) salphens 3a and 3d were synthesised following a previously reported procedure.48,49 The new platinum(II) and cobalt(III) salphens 2, 3b, 3c and 3e, and cobalt(III) salnaphs 4a–4e were synthesised by first reacting 4-(4-formyl-3-hydroxyphenoxy)–N,N,N-trimethylethan-1-ammonium bromide with 1,2-phenylendiamine and 2,3-diaminonaphthalene respectively (Scheme 1). Platinum(II) complex 2 was obtained by reacting the salphen ligand with K2PtCl4. To form the desired octahedral cobalt complexes, the salphen or salnaph ligand was added to Co(OAc)2, followed by the axial ligand of choice (i.e. MeNH2, pyridine, pyrrolidine, NH3 or BzNH2). Reactions were conducted in air to oxidise the Co2+ centre to Co3+.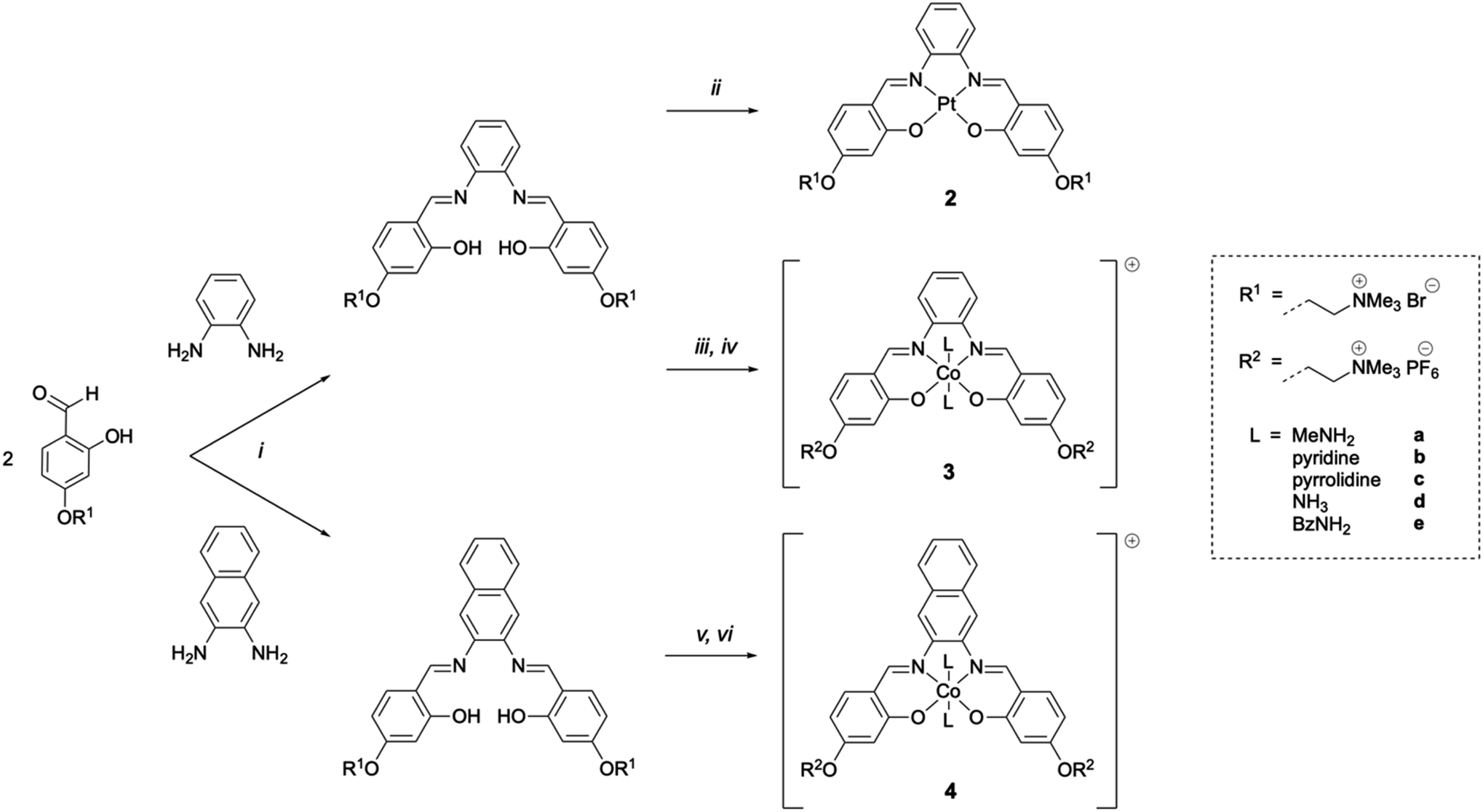 | ||
| Scheme 1 Synthesis routes to complexes 2, 3a–3e and 4a–4e. [i] EtOH, reflux, 5 h, N2; [ii] K2PtCl4, DMSO, 65 °C, 18 h, N2; [iii] Co(OAc)2·4H2O, MeOH, r.t., 1 h, N2; [iv] 10 eq. axial ligand L, aq. sat. NH4PF6, MeOH, r.t., 3 h, air; [v] Co(OAc)2·4H2O, MeOH, r.t., 1 min, air; [vi] 10 eq. axial ligand L, aq. sat. NH4PF6, MeOH, r.t., 5 h, air. | ||
On successful formation of the desired platinum(II) and cobalt(III) complexes, shifts of the aromatic proton resonances in the 1H NMR spectra were observed. Furthermore, the –O![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) resonance of the ligand disappeared upon coordination of the metal and, in the case of the cobalt complexes, new peaks corresponding to the attached axial ligands appeared (Fig. S2†). All complexes were fully characterised by 1H NMR spectroscopy, mass spectrometry and elemental analyses.
resonance of the ligand disappeared upon coordination of the metal and, in the case of the cobalt complexes, new peaks corresponding to the attached axial ligands appeared (Fig. S2†). All complexes were fully characterised by 1H NMR spectroscopy, mass spectrometry and elemental analyses.
To investigate the stability of the cobalt(III) complexes (particularly the dissociation of the axial ligand, L), the 1H NMR spectra of complexes 3a–3e and 4a–4e were determined in aqueous buffer over time. As shown in Fig. S3,† varying the axial ligand allows for a range of stabilities to be obtained, with complexes 3a, 3b, 3d and 4a, 4b, 4d remaining largely unchanged in aqueous buffer up to 8 h. For complexes 3c, 3e, 4c and 4e, the axial ligand is lost more rapidly.
Inhibition of Aβ1-42 aggregation
Metal complexes 1, 2, 3a–3e and 4a–4e were assessed for their ability to inhibit Aβ1-42 aggregation using the Thioflavin-T (ThT) assay. ThT is a benzothiazole dye routinely used to monitor Aβ aggregation that shows strong fluorescence emission when bound to fibrillar structures but low fluorescence emission when free in solution.50 Complexes were incubated in the presence of 5 μM monomeric Aβ at [Aβ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :complex] concentrations ratios = 25:1, 20:1, 15:1, 10:1 and 5:1 at pH 7.4 and 37 °C for 24 h. The % inhibition by the complexes was determined by measuring the ThT fluorescence at the endpoint of aggregations assuming that decreased fluorescence values indicate a reduced final load of fibrils. As shown in Fig. 2 and S4,† all investigated complexes show a concentration-dependent inhibitory effect. Moreover, this inhibition was greater for octahedral complexes 3a–3e and 4a–4e compared to square planar complexes 1 and 2. This supports the hypothesis that the octahedral cobalt(III) complexes do not interact solely through non-covalent interactions but can also coordinate to Aβ upon release of their axial ligands. Furthermore, the fact that greater than 50% inhibitory activity is observed for all complexes at a [Aβ1-42]:[complex] ratio of 5:1 indicates that their interaction with Aβ is strong enough to affect neighbouring Aβ monomers, as well as the Aβ monomer being directly bound to. Similar conclusions were made by Meade et al. on the binding of their cobalt(III) Schiff base complexes to Aβ, in which successful inhibition of Aβ aggregation was observed at sub-equimolar ratios in experimental and theoretical studies.35
:complex] concentrations ratios = 25:1, 20:1, 15:1, 10:1 and 5:1 at pH 7.4 and 37 °C for 24 h. The % inhibition by the complexes was determined by measuring the ThT fluorescence at the endpoint of aggregations assuming that decreased fluorescence values indicate a reduced final load of fibrils. As shown in Fig. 2 and S4,† all investigated complexes show a concentration-dependent inhibitory effect. Moreover, this inhibition was greater for octahedral complexes 3a–3e and 4a–4e compared to square planar complexes 1 and 2. This supports the hypothesis that the octahedral cobalt(III) complexes do not interact solely through non-covalent interactions but can also coordinate to Aβ upon release of their axial ligands. Furthermore, the fact that greater than 50% inhibitory activity is observed for all complexes at a [Aβ1-42]:[complex] ratio of 5:1 indicates that their interaction with Aβ is strong enough to affect neighbouring Aβ monomers, as well as the Aβ monomer being directly bound to. Similar conclusions were made by Meade et al. on the binding of their cobalt(III) Schiff base complexes to Aβ, in which successful inhibition of Aβ aggregation was observed at sub-equimolar ratios in experimental and theoretical studies.35
 | ||
| Fig. 2 Inhibition of Aβ1-42 aggregation (%) induced by metal complexes 1, 2, 3c and 4c determined using the thioflavin-T assay. Complexes were tested at [Aβ42]:[complex] = 25:1, 20:1, 15:1, 10:1 and 5:1, and all samples were incubated at 37 °C with shaking for 24 h. The dotted line represents the half-inhibition point and all data is reported as the mean ± standard deviation of three independent experiments (see Fig. S4† for inhibition studies with all other metal complexes 3a, 3b, 3d, 3e and 4a, 4b, 4d, 4e). | ||
Ideally, the axial ligands attached to cobalt(III) complexes 3a–3e and 4a–4e should remain in place until the complex reaches the target location in the brain, but yet be labile enough to allow for efficient coordination to Aβ. As discussed above, we observe a degree of axial ligand dissociation for all our complexes when dissolved in aqueous buffer (Fig. S3†). The dissociation rate appears to be dependent on the axial ligands attached, with some complexes remaining largely stable for up to 8 h. However, in the presence of Aβ, all complexes 3a–3e and 4a–4e show similar behaviour against Aβ aggregation, suggesting that they all undergo axial ligand dissociation and subsequent coordination in the presence of Aβ (Fig. S4†). Due to the similarities in activity between complexes 3a–3e and 4a–4e, complexes 3c and 4c were selected as representative examples in all subsequent biophysical characterisation.
To confirm that the reduction in fluorescence in Fig. 2 and S4† is linked to the inhibitory strength of the metal complexes, we performed aggregation kinetics at different complex concentrations (using 1, 3c and 4c as representative examples of the complexes under investigation – Fig. 3 and S5†). We normalized the fluorescence data to remove any bias due to potential quenching and only focus on the kinetic aspects of the inhibition (Fig. 3). We found a significant inhibition of the aggregation kinetics for complexes 3c and 4c. Noteworthily, this effect is present at sub-stoichiometric concentrations of the metal complexes. On the contrary, complex 1 did not show a significant inhibition at these concentrations, in agreement with the endpoint analyses (Fig. 2).
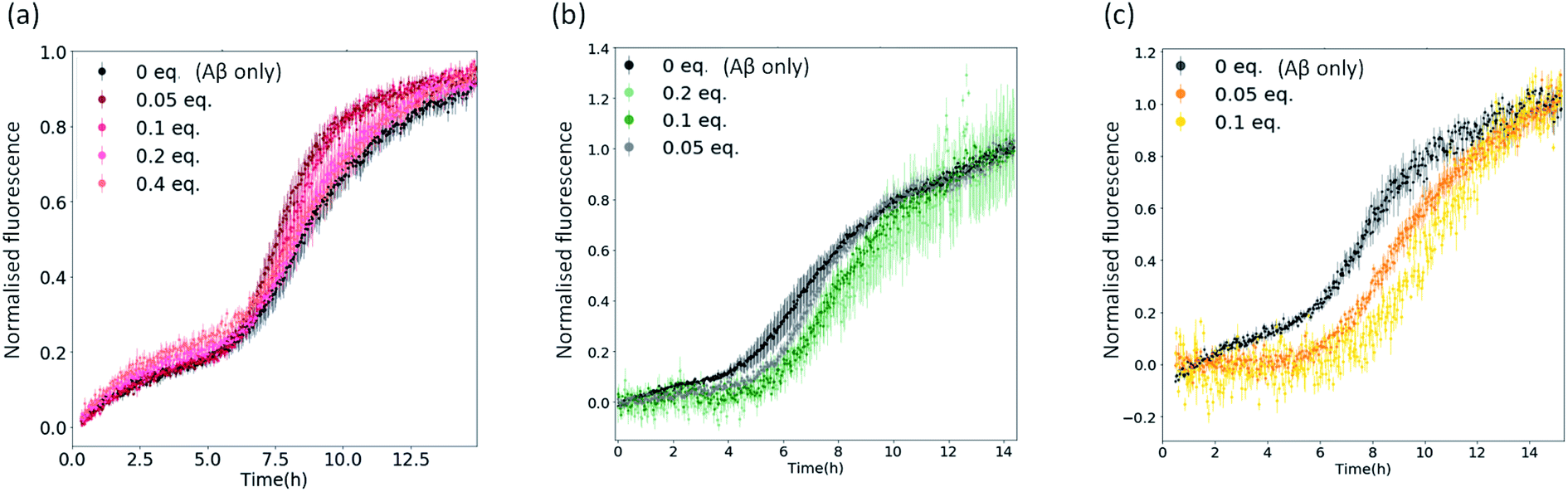 | ||
| Fig. 3 Plots showing normalised ThT fluorescence over time incubated with 5 μM Aβ and increasing amounts of (a) metal complex 1; (b) metal complex 3c; (c) metal complex 4c. | ||
Regardless of the effects on the kinetics of aggregation, the delta fluorescence values (Fig. S5†) corresponding to the normalized aggregation kinetics in Fig. 3 show that all metal complexes, including compound 1, lower the ThT fluorescence at the endpoint of aggregation. This result could be because the complexes decrease the final yield of ThT-positive fibrils and/or displace/quench ThT. To assess the extent to which the complexes displace and/or quench ThT, we generated Aβ1-42 fibrils, added ThT, and recorded their fluorescence in the absence and upon addition of 0.1 equivalents of the complexes 1, 3c, and 4c (Fig. S6†). A ∼20% ThT fluorescence reduction by all complexes was observed, indicating that these molecules can interfere with ThT. Nevertheless, this decrease was lower than the one observed in the aggregation kinetics of complexes 3c and 4c (∼50%), indicating that the complexes also reduce the final fibril yield. The decrease in fluorescence associated with complex 1 was found to be comparable to ThT displacement/quenching (Fig. S6†).
To verify whether the metal complexes also lower the final yield of fibrils (despite some ThT displacement/quenching), we incubated Aβ1-42 with and without a selection of metal complexes (i.e., 1, 2, 3c or 4c) at 1:1 molar ratio for 48 h at 37 °C and then imaged these samples using negative-stain transmission electron microscopy (TEM) at 52000× magnification (Fig. S7†). We observed significantly reduced or no fibril formation for all metal complexes which indicates that, at high concentrations (≥ equimolar), the metal complexes can suppress Aβ aggregation in vitro. This is true also for complexes 1 and 2, despite being less effective than 3c and 4c at sub-stoichiometric concentrations. These results are consistent with the observations recently reported by Meade et al. where they also observed by TEM significant reduction of fibril formation upon addition of cobalt(III) complexes.37
Probing binding mode to Aβ
To probe the interactions of the complexes with Aβ, we conducted a series of fluorescence titrations and 1H NMR spectroscopy experiments. The truncated peptide Aβ1-16 is intrinsically fluorescent due to its Tyr10 residue and complexes that interact non-covalently with Aβ via π–π stacking are expected to be able to quench this fluorescence.32,51 To investigate this, the fluorescence emission of the truncated peptide Aβ1-16 was recorded as increasing concentrations of 1, 3c and 4c (as selected examples of the complexes under investigation) were added (0 to 0.75 eq.).Cisplatin – which can bind to Aβ1-16 via coordination with the histidine residues – was included as a negative control.32 As seen in Fig. 4, minimal quenching effects were observed upon titration with cisplatin which cannot π–π stack with Aβ. In contrast, significant quenching was observed with 1, 3c and 4c, indicating that all these complexes can π–π stack with Aβ as would be expected from their structures. The quenching effect seen is greatest for complex 4c which has an extended aromatic scaffold.
 | ||
| Fig. 4 Normalised fluorescence emission of the Tyr10 residue of Aβ1-16 on addition of cisplatin (negative control), 1, 3c and 4c (0 to 0.75 eq., phosphate buffer, pH 7.4). Quenching is observed with both square planar 1 (blue) and octahedral 3c and 4c (orange) complexes. The highest degree of quenching is seen with Co-salnaph complex 4c (square). All data is reported as the mean ± standard deviation of three independent experiments. | ||
1H NMR experiments were conducted to probe the interactions between Aβ and the metal complexes further as coordination to Aβ would be expected to strongly perturb the 1H NMR spectrum of Aβ.32,33 Thus, the 1H NMR spectra of Aβ1-28 with and without our complexes were recorded after 24 h incubation at 37 °C in D2O:phosphate buffer (1:9, pH 7.4). The truncated Aβ1-28 peptide was used instead of the full length peptide as it retains the N-terminal metal ion binding site of interest, whilst being less aggregation-prone.32,33 On addition of square-planar complexes 1 and 2, minor perturbations and broadening of the aromatic signals of Aβ1-28 were observed, which are likely due to π–π stacking effects (Fig. 5; top). In contrast, greater signal broadening was observed when Aβ1-28 was incubated with octahedral complexes 3c and 4c, indicating the existence of additional coordination interactions. The Aβ1-28 His signals (see Fig. 5; top) are greatly affected by addition of these cobalt complexes. In addition, new broad signals appear in the region around 6.5 ppm which has been previously associated to Co-His coordination34 (although it should be pointed out that the complexes 3c and 4c have a signal in this region and therefore these resonances cannot be unambiguously assigned).
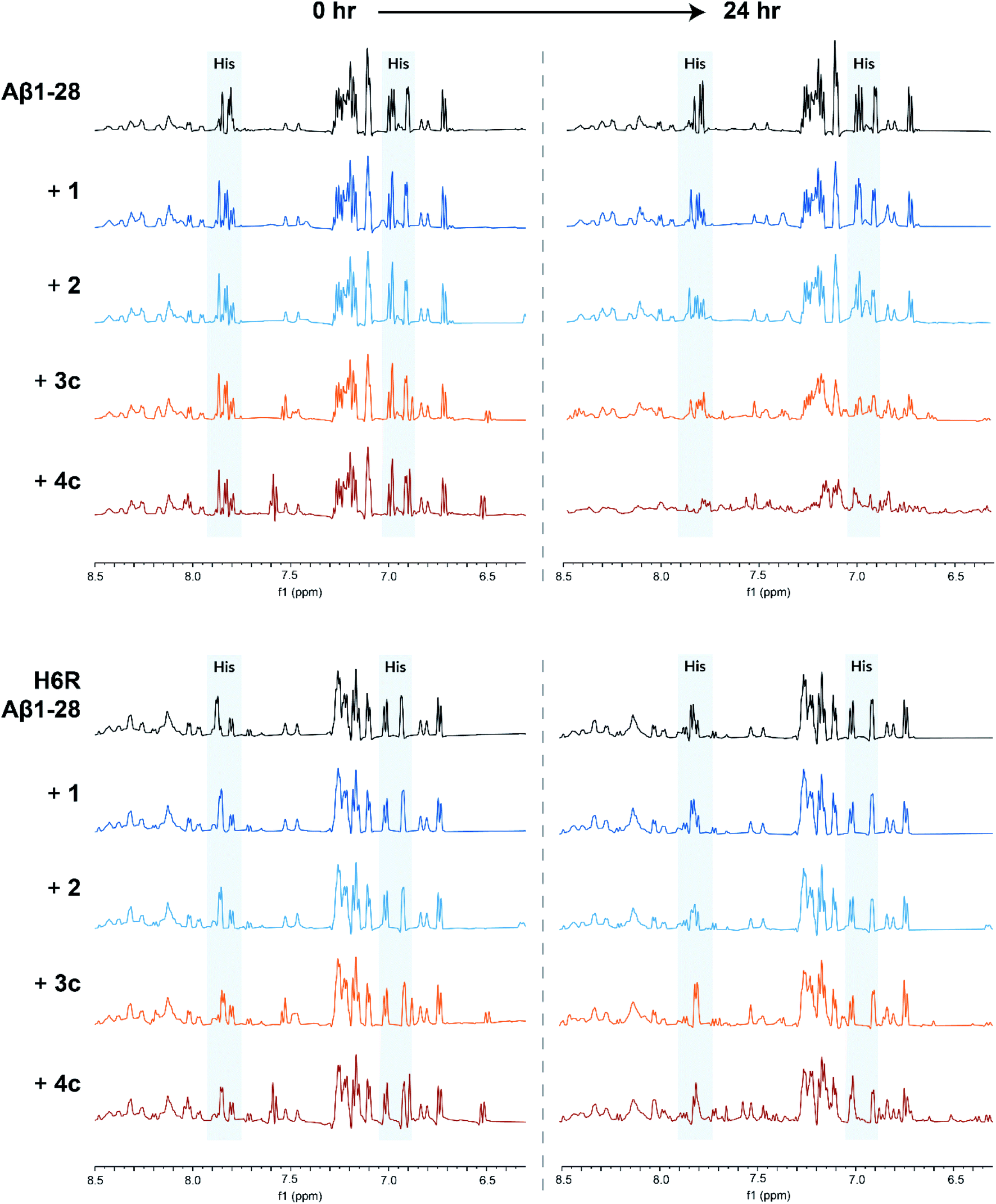 | ||
| Fig. 5
1H NMR spectra of Aβ1-28 (top) or H6R Aβ1-28 (bottom) incubated by itself (black), with square planar 1 and 2 (blue) or octahedral 3c and 4c (orange) complexes (in a 1-to-1 Aβ-to-complex ratio). All spectra were recorded in D2O:phosphate buffer (1:9, pH 7.4) at 37 °C and the spectra at 0 h and 24 h incubation time are shown. Peaks corresponding to the Aβ histidine residues are highlighted in blue. | ||
Computational studies conducted by Heffern et al. and Iscen et al. on the binding of other octahedral cobalt(III) complexes to Aβ predicted that the most thermodynamically favourable coordination mode to Aβ is coordination to His6 and either His13 or His14.34,35 To investigate if this was also the case for our complexes, we repeated the 1H NMR experiments with a point-mutated Aβ1-28 peptide in which the His6 residue was replaced by an arginine (H6R mutation). For H6R Aβ1-28, only minor perturbations of the aromatic signals were observed when incubated with any of the metal complexes, suggesting that the His6 residue is key for the binding of the cobalt(III) complexes to Aβ (Fig. 5; bottom).
Cytotoxicity studies
Any compound developed to interact with Aβ should be non-cytotoxic to mammalian cells. To investigate the cytotoxicity in vitro, human neuroblastoma cells (SH-SY5Y) were incubated with complex 4b for 24 h at concentrations up to 100 μM and the resulting cell viability was assessed using the MTS assay. An IC50 of >100 μM was observed in SH-SY5Y cells, making complex 4b a promising candidate for in vivo studies. For comparison, the MTS assay was also performed with complexes 1, 2, 3c and 4c; the results showed that, like for complex 4b, these complexes have negligible cytotoxic effects towards SH-SY5Y cells at concentrations up to 20 μM (see Fig. S8†).In vivo delivery and safety evaluation
For molecules to efficiently cross the BBB and enter the brain, characteristics such as moderate lipophilicity (logP of ∼2.1), a molecular weight of below 400 Da and a low propensity for hydrogen bond formation are thought to be crucial.42 Like the majority of the metal complexes previously developed for this application, our complexes have molecular weights greater than 400 Da, making them unlikely to cross the BBB by themselves; thus, in order to ensure that a therapeutic dose of our complexes were delivered to the brain, ultrasound-mediated BBB opening methods were employed.52 This method was chosen over other BBB delivery methods because drug delivery to the brain can be achieved in a non-invasive, targeted and reversible manner. Since the hippocampal regions of the brain are known to be particularly affected during the early stages of Alzheimer's disease, metal complexes were delivered specifically to the left hippocampus of mice brains in all experiments, leaving the right hippocampus as our non-ultrasound control.53
To prove that we were able to deliver our complexes into the brain using focused ultrasound and microbubbles, the left hippocampus of wild-type C57BL/6J mice (a very commonly used type of laboratory mouse for in vivo studies) was sonicated using the ultrasound sequences developed by Morse et al. (frequency = 1 MHz; peak-negative pressure = 350 kPa; pulse length = 5 cycles; pulse repetition frequency = 1.25 kHz; burst length = 10 ms; burst repetition frequency = 0.5 Hz).44 After five ultrasound bursts had been emitted, SonoVue® microbubbles followed by the metal complex of interest (30 mg kg−1) were administered intravenously into the bloodstream. Following transcardial perfusion and fixation, brains were extracted, sectioned, and analysed using laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS). For these experiments, the platinum(II)–salphen complex 2 and cobalt(III)–salnaph complex 4c were used. It should be noted that in the case of the cobalt(III) complex, background detection (due to the endogenous presence of this metal) was expected. The LA-ICP-MS scans show clear 195Pt signal in the left hippocampus (Fig. 6a), corresponding to the injected platinum complex, while no 195Pt signal was observed in the right hippocampus. This shows that complex 2 is unable to cross the BBB by itself as expected, but it can be delivered into the brain using focused ultrasound and microbubbles. Similarly, LA-ICP-MS scans indicate higher accumulation of 59Co (vs. background levels of endogenous cobalt) in the sonicated region of the brain (Fig. 6b).
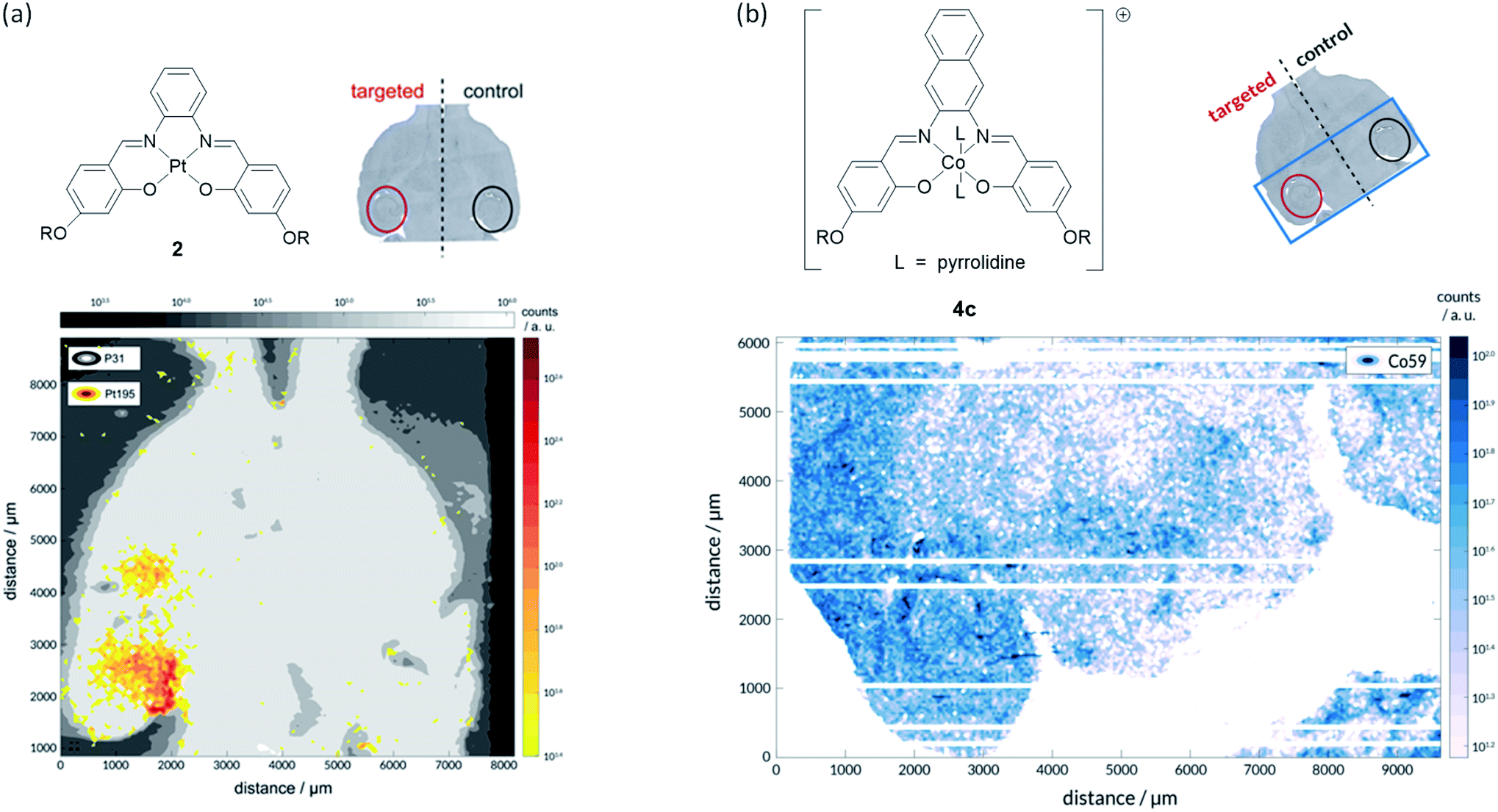 | ||
| Fig. 6 LA-ICP-MS studies with PtII and CoIII complexes 2 and 4c respectively. (a) Successful delivery of platinum(II) salphen 2 across the BBB to the left hippocampus is shown by the element distribution map for 195Pt (yellow/red); the image was overlaid on 31P map (grey) to provide brain outline. No evidence of complex accumulation in the right hippocampus (no-ultrasound control), indicating that it does not cross the BBB by itself; (b) successful delivery of cobalt(III) salphen 4c across the BBB to the left hippocampus is shown by the element distribution map for 59Co (white/blue). A significantly higher concentration of 59Co was detected on the sonicated area. | ||
Next, we sought to establish the safety of repeated complex 4b + ultrasound treatments as Alzheimer's disease patients typically survive for eight to ten years after symptoms begin to show and are therefore likely to require multiple treatments. To assess this, the left hippocampus of wild-type C57BL/6J and transgenic 5xFAD mice (4–5 months old; 5xFAD is a mouse model of Alzheimer's disease which develops amyloid pathology starting from 2 months in age) were sonicated using the same ultrasound sequences as above.44 After five ultrasound bursts had been emitted, SonoVue® microbubbles followed by complex 4b (20 mg kg−1; number of mice = 3) were administered intravenously into the bloodstream. Treatment was conducted on a weekly basis over a four-week period with behaviour and body weight monitored throughout. At the study end-point, brains were harvested for post-mortem histological analyses. No changes in behaviour or body weight were detected upon weekly treatment with complex 4b in either mouse model and no signs of tissue damage were observed in haematoxylin and eosin-stained (H&E) brain sections, indicating that this treatment protocol was well-tolerated (Fig. 7 and S9†).
 | ||
| Fig. 7 No signs of tissue damage were observed in H&E-stained sections of brains in which complex 4b was delivered by sonication. | ||
Conclusions
We have designed and synthesised a series of metal salphen and salnaph complexes that are able to inhibit amyloid beta aggregation in vitro. These complexes were designed to overcome some of the limitations of previously reported metal complexes. For example, we report that octahedral cobalt(III) complexes with labile axial ligands have high inhibitory activity against Aβ aggregation due to their dual binding mode involving π–π stacking interactions and direct metal coordination. All our cobalt complexes show a significant inhibition of Aβ aggregation at just 0.1 equivalents of the complex, with the use of 0.2 equivalents completely inhibiting aggregation in most cases. Our metal complexes have minimal cytotoxicity in SH-SY5Y human neuroblastoma cells, making them promising candidates for in vivo studies. We also show that these complexes can be successfully delivered across the blood–brain barrier to specific locations in the brains of mice using focused ultrasound and are well-tolerated. To the best of our knowledge, this is the first time that a metal-based compound with in vitro inhibitory activity towards Aβ aggregation has been non-invasively delivered across the blood–brain barrier in vivo.Ethical statement
All animal procedures were performed in compliance with the European Directive 63/2010/EU and the UK Animals (Scientific Procedures) Act 1986. All protocols were approved by Imperial College London's Animal Welfare and Ethical Review Body.Author contributions
T. G. C., C. L. R. and V. R. synthesised the compounds. T. G. C. and M. S. performed spectroscopic and biophysical studies with the compounds and Aβ peptide. T. G. C. and S. V. M. performed the sonication experiments. T. G. C., S. V. M. and H. W. sectioned mice brains and performed histology. T. G. C., F. A. A, J. J. C. and R. V. analysed the data and wrote the manuscript. J. J. C. and R. V. designed the project and raised the funding.Conflicts of interest
There are no conflicts of interest to declareAcknowledgements
PhD studentships for T. G. Chan and S. V. Morse were provided by the EPSRC Centre for Doctoral Training in Neurotechnology (EP/L016737/1) and Medical Imaging (EP/L015226/1) respectively. PhD studentships for C. L. Ruehl and V. Rakers were provided by the Department of Chemistry, Imperial College London. The authors would also like to thank Mr P. Haycock (Imperial College London) for NMR assistance, Dr J. Barrit (Imperial College London) for transmission electron microscopy, Mr Barry Sampson (Imperial College London) for LA-ICP-MS assistance. F. A. Aprile thanks UK Research and Innovation (Future Leaders Fellowship MR/S033947/1) and the Alzheimer's Society, UK (grant 511) for support.References
- B. R. Roberts, M. Lind, A. Z. Wagen, A. Rembach, T. Frugier, Q. X. Li, T. M. Ryan, C. A. McLean, J. D. Doecke, C. C. Rowe, V. L. Villemagne and C. L. Masters, Brain, 2017, 140, 1486–1498 CrossRef PubMed.
- G. F. Chen, T. H. Xu, Y. Yan, Y. R. Zhou, Y. Jiang, K. Melcher and H. E. Xu, Acta Pharmacol. Sin., 2017, 38, 1205–1235 CrossRef CAS PubMed.
- W. Xia, T. Yang, G. Shankar, I. M. Smith, Y. Shen, D. M. Walsh and D. J. Selkoe, Arch. Neurol., 2009, 66, 190–199 CrossRef PubMed.
- P. M. Murphy and H. LeVine, J. Alzheimer's Dis., 2010, 19, 311–323 Search PubMed.
- L. Pini, M. Pievani, M. Bocchetta, D. Altomare, P. Bosco, E. Cavedo, S. Galluzzi, M. Marizzoni and G. B. Frisoni, Ageing Res. Rev., 2016, 30, 25–48 CrossRef PubMed.
- L. Crews and E. Masliah, Hum. Mol. Genet., 2010, 19, R12–R20 CrossRef CAS PubMed.
- M. A. Deibel, W. D. Ehmann and W. R. Markesbery, J. Neurol. Sci., 1996, 143, 137–142 CrossRef CAS PubMed.
- M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, J. Neurol. Sci., 1998, 158, 47–52 CrossRef CAS PubMed.
- L. M. Miller, Q. Wang, T. P. Telivala, R. J. Smith, A. Lanzirotti and J. Miklossy, J. Struct. Biol., 2006, 155, 30–37 CrossRef CAS PubMed.
- J. Dong, C. S. Atwood, V. E. Anderson, S. L. Siedlak, M. A. Smith, G. Perry and P. R. Carey, Biochemistry, 2003, 42, 2768–2773 CrossRef CAS PubMed.
- M. W. Bourassa, A. C. Leskovjan, R. V. Tappero, E. R. Farquhar, C. A. Colton, W. E. Van Nostrand and L. M. Miller, Biomed. Spectrosc. Imaging, 2013, 2, 129–139 CAS.
- D. P. Smith, G. D. Ciccotosto, D. J. Tew, M. T. Fodero-Tavoletti, T. Johanssen, C. L. Masters, K. J. Barnham and R. Cappai, Biochemistry, 2007, 46, 2881–2891 CrossRef CAS PubMed.
- M. C. Lee, W. C. Yu, Y. H. Shih, C. Y. Chen, Z. H. Guo, S. J. Huang, J. C. C. Chan and Y. R. Chen, Sci. Rep., 2018, 8, 4772 CrossRef PubMed.
- F. Hane, G. Tran, S. J. Attwood and Z. Leonenko, PLoS One, 2013, 8, e59005 CrossRef CAS PubMed.
- K. Hensley, N. Hall, R. Subramaniam, P. Cole, M. Harris, M. Aksenov, M. Aksenova, S. P. Gabbita, J. F. Wu, J. M. Carney, M. Lovell, W. R. Markesbery and D. A. Butterfield, J. Neurochem., 1995, 65, 2146–2156 CrossRef CAS PubMed.
- D. A. Butterfield, T. Reed, M. Perluigi, C. De Marco, R. Coccia, C. Cini and R. Sultana, Neurosci. Lett., 2006, 397, 170–173 CrossRef CAS PubMed.
- E. Tamagno, M. Guglielmotto, M. Aragno, R. Borghi, R. Autelli, L. Giliberto, G. Muraca, O. Danni, X. Zhu, M. A. Smith, G. Perry, D. G. Jo, M. P. Mattson and M. Tabaton, J. Neurochem., 2008, 104, 683–695 CAS.
- C. Hureau, V. Balland, Y. Coppel, P. L. Solari, E. Fonda and P. Faller, J. Biol. Inorg Chem., 2009, 14, 995–1000 CrossRef CAS PubMed.
- T. R. Young, A. Kirchner, A. G. Wedd and Z. Xiao, Metallomics, 2014, 6, 505–517 CrossRef CAS PubMed.
- N. Yako, T. R. Young, J. M. Cottam Jones, C. A. Hutton, A. G. Wedd and Z. Xiao, Metallomics, 2017, 9, 278–291 CrossRef CAS PubMed.
- C. Hureau and P. Dorlet, Coord. Chem. Rev., 2012, 256, 2175–2187 CrossRef CAS.
- B. Alies, H. Eury, C. Bijani, L. Rechignat, P. Faller and C. Hureau, Inorg. Chem., 2011, 50, 11192–11201 CrossRef CAS PubMed.
- B. Alies, A. Conte-Daban, S. Sayen, F. Collin, I. Kieffer, E. Guillon, P. Faller and C. Hureau, Inorg. Chem., 2016, 55, 10499–10509 CrossRef CAS PubMed.
- F. Bousejra-Elgarah, C. Bijani, Y. Coppel, P. Faller and C. Hureau, Inorg. Chem., 2011, 50, 9024–9030 CrossRef CAS PubMed.
- P. Faller, C. Hureau and O. Berthoumieu, Inorg. Chem., 2013, 52, 12193–12206 CrossRef CAS PubMed.
- Y. Miller, B. Ma and R. Nussinov, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9490–9495 CrossRef CAS PubMed.
- D. J. Hayne, S. Lim and P. S. Donnelly, Chem. Soc. Rev., 2014, 43, 6701–6715 RSC.
- J. M. Suh, G. Kim, J. Kang and M. H. Lim, Inorg. Chem., 2019, 58, 8–17 CrossRef CAS PubMed.
- H. Liu, Y. Qu and X. Wang, Future Med. Chem., 2018, 10, 697–701 Search PubMed.
- L. M. F. Gomes, J. C. Bataglioli and T. Storr, Coord. Chem. Rev., 2020, 412, 213255 CrossRef CAS.
- K. J. Barnham, V. B. Kenche, G. D. Ciccotosto, D. P. Smith, D. J. Tew, X. Liu, K. Perez, G. A. Cranston, T. J. Johanssen, I. Volitakis, A. I. Bush, C. L. Masters, A. R. White, J. P. Smith, R. A. Cherny and R. Cappai, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6813–6818 CrossRef CAS PubMed.
- G. Ma, F. Huang, X. Pu, L. Jia, T. Jiang, L. Li and Y. Liu, Chem.–Eur. J., 2011, 17, 11657–11666 CrossRef CAS PubMed.
- I. Sasaki, C. Bijani, S. Ladeira, V. Bourdon, P. Faller and C. Hureau, Dalton Trans., 2012, 41, 6404–6407 RSC.
- M. C. Heffern, P. T. Velasco, L. M. Matosziuk, J. L. Coomes, C. Karras, M. A. Ratner, W. L. Klein, A. L. Eckermann and T. J. Meade, ChemBioChem, 2014, 15, 1584–1589 CrossRef CAS PubMed.
- A. Iscen, C. R. Brue, K. F. Roberts, J. Kim, G. C. Schatz and T. J. Meade, J. Am. Chem. Soc., 2019, 141, 16685–16695 CrossRef CAS PubMed.
- G. S. Yellol, J. G. Yellol, V. B. Kenche, X. M. Liu, K. J. Barnham, A. Donaire, C. Janiak and J. Ruiz, Inorg. Chem., 2015, 54, 470–475 CrossRef CAS PubMed.
- K. F. Roberts, C. R. Brue, A. Preston, D. Baxter, E. Herzog, E. Varelas and T. J. Meade, J. Inorg. Biochem., 2020, 213, 111265 CrossRef CAS PubMed.
- M. C. Heffern, V. Reichova, J. L. Coomes, A. S. Harney, E. A. Bajema and T. J. Meade, Inorg. Chem., 2015, 54, 9066–9074 CrossRef CAS PubMed.
- K. Hynynen, N. McDannold, N. Vykhodtseva and F. A. Jolesz, Radiology, 2001, 220, 640–646 CrossRef CAS PubMed.
- J. J. Choi, K. Selert, F. Vlachos, A. Wong and E. E. Konofagou, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16539–16544 CrossRef CAS PubMed.
- J. Park, Y. Zhang, N. Vykhodtseva, F. A. Jolesz and N. J. McDannold, J. Controlled Release, 2012, 162, 134–142 CrossRef CAS PubMed.
- L. H. Treat, N. McDannold, Y. Zhang, N. Vykhodtseva and K. Hynynen, Ultrasound Med. Biol., 2012, 38, 1716–1725 CrossRef PubMed.
- K. C. Wei, P. C. Chu, H. Y. J. Wang, C. Y. Huang, P. Y. Chen, H. C. Tsai, Y. J. Lu, P. Y. Lee, I. C. Tseng, L. Y. Feng, P. W. Hsu, T. C. Yen and H. L. Liu, PLoS One, 2013, 8, e58995 CrossRef CAS.
- S. V. Morse, A. N. Pouliopoulos, T. G. Chan, M. J. Copping, J. Lin, N. J. Long and J. J. Choi, Radiology, 2019, 291, 459–466 CrossRef PubMed.
- T. G. Chan, S. V. Morse, M. J. Copping, J. J. Choi and R. Vilar, ChemMedChem, 2018, 13, 1311–1314 CrossRef CAS PubMed.
- A. Abrahao, Y. Meng, M. Llinas, Y. Huang, C. Hamani, T. Mainprize, I. Aubert, C. Heyn, S. E. Black, K. Hynynen, N. Lipsman and L. Zinman, Nat. Commun., 2019, 10, 4373 CrossRef PubMed.
- N. Lipsman, Y. Meng, A. J. Bethune, Y. Huang, B. Lam, M. Masellis, N. Herrmann, C. Heyn, I. Aubert, A. Boutet, G. S. Smith, K. Hynynen and S. E. Black, Nat. Commun., 2018, 9, 2336 CrossRef PubMed.
- V. Rakers, P. Cadinu, J. B. Edel and R. Vilar, Chem. Sci., 2018, 9, 3459–3469 RSC.
- C. L. Ruehl, A. H. M. Lim, T. Kench, D. J. Mann and R. Vilar, Chem.–Eur. J., 2019, 25, 9691–9700 CrossRef CAS PubMed.
- H. Naiki, K. Higuchi, M. Hosokawa and T. Takeda, Anal. Biochem., 1989, 177, 244–249 CrossRef CAS.
- C. D. Syme, R. C. Nadal, S. E. J. Rigby and J. H. Viles, J. Biol. Chem., 2004, 279, 18169–18177 CrossRef CAS PubMed.
- W. M. Pardridge, J. Cereb. Blood Flow Metab., 2012, 32, 1959–1972 CrossRef CAS PubMed.
- Y. Mu and F. H. Gage, Mol. Neurodegener., 2011, 6, 85 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02273c |
| This journal is © The Royal Society of Chemistry 2021 |