
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMass spectrometric detection of fleeting neutral intermediates generated in electrochemical reactions†
Jilin
Liu‡
abc,
Kai
Yu‡
 ab,
Hong
Zhang
ab,
Jing
He
abc,
Jie
Jiang
*abc and
Hai
Luo
*d
ab,
Hong
Zhang
ab,
Jing
He
abc,
Jie
Jiang
*abc and
Hai
Luo
*d
aSchool of Environment, School of Marine Science and Technology (Weihai), Harbin Institute of Technology, Weihai, Shandong 150090, China. E-mail: jiejiang@hitwh.edu.cn
bState Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology, Harbin, Heilongjiang 150090, China
cSchool of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin, Heilongjiang 150001, China
dBeijing National Laboratory for Molecular Sciences, College of Chemistry and Molecular Engineering, Peking University, Beijing, 100871, China. E-mail: hluo@pku.edu.cn
First published on 12th June 2021
Abstract
Towards the goal of on-line monitoring of transient neutral intermediates during electrochemical reactions, an electrochemistry-neutral reionization-mass spectrometry (EC-NR-MS) technique was developed in this work. The EC-NR setup consisted of a customized EC flow cell, a sonic spray ionization source, a heating tube, an ion deflector and an electrospray ionization source, which were respectively used for the precise control of the electrochemical reaction, solution nebulization, droplet desolvation, ion deflection and neutral intermediate ionization. Based on the EC-NR-MS approach, some long-sought neutral radicals including TPrA˙, DBAE˙ and TEOA˙, which belong to important reductive intermediates in electrochemiluminescence (ECL) reactions, were successfully identified which helps to clarify the previously unproven ECL reaction mechanism. These findings were also supported by spin-trapping experiments and the tandem MS technique. Accordingly, the EC-NR-MS method provides a direct solution for studying complicated electrochemical reactions, especially for detecting short-lived neutral radicals as well as ionic intermediates.
Introduction
Neutral radicals play crucial roles in electrosynthesis,1–3 electrochemiluminescence4,5 and biological redox reactions.6,7 Accurate identification of such intermediates is essential for deriving the reaction mechanisms and further developing efficient reaction systems or novel materials. The most commonly used characterization techniques include density functional theory (DFT) calculation,8,9 cyclic voltammetry (CV),10,11 electron paramagnetic resonance spectroscopy (EPR)12–14 and spin trapping;2 however, these methods are either off-line or unable to directly detect neutral intermediates.Recently, our group reported a neutral-reionization (NR) strategy, which can isolate the neutral serine octamer from other ions for mass spectrometric analysis.15 Herein, this setup was modified to study electrogenerated neutral intermediates, such as those in electrochemiluminescence reactions involving tertiary amine co-reactants. Considering that electrochemical reactions are generally ultrafast and neutral radicals are short-lived, an on-line and direct characterization method should be recommended. Due to the significant advantages of on-line analysis, high sensitivity and selectivity, and the ability to provide the molecular weight and structure information of the analyte, electrochemistry-mass spectrometry (EC-MS) was employed for this research.16,17 As illustrated in Fig. 1, the established technique, named electrochemistry-neutral reionization-mass spectrometry (EC-NR-MS), consists of a custom designed EC flow cell, an easy ambient sonic-spray ionization (EASI) source, a heating tube, an ion deflector and an electrospray ionization source (see ESI, Fig. S1† for more details).15,18–20 During the measurement, the electrooxidation can be precisely controlled in the EC flow cell, and the solution containing electrochemical products was pumped out from the EC cell and subsequently nebulized with the aid of a high-flow gas stream (the EASI specialty). The droplet desolvation and ion deflection can be achieved when the mist traversed the heating tube and the deflector successively. Finally, the electrochemically generated neutral species were ionized by ESI for mass spectrometric analysis.
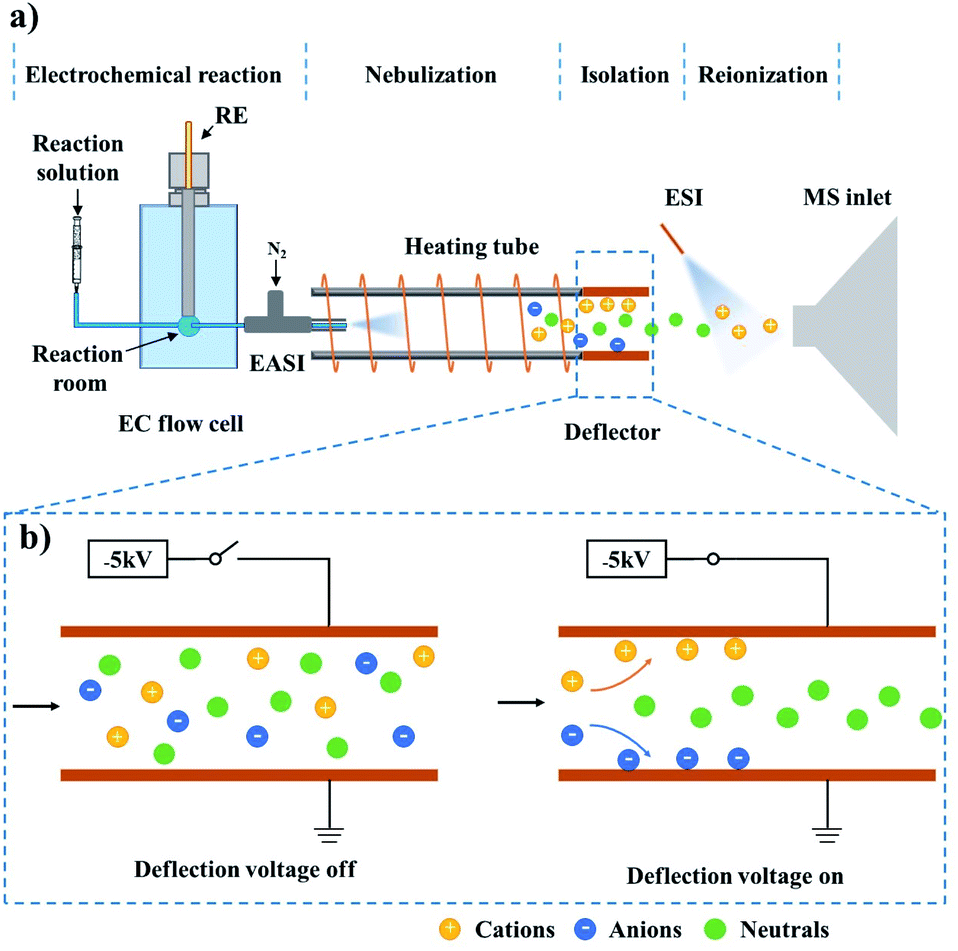 | ||
| Fig. 1 Schematic diagram of (a) the basic components of the EC-NR-MS setup, and (b) the working principle of the deflector. When the deflection voltage is on, ionic species are deflected; only neutral species can pass through the deflector and are subsequently ionized by the ESI plume and detected by MS. | ||
Based on the EC-NR-MS platform, the electrooxidation processes of tri-n-propylamine (TPrA), 2-(dibutylamino)ethanol (DBAE) and triethanolamine (TEOA) were monitored in real time. Although these tertiary amines have been widely used as co-reactants in electrochemiluminescence21–23 and electron donors in photocatalysis,24,25 their neutral radical intermediate species cannot be directly detected with traditional techniques.11,26
Experimental section
Chemicals
Methanol (MS grade), triethanolamine (98%), tri-n-propylamine (98%), 2-(dibutylamino)ethanol (99%), tris(2,2′-bipyridine)ruthenium(II) dichloride hexahydrate and 5,5-dimethyl-1-pyrroline N-oxide (98%) were purchased from Sigma Aldrich (Darmstadt, Germany). PBS buffer solution (pH = 7.5) was purchased from Aladdin (Shanghai, China). The glassy carbon (GC) disk electrode (diameter 0.3 mm), platinum (Pt) plate and Ag/AgCl reference electrode were from Gaoss Union (Wuhan, China). Ultrapure water was produced by using a Milli-Q water purification system (Milford, MA). The solvent was bubbled with pure nitrogen for one hour to deoxygenate and all sample solutions were used immediately after preparation.Electrochemistry-neutral reionization-mass spectrometry
As shown in Fig. S1,† the electrochemistry-neutral reionization-mass spectrometry (EC-NR-MS) setup consisted of an EC flow cell, an easy ambient sonic-spray ionization (EASI) source, a heating tube (i.d. 5.8 mm, o.d. 7.4 mm, and length 10 cm), an ion deflector and an electrospray ionization (ESI) source. The EC flow cell was made of PEEK and the internal volume for sample solution was 100 mL. A typical three-electrode unit includes a GC working electrode, Pt plate counter electrode and Ag/AgCl reference electrode connected to a CHI660E AutoLab (CH Instruments, Shanghai, China) for achieving the electrochemical reaction. The WE and CE were placed in parallel on the left and right sides of the EC flow cell while the RE was fixed on the top (see Fig. S2† for more details). The inlet of the EASI capillary was in the EC flow cell and close to the WE, and the capillary outlet was inserted into the heating tube to maximize the collection of droplets. During the experiment, the sample solution was pumped into the EC flow cell at a flow rate of 25 μL min−1. The electrooxidation products in solution were then sprayed out from the other side of the EC flow cell under a gas pressure of 60 psi (the EASI specialty). The formed droplets completely entered the heating tube (made of glass and its temperature was kept at 100 °C) for desolvation. A pair of parallel copper plates (10 × 10 mm2) integrated into a PEEK scaffold were utilized as an ion deflector. The distance between the two plates was 10 mm. One of the plates was grounded while the other plate was input a voltage of −5 kV, helping to trap the ions emerging from the heating tube. The other side of the ion deflector was facing the MS inlet and their distance was 10 mm. In order to ionize the remaining neutral products for mass spectrometric analysis, the ESI source was placed in front of the MS inlet at a distance of 4 mm. The angle between the tip of the ESI sprayer and the MS inlet was ca. 60°, the spray voltage was set to 5 kV, and methanol was used as the spray solvent.Mass spectrometry
All MS data were acquired in positive-ion mode by using an LTQ mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). The experimental parameters were capillary temperature (275 °C), capillary voltage (35 V) and tube lens voltage (110 V). The Xcalibur software package (Version 2.0.7, Thermo Fisher Scientific, USA) was used for data analysis.The tandem mass spectrometry (MS2) data were obtained in positive ion mode by using an LTQ mass spectrometer. The collision induced dissociation (CID) voltage was set to 30 V or 35 V depending on the analytes and the maximum ion injection time was 30 ms.
Results and discussion
Electrooxidation of tri-n-propylamine
To evaluate the usability of EC-NR-MS, tri-n-propylamine (TPrA), a well-known co-reactant for electrochemiluminescence (ECL) reactions, was chosen as the proof of concept.5,27–30 One famous ECL system based on this reagent is tris(2,2′-bipyridine)ruthenium(II) (Ru(bpy)32+)–TPrA. As proposed by Bard et al., TPrA could be electrooxidized at 0.8 V, generating a radical cation (TPrA˙+) and a neutral radical (TPrA˙). Using TPrA˙ as the reducing agent for Ru(bpy)32+ would form Ru(bpy)3+, which then oxidized by TPrA˙+ to produce the excited state of Ru(bpy)32+ (see Scheme S1†).26,31–33 Although this ECL mechanism has been widely accepted, the generation and evolution details of TPrA˙+ and TPrA˙ are still unknown. In 2016, Shao et al. found two relatively stable TPrA electrooxidation products ([Pr2N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2CH3]+ and NHPr2, see Scheme S1†) by employing an online EC-MS method.34 A more recent report from Xu et al. presented another in situ EC-MS technique, which successfully captured the electrogenerated TPrA˙+.35 Unfortunately, these endeavours have not achieved the detection of TPrA˙ and thus cannot confirm the TPrA electrooxidation process completely.
CHCH2CH3]+ and NHPr2, see Scheme S1†) by employing an online EC-MS method.34 A more recent report from Xu et al. presented another in situ EC-MS technique, which successfully captured the electrogenerated TPrA˙+.35 Unfortunately, these endeavours have not achieved the detection of TPrA˙ and thus cannot confirm the TPrA electrooxidation process completely.
Considering that the electrochemical reaction is generally ultrafast, a shorter distance between the EC flow cell and MS inlet may collect more information of the reaction products. Therefore, we first chose EC/EASI coupled with MS to study the electrooxidation of TPrA (Fig. S3†). Due to the soft ionization of EASI (which does not require the application of heat, high voltages, laser beams or UV light), the reaction products remain intact during the ionization process.36–39 As shown in Fig. S4–S6,† the fleeting intermediates of TPrA, including TPrA˙+ (its half-life was estimated to 200 μs)26 and [Pr2NCHCH2CH3]+, were detected and characterized. In addition, EC/EASI-MS was applied to monitor the interaction between Ru(bpy)32+ and TPrA (see Fig. S7†). The key intermediates including TPrA˙+ and Ru(bpy)3+ can be observed simultaneously on the MS spectrum, partially evidencing the mechanism of Scheme S1.†
Encouraged by the EC/EASI-MS results, we aimed at a more challenging goal of TPrA˙ detection. Since the m/z value of protonated TPrA˙ is equal to that of TPrA˙+ in the MS spectrum, it is impossible to distinguish between them by MS. To overcome the drawback, the neutral reionization (NR) setup15,19 was coupled with EC/EASI (see Fig. S1†) for extracting the electrochemically generated TPrA˙. Prior to the electrooxidation experiments, the performance of the EC/EASI-NR-MS setup was tested by using TPrA solution (100 ppm TPrA, 1 mM PBS buffer, and pH = 7.5). Under a gas pressure of 60 psi, the TPrA solution was rapidly sprayed out from the tee with the molecules being ionized via the EASI source, and these ionic species were further transferred to the NR part. When there was no deflection voltage (V) applied, the resulting mass spectrum was dominated by the peak at m/z 144 (Fig. S8a†). As expected, MS cannot provide any sampling signal as the deflection voltage was set to −5 kV, (Fig. S8b†), indicating that the ionic species were completely trapped by the deflector. Meanwhile, if the ESI at the end of the deflector was applied with a voltage of 5 kV, the remaining neutral molecules in the gas stream were “re-ionized” and thus detected again by MS. The obtained mass spectrum is shown in Fig. S8c† and is similar to the undeflected result of Fig. S8a.† Such results demonstrate that our EC-NR-MS method is qualified for studying electrochemically generated neutral molecules.
With the successful isolation and detection of neutral molecules, EC-NR-MS was subsequently devoted to capturing TPrA˙. The oxidation potential (E) and deflection voltage (V) were set at 0.8 V and −5 kV, respectively. As shown in Fig. 2a, none of the ionic oxidation products such as [Pr2NCHCH2CH3]+ (m/z 142) and TPrA˙+ (m/z 143) can be observed in the absence of ESI, illustrating that the intermediate ions along with those initial EASI-generated molecular ions (e.g. m/z 144 and 102, see Fig. S8a†) have been removed by the deflector. With the ESI high voltage (5 kV) on, several peaks owing to the post-deflector ESI “reionization” of the remaining neutral species appeared in Fig. 2b. Obviously, due to the occurrence of electrooxidation here, the relative intensity of m/z 102 has significantly increased in comparison with that in Fig. S8c.† Meanwhile, the peak at m/z 142 attributed to [Pr2NCHCH2CH3]+ is absent, suggesting that all the ionic electrooxidation intermediates have been removed by the deflector. It is worth emphasizing that compared with Fig. S8c,† there is a new peak at m/z 143 in Fig. 2b. Considering that the ionic electrooxidation intermediates have been removed by the deflector, this new MS signal should originate from the electrogenerated neutral species, which is most likely to be protonated TPrA˙ (m/z 143). As far as we know, this is the first mass spectrometric evidence of TPrA˙.
 | ||
| Fig. 2 EC-NR-MS spectra of 100 ppm TPrA with 1 mM PBS (pH = 7.5), (a) without ESI and (b) with ESI. | ||
To further confirm the mass spectrometric detection of TPrA˙, a common radical trapping agent DMPO40–43 was used to capture this neutral radical. The proposed reaction pathway between TPrA˙ and DMPO to form a neutral spin adduct (DMPO/TPrA˙) is described in Scheme S2 (ESI†). The generation of DMPO/TPrA˙ can be checked online by EC-NR-MS as well. To this end, a solution containing 100 ppm TPrA, 1 mM PBS, and 0.1 mM DMPO was used in the experiments with the deflector voltage (V) set at −5 kV. Fig. 3a and c were obtained in the absence of ESI, and no signal appeared on the spectra, consistent with the fact that all the generated ionic species were removed by the deflector. Fig. 3b and d were acquired by inputting 5 kV to the ESI. The mass peaks observed at m/z 144, 114 and 102 in Fig. 3b correspond to the protonated TPrA, DMPO and NHPr2, respectively. When a potential (E) of 0.8 V was applied to the working electrode of the EC flow cell, a new peak (m/z 256) which should be assigned to the protonated spin adduct [DMPO/TPrA˙ + H]+ is shown in Fig. 3d. Note that the formation of DMPO/TPrA˙ significantly decreased the amount of free TPrA˙, and thus the protonated TPrA˙ peak (m/z 143) is too low in intensity to be seen in Fig. 3d (compared with that in Fig. 2b). The protonated spin adduct [DMPO/TPrA˙ + H]+ was further characterized by MS2 (see Fig. S10, ESI†). Evidently, the formation of the protonated spin adduct [DMPO/TPrA˙ + H]+ corroborates the previous finding that the neutral radical (TPrA˙) was indeed generated during the electrooxidation of TPrA under 0.8 V.
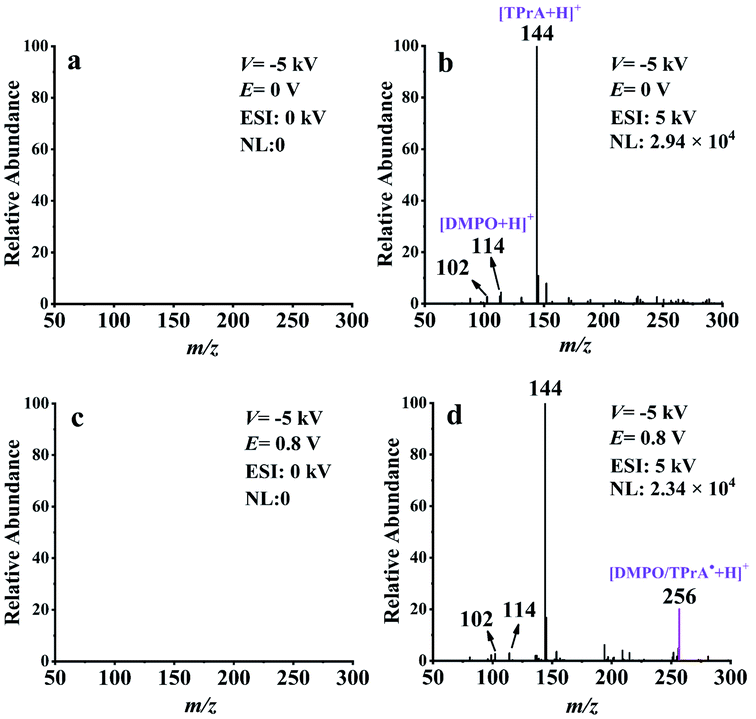 | ||
| Fig. 3 EC-NR-MS spectra of 100 ppm TPrA solution containing 0.1 mM DMPO and 1 mM PBS (pH = 7.5): (a) E = 0 V without ESI, (b) E = 0 V with ESI, (c) E = 0.8 V without ESI, and (d) E = 0.8 V with ESI. E is the applied electrooxidation potential for TPrA and the ionization voltage of ESI is 5 kV. | ||
Additionally, we have verified the role of TPrA˙ in the ECL process by adding DMPO to remove TPrA˙ from the Ru(bpy)32+/TPrA reaction system (see the ESI†). Comparing the patterns of Fig. S11 and S7,† one can realize that the neutral radical TPrA˙ is indeed functioned as the reducing agent for Ru(bpy)32+ to produce Ru(bpy)3+.
Electrooxidation of 2-(dibutylamino)ethanol
The feasibility of EC-NR-MS was then investigated by capturing the neutral intermediate generated in the electrooxidation of 2-(dibutylamino)ethanol (DBAE). Different from TPrA, DBAE is believed to be a more environmentally friendly ECL co-reactant and is more efficient than TPrA on Pt and Au electrodes. However, direct experimental observation on the electrogenerated active products of DBAE has not been achieved.44 The same as the previous experiment, the EC/EASI device was selected to monitor the electrooxidation process of DBAE. Two reaction intermediates include an iminium ion (DBAE-I) and radical cation DBAE˙+ were detected and charactered by MS for the first time (see ESI Fig. S12†).Thereafter, EC-NR-MS was used to capture the electrogenerated neutral radical DBAE˙. As shown in Fig. 4a, no MS signal can be observed when the deflection voltage was set to −5 kV, indicating the complete deflection of the ions. When the ESI voltage was set at 5 kV, the peak of protonated DBAE (m/z 174) was observed (see Fig. 4b). Under an oxidation potential (E) of 1.0 V, two new peaks appeared at m/z 118 and 173 in Fig. 4d. It is worth noting that the ion at m/z 173 is most likely the protonated DBAE˙. To verify this inference, DMPO was used to capture DBAE˙ and the formation of a spin adduct [DMPO/DBAE˙ + H]+ can be seen in Fig. S14.† As far as we know, this is the first MS evidence of DBAE˙. The proposed electrooxidation steps based on above results are in Scheme S4.†
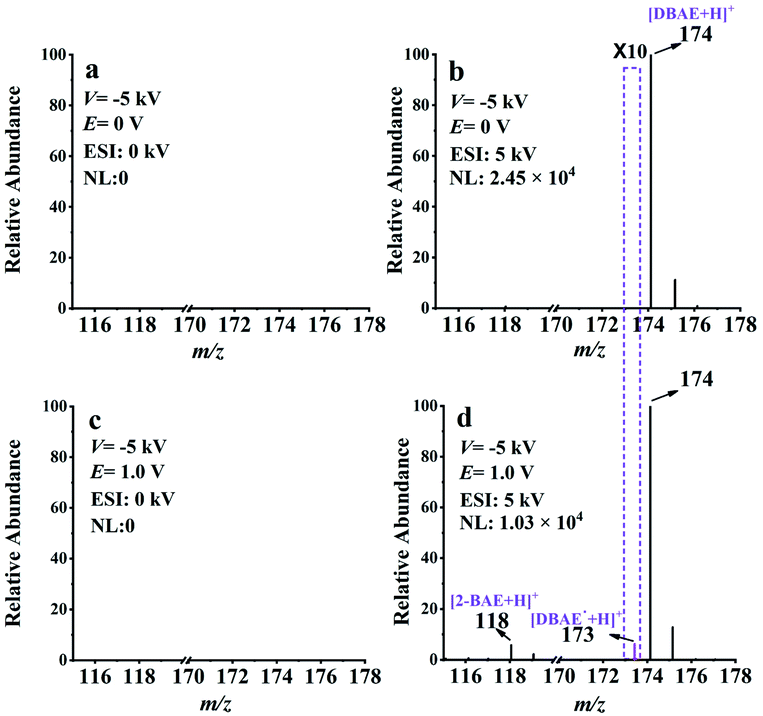 | ||
| Fig. 4 EC-NR-MS spectra of 100 ppm DBAE with 1 mM PBS (pH = 7.5), (a) E = 0 V without ESI, (b) E = 0 V with ESI, (c) E = 1.0 V without ESI, and (d) E = 1.0 V with ESI. E is the applied electrooxidation potential for DBAE and the ionization voltage of ESI is 5 kV. | ||
Electrooxidation of triethanolamine
EC-NR-MS was additionally applied to study the electrooxidation of triethanolamine (TEOA). This chemical is commonly used as an electron donor in photocatalysis24,45 or a co-reactant of ECL.4,21 As proposed, TEOA can be electrooxidized to generate the radical cation (TEOA˙+) and then deprotonates to form TEOA˙.24,46,47 With EC/EASI, we successfully detected and charactered the electrooxidation intermediates of TEOA for the first time, including the iminium ion (TEOA-I) and radical cation TEOA˙+.Then the generation of TEOA˙ was confirmed by EC-NR-MS. With the voltage of the deflector and ESI set separately at −5 kV and 5 kV, the dominate peak (at m/z 150) of the spectrum in Fig. 5b corresponded to protonated TEOA. When 1.0 V was applied to the WE, several peaks that should be related to the oxidation products of TEOA appeared on the spectrum of Fig. 5d. The ion at m/z 106 is associated with the oxidation intermediate DEA, while that at m/z 149 is most likely the protonated neutral radical TEOA˙. The generation of TEOA˙ was further verified with DMPO (check Fig. S18† for more details). This protonated spin adduct [DMPO/TEOA˙ + H]+ was further characterized by MS2 (see Fig. S19†). Thus, the electrooxidation mechanism of TEOA was achieved and is presented in Scheme S6.†
 | ||
| Fig. 5 EC-NR-MS spectra of 100 ppm TEOA with 1 mM PBS (pH = 7.5), (a) E = 0 V without ESI, (b) E = 0 V with ESI, (c) E = 1.0 V without ESI, and (d) E = 1.0 V with ESI. E is the applied electrooxidation potential for TEOA and the ionization voltage of ESI was 5 kV. | ||
Conclusions
In summary, a novel EC-NR-MS platform was established for on-line monitoring of short-lived neutral intermediates generated in electrochemical reactions. Based on the ion deflector of EC-NR setup, the electrogenerated neutral radicals can be successfully separated from the ionic products, allowing the transient neutral radicals TPrA˙, DBAE˙ and TEOA˙ to be detected by MS for the first time. The formation of these neutral radicals was further confirmed by using DMPO as the spin-trap agent and the MS2 method. Other reaction products such as iminium ions, NHPr2, 2-BAE and DEA were determined as well. Based on the data acquired, we have confirmed the low-oxidation potential ECL mechanism of the Ru(bpy)32+–TPrA system and the electrooxidation processes of DBAE and TEOA. Moreover, this EC-NR-MS technique provides a direct solution for elucidating complicated electrochemical reactions, especially for identifying short-lived neutral radicals as well as ionic intermediates.Author contributions
Jie Jiang designed the experiment, Hai Luo designed the experimental setup, Jilin Liu and Kai Yu performed the experiment and wrote the manuscript, Jing He and Hong Zhang modified the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21804027, 22074026, and 21874002).References
- Y. Ma, X. Yao, L. Zhang, P. Ni, R. Cheng and J. Ye, Angew. Chem., Int. Ed., 2019, 58, 16548–16552 CrossRef CAS PubMed.
- L. Tang, Z. Wang, Y. He and Z. Guan, ChemSusChem, 2020, 13, 4929–4936 CrossRef CAS PubMed.
- D. Wang, L. Zhang and S. Luo, Org. Lett., 2017, 19, 4924–4927 CrossRef CAS PubMed.
- X. Qin, C. Gu, M. Wang, Y. Dong, X. Nie, M. Li, Z. Zhu, D. Yang and Y. Shao, Anal. Chem., 2018, 90, 2826–2832 CrossRef CAS PubMed.
- Y. Dong, G. Chen, Y. Zhou and J. Zhu, Anal. Chem., 2016, 88, 1922–1929 CrossRef CAS PubMed.
- E. Monzani, S. Nicolis, S. Dell'Acqua, A. Capucciati, C. Bacchella, F. A. Zucca, E. V. Mosharov, D. Sulzer, L. Zecca and L. Casella, Angew. Chem., Int. Ed., 2019, 58, 6512–6527 CrossRef CAS PubMed.
- T. A. Enache and A. M. Oliveira-Brett, J. Electroanal. Chem., 2011, 655, 9–16 CrossRef CAS.
- L. Song, N. Fu, B. G. Ernst, W. H. Lee, M. O. Frederick, R. A. DiStasio Jr and S. Lin, Nat. Chem., 2020, 12, 747–754 CrossRef CAS PubMed.
- D. Lehnherr, Y. H. Lam, M. C. Nicastri, J. Liu, J. A. Newman, E. L. Regalado, D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2020, 142, 468–478 CrossRef CAS PubMed.
- V. Regnier, E. A. Romero, F. Molton, R. Jazzar, G. Bertrand and D. Martin, J. Am. Chem. Soc., 2019, 141, 1109–1117 CrossRef CAS PubMed.
- M. Zhou, J. Heinze, K. Borgwarth and C. P. Grover, ChemPhysChem, 2003, 4, 1241–1243 CrossRef CAS PubMed.
- K. Zhang, J. Liu, L. Wang, B. Jin, X. Yang, S. Zhang and J. H. Park, J. Am. Chem. Soc., 2020, 142, 8641–8648 CrossRef CAS PubMed.
- C. M. Hindson, P. S. Francis, G. R. Hanson and N. W. Barnett, Chem. Commun., 2011, 47, 7806–7808 RSC.
- C. M. Hindson, G. R. Hanson, P. S. Francis, J. L. Adcock and N. W. Barnett, Chem.–Eur. J., 2011, 17, 8018–8022 CrossRef CAS.
- K. Yu, H. Zhang, J. He, R. N. Zare, Y. Wang, L. Li, N. Li, D. Zhang and J. Jiang, Anal. Chem., 2018, 90, 7154–7157 CrossRef CAS PubMed.
- U. Karst, Angew. Chem., Int. Ed., 2004, 116, 2530–2532 CrossRef.
- H. Zhang, L. Qiao, W. Wang, J. He, K. Yu, M. Yang, H. You and J. Jiang, Anal. Chim. Acta, 2020, 1107, 107–112 CrossRef CAS PubMed.
- P. Liu, P. Zhao, R. G. Cooks and H. Chen, Chem. Sci., 2017, 8, 6499–6507 RSC.
- H. Zhang, Z. Wei, J. Jiang and R. G. Cooks, Angew. Chem., Int. Ed., 2018, 57, 17141–17145 CrossRef CAS PubMed.
- H. Chen, L. S. Eberlin and R. G. Cooks, J. Am. Chem. Soc., 2007, 129, 5880–5886 CrossRef CAS PubMed.
- X. Qin, Y. Dong, M. Wang, Z. Zhu, M. Li, D. Yang and Y. Shao, ACS Sens., 2019, 4, 2351–2357 CrossRef CAS PubMed.
- L. Yang, A. D. Hendsbee, Q. Xue, S. He, C. R. De-Jager, G. Xie, G. C. Welch and Z. Ding, ACS Appl. Mater. Interfaces, 2020, 12, 51736–51743 CrossRef CAS PubMed.
- S. Kirschbaum-Harriman, M. Mayer, A. Duerkop, T. Hirsch and A. J. Baeumner, Analyst, 2017, 142, 2469–2474 RSC.
- Y. Kuramochi, O. Ishitani and H. Ishida, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS.
- M. Abdellah, A. M. El-Zohry, L. J. Antila, C. D. Windle, E. Reisner and L. Hammarstrom, J. Am. Chem. Soc., 2017, 139, 1226–1232 CrossRef CAS PubMed.
- W. Miao, J. P. Choi and A. J. Bard, J. Am. Chem. Soc., 2002, 124, 14478–14485 CrossRef CAS PubMed.
- X. Tang, D. Zhao, J. He, F. Li, J. Peng and M. Zhang, Anal. Chem., 2013, 85, 1711–1718 CrossRef CAS PubMed.
- L. Chen, D. J. Hayne, E. H. Doeven, J. Agugiaro, D. J. D. Wilson, L. C. Henderson, T. U. Connell, Y. Nai, R. Alexander, S. Carrara, C. F. Hogan, P. S. Donnelly and P. S. Francis, Chem. Sci., 2019, 10, 8654–8667 RSC.
- Z. Guo and S. Dong, Anal. Chem., 2004, 76, 2683–2688 CrossRef CAS PubMed.
- Z. Liu, W. Qi and G. Xu, Chem. Soc. Rev., 2015, 44, 3117–3142 RSC.
- L. Hu and G. Xu, Chem. Soc. Rev., 2010, 39, 3275–3304 RSC.
- Z. Han, Y. Zhang, Y. Wu, Z. Li, L. Bai, S. Huo and X. Lu, Anal. Chem., 2019, 91, 8676–8682 CrossRef CAS PubMed.
- W. Miao, Chem. Rev., 2008, 108, 2506–2553 CrossRef CAS PubMed.
- R. Qiu, X. Zhang, H. Luo and Y. Shao, Chem. Sci., 2016, 7, 6684–6688 RSC.
- J. Hu, N. Zhang, P. K. Zhang, Y. Chen, X. H. Xia, H. Y. Chen and J. J. Xu, Angew. Chem., Int. Ed., 2020, 59, 18244–18248 CrossRef CAS PubMed.
- S. F. Teunissen, A. M. A. P. Fernandes, M. N. Eberlin and R. M. Alberici, TrAC, Trends Anal. Chem., 2017, 90, 135–141 CrossRef CAS.
- R. M. Alberici, R. C. Simas, G. B. Sanvido, W. Romao, P. M. Lalli, M. Benassi, I. B. Cunha and M. N. Eberlin, Anal. Bioanal. Chem., 2010, 398, 265–294 CrossRef CAS PubMed.
- E. T. Jansson, M. T. Dulay and R. N. Zare, Anal. Chem., 2016, 88, 6195–6198 CrossRef CAS PubMed.
- L. M. Wingen and B. J. Finlayson-Pitts, Chem. Sci., 2019, 10, 884–897 RSC.
- A. Reis, M. Rosário, M. Domingues, F. M. L. Amado, A. J. V. Ferrer-Correia and P. Domingues, J. Am. Soc. Mass Spectrom., 2003, 14, 1250–1261 CrossRef CAS PubMed.
- H. Taniguchi and K. P. Madden, J. Am. Chem. Soc., 1999, 121, 11875–11879 CrossRef CAS.
- M. Yan, J. Xin, L. Fan, J. Ye, T. Xiao, J. Huang and X. Yang, Anal. Chem., 2021, 3461–3469 CrossRef CAS PubMed.
- A. Zanut, A. Fiorani, S. Canola, T. Saito, N. Ziebart, S. Rapino, S. Rebeccani, A. Barbon, T. Irie, H. P. Josel, F. Negri, M. Marcaccio, M. Windfuhr, K. Imai, G. Valenti and F. Paolucci, Nat. Commun., 2020, 11, 2668 CrossRef CAS PubMed.
- X. Liu, L. Shi, W. Niu, H. Li and G. Xu, Angew. Chem., Int. Ed., 2007, 46, 421–424 CrossRef CAS PubMed.
- T. M. McCormick, B. D. Calitree, A. Orchard, N. D. Kraut, F. V. Bright, M. R. Detty and R. Eisenberg, J. Am. Chem. Soc., 2010, 132, 15480–15483 CrossRef CAS PubMed.
- T. Ghosh, T. Slanina and B. Konig, Chem. Sci., 2015, 6, 2027–2034 RSC.
- X. Xu, X. Qin, L. Wang, X. Wang, J. Lu, X. Qiu and Y. Zhu, Analyst, 2019, 144, 2359–2366 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01385h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |