
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnabling highly (R)-enantioselective epoxidation of styrene by engineering unique non-natural P450 peroxygenases†
Panxia
Zhao‡
ab,
Jie
Chen‡
ab,
Nana
Ma
ab,
Jingfei
Chen
a,
Xiangquan
Qin
ac,
Chuanfei
Liu
a,
Fuquan
Yao
a,
Lishan
Yao
a,
Longyi
Jin
c and
Zhiqi
Cong
 *ab
*ab
aCAS Key Laboratory of Biofuels, Shandong Provincial Key Laboratory of Synthetic Biology, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao, Shandong 266101, China. E-mail: congzq@qibebt.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
cDepartment of Chemistry, Yanbian University, Yanji, Jilin 133002, China
First published on 22nd March 2021
Abstract
Unlike the excellent (S)-enantioselective epoxidation of styrene performed by natural styrene monooxygenases (ee > 99%), the (R)-enantioselective epoxidation of styrene has not yet achieved a comparable efficiency using natural or engineered oxidative enzymes. This report describes the H2O2-dependent (R)-enantioselective epoxidation of unfunctionalized styrene and its derivatives by site-mutated variants of a unique non-natural P450BM3 peroxygenase, working in tandem with a dual-functional small molecule (DFSM). The observed (R)-enantiomeric excess (ee) of styrene epoxidation is up to 99% with a turnover number (TON) of 918 by the best enantioselective mutant F87A/T268I/L181Q, while the best active mutant F87A/T268I/V78A/A184L (with 98% ee) gave a catalytic TON of 4350, representing the best activity of a P450 peroxygenase towards styrene epoxidation to date. Following this approach, a set of styrene derivatives, such as o-, m-, p-chlorostyrenes and fluorostyrenes, could also be epoxidized with modest to very good TONs (362–3480) and high (R)-enantioselectivities (95–99% ee). The semi-preparative scale synthesis of (R)-styrene oxide performed at 0 °C with high conversion, maintaining enantioselectivity, and moderate isolated yields, further suggests the potential application of the current P450 enzymatic system in styrene epoxidation. This study indicates that the synergistic use of protein engineering and an exogenous DFSM constitutes an efficient strategy to control the enantioselectivity of styrene epoxidation, thus substantially expanding the chemical scope of P450 enzymes as useful bio-oxidative catalysts.
Introduction
Optically-pure styrene oxides are extremely useful building blocks for synthesizing chiral organic compounds and functional polymers.1 The asymmetric epoxidation of styrenes represents one of the most direct ways to prepare such compounds, and thus, it has attracted significant attention in recent years.2 However, despite critical progress in the development of asymmetric epoxidations based on synthetic molecular catalysts or biocatalysts, there is limited understanding regarding the highly-enantioselective epoxidation of terminal olefins, including styrene. Most catalytic systems demonstrate excellent enantioselective excess (ee) for (S)-styrene oxide generation, but not for the (R)-configuration.3–13 For example, the proline-derived C1-symmetric salen ligand with a Ti(OiPr)4 catalyst effectively converted several styrene derivatives to (S)-epoxides with 96–98% ee.3 The famous Jacobsen catalyst only generated 57% ee for (R)-styrene oxide, but that value improved to 86% when the reaction was performed at −78 °C.4 Similarly, a modified Shi epoxidation produced (R)-styrene oxide with decent enantioselectivity (71–85% ee).5 Some biomimetic porphyrin-ligand-based metal catalysts also suffer from insufficient enantioselectivity issues.6In nature, styrene monooxygenases (SMOs) are the most notable biocatalysts performing styrene epoxidation reactions; however, their outstanding selectivity produces only (S)-styrene oxide (>99% ee).7 Recently, the first natural SMO to generate (R)-styrene oxide (with 91% ee) was identified from the genome of Streptomyces sp. NRRL S-31.8 However, the highest known (R)-enantioselectivity (98% ee) for styrene oxidation was achieved by a strain of Mycobacterium sp. containing an ethene monooxygenase that interacts with a limited substrate scope.9 Certain engineered enzymes, such as cytochrome P450 monooxygenases and the Agrocybe aegerita unspecific peroxygenase (AaeUPO), have also been designed for the asymmetric epoxidation of olefins.10,11 Although some evolved variants exhibit excellent (R)-enantioselectivities for aromatic ring-substituted styrene derivatives (up to 99% ee),12 to the best of our knowledge, the highest (R)-enantioselectivity toward styrene oxide (96% ee) was observed by Reetz and Wang using a SO5 mutant of the NADPH-dependent P450BM3 enzyme.13 However, in terms of potential synthetic applications as chiral pharmaceutical intermediates, the ee value typically must exceed 98%.14 Considering the very high (S)-enantioselectivity achieved by native SMO enzymes, it remains a significant challenge to achieve comparable (R)-enantioselectivity in the epoxidation of unfunctionalized styrene using native enzymes or rationally-designed non-natural enzymes.
Recently, peroxygenase enzymes have garnered increasing interest related to the development of novel biocatalysts.15 These enzymes can promote P450-like oxyfunctionalization reactions without a cofactor (i.e., NADPH) or reduced partner proteins, thus exhibiting potential for practical synthetic applications.16 By utilizing the known “shunt pathway” in the catalytic cycle of P450 enzymes, an NADPH-dependent P450 monooxygenase from Balillus megaterium (P450BM3) can be repurposed for peroxygenase activity via site-directed mutagenesis or directed evolution.17 We recently constructed a unique artificial P450BM3 peroxygenase system by introducing an external dual-functional small molecule (DFSM),18 such as N-(ω-imidazolyl)-hexanoyl-L-phenylalanine (Im-C6-Phe). The DFSM binds to P450BM3 using an acyl amino acid as the anchoring group, and its imidazolyl moiety serves as a built-in acid–base catalyst to facilitate H2O2 activation. Given the fact that the ee of (R)-styrene oxide increased from 7% to 84% by the P450BM3_F87A mutant upon addition of Im-C6-Phe,18a we reasoned that the DFSM may also participate in tuning substrate orientation in active site pockets to influence the enantioselectivity of epoxidation (Fig. 1). Encouraged by the valuable contribution of the DFSM, herein we report further improvements in the enantioselectivity by protein engineering of the P450 peroxygenase system. This approach leads to excellent (R)-enantioselectivity of styrene epoxidation (up to 99% ee), relative to enzymatic and chemical catalysts reported to date.
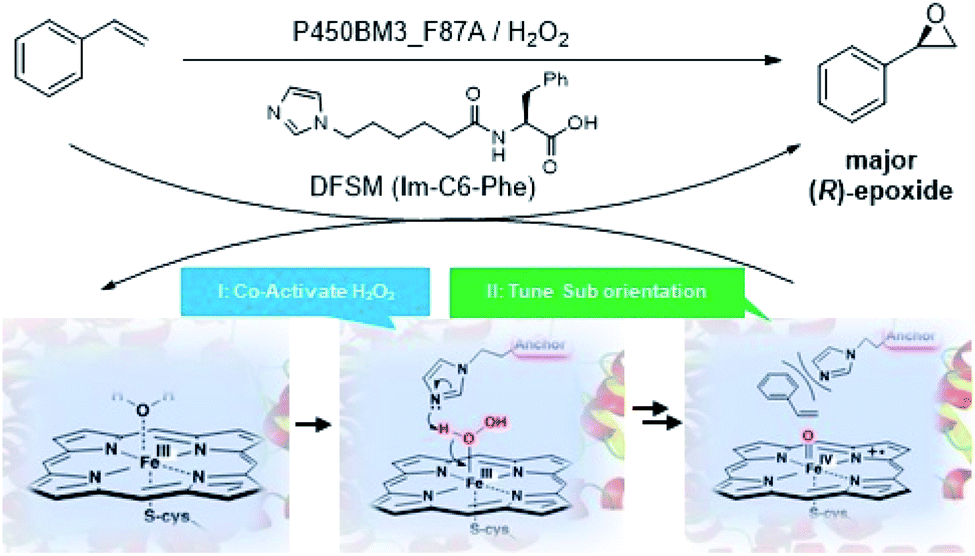 | ||
| Fig. 1 Asymmetric epoxidation of unfunctionalized styrenes catalyzed by DFSM-facilitated P450BM3 peroxygenases. The DFSM, a dual-functional small molecule, binds with P450 through an anchor group, and is proposed to play a role in activating H2O2 and tuning substrate orientation by using the other inbuilt base group (imidazolyl group). | ||
Results and discussion
Effect of T268 mutation on the activity and enantioselectivity of styrene epoxidation by NADPH- and H2O2-dependent P450BM3 systems
It is well known that key residues located near the active site of enzymes significantly affect their catalytic activity and selectivity. However, among these residues, some may be highly conserved so that they are generally untouchable in protein engineering because the mutation of highly conserved residues may lead to the possible decrease or even loss of their intrinsic functions. T268 located close to the heme center in P450BM3 belongs to one of such residues mentioned above. Indeed, the highly conserved threonine residue at the proximal position of heme is believed to play a versatile role in the formation of active oxygen species, compound I, in the catalytic cycle of most NADPH-dependent P450 enzymes (Fig. 2A, Path A).19 Many studies have shown that mutating the conserved threonine decreases or even eliminates the catalytic activity of native NADPH-dependent P450 enzymes.20 However, we have demonstrated that the introduction of a DFSM could partially rescue the peroxygenase activity of the DFSM-facilitated P450BM3-H2O2 system even when the T268 residue was replaced by valine.18a This provided us a chance to construct a novel H2O2-dependent P450 biocatalyst based on its T268 mutant.18b,d,e More importantly, it is worthy to note that T268 mutation also showed a positive effect on regio- or stereoselectivity in P450-catalyzed non-native reactions. For instance, Arnold, Fasan, and Shoji groups have respectively reported that the T268A mutant of P450BM3 showed a drastic effect on the enantioselectivity in intramolecular C–H amination or cyclopropanation reactions.21 We observed the substantial contribution of T268 mutations in regioselective demethylation for the oxidation of aromatic ethers by the DFSM-facilitated P450BM3 peroxygenase.18e Therefore, we hypothesized that merging the positive effect of Im-C6-Phe and the potential positive effect of T268 mutation will improve the (R)-enantioselectivity of styrene epoxidation.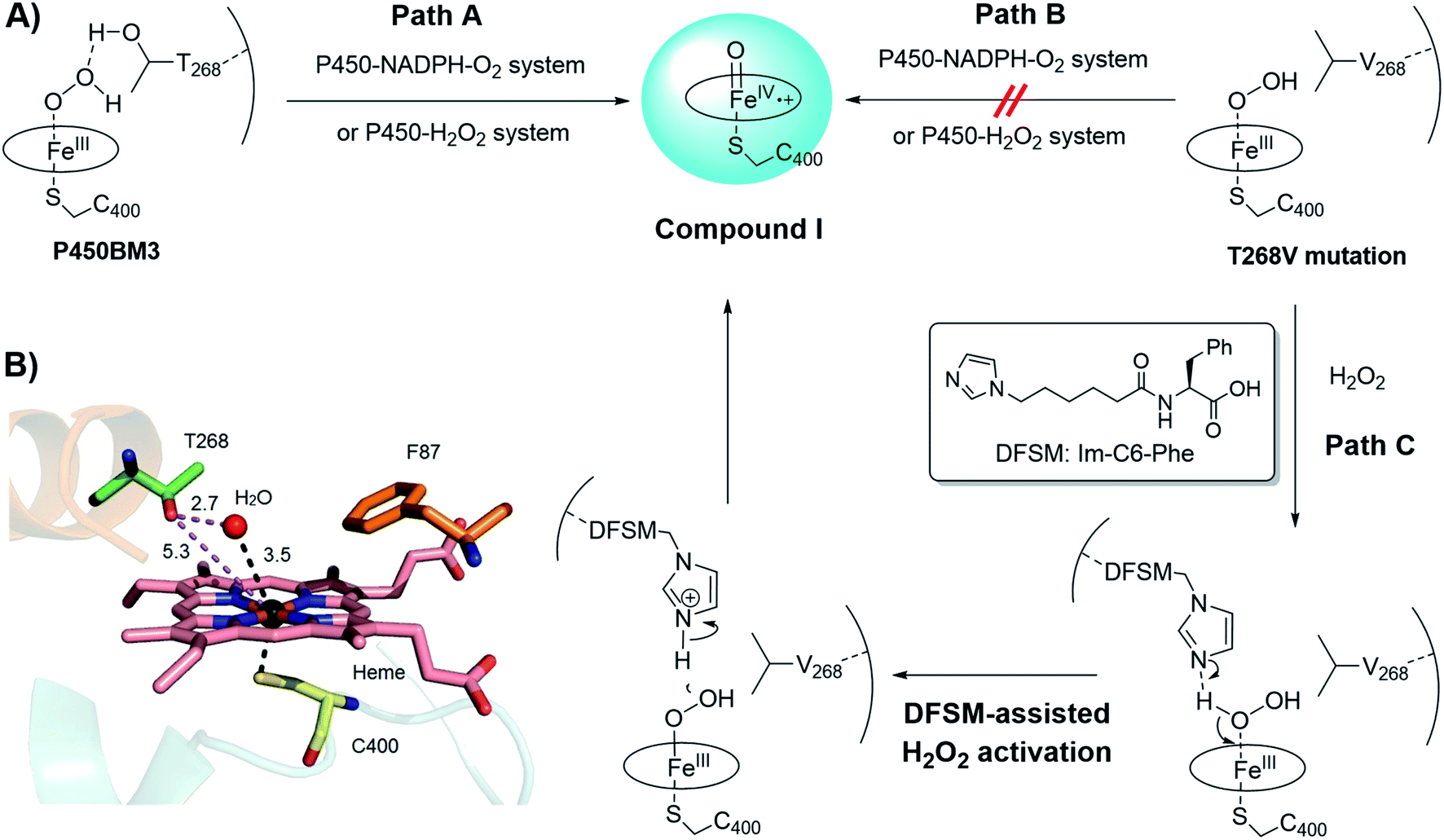 | ||
| Fig. 2 (A) Proposed mechanistic role of the T268 residue in the formation of compound I in the catalytic cycle of NADPH-dependent P450BM3 monooxygenase and DFSM-facilitated P450BM3 peroxygenase (Path A), the mutation of T268 impeding the formation of compound I in both NADPH- and H2O2-dependent P450BM3 systems (Path B), and the plausible role of DFSM in the activation of to induce the formation of compound I in the case of the T268V mutation in the DFSM-facilitated P450BM3 peroxygenase (Path C). (B) Active site of wild-type P450BM3 (PDB no. 1JPZ), highlighting the relative positions of T268 and the axially-coordinated H2O. | ||
We initially evaluated the effect of T268 mutation on the activity and enantioselectivity of styrene epoxidation by NADPH- and H2O2-dependent P450BM3 systems, respectively. The epoxidation of styrene was firstly examined by using full-length P450BM3 enzymes upon addition of NADPH. The catalytic turnover numbers (TONs) of variants with single mutations of phenylalanine (F87A, F87G, and F87V) reached 142, 628, and 788 together with poor enantioselectivity, respectively (Table 1, entries 1–3 and Fig. S3†). The double mutants (F87A/T268V, F87G/T268V, and F87V/T268V) exhibited drastically weakened catalytic activities, as evidenced by their 8-, 23-, and 11-fold decreases in TONs relative to the corresponding single mutants (Table 1, entries 4–6). These results are consistent with previous reports that the mutation of the threonine residue leads to the decrease of catalytic activity. We reasoned that the lower epoxidation activity may be due to the sluggish formation of compound I when the T268 residue is substituted with valine as shown in the proposed catalytic mechanism (Fig. 2A, Path B). Although protein engineering can efficiently improve the catalytic activity of enzymes in most cases, we concluded that it is difficult to further improve the catalytic activity of the T268 mutants of P450BM3 by this general approach due to the above mechanism. Therefore, it is difficult to control the enantioselectivity of epoxidation by the approach of mutating the T268 residue in the NADPH-dependent P450BM3 system.
| Entry | P450s | Im-C6-Phe | Oxidant | TONc | eed |
|---|---|---|---|---|---|
| a Reaction conditions: full_F87X (0.5 μM), styrene (4 mM), and NADPH (5 mM) in 0.1 M pH 8.0 phosphate buffer at 25 °C. b Reaction conditions: H_F87X/T268V (0.5 μM), styrene (4 mM), Im-C6-Phe (500 μM), and H2O2 (20 mM) in pH 8.0 phosphate buffer. c TON: turnover numbers estimated for 30 minute reactions. d ee: enantiomeric excess (%) of (R)-styrene oxide determined by chiral GC and calculated from (R)-styrene oxide/((R)-styrene oxide + (S)-styrene oxide) according to the corresponding peak area. e Previously-reported results.18a For entries 1–6, “full” denoted mutants of full-length P450BM3; for entries 7–12, “H” denoted mutants of the P450BM3 heme domain; nd: not detected. | |||||
| 1 | full_F87A | ✗ | O2/NADPH | 142 ± 20 | 14 |
| 2 | full_F87G | ✗ | O2/NADPH | 628 ± 24 | 63 |
| 3 | full_F87V | ✗ | O2/NADPH | 788 ± 3 | −58 |
| 4 | full_F87A/T268V | ✗ | O2/NADPH | 18 ± 1 | nd |
| 5 | full_F87G/T268V | ✗ | O2/NADPH | 27 ± 1 | nd |
| 6 | full_F87V/T268V | ✗ | O2/NADPH | 72 ± 1 | −21 |
| 7e | H_F87A/T268V | ✗ | H2O2 | nd | nd |
| 8 | H_F87G/T268V | ✗ | H2O2 | nd | nd |
| 9 | H_F87V/T268V | ✗ | H2O2 | nd | nd |
| 10e | H_F87A/T268V | ✓ | H2O2 | 382 ± 2 | 94 |
| 11 | H_F87G/T268V | ✓ | H2O2 | 767 ± 2 | 83 |
| 12 | H_F87V/T268V | ✓ | H2O2 | 161 ± 3 | 52 |
We then scrutinized the effect of the T268V mutation on the H2O2-driven P450BM3 system. Double mutations (F87A/T268V, F87G/T268V, and F87V/T268V) in the P450BM3 heme domain led to complete loss of catalytic activity towards styrene epoxidation in the absence of Im-C6-Phe (Table 1, entries 7–9). Comparing these results to the catalytic TONs achieved by the heme domain with single mutants, F87A (42), F87G (53), and F87V (120), under identical reaction conditions,18a suggested that the T268 residue may also play a key role in the formation of compound I in the H2O2-driven P450BM3 system (Fig. 2A, Path A and B). Additionally, the catalytic TONs of the double mutants (F87X/T268V) reached 382, 767, and 161 following the addition of Im-C6-Phe (entries 10–12). This indicated that the DFSM could act as a built-in base to assist the activation of H2O2, thus reviving the peroxygenase activity of P450BM3, even when the highly-conserved T268 residue was mutated (Fig. 2A, Path C). Unlike the T268 mutation in the NADPH-dependent P450BM3 monooxygenase, which almost irreversibly blocks the formation of compound I, this is a mechanistic advantage of the DFSM-facilitated P450BM3 peroxygenase that allows us to further improve the catalytic activity of this peroxygenase system by protein engineering. Moreover, double-mutants containing T268V demonstrated enhanced (R)-enantioselectivity for styrene epoxidation (entries 10–12) relative to previously-reported results regarding single mutants.18a,22 This result indicated the importance of mutating T268 to further tune the enantioselectivity of styrene epoxidation in the DFSM-facilitated P450BM3-H2O2 system. Therefore, we reasoned that both the catalytic activity and (R)-enantioselectivity for styrene epoxidation by this unique non-natural P450 peroxygenase system can be well improved by mutations of T268 and further protein engineering.
Protein engineering for improving the activity and (R)-enantioselectivity of styrene epoxidation
We firstly constructed a library of double mutants probing only two selected positions (F87 and T268) to avoid excess screening efforts (Table S1 and Fig. S4†). None of the examined variants exhibited epoxidation activity in the absence of Im-C6-Phe. However, certain mutants (F87A/T268V, F87A/T268I, F87G/T268V, F87G/T268I, F87V/T268I, and F87I/T268I) demonstrated the formation of styrene oxide with moderate to good TONs (318 to 767) and good to excellent (R)-enantioselectivity (83% to 97% ee), in the presence of Im-C6-Phe. These results demonstrated the importance of T268 mutation for tuning the enantioselectivity. It is worth noting that only T268V and T268I mutations showed positive effects for the ee enhancement when combined with mutations at the 87 position, and the TONs changed with the increase or decrease of the residue size of mutations at the 268 position. On the other hand, smaller mutations seemed beneficial for TON improvement, while both bigger and smaller mutations led to decreasing ee values when compared with F87A (Table S1†). These results suggested that the nature (or the suitable combination) of double mutants is quite significant for creating a suitable space for accommodating substrates near the active site.Despite its relatively low TON (335), the double mutant F87A/T268I (AI) was selected as the parent enzyme on which to perform the next round of mutations because of its excellent enantioselectivity (97% ee). With the aim of further improving the catalytic activity and enantioselectivity, we introduced additional mutations at ten other key residues around the substrate pocket highlighted in Fig. 3A (L75, V78, A82, L181, A184, I263, A264, E267, A328, R255).19 Residues L75, V78, A82, I263, A264, E267, and A328 are located around the hydrophobic access channel for the native substrate of P450BM3. Residues L181 and A184 are situated near the top of the active site, and A184 has specifically been reported to affect the enantioselectivity of styrene epoxidation by NADPH-dependent P450BM3.10a Residue R255 is located on the surface of the protein along helix I and is known to be a site that influences the catalytic activity of P450PMO.23 These residues were selectively mutated to several other amino acids with structurally- and functionally-diverse side chains to tune substrate pocket properties. Similar approaches have been applied to construct small and smart libraries, which efficiently decrease the effort involved in developing stereoselective isozymes or multi-functional enzyme variants by directed evolution.24 Therefore, a library containing approximately 60 combinations of these ten mutations in AI was created and screened for styrene epoxidation activity and enantioselectivity (Table S2 and Fig. S5†).
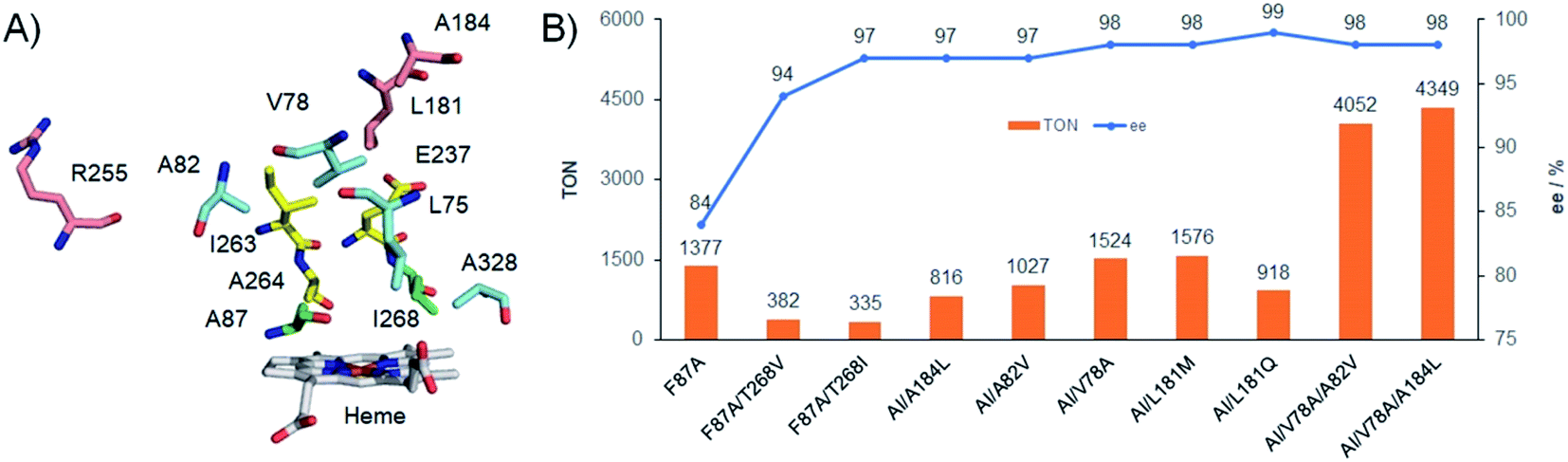 | ||
| Fig. 3 (A) The residues around the active site of P450BM3 (PDB no. 1JPZ), and the residues F87 and T268 were mutated to A87 and I268, respectively. (B) Protein engineering of P450BM3 for styrene epoxidation. Experiments were performed using P450BM3 variants (μM), 4 mM styrene, 20 mM (or 80 mM) H2O2, and 2 mM Im-C6-Phe in pH 8.0 phosphate buffer at 4 °C (or 25 °C) for 30 min. “AI” denoted the double mutant F87A/T268I. *In some cases, trace amounts of phenyl acetaldehydes were detected by GC. | ||
Most of the examined triple mutants involving the 75, 263, 264, 267, and 328 positions led to generally negative effects, i.e., reducing enantioselectivity, catalytic TONs, or both. Triple mutants involving a change at R255 increased the proportion of R-styrene oxide produced (up to >99% ee), but significantly decreased the catalytic TONs to 120 (Fig. S5 and Table S2†). Fortunately, some triple mutants modified at the 78, 82, 181, and 184 positions, including AI/V78A, AI/A82V, AI/L181Q, AI/L181M, and AI/A184L, exhibited improved enantioselectivities and catalytic TONs compared with the parent AI (Fig. 3B). Among them, AI/L181Q demonstrated the best (R)-enantioselectivity (99% ee) with a good TON of 918. The catalytic TON of AI/L181Q is close to 1000, which is considered an important benchmark for potential practical applicability. To further improve the enantioselectivity and catalytic TONs, we prepared several quadruple mutants by combining these beneficial mutation sites on the basis of AI. Although more than half of them did not show positive effects on either enantioselectivities or catalytic TONs, combining them with the most active triple mutant AI/V78A resulted in four highly-active quadruple mutants (Fig. S6 and Table S3†). Unfortunately, the mutant AI/V78A/L181Q increased the catalytic TON to 2060 but accompanied by decreased enantioselectivity (97% ee) when compared with AI/L181Q. Interestingly, both other mutants containing V78A, AI/V78A/A82V (TON = 4052) and AI/V78A/A184L (TON = 4349) exhibited excellent catalytic activity together with the same enantioselectivity relative to AI/V78A, each achieving 98% ee (Fig. 3B, S6 and Table S3†).
Substrate scope
Encouraged by these excellent results, we examined the scope of olefinic substrates that could be epoxidized using the DFSM-facilitated P450BM3 peroxygenase system. As discussed above, appropriate spaces near the active site have an important influence on the activity and selectivity of the substrate. Considering the size difference between styrene and its derivatives, it is reasonable that mutants favourable for styrene are not necessarily optimal mutants for other styrene derivatives. In theory, a full scan of all established mutants would help us find the most beneficial mutant for each substrate, but this requires too much screening work. Therefore, we respectively selected several mutants with different mutation sizes at key sites to examine different substrates. The advantage of this approach is that if we find mutants that have relatively good activity and selectivity for a substrate, even if it is not the best, we can achieve our aim of demonstrating that the current system is effective for that substrate.By employing different P450BM3 mutants, we discovered that the catalytic system has a broad substrate scope, with the capability to catalyze epoxidations of ortho-, meta-, and para-substituted chlorostyrenes (1b–d) and fluorostyrenes (1e–g; Fig. 4, S7–S11 and Tables S4–S9†) to generate the corresponding (R)-enantiomeric epoxide products (2d–g) with moderate to excellent TONs (up to 3480) and excellent ee (>99%). It is worthwhile to note that the F87A/T268A mutant achieved better ee (87%) and excellent TON (1724) compared to F87A/T268I and F87A/T268V mutants, the latter only provided moderate ee (50% and 51%) and TONs (153 and 514) for the epoxidation of o-chlorostyrene (Fig. S7 and Table S4†). The (R)-enantioselectivity of 2b can be improved to 95% by the triple mutant F87A/T268A/V78A. In addition, the best ee of 2c reached 96% by F87A/T268I/A82I with a moderate TON (362). This indicated that the size of the amino acid side chain at the 268 position substantially affected the enantioselectivity and catalytic activity of certain substrates having larger Cl-substituted groups. This is also confirmed by the reaction results of 2f–2g, in which substrates with smaller F-substituted groups (2e–2g) or less steric hindrance 2f (para-chlorostyrene) gave excellent ee (97–99%) and catalytic TONs.
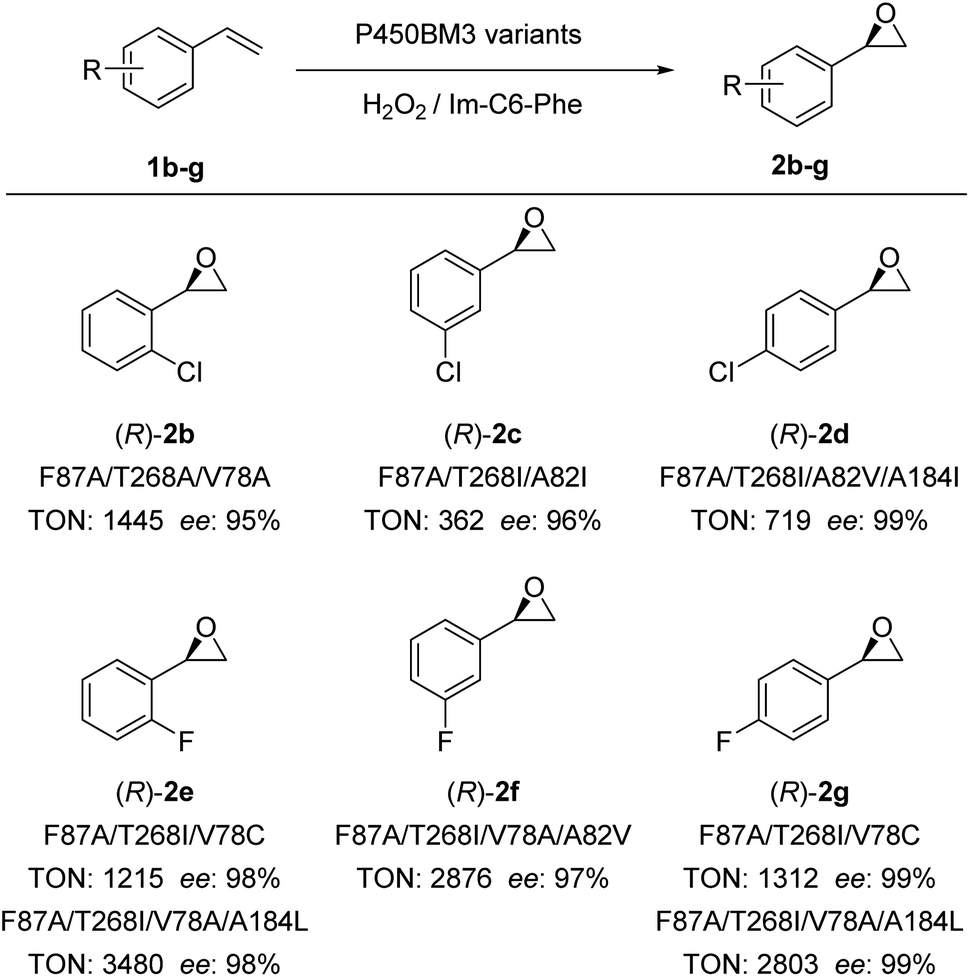 | ||
| Fig. 4 Substrate scope of epoxidation reactions catalyzed by P450BM3 variants using H2O2 in the presence of Im-C6-Phe. *In some cases, trace amounts of phenyl acetaldehyde were detected by GC. | ||
Semi-preparative scale synthesis of (R)-styrene oxide
Having achieved highly (R)-enantioselective epoxidation of styrenes, we next investigated the semi-preparative scale synthesis of (R)-styrene oxide by using the DFSM-facilitated P450BM3 peroxygenase system to further examine its practical applicability. The reactions were performed with a higher styrene concentration (10 mM) by using the most active F87A/T268I/V78A/A82V mutant (2.5 μM) and the most enantioselective F87A/T268I/L181Q mutant (6 μM) in the presence of optimized amounts of Im-C6-Phe (2 mM) and H2O2 (80 mM) (Fig. S13 and S14†) in 20 mL 0.1 M pH 8.0 phosphate buffer at 0 °C, respectively. As shown in Table 2, the starting styrene is almost consumed by both mutants of F87A/T268I/L181Q (consumption ratio: 84%) and F87A/T268I/V78A/A82V (consumption ratio: 97%), while enantioselectivities were almost maintained (Fig. S15–S17†). Styrene oxide was isolated as a colourless liquid (10.5 mg, 43.8% for F87A/T268I/L181Q and 13 mg, 54.2% for F87A/T268I/V78A/A82V, respectively). The moderate yields relative to high styrene consumption may be due to the volatilization of the substrate, and a similar styrene volatilization phenomenon was also observed by Hollmann et al. in their previous reports.16a Although these results indicated the potential of the DFSM-facilitated P450BM3 peroxygenase system in the (R)-enantioselective epoxidation of styrene and its derivatives, there are still some challenges to develop a practical biocatalytic system for synthetic application. Firstly,it is worth noting that developing a more efficient DFSM is crucial for achieving this aim because of the larger loading amount. Moreover, it is also an issue for the stability of P450 enzymes and their reusability under the reaction conditions, e.g. low temperature (0 °C). The stability issue may be solved by further protein engineering. Considering the difficulty to recover trace amounts of P450 enzyme catalysts, further development of immobilized P450s may be a solution to improve the practical application capacity of the current catalytic system.| Mutants | Consumptionc/% | Yieldd/% | eee/% |
|---|---|---|---|
| a Reaction conditions: heme domain of P450BM3 mutants (2.5 μM), styrene (10 mM, dissolved in 2% methanol), H2O2 (80 mM), Im-C6-Phe (2 mM) in 20 mL 0.1 M pH 8.0 phosphate buffer at 0 °C. b Trace amount of phenyl acetaldehyde was detected by GC but not isolated (Fig. S15). c Consumption ratio (%) = consumed styrene/initial styrene × 100%. d Isolated yields based on starting styrene. e ee: enantiomeric excess (%) of (R)-styrene oxide determined by chiral GC. f 6.0 μm enzyme was used. | |||
| F87A/T268I/L181Qf | 84 | 43.8 | 99 |
| F87A/T268I/V78A/A82V | 97 | 54.2 | 97 |
Conclusions
In summary, we have achieved high (R)-enantioselectivity and activity in the H2O2-dependent epoxidation of unfunctionalized styrene and its derivatives through protein engineering of a unique DFSM-facilitated P450BM3 peroxygenase system. The enantiomeric excess of (R)-styrene oxide (99%) is comparable to or even higher than that of reported native or engineered oxidative enzymes. This enantioselectivity is comparable but inverse to the well-known styrene monooxygenase (styA) that oxidizes styrene to (S)-styrene oxide with >99% ee.2a,8 Furthermore, a catalytic TON of ∼4500 (with 98% ee) was achieved by the most active mutants in our system, and following this approach, a set of styrene derivatives, such as o-, m-, p-chlorostyrenes and fluorostyrenes could also be epoxidized with modest to very good TONs (362–3480) and high (R)-enantioselectivities (95–99% ee). Finally, the semi-preparative scale synthesis of (R)-styrene oxide gave high conversion, maintaining enantioselectivity, and moderate isolated product yields. These results suggested the potential application of the current P450 enzymatic system in styrene epoxidation. The crucial mutation of T268 and addition of a DFSM comprise an effective, novel approach for controlling enantioselectivity, which cannot be applied to natural NADPH-dependent P450BM3 monooxygenases because they lose their monooxygenation activity following T268 mutation. This work substantially expands the catalytic potential of P450 enzymes as (R)-enantioselective epoxygenases, further indicating that the discussed P450 peroxygenase system may have unique advantages for asymmetric epoxidation.Author contributions
Z. C. conceived the project. Z. C., L. Y. and L. J. supervised the investigation. Z. C., P. Z. and J. C. designed the experiments. P. Z. and J. C. performed majority of the experiments and data analysis. N. M. and F. Y. participated in construction of mutant library and protein purification. J. C. participated in data analysis. X. Q. and C. L. prepared the small molecule materials. Z. C. and P. Z. wrote the manuscript. All the authors edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21778060 and 21977104), the Qingdao Innovative Leading Talent Project (18-1-2-9-zhc), QIBEBT (I201901 and Y872361901), and the Director Innovation Fund of Key Laboratory of Biofuels, Chinese Academy of Sciences (Y872131901). We also thank Dr Suzanne Adam (Liwen Bianji, Edanz Editing China) for editing the English text of the draft of this manuscript.Notes and references
- (a) G. Xu, Q. Mahmood, C. Lv, R. Yang, L. Zhou and Q. Wang, Asymmetric kinetic resolution polymerization, Coord. Chem. Rev., 2020, 414, 213–296 CrossRef; (b) W. Ren, T. Yue, X. Zhang, G. Gu, Y. Liu and X. Lu, Stereoregular CO2 Copolymers from Epoxides with an Electron-Withdrawing Group: Crystallization and Unexpected Stereocomplexation, Macromolecules, 2017, 50, 7062–7069 CrossRef CAS; (c) G. Wu, S. Wei, W. Ren, X. Lu, B. Li, Y. Zu and D. Darensbourg, Alternating copolymerization of CO2 and styrene oxide with Co(iii)-based catalyst systems: differences between styrene oxide and propylene oxide, Energy Environ. Sci., 2011, 4, 5084–5092 RSC.
- (a) H. Lin, M.-Y. Xu, Y. Liu, Z.-L. Wu, Biocatalytic epoxidation for green synthesis in green biocatalysis, ed. Patel R. N., John Wiley & Sons, Inc., 1st edn, 2016 Search PubMed; (b) W. Sun and Q. Sun, Bioinspired manganese and iron complexes for enantioselective oxidation reactions: ligand design, catalytic activity, and beyond, Acc. Chem. Res., 2019, 52, 2370–2381 CrossRef CAS PubMed; (c) S. Wu, Y. Zhou and Z. Li, Biocatalytic selective functionalisation of alkenes via single-step and one-pot multi-step reactions, Chem. Commun., 2019, 55, 883–896 RSC; (d) E. Rose, B. Andrioletti, S. Zrig and M. Quelquejeu-Ethéve, Enantioselective epoxidation of olefins with chiral metalloporphyrin catalysts, Chem. Soc. Rev., 2005, 34, 573–583 RSC.
- K. Matsumoto, T. Oguma and T. Katsuki, Highly enantioselective epoxidation of styrenes catalyzed by proline-derived C1-symmetric titanium(salan) complexes, Angew. Chem., Int. Ed., 2009, 48, 7432–7435 CrossRef CAS PubMed.
- (a) W. Zhang, J. L. Loebach, S. R. Wilson and E. N. Jacobsen, Enantioselective epoxidation of unfunctionalized olefins catalyzed by (salen)manganese complexes, J. Am. Chem. Soc., 1990, 112, 2801–2803 CrossRef CAS; (b) M. Palucki, P. J. Pospisil, W. Zhang and E. N. Jacobsen, Highly enantioselective, low-temperature epoxidation, J. Am. Chem. Soc., 1994, 116, 9333–9334 CrossRef CAS.
- H. Tian, X. She, J. Xu and Y. Shi, Enantioselective epoxidation of terminal olefins by chiral dioxirane, Org. Lett., 2001, 3, 1929–1931 CrossRef CAS PubMed.
- C. Wang and H. Yamamoto, Asymmetric epoxidation using hydrogen peroxide as oxidant, Chem.–Asian J., 2015, 10, 2056–2068 CrossRef CAS PubMed.
- (a) T. Heine, A. Scholtissek, S. Hofmann, R. Koch and D. Tischler, Accessing enantiopure epoxides and sulfoxides: related flavin-dependent monooxygenases provide reversed enantioselectivity, ChemCatChem, 2020, 12, 199–209 CrossRef CAS; (b) H. Lin, Y. Tang, S. Dong, R. Lang and H. Chen, A new monooxygenase from Herbaspirillum huttiense catalyzed highly enantioselective epoxidation of allylbenzenes and allylic alcohols, Catal. Sci. Technol., 2020, 10, 2145–2151 RSC; (c) M. M. C. H. van Schie, C. E. Paul, I. W. C. E. Arends and F. Hollmann, Photoenzymatic epoxidation of styrenes, Chem. Commun., 2019, 55, 1790–1792 RSC.
- C. Cui, C. Guo, H. Lin, Z.-Y. Ding, Y. Liu and Z.-L. Wu, Functional characterization of an (R)-selective styrene monooxygenase from streptomyces sp. NRRL S-31, Enzyme Microb. Technol., 2019, 109391 Search PubMed.
- C. Nothe and S. Hartmans, Formation and Degradation of Styrene Oxide Stereoisomers by Different Microorganisms, Biocatalysis, 1994, 10, 219–225 CrossRef CAS.
- (a) K. L. Tee and U. Schwaneberg, A screening system for the directed evolution of epoxygenases: importance of position 184 in P450BM3 for stereoselective styrene epoxidation, Angew. Chem., Int. Ed., 2006, 45, 5380–5383 CrossRef CAS PubMed; (b) A. Shehzad, S. Panneerselvam, M. Linow, M. Bocola, D. Roccatano, J. Mueller-Dieckmann, M. Wilmanns and U. Schwaneberg, P450 BM3 crystal structures reveal the role of the charged surface residue Lys/Arg184 in inversion of enantioselective styrene epoxidation, Chem. Commun., 2013, 49, 4694–4696 RSC; (c) L. Wang, S. Wei, X. Pan, P. Liu, X. Du, C. Zhang, L. Pu and Q. Wang, Enhanced turnover for the P450 119 peroxygenase-catalyzed asymmetric epoxidation of styrenes by random mutagenesis, Chem. - Eur. J., 2018, 24, 2741–2749 CrossRef CAS PubMed; (d) C. Zhang, P.-X. Liu, L.-Y. Huang, S.-P. Wei, L. Wang, S.-Y. Yang, X.-Q. Yu, L. Pu and Q. Wang, Engineering P450 peroxygenase to catalyze highly enantioselective epoxidation of cis-β-ethylstyrenes, Chem. - Eur. J., 2016, 22, 10969–10975 CrossRef CAS PubMed.
- (a) G. Xu, M. Crotti, T. Saravanan, K. M. Kataja and G. J. Poelarends, Enantiocomplementary epoxidation reactions catalyzed by an engineered cofactor-independent non-natural peroxygenase, Angew. Chem., Int. Ed., 2020, 59, 10374–10378 CrossRef CAS PubMed; (b) M. C. R. Rauch, F. Tieves, C. E. Paul, I. W. C. E. Arends, M. Alcalde and F. Hollmann, Peroxygenase-catalysed epoxidation of styrene derivatives in neat reaction media, ChemCatChem, 2019, 11, 4519–4523 CrossRef CAS PubMed; (c) S. Peter, M. Kinne, R. Ullrich, G. Kayser and M. Hofrichter, Epoxidation of linear, branched and cyclic alkenes catalyzed by unspecific peroxygenase, Enzyme Microb. Technol., 2013, 52, 370–376 CrossRef CAS PubMed; (d) M. Kluge, R. Ullrich, K. Scheibnerb and M. Hofrichter, Stereoselective benzylic hydroxylation of alkylbenzenes and epoxidation of styrene derivatives catalyzed by the peroxygenase of Agrocybe aegerita, Green Chem., 2012, 14, 440–446 RSC.
- (a) A. Li, J. Liu, S. Q. Pham and Z. Li, Engineered P450pyr monooxygenase for asymmetric epoxidation of alkenes with unique and high enantioselectivity, Chem. Commun., 2013, 49, 11572–11574 RSC; (b) A. Li, S. Wu, J. P. Adams, R. Snajdrova and Z. Li, Asymmetric epoxidation of alkenes and benzylic hydroxylation with P450tol monooxygenase from Rhodococcus coprophilus TC-2, Chem. Commun., 2014, 50, 8771–8774 RSC; (c) Q.-S. Li, J. Ogawa, R. D. Schmid and S. Shimizu, Residue size at position 87 of cytochrome P450 BM-3 determines its stereoselectivity in propylbenzene and 3-chlorostyrene oxidation, FEBS Lett., 2001, 508, 249–252 CrossRef CAS PubMed.
- D. Yu, J.-b. Wang and M. T. Reetz, Exploiting designed oxidase-peroxygenase mutual benefit system for asymmetric cascade reactions, J. Am. Chem. Soc., 2019, 141, 5655–5658 CrossRef CAS PubMed.
- S. Q. Pham, G. Pompidor, J. Liu, X.-D. Li and Z. Li, Evolving P450pyr hydroxylase for highly enantioselective hydroxylation at non-activated carbon atom, Chem. Commun., 2012, 48, 4618–4620 RSC.
- (a) M.-C. Sigmund and G. J. Poelarends, Current state and future perspectives of engineered and artificial peroxygenases for the oxyfunctionalization of organic molecules, Nat. Catal., 2020, 3, 690–702 CrossRef CAS; (b) Y. Wang, D. Lan, R. Durrani and F. Hollmann, Peroxygenases en route to becoming dream catalysts. What are the opportunities and challenges?, Curr. Opin. Chem. Biol., 2017, 37, 1–9 CrossRef PubMed.
- (a) F. Tieves, S. J.-P. Willot, M. Martinus, M. M. C. H. van Schie, M. C. R. Rauch, S. H. H. Younes, W. Zhang, J. Dong, P. G. de Santos, J. M. Robbins, B. Bommarius, M. Alcalde, A. S. Bommarius and F. Hollmann, Formate oxidase (FOx) from Aspergillus oryzae: one catalyst enables diverse H2O2-dependent biocatalytic oxidation reactions, Angew. Chem., Int. Ed., 2019, 58, 7873–7877 CrossRef CAS PubMed; (b) W. Zhang, E. Fernández-Fueyo, Y. Ni, M. van Schie, J. Gacs, R. Renirie, R. Wever, F. G. Mutti, D. Rother, M. Alcalde and F. Hollmann, Selective aerobic oxidation reactions using a combination of photocatalytic water oxidation and enzymatic oxyfunctionalizations, Nat. Catal., 2018, 1, 55–62 CrossRef CAS PubMed.
- (a) O. Shoji, T. Fujishiro, K. Nishio, Y. Kano, H. Kimoto, S. C. Chien, H. Onoda, A. Muramatsu, S. Tanaka, A. Hori, H. Sugimoto, Y. Shirob and Y. Watanabe, A substrate-binding-state mimic of H2O2-dependent cytochrome P450 produced by one-point mutagenesis and peroxygenation of non-native substrates, Catal. Sci. Technol., 2016, 6, 5806–5811 RSC; (b) P. C. Cirino and F. H. Arnold, A self-sufficient peroxide-driven hydroxylation biocatalyst, Angew. Chem., Int. Ed., 2003, 42, 3299–3301 CrossRef CAS PubMed.
- (a) N. Ma, Z. Chen, J. Chen, J. Chen, C. Wang, H. Zhou, L. Yao, O. Shoji, Y. Watanabe and Z. Cong, Dual-functional small molecules for generating an efficient cytochrome P450BM3 peroxygenase, Angew. Chem., Int. Ed., 2018, 57, 7628–7633 CrossRef CAS PubMed; (b) Z. Chen, J. Chen, N. Ma, H. Zhou and Z. Cong, Selective hydroxylation of naphthalene using the H2O2-dependent engineered P450BM3 driven by dual-functional small molecules, J. Porphyrins Phthalocyanines, 2018, 22, 831–836 CrossRef CAS; (c) J. Xu, C. Wang and Z. Cong, Strategies for substrate-regulated P450 catalysis: from substrate engineering to co-catalysis, Chem. - Eur. J., 2019, 25, 6853–6863 CrossRef CAS PubMed; (d) J. Chen, F. Kong, N. Ma, P. Zhao, C. Liu, X. Wang and Z. Cong, Peroxide-driven hydroxylation of small alkanes catalyzed by an artificial P450BM3 peroxygenase system, ACS Catal., 2019, 9, 7350–7355 CrossRef CAS; (e) Y. Jiang, C. Wang, N. Ma, J. Chen, C. Liu, F. Wang, J. Xu and Z. Cong, Regioselective aromatic O-demethylation with an artificial P450BM3 peroxygenase system, Catal. Sci. Technol., 2020, 10, 1219–1223 RSC; (f) X. Wang, J. Chen, N. Ma and Z. Cong, Selective hydroxylation of alkanes catalyzed by cytochrome P450 enzymes, Acta Chim. Sin., 2020, 78, 490–503 CrossRef.
- C. J. Whitehouse, S. G. Bell and L.-L. Wong, P450BM3 (CYP102A1): connecting the dots, Chem. Soc. Rev., 2012, 41, 1218–1260 RSC and reference cited therein.
- (a) S. A. Martinis, W. M. Atkins, P. S. Stayton and S. G. Sligar, A conserved residue of cytochrome P450, is involved in heme-oxygen stability and activation, J. Am. Chem. Soc., 1989, 111, 9253–9254 CrossRef; (b) G. Truan and J. A. Peterson, Thr268 in substrate binding and catalysis in P450BM3, Arch. Biochem. Biophys., 1998, 349, 53–64 CrossRef CAS PubMed.
- (a) J. A. McIntosh, P. S. Coelho, C. C. Farwell, Z. J. Wang, J. C. Lewis, T. R. Brown and F. H. Arnold, Enantioselective intramolecular C-H amination catalyzed by engineered cytochrome P450 enzymes in vitro and in vivo, Angew. Chem., Int. Ed., 2013, 52, 9309–9312 CrossRef CAS PubMed; (b) R. Singh, M. Bordeaux and R. Fasan, P450-catalyzed intramolecular sp3 C-H amination with arylsulfonyl azide substrates, ACS Catal., 2014, 4, 546–552 CrossRef CAS PubMed; (c) K. Suzuki, Y. Shisaka, J. K. Stanfield, Y. Watanabe and O. Shoji, Enhanced cis- and enantioselective cyclopropanation of styrene catalysed by cytochrome P450BM3 using decoy molecules, Chem. Commun., 2020, 56, 11026–11029 RSC.
- The values of (R)-enantiomeric excess percent are respectively 84% (F87A), 73% (F87G), and -62% (F87V).
- R. Fasan, M. M. Chen, N. C. Crook and F. H. Arnold, Engineered alkane-hydroxylating cytochrome P450(BM3) exhibiting native-like catalytic properties, Angew. Chem., Int. Ed., 2007, 46, 8414–8418 CrossRef CAS PubMed.
- (a) J. Xu, Y. Cen, W. Singh, J. Fan, L. Wu, X. Lin, J. Zhou, M. Huang, M. T. Reetz and Q. Wu, Stereodivergent protein engineering of a lipase to access all possible stereoisomers of chiral esters with two stereocenters, J. Am. Chem. Soc., 2019, 141, 7934–7945 CrossRef CAS PubMed; (b) Y. Li and L. L. Wong, Multi-Functional Oxidase Activity of CYP102A1 (P450BM3) in the Oxidation of Quinolines and Tetrahydroquinolines, Angew. Chem., Int. Ed., 2019, 58, 9551–9555 CrossRef CAS PubMed; (c) S. Grobe, C. Badenhorst, T. Bayer, E. Hamnevik, S. Wu, C. Grathwol, A. Link, S. Koban, H. Brundiek, B. Großjohann and U. Bornscheuer, Engineering Regioselectivity of a P450 Monooxygenase Enables the Synthesis of Ursodeoxycholic Acid via 7β-Hydroxylation of Lithocholic Acid, Angew. Chem., Int. Ed., 2021, 60, 753–757 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00317h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |