
A pyridinium-pended conjugated polyelectrolyte for efficient photocatalytic hydrogen evolution and organic solar cells†
Muhammad
Rafiq
,
Jianhua
Jing
,
Yuanying
Liang
,
Zhicheng
Hu
,
Xi
Zhang
,
Haoran
Tang
,
Li
Tian
,
Yingwei
Li
 * and
Fei
Huang
*
* and
Fei
Huang
*
Institute of Polymer Optoelectronic Materials and Devices, State Key Laboratory of Luminescent Materials and Devices, State Key Laboratory of Pulp and Paper Engineering, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, P. R. China. E-mail: liyw@scut.edu.cn; msfhuang@scut.edu.cn
First published on 18th December 2020
Abstract
In this contribution, we report a novel pyridinium-pended conjugated polyelectrolyte (DPTFBr) for application in photocatalytic hydrogen evolution and organic solar cells. DPTFBr contains pyridinium salts attached to its backbone, which enable its transition from the pristine state to a radical state in the presence of amines and under illumination via a photo-induced amine doping process. The transition results in apparent shifts in the UV-vis absorption spectrum, electrochemical redox potential, and electron spin resonance of the resulting polymer. The unique properties of DPTFBr facilitate its application in photocatalytic hydrogen evolution with a high photocatalytic hydrogen evolution rate of 7.33 mmol g−1 h−1, which is 63- and 12-fold higher than those of its neutral and oxidized analogues, respectively. Additionally, the excellent interface modification capabilities of the pyridinium side chains in DPTFBr enable efficient electron transport/collection, thus giving rise to high-performance organic solar cells with a power conversion efficiency of over 16%.
Introduction
Organic semiconductors have received considerable attention due to their versatile potential for various applications. The varied chemical structures of organic semiconductors give rise to versatile electronic properties, which enable their use in multiple applications, such as organic light emitting diodes, polymer solar cells, organic thin-film transistors, and photocatalysis.1–10 Conjugated polyelectrolytes (CPEs), which are a class of organic semiconductors, possess hydrophilic functionalities as side chains, which provide unique solubility and processability in water/alcohol-like polar solvents and allow the realization of multiple applications.11–14 For example, CPEs can be used as electron transport materials to improve the electron collection of organic solar cells by inducing interface doping, modulating the work function of the electrode, etc.12,13,15–17 In photocatalysis, CPEs exhibit much better dispersity in water than conjugated polymers with hydrophobic side chains. The hydrophilic side chains of CPEs are capable of improving the interface contact for water reduction and facilitating co-catalyst loading, resulting in improved photocatalytic performance.18–23Doping has proven to be an efficient strategy to tune the electronic properties of organic semiconductors.24–27 The introduction of dopants into the organic semiconductor matrix can result in efficient doping in which the dopants and semiconductors are in close contact with each other. To realize efficient doping, both p-type dopants (such as I2, metal complexes, inorganic salts, tetrafluorotetracyanoquinodimethane and its derivatives, etc.)28–32 and n-type dopants (such as alkali metals, strong electron donors, organometallic complexes, mono- and multivalent anions, etc.)33–37 have been widely developed to improve the electronic properties of organic semiconductors. Efficient doping of semiconductors can minimize charge transport loss, enhance conductivity, and facilitate ohmic contact at the semiconductor/metal (or metal oxide) interface, and thus contribute to enhanced performance in organic electronics.34,38–45 An alternative strategy for realizing efficient doping is to develop CPEs with dopant groups tethered on the conjugated backbones.17,46–48 This design offers the advantages of intimate contact between the dopants and conjugated backbones, large structural variation to achieve versatile doping behaviours, and excellent solution processability, thus making it highly feasible to design novel materials for application in organic electronics.42,43,49–51
Ammonium groups have been widely used to construct high-performance CPEs.11,12,14 Ammonium groups provide the advantages of easy synthesis and good solubility in alcohol-like solvents, making these CPEs promising candidates in organic solar cell and photocatalysis applications. However, compared to aliphatic ammonium groups and their CPEs, pyridinium salts and their derived CPEs are less reported.52–55 Pyridinium groups could be either tethered to the conjugated backbones as side chains52,53,55 or incorporated into the conjugated backbones of the CPEs54 to provide good water/alcohol solubility and enable versatile applications in organic electronics, sensing, etc.52,54,55 Thus, it is highly desirable to design novel pyridinium CPEs with unique properties to realize more applications.
In this contribution, we have designed a novel pyridinium-pended conjugated polyelectrolyte (DPTFBr, Fig. 1) and investigated its application as a photocatalyst for hydrogen evolution and as a cathode interlayer for non-fullerene polymer solar cells (NF-PSCs). Inspired by previous molecular designs,52–55 the pyridinium was attached to the CPE backbone as an antenna for the electron donors, resulting in the formation of a delocalized radical, which is stabilized by the CPE backbone. In the presence of irradiation and amines, DPTFBr could undergo a transition from the pristine state to a radical state via a photo-induced amine doping process, resulting in apparent shifts in its UV-vis absorption spectrum, electrochemical redox properties, and electron spin resonance. The photo-induced amine doping process of DPTFBr also implies that the hole in DPTFBr could be easily consumed by amines upon irradiation, which is an attractive feature in photocatalytic hydrogen evolution using organic photocatalysts (which requires hole scavengers and irradiation for hydrogen evolution). Compared to its neutral and oxidized analogues, DPTFBr exhibited a 63- and 12-fold higher photocatalytic hydrogen evolution rate due to the enhanced hole scavenging process and improved dispersity. Moreover, the pyridinium side chains of DPTFBr can provide good interface modification capability to enable better electron transport/collection properties for NF-PSCs. PM6:Y6-based photovoltaic devices with DPTFBr as the cathode interlayer showed a PCE of 16.14%, which was higher than that with DPTFNO (15.47%). These results demonstrated a novel pyridinium-pended CPE, and may be useful for designing novel CPEs for various applications.
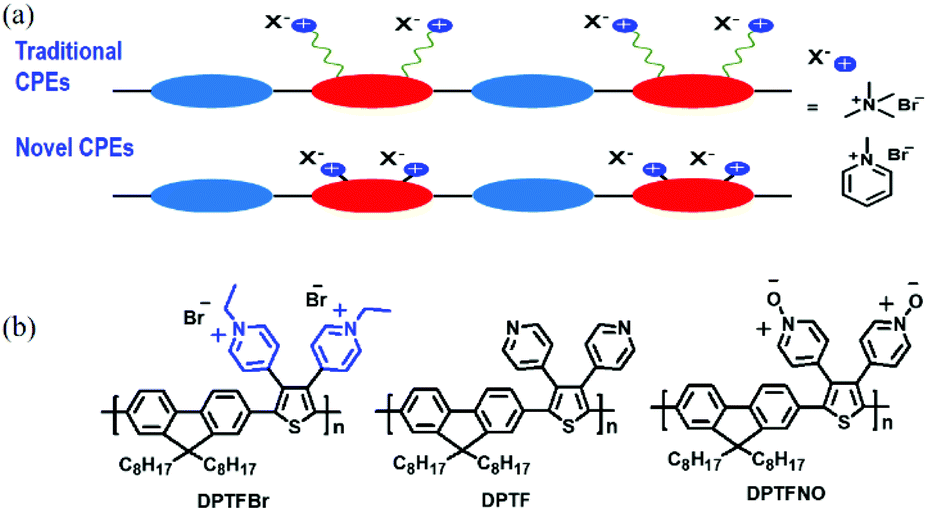 | ||
| Fig. 1 (a) Schematic depictions of traditional and novel CPEs, and (b) the chemical structures of DPTFBr, DPTF, and DPTFNO. | ||
Experimental section
Synthesis of 3,4-dipyridylthiophene
4-Pyridylboronic acid pinacol ester (2.01 g, 9.80 mmol), tetrakis(triphenylphosphine) palladium (0) (407 mg, 0.352 mmol), and potassium phosphate (5.95 g, 28.0 mmol) were added to a 250 mL two-necked flask. Under an argon atmosphere, 1,4-dioxane (70 mL) and 3,4-dibromothiophene (847 mg, 3.61 mmol) were added successively, and the suspension was stirred at 90 °C for 72 h. After cooling to room temperature, the mixture was extracted with ethyl acetate, and the organic layer was collected and dried with anhydrous magnesium sulfate. The organic layer was then evaporated to give a dark brown residue, which was further purified by silica gel column chromatography using a mixture of ethyl acetate and petroleum ether (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume) as the eluent to give 3,4-dipyridylthiophene as a light-yellow solid (575 mg, 2.49 mmol) in 69% yield. 1H NMR (500 MHz, CDCl3) δ (ppm): 8.54 (d, 4H), 7.51 (s, 2H), 7.11 (d, 4H). 13C NMR (100 MHz, CDCl3), 149.88, 141.69, 138.92, 124.66, 111.83.
:1 by volume) as the eluent to give 3,4-dipyridylthiophene as a light-yellow solid (575 mg, 2.49 mmol) in 69% yield. 1H NMR (500 MHz, CDCl3) δ (ppm): 8.54 (d, 4H), 7.51 (s, 2H), 7.11 (d, 4H). 13C NMR (100 MHz, CDCl3), 149.88, 141.69, 138.92, 124.66, 111.83.
Synthesis of 4-(2,5-dibromo-4-phenyl-thiophen-3-yl)-pyridine (M1)
3,4-Dipyridylthiophene (1.5 g, 2 mmol) was dissolved in dry DMF. After cooling to 0 °C, N-bromosuccinamide (3 g, 4.6 mmol) was added dropwise over 30 minutes. Afterwards, the reaction mixture was stirred at room temperature for 20 hours. When the reaction was completed, 50 mL of water was added to the reaction and stirred for 5 minutes. The mixture was then extracted with ethyl acetate, followed by collecting the organic layer and drying it with anhydrous magnesium sulfate. The organic layer was then evaporated to give a brownish-yellow residue, which was further purified by silica gel column chromatography using ethyl acetate and petroleum ether (1:2 ratio) as the eluent to give a light-yellow solid. 1H NMR (500 MHz, CDCl3) δ (ppm): 8.54 (d, 4H), 7.01 (d, 4H). 13C NMR (100 MHz, CDCl3), 149.95, 143.42, 138.73, 126.80, 123.51.
Synthesis of DPTF
M1 (100.2 mg, 0.25 mmol), M2 (162.5 mg, 0.25 mmol), toluene (4 mL), and 1 mL of an aqueous solution of CS2CO3 (329.7 mg, 1.01 mmol) were added to a 20 mL flask. The mixture was degassed for 20 minutes to remove oxygen. Pd(PPh3)4 (2.0 mg) was then added and the reaction mixture was degassed again for 10 minutes, followed by heating at 100 °C for 18 hours. The polymer was precipitated in 150 mL of methanol. The dissolution–precipitation process was repeated two more times. The solid was then dissolved, collected, and dried under vacuum according the processes described above, leading to a yield of 85%. 1H NMR (500 MHz, CDCl3) δ (ppm): 8.39–8.38 (m, 4H), 7.61–7.59 (m, 2H), 7.30–7.28 (m, 2H), 7.02 (m, 2H), 6.91–6.89 (m, 4H), 1.70–1.52(m, 4H), 1.3–0.97(m, 24H), 0.86–0.83 (m, 6H). Mn = 14.5 kDa, Mw = 27.5 kDa.Synthesis of DPTFBr
DPTFBr was synthesized from the neutral polymer DPTF. Briefly, 100 mg of DPTF was dissolved in 15 mL of toluene containing 1.5 mL bromoethane. The reaction mixture was stirred at 60 °C for 72 hours with the addition of 5 mL of methanol every 8 hours. Subsequently, the polymer solution was concentrated and dissolved in methanol, and then precipitated in a mixture of ethyl acetate and hexane. The solid product was collected and dissolved in methanol and again precipitated in a mixture of ethyl acetate and hexane. Finally, the polymer was collected and dried under vacuum to obtain a brown-yellow solid in a yield of 93%. 1H NMR (500 MHz, MeOD) δ (ppm): 8.87–8.85 (m, 4H), 7.82–7.74 (m, 6H), 7.53–7.47 (m, 2H), 7.33–7.22 (m, 2H), 4.66–4.58 (m, 4H), 2.06–1.92 (m, 4H), 1.68 (m, 6H), 1.33–0.99 (m, 24H), 0.87–0.81 (m, 6H).Synthesis of DPTFNO
DPTFNO was synthesized from the neutral polymer DPTF. Typically, 100 mg DPTF was dissolved in 15 mL of acetic acid and 5 mL of tetrahydrofuran. To the reaction mixture, 2.5 mL of hydrogen peroxide (H2O2) was added, and the reaction mixture was stirred at 80 °C for 72 hours. 5 mL of methanol was added to the reaction mixture every 8 hours. Subsequently, the polymer solution was concentrated and precipitated in acetone. The solid product was collected and dried under vacuum to obtain a yellow solid (85% yield). 1H NMR (500 MHz, MeOD) δ (ppm): 8.50–7.75 (m, 8H), 7.50–6.90 (m, 6H), 2.05–1.87 (m, 4H), 1.27–0.98 (m, 24H), 0.84–0.78 (m, 6H).Results and discussion
The chemical structure of DPTFBr is shown in Fig. 1b and the detailed synthetic procedure for DPTFBr is outlined in Scheme 1. The parent neutral polymer (DPTF) was prepared by standard Suzuki coupling polymerization from 4-(2,5-dibromo-4-phenyl-thiophen-3-yl)-pyridine (M1) and 2,2′-(9,9-dioctyl-9H-fluorene-2,7-diyl)bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane) (M2). The number average molecular weight (Mn) and weight average molecular weight (Mw) of DPTF were determined to be 14.5 kg mol−1 and 27.5 kg mol−1, respectively, using gel permeation chromatography (Fig. S1†). DPTFBr was prepared from DPTF and bromoethane via a quaternization reaction. The chemical structure of DPTFBr was confirmed using 1H NMR spectroscopy (Fig. S2†). DPTFBr showed good solubility in polar solvents, such as methanol, N,N-dimethylformamide, dimethylsulfoxide, etc. The orthogonal solubility of DPTFBr provides a good opportunity to increase the photocatalytic performance for hydrogen evolution and allow good interface contact at the cathode electrode to improve the photovoltaic performance.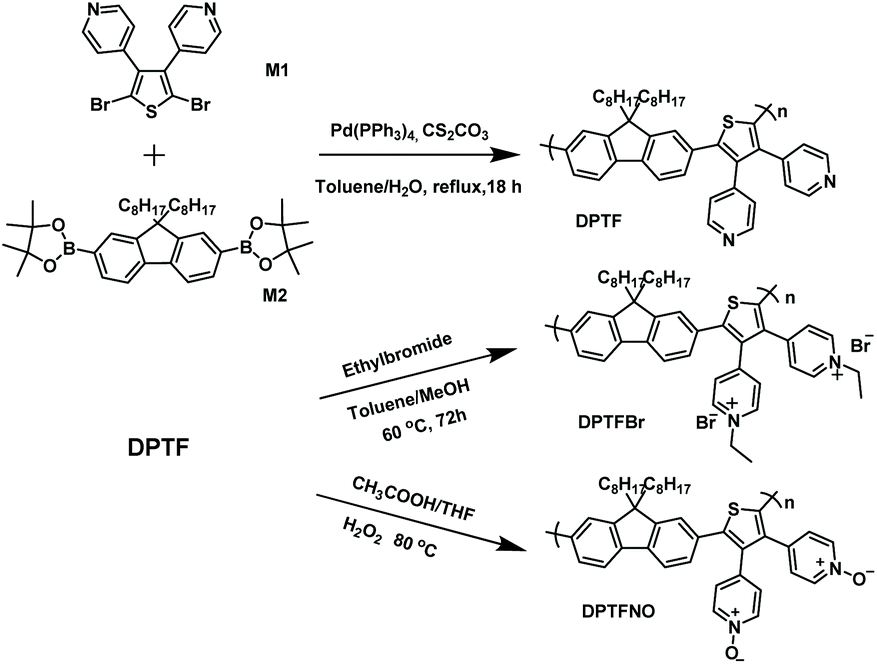 | ||
| Scheme 1 Synthetic routes for the conjugated polymers DPTF, DPTFBr, and DPTFNO. | ||
The absorption spectra of DPTFBr both in solution and as a film were first investigated. As shown in Fig. 2a and Fig. S3,† the absorption spectrum of DPTFBr in methanol extends from 250 nm to 550 nm with a peak at 328 nm and a shoulder peak at 387 nm. A similar but slightly red-shifted spectrum was observed for the thin-film DPTFBr. To study the photo-induced amine doping process of DPTFBr, DPTFBr samples with triethanolamine (TEOA, 25 wt%) were prepared and tested with/without illumination. The DPTFBr solution with TEOA showed a similar absorption spectrum to that of the DPTFBr solution. However, after illumination for 30 min, the colour of the DPTFBr solution with TEOA turned from yellow to deep brown (inset of Fig. 2a). Correspondingly, the illumination led to a red shift of the absorption peak at 328 nm to 342 nm and the disappearance of the shoulder peak for the sample in solution. The shift in the absorption spectrum indicates that the conjugated backbone is strongly affected by TEOA and illumination. The change in the absorption spectra of DPTFBr was also confirmed by the thin-film absorption of these samples (Fig. 2b). The film prepared from a solution containing TEOA (red curve) showed only a slight difference from that prepared using pristine DPTFBr solution, and the application of illumination (blue curve) resulted in the appearance of a peak at 336 nm instead of the shoulder peak, indicating a significant change in the conjugated backbone of DPTFBr. Surprisingly, after the above solution was stored in the ambient environment (recovered sample) for over 20 days, it showed almost the same absorption spectrum as the pristine DPTFBr solution. The deep colour also returned to light yellow for the recovered sample, indicating that the effect caused by TEOA and illumination gradually diminishes.
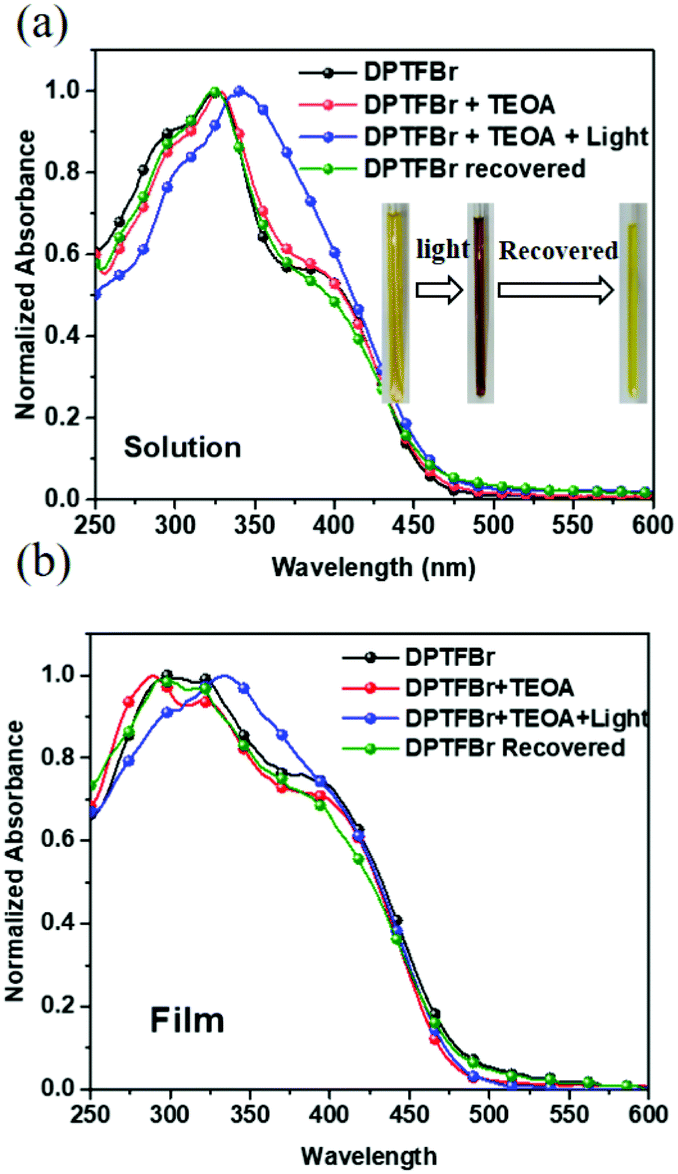 | ||
| Fig. 2 UV-vis absorption spectra of DPTFBr in solution (a) and in a thin film (b) in response to 25% TEOA and illumination. | ||
The significant change in the absorption spectra of DPTFBr indicates that TEOA and illumination could synergistically affect the electronic properties of DPTFBr. DPTFBr has pyridinium salts attached to its conjugated backbone. Pyridine-salt-based aromatic rings (such as viologen and its derivatives) are prone to form radical or quinoid-type analogues in the presence of reducing agents.56 In our case, when DPTFBr is photoexcited, the electrons donated by the reducing agent TEOA will be transferred to the pyridinium unit, triggering the formation of the radical state of DPTFBr (Scheme 2). The formed radical could further delocalize along the backbone of DPTFBr, thus affecting the intramolecular charge transfer and resulting in the apparent absorption change.57,58 It should be noted that conjugated polymers with stable radicals in their backbones are rarely reported. In our case, the quinoid-type pyridinium was utilized to stabilize the radicals, thus providing more chances for the radical to move along the conjugated backbone. Moreover, the recovered samples showed almost the same spectra as the pristine DPTFBr, suggesting that the radical could be slowly quenched by ambient O2. Nevertheless, the design of DPTFBr could provide a convenient strategy to control the radical formation and delocalization along the conjugated skeleton, thus resulting in various electronic properties.
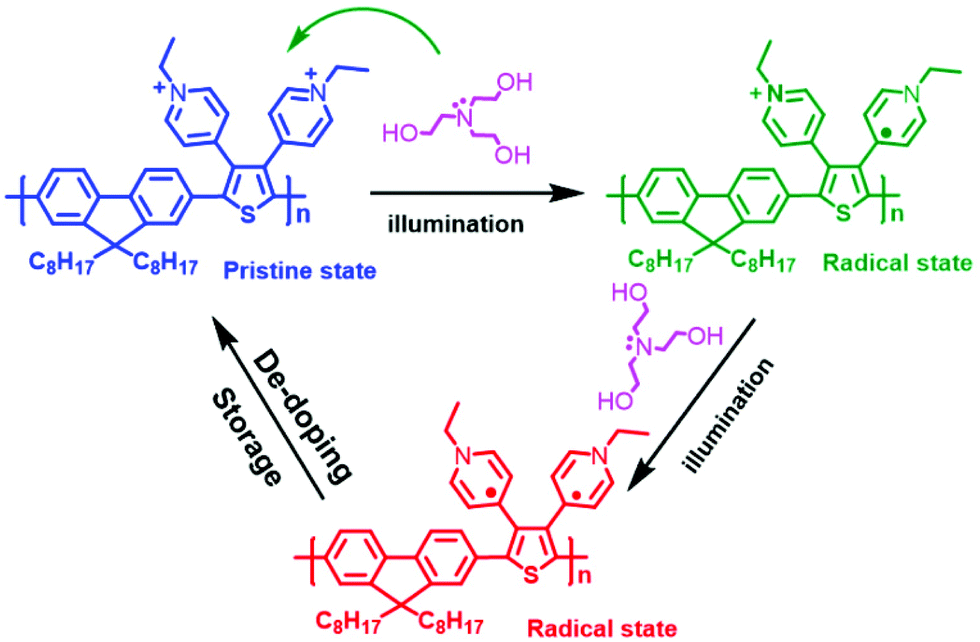 | ||
| Scheme 2 Photo-induced amine doping and de-doping processes involving DPTFBr. | ||
To verify the radical state of DPTFBr in the presence of TEOA and illumination, electron spin resonance (ESR) spectra of the DPTFBr samples were recorded. As shown in Fig. 3a, the pristine DPTFBr solution illuminated for 30 min showed very weak ESR signals, indicating the low radical concentration in the backbone of DPTFBr. After adding 10 wt% TEOA to the DPTFBr sample, the corresponding ESR intensity showed a slight enhancement, suggesting weak radical formation at this low TEOA concentration. Further increasing the concentration of TEOA to 25 wt% in the DPTFBr solution resulted in an apparent enhancement in the ESR signal with a g value of 2.005, demonstrating the formation of radicals along the conjugated backbone of DPTFBr. These results coincide well with the above absorption results, indicating that the addition of TEOA induced the formation of radicals in the conjugated backbone of DPTFBr. Interestingly, the recovered samples (stored for over 20 days) exhibited almost the same ESR spectrum as pristine DPTFBr, indicating that the radicals generated by TEOA and illumination were gradually quenched. The reversible stimulation of the radical formation and quenching in DPTFBr provides a feasible tool to tune the radical properties of conjugated polymers.59–61
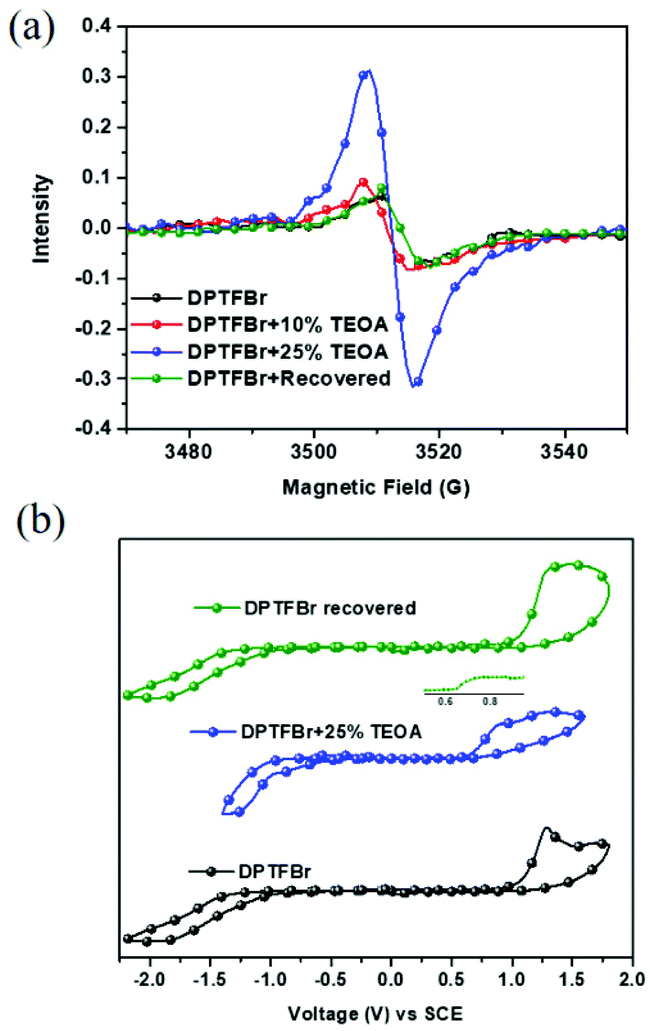 | ||
| Fig. 3 (a) ESR spectra of DPTFBr samples with 0 wt%, 10 wt%, and 25 wt% TEOA (illuminated for 30 min) and a recovered sample. (b) CV curves of pure DPTFBr, DPTFBr with 25% TEOA, and recovered DPTFBr samples. | ||
This radical formation and quenching process in DPTFBr was further evidenced by its electrochemical redox properties. Cyclic voltammetry (CV) tests of DPTFBr with/without 25% TEOA and illumination were conducted, and the electrochemical redox properties are summarized in Table 1. The highest occupied molecular orbital energy levels (EHOMO) were calculated using the equation EHOMO = −e(Eox + 4.80) eV from the potential of the onset of oxidation. The lowest unoccupied molecular orbital energy levels (ELUMO) were calculated using ELUMO = −e(Ere + 4.80) eV. As shown in Fig. 3b, DPTFBr and irradiated DPTFBr (25% TEOA + light) exhibited different cyclic voltammetric behaviours. The Eox and Ere for DPTFBr were measured to be 1.09 and −0.96 V, respectively, while the corresponding EHOMO and ELUMO of DPTFBr were calculated to be −5.89 and −3.84 eV, respectively. The irradiated sample of DPTFBr with 25% TEOA showed an Eox at 0.65 V, which originates from the oxidation of TEOA (Fig. S4†).62 The Ere of the irradiated sample of DPTFBr with 25% TEOA was −0.48 V, corresponding to an ELUMO of −4.3 eV. The lower LUMO energy level indicates that the irradiated DPTFBr is more easily reduced, which is similar to the results in a previous report.63,64 The irradiated DPTFBr sample, which was stored for over 20 days, showed electrochemical redox potentials similar to those of pristine DPTFBr, indicating that DPTFBr could return to the original cationic state in the ambient environment. These observations suggest that DPTFBr is a promising candidate to realize stable reversible doping and de-doping.
| Polymer | λ abs [nm] | E ox [V] | E re [V] | E HOMO [eV] | E LUMO [eV] | E optg [eV] |
|---|---|---|---|---|---|---|
| a Absorption peaks in solution. | ||||||
| DPTFBr | 328, 387 | 1.09 | −0.96 | −5.89 | −3.84 | 2.56 |
| DPTF | 384 | 1.23 | −1.05 | −6.03 | −3.75 | 2.66 |
| DPTFNO | 294, 370 | 1.45 | −1.25 | −6.25 | −3.55 | 2.72 |
To understand the photo-induced amine doping process of DPTFBr, we also performed 1HNMR analysis of DPTFBr with/without the addition of TEOA and illumination in CD3OD. The chemical shifts of the H atoms in the pyridinium ring of DPTFBr were 8.86 and 7.79 ppm (Fig. S2†). However, as a result of the synergistic effects of TEOA and illumination, additional chemical shifts were observed at 8.45–8.25 and 7.20–6.80 ppm,65 indicating that a fraction of the pyridinium rings were transformed into the radical-type or quinoid-type rings. However, these additional chemical shifts disappeared in the irradiated samples stored for over 20 days. The broad 1HNMR spectrum might be ascribed to the aggregation of DPTFBr after adding TEOA. Nevertheless, these results clearly confirm the doping and de-doping process of DPTFBr.
To further understand the photo-induced amine doping phenomenon in DPTFBr, we also prepared two other polymers (DPTF and DPTNO) with the same conjugated backbone for comparison. The absorption spectra of DPTF and DPTNO with/without TEOA and illumination in solution were first collected. The oxidation/reduction potentials (Eox/Ere), EHOMO, and ELUMO values of DPTF and DPTFNO are also summarized in Table 1 and shown in Fig. 4a and b. As shown in Fig. 4a, the absorption of DPTF in solution extends from 300–450 nm with an ICT peak at 385 nm. The double stimuli of TEOA and illumination did not trigger any changes in the solution of DPTF, indicating that no transition state exists in DPTF. The absorption spectra of DPTFNO also showed almost no difference in response to the addition of TEOA or application of illumination. These results indicate the importance of pyridinium salts for radical formation in the conjugated backbones. Compared to pyridine and oxidized pyridine, the pyridinium unit is more prone to form the quinoid-type structure, thus triggering the apparent change in its electronic properties. The optical band gaps (Eoptg) of these polymers were calculated using the equation Eoptg = 1240/λonset, and the resultant values were 2.66 eV for DPTF and 2.72 eV for DPTFNO, respectively. The CV curves of DPTF and DPTFNO with/without TEOA and illumination were recorded, but no differences were observed with respect to the parent spectra.
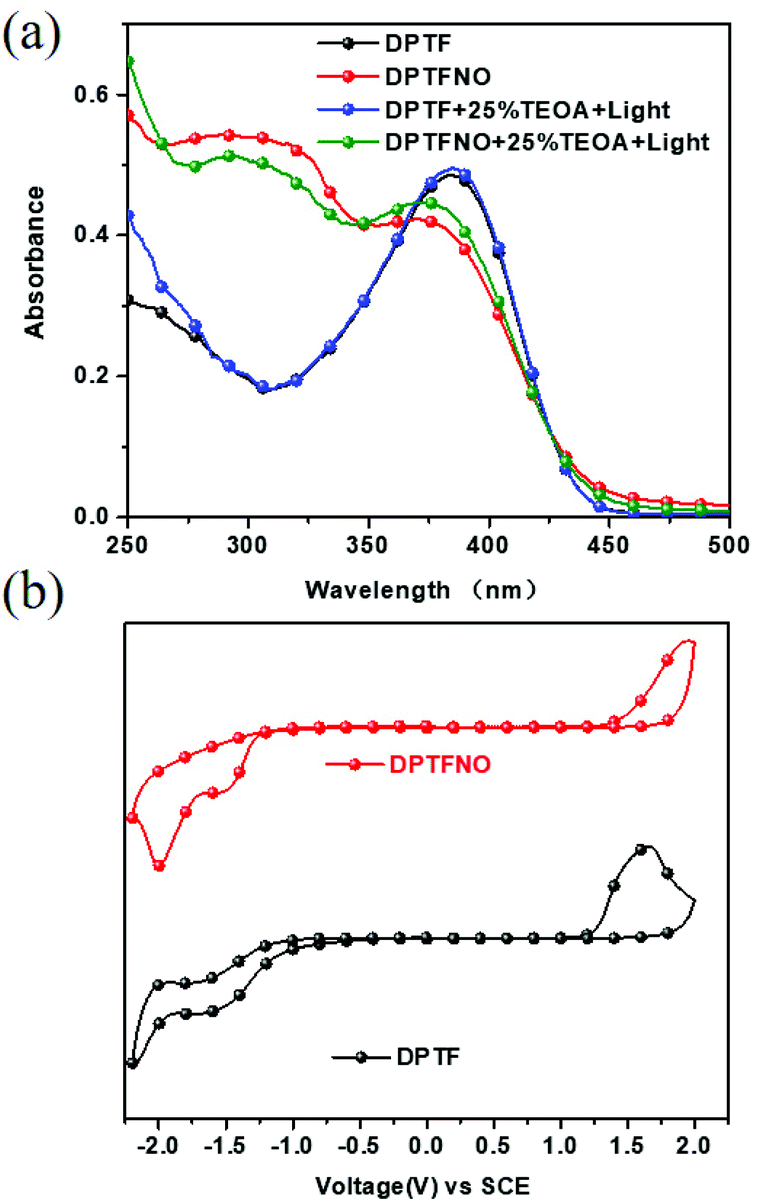 | ||
| Fig. 4 Absorption spectra in solution (a) and CV curves (b) of DPTF and DPTFNO. | ||
The chemical structure of conjugated polymers is of critical importance to their photocatalytic performance.66–73 Herein, these polymers were first applied for photocatalytic hydrogen evolution. Among the three polymers, DPTFBr and DPTFNO could be dispersed well in water, which could potentially facilitate good interface contact with both metal cocatalysts and water.74–77 In contrast, DPTF, with its low-polarity pyridine groups, showed poor solubility in polar solvents and could only form an opaque solution in water. The photocatalytic experiments for hydrogen evolution were carried out in the presence of TEOA as a sacrificial hole scavenger and Pt as a cocatalyst (3 wt% relative to the polymers). The H2 generated using the each of the three polymers as a function of time is presented in Fig. 5a, which reveals their diverse photocatalytic activities. The strategy of using DPTF as a photocatalyst produced 1.46 μmol of H2 in 5 hours, resulting in a hydrogen rate (HER) of 0.29 μmol h−1 (0.12 mmol g−1 h−1), while employing DPTFNO as the photocatalyst produced 7.16 μmol of H2 in 5 hours, corresponding to a HER of 1.43 μmol h−1 (0.57 mmol g−1 h−1). The much higher HER values of DPTFNO compared to that of DPTF could be attributed to the better dispersity of DPTFNO, which significantly reduces charge recombination. Surprisingly, an extremely high HER of 18.33 μmol h−1 (7.33 mmol g−1 h−1) was obtained using DPTFBr as the photocatalyst, which is 63- and 12-fold higher than those of DPTF and DPTFNO, respectively. The remarkable improvement in the photocatalytic performance in the presence of DPTFBr demonstrates that the role of the side chain of the conjugated polymer is critical in the photocatalytic process. Since TEOA could interact strongly with DPTFBr, the sacrificial hole process could be considerably promoted, resulting in enhanced photocatalytic hydrogen evolution rates. Moreover, the pyridinium groups in DPTFBr may behave as binding cores (similar to the situation in ammonium-based CPEs) to interact with cocatalysts, contributing to much better charge transfer between the CPEs and Pt co-catalysts.20,21,77,78
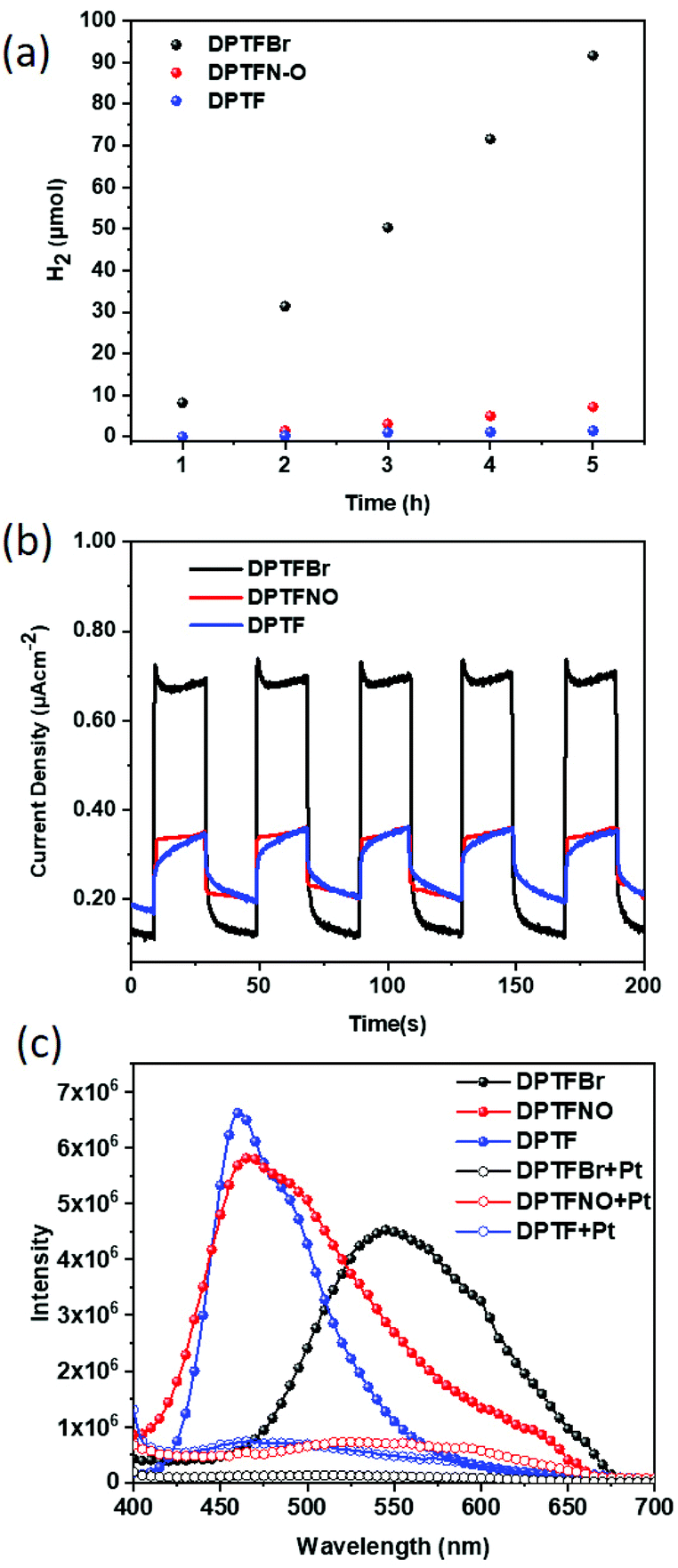 | ||
| Fig. 5 (a) Photocatalytic hydrogen evolution data from DPTF, DPTFBr, and DPTFNO. (b) The photocurrent responses of the conjugated polymers at an applied voltage of 0 V (with 0.1 M Na2SO4 as the electrolyte). (c) PL spectra of CPEs with or without 3% Pt cocatalyst. | ||
Additionally, ascorbic acid was utilized as a hole scavenger to investigate the photocatalytic hydrogen evolution of these polymers, as presented in Fig. S5.† The results demonstrated that DPTFBr incorporating ascorbic acid as the hole scavenger showed moderate HER performance (0.90 mmol g−1 h−1), which was much lower than that achieved with TEOA as the sacrificial donor. However, a negligible amount of hydrogen was detected in the cases of DPTF and DPTFNO.
Transmission electron microscope (TEM) characterization was performed to investigate the morphologies of DPTF, DPTFNO, and DPTFBr in aqueous solution, as shown in Fig. S6 and S7.† Generally, good dispersity of a conjugated polyelectrolyte in water can result in high photocatalytic performance.21 It can be observed from Fig. S6† that solid particles with diameters ranging from 100 nm to 200 nm were formed in the DPTF samples, indicating that DPTF was dispersed in water as particles. DPTFNO samples formed sheet-like aggregates with particles, suggesting that DPTFNO may be partly soluble in aqueous solution. In contrast, smaller sheet-like aggregates with particle diameters between 30 nm and 50 nm were formed in DPTFBr samples, verifying the better dispersity of DPTFBr in aqueous solution. The diverse dispersities of these polymers coincides well with the hydrophilic structures in these polymers.
In order to further understand the different performances of these conjugated polymers, the photocurrent responses of these polymers as thin films were explored. The polymers were coated onto indium tin oxide electrodes and voltages of 0 V and −0.2 V (with 0.1 M Na2SO4 as the electrolyte) were applied. As shown in Fig. 5b, DPTFBr showed a much higher photocurrent response (nearly 2.5 times greater) than those of DPTF and DPTFNO at 0 V, which might be attributed to the much higher concentration of photoexcited charge carriers in DPTFBr than that in DPTF and DPTFBr.79 A similar phenomenon was also observed when a voltage of −0.2 V was applied (Fig. S8†). These results indicate that the design of DPTFBr represents a promising strategy to realize high-performance organic photocatalysts for hydrogen evolution. Photoluminescence (PL) spectroscopy characterizations of these three CPEs (DPTFBr, DPTFNO, and DPTF) in solution state with/without a Pt cocatalyst were conducted to investigate the charge recombination of the CPEs and the charge transfer between the CPEs and the Pt cocatalyst. As shown in Fig. 5c, their PL intensities in aqueous solution gradually decreased in the order DPTF > DPTFNO > DPTFBr, indicating the reduced charge recombination in the corresponding polymers during the photocatalytic process. Moreover, after adding 3% Pt cocatalyst to these polymers, apparent PL quenching was observed in the DPTFNO and DPTF samples, while almost complete PL quenching was observed in the DPTFBr sample, implying that the charge transfer from DPTFBr to the Pt cocatalyst is more efficient. The PL results of these polymers are consistent with their photocatalytic results.
Ammonium-functionalized CPEs have good interface modification capability, which can improve the photovoltaic performance of PSCs. In contrast, pyridinium-based CPEs for high-performance photovoltaic cells are seldom reported. Herein, we investigated the potential of DPTFBr as a cathode interface material for PSCs. Devices with the architecture ITO/PEDOT:PSS/PBDB-T-2F:BTPTT-4F/CPE/Ag were fabricated,80 in which PBDB-T-2F and BTPTT-4F were used as the donor and acceptor, respectively. DPTFBr and DPTFNO have good solubility in methanol, thus allowing orthogonal processing (with respect to the active layer) and the formation of a high-quality film. The poor solubility of DPTF in alcohol-like solvents limits its application as a cathode interlayer in NF-PSCs. A device with the widely used cathode interlayer PFN-Br was also fabricated for comparison. As shown in Fig. 6 and Table 2, DPTFBr provided efficient NF-PSCs with a PCE of 16.14%, short-circuit current density (Jsc) of 26.23 mA cm−2, open circuit voltage (Voc) of 0.84, and fill factor (FF) of 73.72%. Notably, the photovoltaic performance of the device with DPTFBr is higher than that with PFN-Br as the cathode interlayer. Devices with DPTFNO showed a moderate PCE of 15.29% (Jsc = 26.19 mA cm−2, Voc = 0.83 V, FF = 70.30%.), which is comparable to that of the device with PFN-Br. The higher PCE of the DPTFBr-based photovoltaic devices compared to that of the DPTFNO-based devices mainly originates from the higher FF, indicating that DPTFBr possesses better interface modification capability. The better interface modification capability of DPTFBr could be attributed to its pyridinium side chains, which could lower the work function of metal electrode, induce interface doping, etc.17,81–86 The EQE spectra of the devices were also collected to confirm the obtained Jsc values of the DPTFBr-based devices. The calculated Jsc values from the EQE curves were compared with the measured values, as summarized in Table 2. In the DPTFBr-based devices, the calculated Jsc is similar to the measured one. These results suggest that pyridinium-based CPEs are promising candidates as cathode interface materials to achieve high-performance NF-PSCs.
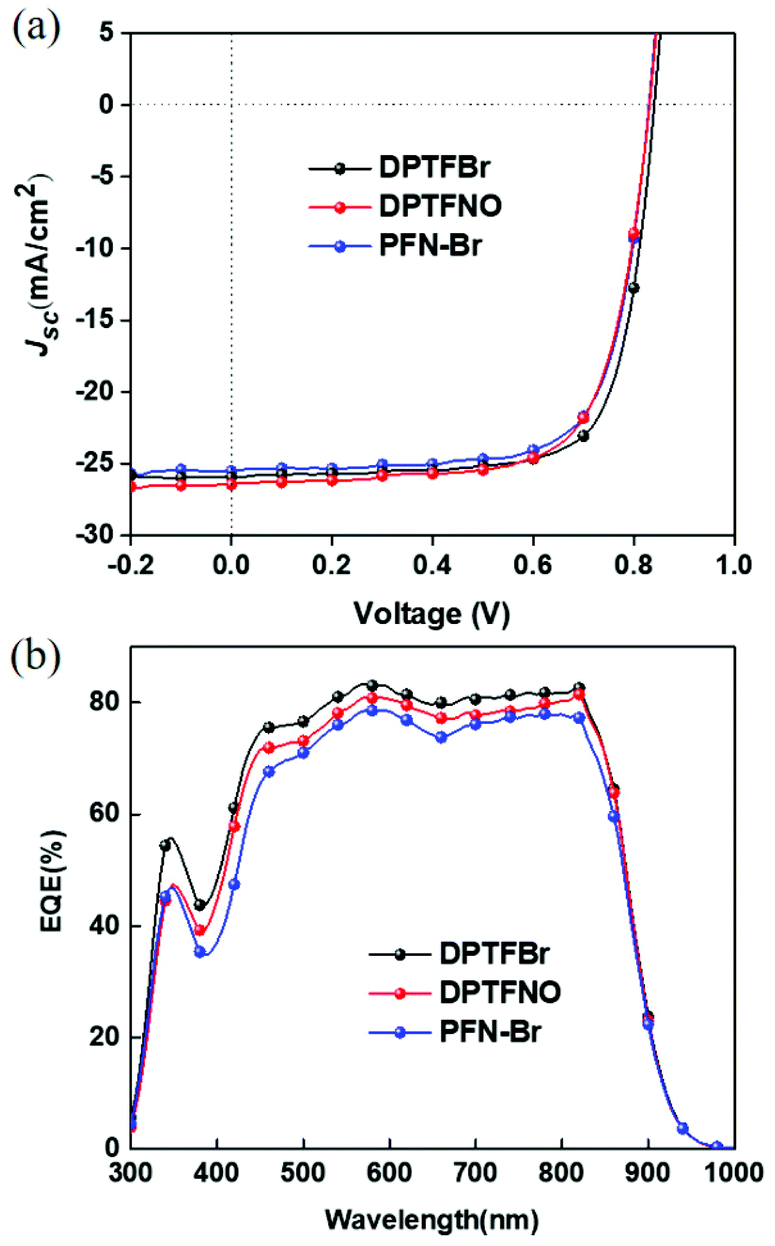 | ||
| Fig. 6 J–V and EQE curves of devices with DPTFBr, DPTFNO, and PFN-Br as the cathode interlayer. | ||
| Interlayer | V oc (V) | J sc (mA cm−2) | J sc (mA cm−2) | FF (%) | PCE/PCEmax (%) |
|---|---|---|---|---|---|
| a Calculated Jsc from EQE. | |||||
| PFN-Br | 0.83 ± 0.00 | 25.33 ± 0.14 | 23.68 | 72.33 ± 0.37 | 15.23 ± 0.07 (15.28) |
| DPTFBr | 0.84 ± 0.00 | 26.23 ± 0.07 | 25.51 | 73.27 ± 0.88 | 16.00 ± 0.15 (16.14) |
| DPTFNO | 0.83 ± 0.01 | 26.19 ± 0.36 | 24.68 | 70.30 ± 0.31 | 15.29 ± 0.27 (15.47) |
Conclusions
In conclusion, we have presented a novel pyridinium-pended conjugated polyelectrolyte, which exhibited photo-induced amine doping behaviour. The pyridinium groups attached to the conjugated backbone provided unique sites for the formation of radicals and made the photo-induced amine doping process feasible. The absorption spectra, electrochemical properties, and doping behaviour of DPTFBr could be easily tuned via this process. Compared to its neutral and oxidized analogues, DPTFBr showed much better photocatalytic and photovoltaic performance. DPTFBr enabled a high photocatalytic hydrogen evolution rate of 7.33 mmol g−1 h−1 and high-performance polymer solar cells with a PCE of over 16%. This work presents a novel type of CPE, and the proposed strategy paves the way for the design of functional CPEs for various applications.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was financially supported by the Natural Science Foundation of China (No. 21634004) and the Basic and Applied Basic Research Major Program of Guangdong Province (No. 2019B030302007).References
- A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS.
- Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS.
- C. Duan, K. Zhang, C. Zhong, F. Huang and Y. Cao, Chem. Soc. Rev., 2013, 42, 9071–9104 RSC.
- G. Zhang, Z. A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397–7457 CrossRef CAS.
- G. Zhang, J. Zhao, P. C. Chow, K. Jiang, J. Zhang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Chem. Rev., 2018, 118, 3447–3507 CrossRef CAS.
- F. Huang, Z. Bo, Y. Geng, X. Wang, L. Wang, Y. Ma, J. Hou, W. Hu, J. Pei, H. Dong, S. Wang, Z. Li, Z. Shuai, Y. Li and Y. Cao, Acta Polym. Sin., 2019, 50, 988–1046 Search PubMed.
- X. Wan, C. Li, M. Zhang and Y. Chen, Chem. Soc. Rev., 2020, 49, 2828–2842 RSC.
- Y. Miseki and K. Sayama, Adv. Energy Mater., 2019, 9, 1801294 CrossRef.
- Z. Hu, K. Zhang, F. Huang and Y. Cao, Chem. Commun., 2015, 51, 5572–5585 RSC.
- K. Zhang, Z. Hu, C. Sun, Z. Wu, F. Huang and Y. Cao, Chem. Mater., 2017, 29, 141–148 CrossRef CAS.
- Q. Cui and G. C. Bazan, Acc. Chem. Res., 2018, 51, 202–211 CrossRef CAS.
- Y. Bai, Z. Hu, J. X. Jiang and F. Huang, Chem. – Asian J., 2020, 15, 1780–1790 CrossRef CAS.
- C.-C. Chueh, C.-Z. Li and A. K.-Y. Jen, Energy Environ. Sci., 2015, 8, 1160–1189 RSC.
- Z. A. Page, Y. Liu, V. V. Duzhko, T. P. Russell and T. Emrick, Science, 2014, 346, 441–444 CrossRef CAS.
- Z. Wu, C. Sun, S. Dong, X.-F. Jiang, S. Wu, H. Wu, H.-L. Yip, F. Huang and Y. Cao, J. Am. Chem. Soc., 2016, 138, 2004–2013 CrossRef CAS.
- H. Lu, R. Hu, H. Bai, H. Chen, F. Lv, L. Liu, S. Wang and H. Tian, ACS Appl. Mater. Interfaces, 2017, 9, 10355–10359 CrossRef CAS.
- C. Dai, M. Panahandeh-Fard, X. Gong, C. Xue and B. Liu, Sol. RRL, 2019, 3, 1800255 CrossRef.
- Z. Hu, X. Zhang, Q. Yin, X. Liu, X.-f. Jiang, Z. Chen, X. Yang, F. Huang and Y. Cao, Nano Energy, 2019, 60, 775–783 CrossRef CAS.
- Z. Hu, Z. Wang, X. Zhang, H. Tang, X. Liu, F. Huang and Y. Cao, iScience, 2019, 13, 33–42 CrossRef CAS.
- Y. Wu, X. Zhang, Y. Xing, Z. Hu, H. Tang, W. Luo, F. Huang and Y. Cao, ACS Mater. Lett., 2019, 1, 620–627 CrossRef CAS.
- L.-Y. Ting, J. Jayakumar, C.-L. Chang, W.-C. Lin, M. H. Elsayed and H.-H. Chou, J. Mater. Chem. A, 2019, 7, 22924–22929 RSC.
- K. Walzer, B. Maennig, M. Pfeiffer and K. Leo, Chem. Rev., 2007, 107, 1233–1271 CrossRef CAS.
- I. Salzmann, G. Heimel, M. Oehzelt, S. Winkler and N. Koch, Acc. Chem. Res., 2016, 49, 370–378 CrossRef CAS.
- B. Lussem, C.-M. Keum, D. Kasemann, B. Naab, Z. Bao and K. Leo, Chem. Rev., 2016, 116, 13714–13751 CrossRef CAS.
- H. Tang, Z. Liu, Z. Hu, Y. Liang, F. Huang and Y. Cao, Sci. China: Chem., 2020, 63, 802–809 CrossRef CAS.
- D. Romero, M. Schaer, L. Zuppiroli, B. Cesar and B. Francois, Appl. Phys. Lett., 1995, 67, 1659–1661 CrossRef CAS.
- F. Huang, A. MacDiarmid and B. Hsieh, Appl. Phys. Lett., 1997, 71, 2415–2417 CrossRef CAS.
- E. F. Aziz, A. Vollmer, S. Eisebitt, W. Eberhardt, P. Pingel, D. Neher and N. Koch, Adv. Mater., 2007, 19, 3257–3260 CrossRef CAS.
- P. K. Koech, A. B. Padmaperuma, L. Wang, J. S. Swensen, E. Polikarpov, J. T. Darsell, J. E. Rainbolt and D. J. Gaspar, Chem. Mater., 2010, 22, 3926–3932 CrossRef CAS.
- J. Burschka, A. Dualeh, F. Kessler, E. Baranoff, N.-L. Cevey-Ha, C. Yi, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2011, 133, 18042–18045 CrossRef CAS.
- R. Yamachika, M. Grobis, A. Wachowiak and M. Crommie, Science, 2004, 304, 281–284 CrossRef CAS.
- C.-Z. Li, C.-C. Chueh, F. Ding, H.-L. Yip, P.-W. Liang, X. Li and A. K.-Y. Jen, Adv. Mater., 2013, 25, 4425–4430 CrossRef CAS.
- R. A. Schlitz, F. G. Brunetti, A. M. Glaudell, P. L. Miller, M. A. Brady, C. J. Takacs, C. J. Hawker and M. L. Chabinyc, Adv. Mater., 2014, 26, 2825–2830 CrossRef CAS.
- C. G. Tang, M. N. Syafiqah, Q.-M. Koh, C. Zhao, J. Zaini, Q.-J. Seah, M. J. Cass, M. J. Humphries, I. Grizzi and J. H. Burroughes, Nature, 2019, 573, 519–525 CrossRef CAS.
- C.-Y. Yang, Y.-F. Ding, D. Huang, J. Wang, Z.-F. Yao, C.-X. Huang, Y. Lu, H.-I. Un, F.-D. Zhuang and J.-H. Dou, Nat. Commun., 2020, 11, 1–10 Search PubMed.
- L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481–494 CrossRef CAS.
- M. Zeng, X. Wang, R. Ma, W. Zhu, Y. Li, Z. Chen, J. Zhou, W. Li, T. Liu and Z. He, Adv. Energy Mater., 2020, 2000743 CrossRef CAS.
- N. Cho, H.-L. Yip, J. Davies, P. Kazarinoff, D. Zeigler, M. Durban and Y. Segawa, Adv. Energy Mater., 2011, 1, 1148–1153 CrossRef CAS.
- B. D. Naab, S. Zhang, K. Vandewal, A. Salleo, S. Barlow, S. R. Marder and Z. Bao, Adv. Mater., 2014, 26, 4268–4272 CrossRef CAS.
- Z. Hu, R. Xu, S. Dong, K. Lin, J. Liu, F. Huang and Y. Cao, Mater. Horiz., 2017, 4, 88–97 RSC.
- Z. Chen, Z. Hu, Z. Wu, X. Liu, Y. Jin, M. Xiao, F. Huang and Y. Cao, J. Mater. Chem. A, 2017, 5, 19447–19455 RSC.
- Y. Han, Z. Fei, Y. Lin, J. Martin, F. Tuna, T. Anthopoulos and M. Heeney, npj Flexible Electron., 2018, 2, 11 CrossRef.
- D. Kiefer, A. Giovannitti, H. Sun, T. Biskup, A. Hofmann, M. Koopmans, C. Cendra, S. Weber, L. J. Anton Koster and E. Olsson, ACS Energy Lett., 2018, 3, 278–285 CrossRef CAS.
- A. Patil, Y. Ikenoue, N. Basescu, N. Colaneri, J. Chen, F. Wudl and A. Heeger, Synth. Met., 1987, 20, 151–159 CrossRef CAS.
- Y. Ikenoue, Y. Saida, M.-A. Kira, H. Tomozawa, H. Yashima and M. Kobayashi, J. Chem. Soc., Chem. Commun., 1990, 1694–1695 RSC.
- C.-K. Mai, R. A. Schlitz, G. M. Su, D. Spitzer, X. Wang, S. L. Fronk, D. G. Cahill, M. L. Chabinyc and G. C. Bazan, J. Am. Chem. Soc., 2014, 136, 13478–13481 CrossRef CAS.
- T. H. Reilly III, A. W. Hains, H. Y. Chen and B. A. Gregg, Adv. Energy Mater., 2012, 2, 455–460 CrossRef.
- B. Russ, M. J. Robb, F. G. Brunetti, P. L. Miller, E. E. Perry, S. N. Patel, V. Ho, W. B. Chang, J. J. Urban and M. L. Chabinyc, Adv. Mater., 2014, 26, 3473–3477 CrossRef CAS.
- Z. Wang, N. Zheng, W. Zhang, H. Yan, Z. Xie, Y. Ma, F. Huang and Y. Cao, Adv. Energy Mater., 2017, 7, 1700232 CrossRef.
- L. Ying, P. Zalar, S. D. Collins, Z. Chen, A. A. Mikhailovsky, T. Q. Nguyen and G. C. Bazan, Adv. Mater., 2012, 24, 6496–6501 CrossRef CAS.
- A. Thomas, J. E. Houston, N. Van den Brande, J. De Winter, M. Chevrier, R. K. Heenan, A. E. Terry, S. Richeter, A. Mehdi, B. Van Mele, P. Dubois, R. Lazzaroni, P. Gerbaux, R. C. Evans and S. Clément, Polym. Chem., 2014, 5, 3352–3362 RSC.
- S. Rochat and T. M. Swager, Angew. Chem., 2014, 126, 9950–9954 CrossRef.
- S. Nam, J. Seo, M. Song, H. Kim, M. Ree, Y.-S. Gal, D. D. Bradley and Y. Kim, Org. Electron., 2017, 48, 61–67 CrossRef CAS.
- J. Ding, C. Zheng, L. Wang, C. Lu, B. Zhang, Y. Chen, M. Li, G. Zhai and X. Zhuang, J. Mater. Chem. A, 2019, 7, 23337–23360 RSC.
- P. Wang, S. Lin, Z. Lin, M. D. Peeks, T. Van Voorhis and T. M. Swager, J. Am. Chem. Soc., 2018, 140, 10881–10889 CrossRef CAS.
- C. Franco, P. M. Burrezo, V. Lloveras, R. Caballero, I. Alcon, S. T. Bromley, M. Mas-Torrent, F. Langa, J. T. López Navarrete and C. Rovira, J. Am. Chem. Soc., 2017, 139, 686–692 CrossRef CAS.
- A. Zhang, W. Jiang and Z. Wang, Angew. Chem., 2020, 59, 752–757 CrossRef CAS.
- S. V. Patil and D. S. Argyropoulos, ChemSusChem, 2017, 10, 3284–3303 CrossRef CAS.
- M. Otaki and H. Goto, Macromolecules, 2019, 52, 3199–3209 CrossRef CAS.
- J. Newman and G. Blanchard, Langmuir, 2006, 22, 5882–5887 CrossRef CAS.
- J. Zhou, W. Zhu, M. Zeng, Q. Yang, P. Li, L. Lan, J. Peng, Y. Li, F. Huang and Y. Cao, Sci. China: Chem., 2019, 62, 1656–1665 CrossRef CAS.
- S. Vaddiraju, M. Mathai, E. Kymakis and F. Papadimitrakopoulos, Chem. Mater., 2007, 19, 4049–4055 CrossRef CAS.
- W. Wang, L. Ge, G. Xue, F. Miao, P. Chen, H. Chen, Y. Lin, Y. Ni, J. Xiong and Y. Hu, Chem. Commun., 2020, 56, 1405–1408 RSC.
- R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS.
- L. Li, W.-Y. Lo, Z. Cai, N. Zhang and L. Yu, Macromolecules, 2016, 49, 6903–6909 CrossRef CAS.
- P. B. Pati, G. Damas, L. Tian, D. L. Fernandes, L. Zhang, I. B. Pehlivan, T. Edvinsson, C. M. Araujo and H. Tian, Energy Environ. Sci., 2017, 10, 1372–1376 RSC.
- F. Yu, Z. Wang, S. Zhang, H. Ye, K. Kong, X. Gong, J. Hua and H. Tian, Adv. Funct. Mater., 2018, 28, 1804512 CrossRef.
- P.-J. Tseng, C.-L. Chang, Y.-H. Chan, L.-Y. Ting, P.-Y. Chen, C.-H. Liao, M.-L. Tsai and H.-H. Chou, ACS Catal., 2018, 8, 7766–7772 CrossRef CAS.
- Y. Xu, N. Mao, C. Zhang, X. Wang, J. Zeng, Y. Chen, F. Wang and J.-X. Jiang, Appl. Catal., B, 2018, 228, 1–9 CrossRef.
- C.-L. Chang, W.-C. Lin, C.-Y. Jia, L.-Y. Ting, J. Jayakumar, M. H. Elsayed, Y.-Q. Yang, Y.-H. Chan, W.-S. Wang and C.-Y. Lu, Appl. Catal., B, 2020, 268, 118436 CrossRef CAS.
- Y. Bai, D. J. Woods, L. Wilbraham, C. M. Aitchison, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 8700–8705 RSC.
- M. Rafiq, Z. Chen, H. Tang, Z. Hu, X. Zhang, Y. Xing, Y. Li and F. Huang, ACS Appl. Polym. Mater., 2019, 2, 12–18 CrossRef.
- X. Yang, Z. Hu, Q. Yin, C. Shu, X. F. Jiang, J. Zhang, X. Wang, J. X. Jiang, F. Huang and Y. Cao, Adv. Funct. Mater., 2019, 29, 1808156 CrossRef.
- G. Li, R. Song, W. Ma, X. Liu, Y. Li, B. Rao and G. He, J. Mater. Chem. A, 2020, 8, 12278–12284 RSC.
- W.-H. Wang, L.-Y. Ting, J. Jayakumar, C.-L. Chang, W.-C. Lin, C.-C. Chung, M. H. Elsayed, C.-Y. Lu, A. M. Elewa and H.-H. Chou, Sustainable Energy Fuels, 2020, 4, 5264–5270 RSC.
- A. F. El-Mahdy, A. M. Elewa, S. W. Huang, H. H. Chou and S. W. Kuo, Adv. Opt. Mater., 2020, 8, 2000641 CrossRef CAS.
- Y. Wang, F. Silveri, M. K. Bayazit, Q. Ruan, Y. Li, J. Xie, C. R. A. Catlow and J. Tang, Adv. Energy Mater., 2018, 8, 1801084 CrossRef.
- L. Li, J. Zheng, J. Chen, Z. Luo, Y. Su, W. Tang, X. Gao, Y. Li, C. Cao and Q. Liu, Adv. Mater. Interfaces, 2020, 2000743 CrossRef.
- K. Zhang, C. Zhong, S. Liu, C. Mu, Z. Li, H. Yan, F. Huang and Y. Cao, ACS Appl. Mater. Interfaces, 2014, 6, 10429–10435 CrossRef CAS.
- Z. Zhao, J. He, J. Wang, W. Chen, N. Wang, Y. Zhang and R. Yang, J. Mater. Chem. C, 2015, 3, 4515–4521 RSC.
- W. Chen, W. Jiao, D. Li, X. Sun, X. Guo, M. Lei, Q. Wang and Y. Li, Chem. Mater., 2016, 28, 1227–1235 CrossRef CAS.
- L. Chen, Y. Tan, X. Liu and Y. Chen, Nano Energy, 2016, 27, 492–498 CrossRef CAS.
- K. Yan and C. Z. Li, Macromol. Chem. Phys., 2019, 220, 1900084 CrossRef.
- K. Bini, X. Xu, M. R. Andersson and E. Wang, ACS Appl. Energy Mater., 2018, 1, 2176–2182 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting information, additional experimental details, synthetic method, and characterization data, such as 1H NMR spectra of the polymer, GPC data from DPTF, etc. See DOI: 10.1039/d0py01351j |
| This journal is © The Royal Society of Chemistry 2021 |