
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlycosyl disulfides: importance, synthesis and application to chemical and biological systems
Goreti
Ribeiro Morais
* and
Robert A.
Falconer
 *
*
Institute of Cancer Therapeutics, Faculty of Life Sciences, University of Bradford, Bradford BD7 1DP, UK. E-mail: g.ribeiromorais@bradford.ac.uk; r.a.falconer1@bradford.ac.uk
First published on 10th November 2020
Abstract
The disulfide bond plays an important role in the formation and stabilisation of higher order structures of peptides and proteins, while in recent years interest in this functional group has been extended to carbohydrate chemistry. Rarely found in nature, glycosyl disulfides have attracted significant attention as glycomimetics, with wide biological applications including lectin binding, as key components of dynamic libraries to study carbohydrate structures, the study of metabolic and enzymatic studies, and even as potential drug molecules. This interest has been accompanied and fuelled by the continuous development of new methods to construct the disulfide bond at the anomeric centre. Glycosyl disulfides have also been exploited as versatile intermediates in carbohydrate synthesis, particularly as glycosyl donors. This review focuses on the importance of the disulfide bond in glycobiology and in chemistry, evaluating the different methods available to synthesise glycosyl disulfides. Furthermore, we review the role of glycosyl disulfides as intermediates and/or glycosyl donors for the synthesis of neoglycoproteins and oligosaccharides, before finally considering examples of how this important class of carbohydrates have made an impact in biological and therapeutic contexts.
 Goreti Ribeiro Morais | Goreti Ribeiro Morais graduated in Pharmaceutical Sciences from the University of Coimbra, Portugal (Faculty of Pharmacy). After her PhD in Pharmaceutical Chemistry at University of Lisbon (Portugal) and Kyushu University (Japan), she undertook her first post-doctoral position with Prof. Robert Falconer at the Institute of Cancer Therapeutics, University of Bradford. Following a research fellowship at the Campus Tecnológico e Nuclear, Instituto Superior Tecnico, Portugal focused on radiopharmaceuticals, she returned to the University of Bradford in 2014 as a senior Post-Doctoral Fellow, supporting the Institute of Cancer Therapeutics Drug Discovery programme. Amongst her research interests, she focuses on carbohydrate-based molecules in an effort to target the tumour glycocalyx for anti-metastatic therapy. |
 Robert A. Falconer | Robert Falconer is Professor of Medicinal Chemistry at the Institute of Cancer Therapeutics, University of Bradford. He is a Fellow of the Royal Society of Chemistry. He is additionally co-founder of Incanthera plc, a University of Bradford spin-out company, and is an experienced principal investigator and supervisor of PhD and Masters level students. His research interests are focused on the development of novel targeted cancer therapeutics, including carbohydrate- and peptide-based molecules. |
1. Introduction
Disulfides are important in both biological and in chemical contexts, but are perhaps best known for the essential roles they play in the formation and stabilisation of secondary and tertiary structures in peptides and proteins.1 Although disulfides are relatively stable to harsh biological conditions, including environments rich in gluthathione (G-SH) such as intracellular compartments, they are easily reduced through a thiol-disulfide exchange reaction. This characteristic has been particularly attractive for the design of certain drug molecules, notably including bioreductive prodrugs, and drug delivery systems.2–4 Disulfides are also found in natural products (e.g. polycarpamines, gliotoxin),5 pharmaceuticals (e.g. disulfiram),6 and have multiple other uses, such as in food chemistry (e.g. Z-ajoene),7 amongst others.Although glycosyl disulfides are almost non-existent in nature, there are some notable examples. Interestingly a glucosyl disulfide was found to be part of the structure of glucosinolate (Fig. 1), a plant secondary metabolite found in several foodstuffs that has been shown to possess antioxidant properties.8 Undeniably, carbohydrates play important roles in biological processes, and the development of synthetic glycomimetics has contributed significantly to the understanding of these biomolecules and biological processes.
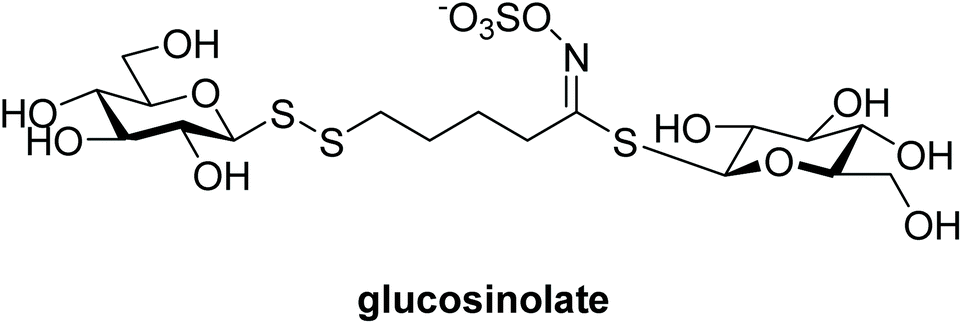 | ||
| Fig. 1 Structure of glucosinolate, a naturally occurring glucosyl disulfide. | ||
Glycomimetics in which a disulfide bond replaces the interglycosidic oxygen have also attracted interest as useful tools in the study of glycobiology. This linkage is relatively easy to access and allows for chemoselective modifications. The increase in the distance between the carbohydrate and aglycone units (3-bond interglycosidic versus 2-bond normally) adds increased flexibility to the molecules with respect to the corresponding natural glycosides. As a result, glycosyl disulfides can thus potentially better adjust to an active conformation (e.g. required for enzyme binding) and can consequently influence biological activity in a positive manner. Simultaneously, replacement of the interglycosidic oxygen by a different atom or functional group is often expected to confer resistance to glycosidase enzymes.9 Synthetic glycosyl disulfides have been studied for several different applications, for example as potential anti-cancer agents, as protein compatible detergents, as lectin binding agents, and as components of dynamic combinatorial libraries. Exploration of mixed disulfides in glycosylation strategies allows for bidirectional cleavage (reductive or hydrolytic). Also, as for the presence of the two sulfur atoms in disulfides, this offers a better coordination of thiophiles, which may offer more favourable reactivity over a single sulfur atom, as found in thioglycosides. In summary, glycosyl disulfides have multiple synthetic applications in glycochemistry, and have been found to be versatile glycosyl donors in the synthesis of glycosides and oligosaccharides.
The growing interest in glycosyl disulfides in glycobiology has triggered the development of several different methods for their preparation. While several reviews have focused on general approaches to the assembly of disulfide bonds,10,11 none has specifically focused on the chemistry of glycosyl disulfides. Herein, we will examine methods available to introduce a disulfide linkage at the glycosyl anomeric carbon (Scheme 1) and will discuss chemical and biological applications of this important class of compounds.
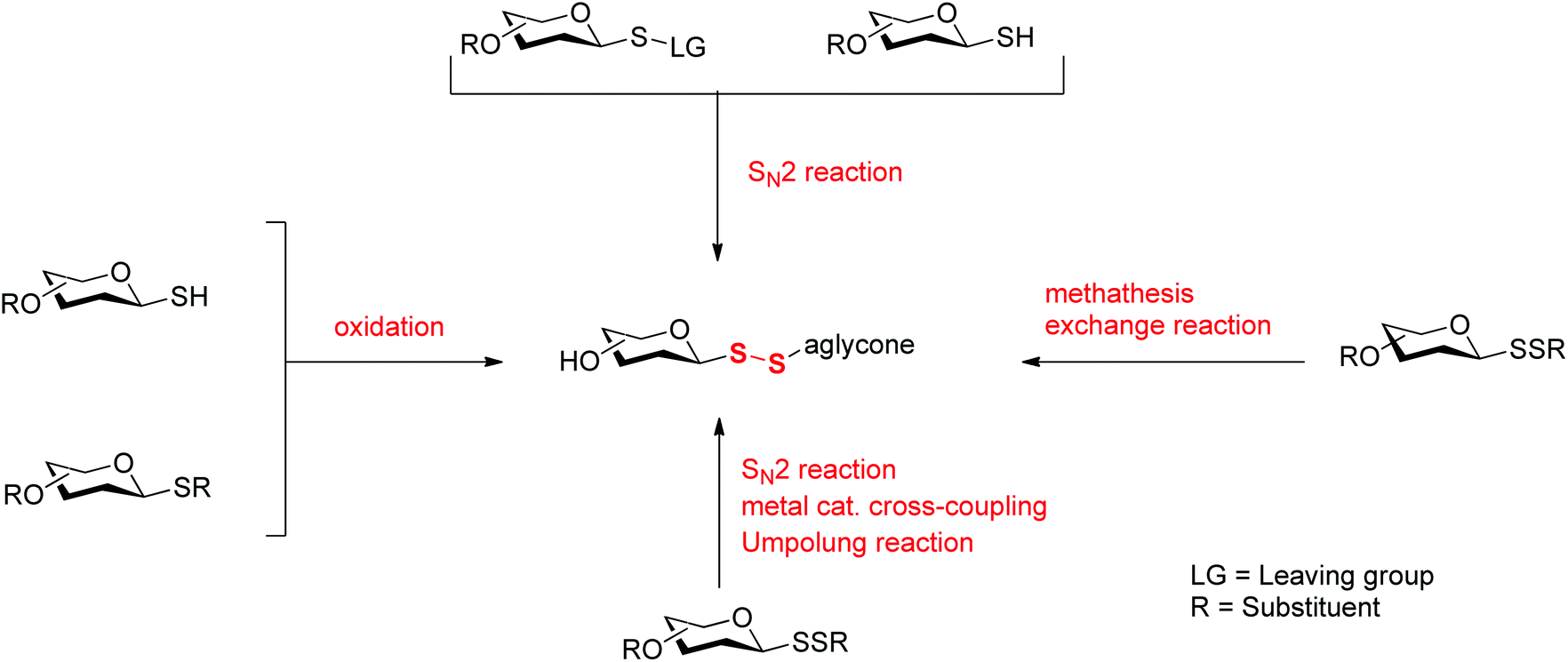 | ||
| Scheme 1 General synthetic strategies for the preparation of glycosyl disulfides. | ||
2. Synthesis of glycosyl disulfides
Synthesis of symmetrical glycosyl disulfides is typically achieved by oxidation of the corresponding glycosyl thiol with iodine,12–15 hydrogen peroxide,15–17 diethylazodicarboxylate (DEAD),18 or m-chloroperoxybenzoic acid.19,20 A number of diverse glycosyl sulfur-based intermediates 2 (thioesters,14,17,20 xanthates,21 dithiocarbamates22 or thiouronium salts23) have been developed to access glycosyl thiols 3 after appropriate chemical modifications (Scheme 2). Alternative methods lead to symmetrical glycosyl disulfides directly from glycosyl bromides 1, for example upon prolonged treatment with tetrathiomolybdate.24 An even more direct method was later described in which symmetrical glycosyl disulfides were obtained from the inexpensive and readily available per-O-acetylated sugars 5 as a starting material, through a sequential approach with no need to isolate intermediates (Scheme 3).25 Formation of the interglycosidic disulfide bond has also been achieved efficiently from the 1-S-acetyl form of glycosides via oxidation using iodine in combination with N-iodosuccinimide.26 Furthermore, the copper(I)-catalysed oxidation of S-trityl-1-thiomannosides directly produced the respective symmetrical mannosyl disulfide in good yields (around 70%).27 It should be noted that the usefulness of m-chloroperoxybenzoic acid as an oxidant is far from ideal since it requires careful control of reaction conditions to avoid over-oxidation products, such as diglycosyl thiosulfinates and thiosulfonates, which are frequently obtained when it is utilised.20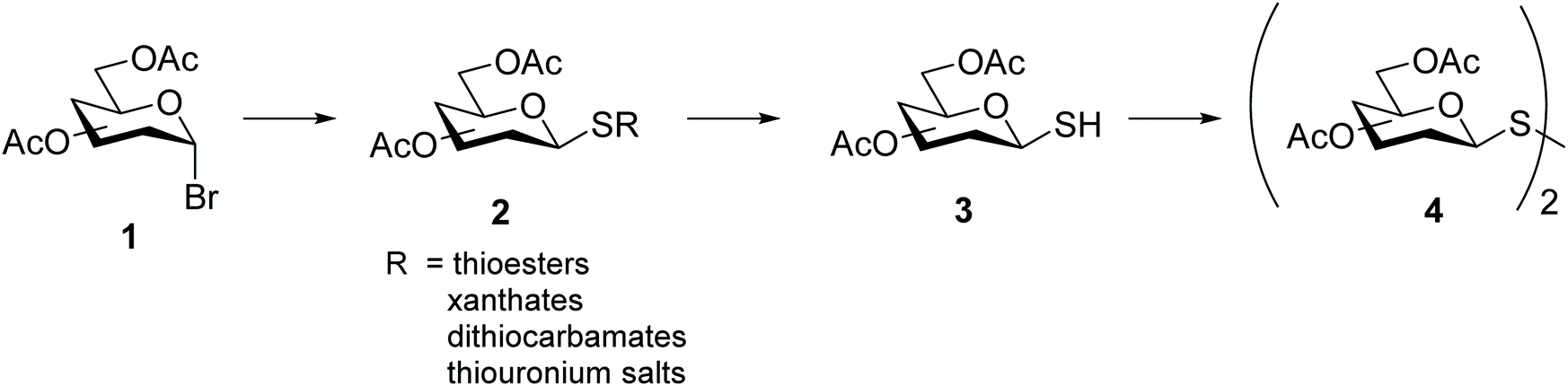 | ||
| Scheme 2 General protocol for the preparation of symmetrical glycosyl disulfides by oxidation. | ||
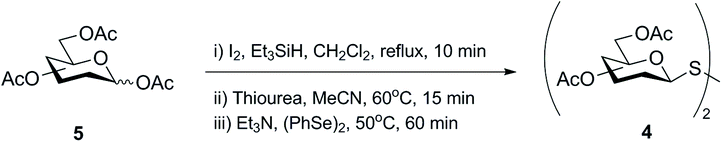 | ||
| Scheme 3 Sequential synthesis of symmetrical glycosyl disulfides directly from per-O-acetylated sugars. | ||
Curiously, the symmetrical mannosyl disulfide 9 was the sole product, even if in low yield, from the attempt to prepare a mixed thiodiglycoside using trichloroacetimidate 6 as glycosyl donor (Scheme 4). Indeed, while thioglycosides have been successfully prepared by the Lewis acid-catalysed reaction of trichloroacetimidate 6 with simple alkyl-thiols, in the case of the reaction with protected thio-mannose 7, the respective disulfide 9 was obtained instead. The authors reasoned that a highly stable S,N acetal intermediate 8 was formed from the reaction of thio-mannose 7 with the imine group of the trichloroacetimidate, and that in this form the glucose moiety was not efficiently activated to react with another thiomannose molecule. Instead, intermediate 8 underwent hydrolysis or oxidation over time (or during purification) to produce the symmetrical disulfide 9. No further investigation was carried out regarding the chemical behaviour of other thioaldoses, or the influence of the glycan protective group, however.28
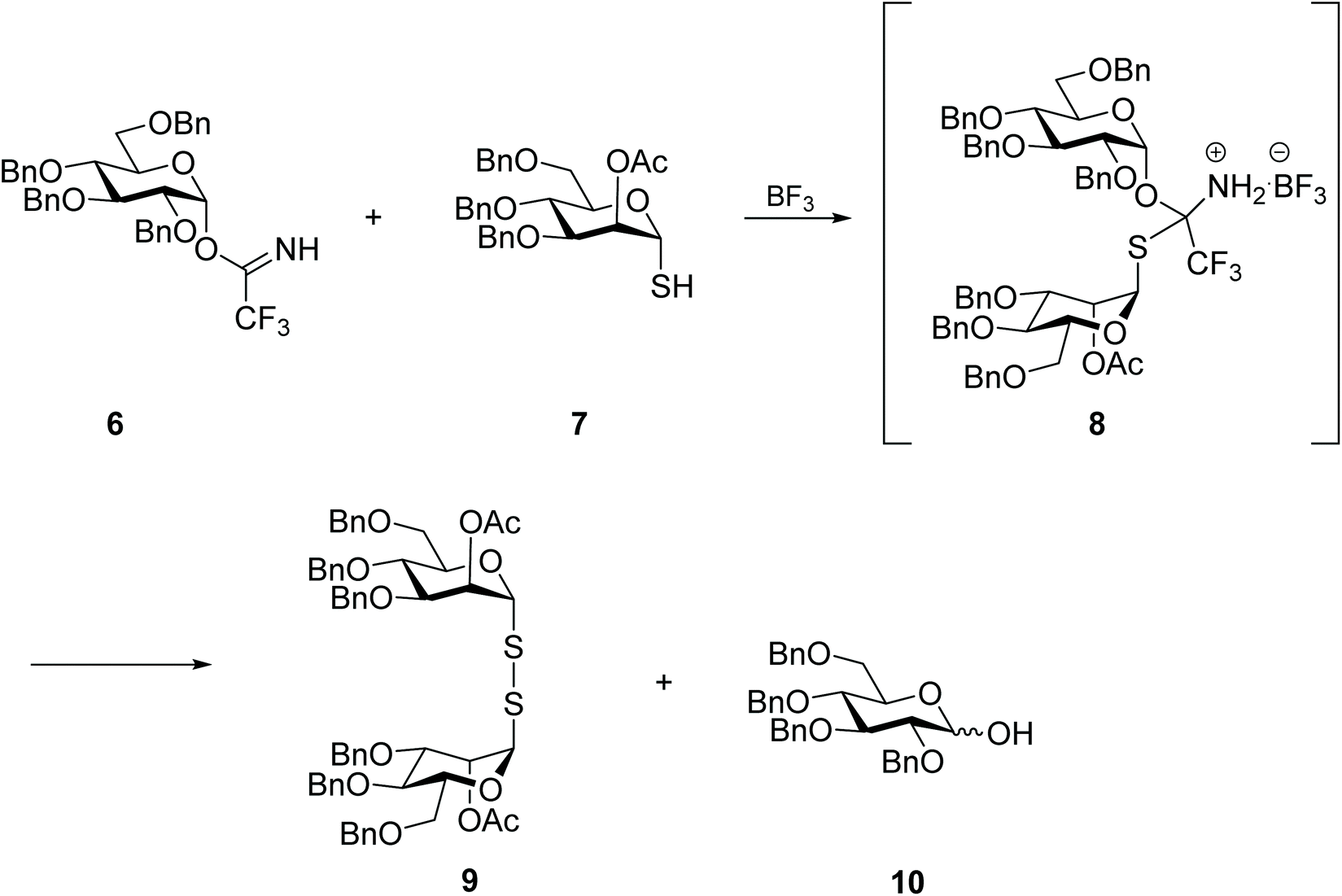 | ||
| Scheme 4 Synthesis of di-mannosyl disulfide via an N,S-acetal intermediate. | ||
An innovative and efficient approach to prepare unsymmetrical glycosyl disulfides was reported by Ramström and co-workers, which consisted of a phosphine-mediated disulfide metathesis (Scheme 5).29 Reactions between disulfides and phosphines are well known. For instance, disulfides can be reduced to the corresponding thiols,30 or undergo desulfurisation into the respective sulfides.31,32 Prior to their work on carbohydrates, the group successfully demonstrated that triethyl phosphoramide (P(NEt2)3) was the most effective of the phosphines for catalysing the exchange of two different symmetrical disulfides in organic solvent.33 This prompted the authors to explore this phosphine-catalyzed reaction to build up a dynamic carbohydrate library, in which three different symmetrical glycosyl disulfides (11–11, 12–12, 13–13) yielded a mixture of unsymmetrical disulfides (11–12, 11–13, 12–13) albeit in an equilibrium with the starting compounds. It seemed that the anomeric configuration played an important role, with the less hindered equatorial aldopyranoses being the most kinetically favoured for the exchange. Addition of a fourth disulfide (14–14) after equilibration of the primary carbohydrate system led to the formation of a secondary dynamic system composed of three further novel disulfides (11–14, 12–14, 13–14) and demonstrated the reversibility of the system. In the absence of phosphine, no exchange product was observed, which confirmed its vital role in the process. The involvement of transient aldopyranosyl-thiolate and aldopyranosyl-thiophosphonium ions was proposed as the mechanism for the exchange process. These species would lead to desulfurisation, however: no desulfurised product was observed under the aqueous conditions explored.29 In their early work with disulfides it was reported that no thiol intermediate was observed during the exchange reaction, even when higher concentrations of catalyst were used, which corroborates a metathesis-type reaction mechanism.33 Despite the debate regarding the mechanism of this phosphine-catalysed disulfide metathesis, the approach enabled successful creation of a carbohydrate library under compatible reaction conditions for 1H STD NMR studies to identify ligand protein interactions.
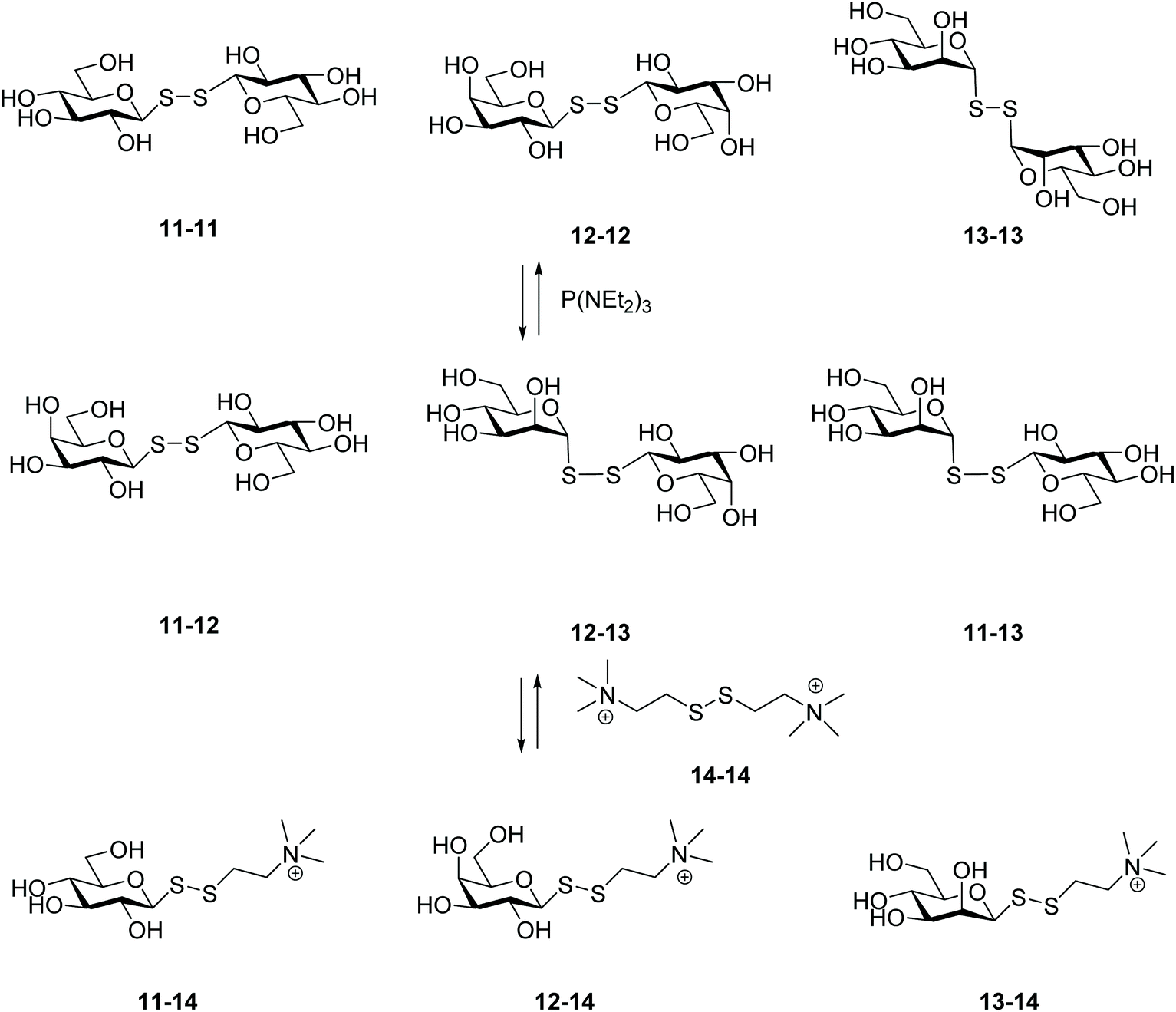 | ||
| Scheme 5 Phosphine-mediated disulfide metathesis approach. | ||
Conventional synthesis of unsymmetric glycosyl disulfides has been elaborated upon, with several interesting approaches being developed over the years. The most common way to access disulfide linkages in mixed glycosyl disulfides involves an SN2 process, in which a glycosulfenyl reagent with a good leaving group is utilised. This approach has the advantage of installing the aglycone without changes to the anomeric configuration. The use of nucleophilic sulfur-based glycosyl donors as an alternative has also been described. These approaches will be now be discussed in more detail.
2.1. Synthesis via electrophilic sulfur-based carbohydrates
Pioneering work on the synthesis of unsymmetrical glycosyl disulfides has relied on a bivalent sulfur atom that exhibits electrophilic character and is susceptible to attack by an electron-rich substituted thiol (Scheme 6).34 The sulfenylbromide 16 was readily prepared in minutes from thioacetate 15, and glycosyl disulfides were produced in good yields (approx. 60–70%). Intermediate 16 proved extremely unstable, however, rapidly leading to the formation of the diglucosyl disulfide, or decomposition into bromoglucose.35 The limited kinetic control of this reaction, which is highly dependent on the solvent, temperature and reaction time, was a major setback which led to this approach being abandoned. Nonetheless, this original work by Horton and co-workers34 established the feasibility of using an electrophilic sulfur-based glycosylsulfenyl transfer agent to access glycosyl disulfides. Other electron-withdrawing substituents were investigated to polarise the anomeric sulfur, resulting in the development of more efficient sulfenyl intermediates.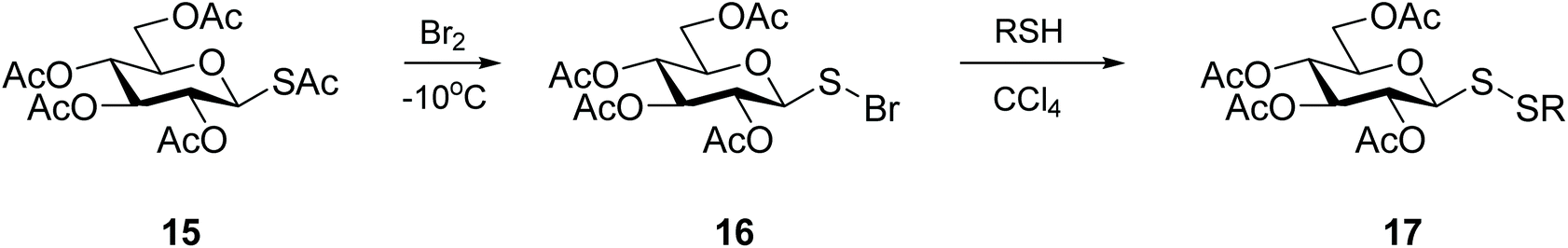 | ||
| Scheme 6 Synthesis of glycosyl disulfides via a sulfenyl halide intermediate. | ||
Motivated by our own previously successful preparation of thioglycosides via a Mitsunobu reaction,36,37 we additionally investigated the usefulness of azo compounds (‘Mitsunobu reagents’) to access disulfide-linked glycosides. The in situ preparation of glycosyl sulfenylhydazine intermediate 18 afforded mixed glycosyl disulfides 19 in a one-pot fashion under mild conditions, with very good yields (Scheme 7). DEAD and DIAD were shown to be the most effective azo compounds, and in the absence of a reacting thiol, the symmetrical diglycosyl disulfide was principally formed. This approach proved to be amenable to a variety of sugar units (monosaccharides and disaccharides) and reacting mercaptans (alkyl, aryl, heteroaryl and glycosyl).18,38
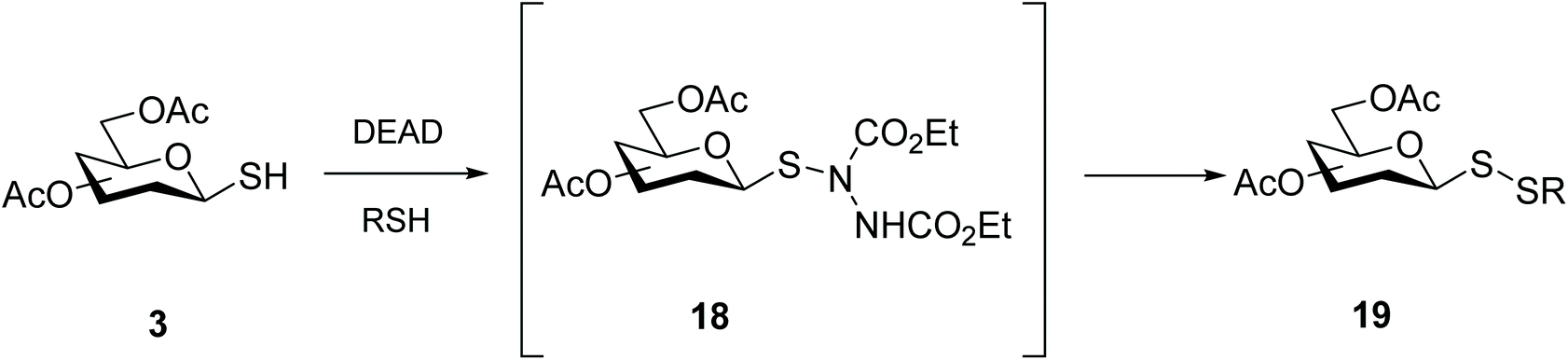 | ||
| Scheme 7 Synthesis of glycosyldisulfides via a sulfenylhydrazine intermediate. | ||
Further one-pot methods for the preparation of unsymmetrical glycosyl disulfides have been reported, for example the in situ oxidation of thioglycosides with 1-chlorobenzotriazole followed by the reaction with alkyl-, aryl- and glycosyl-mercaptans. A feature of this method is the requirement for very low temperatures (−78 °C) to maximise trapping of the respective sulfenyl intermediate and to favour unsymmetrical disulfide over homodimer formation.39 Despite the need for rigorous thermodynamic control, 1-chlorobenzotriazole, like the azo compounds described above, offers the advantage of being an environmentally-friendly oxidant. On the other hand and despite its relative toxicity, Chandrasekaran and co-workers explored thiomolybdate as a reagent to promote a one-pot synthesis of unsymmetrical glycosyl disulfides directly from glycosyl bromides.40 The noteworthy aspect of this method is the sulfur transfer from benzyltriethylammonium tetrathiomolybdate [(BnEt3N)2MoS4] to form symmetrical glycosyl disulfides, which in turn undergo an exchange reaction with other symmetric disulfides when the thiomolybdate reagent is used in excess. Application of this sulfur transfer-dichalcogenide exchange process was also demonstrated for the synthesis of glycosyl selenylsulfides.40
Carbohydrate sulfenamide derivatives, based on phthalimides and succinimides, represent an equally efficient synthon for the preparation of glycomimetics containing an interglycosidic disulfide linkage. The sulfenamides are easily prepared from the silver salts of thiosugars, and their conversion into unsymmetric glycosyl (aryl-, glycosyl-, amino acid and peptide) disulfides is effective under mild conditions.41 The same authors also delved into the use of glycomethanethiosulfonate 20 as a sulfur transfer reagent for the synthesis of unsymmetrical diglycosyl disulfides (Scheme 8).42,43 This glycosulfenyl transfer reagent, designed by Davis and co-workers for the glycosylation of proteins,44 was also explored for the synthesis of various 1,6-disulfide bridged D-hexopyranoses through a nucleophilic attack of the C6 thiolate onto the anomeric thiosulfonate.45 Curiously, while this methodology is adequate for the intramolecular disulfide cyclisation of many monosaccharides, including D-glucose (22), D-mannose, D-allose and D-tallose, in the case of the reaction with galactosyl-methanethiosulfonate 21, the intermolecular reaction was favoured yielding diglycosyl bis(disulfide) 23 (Scheme 8).45
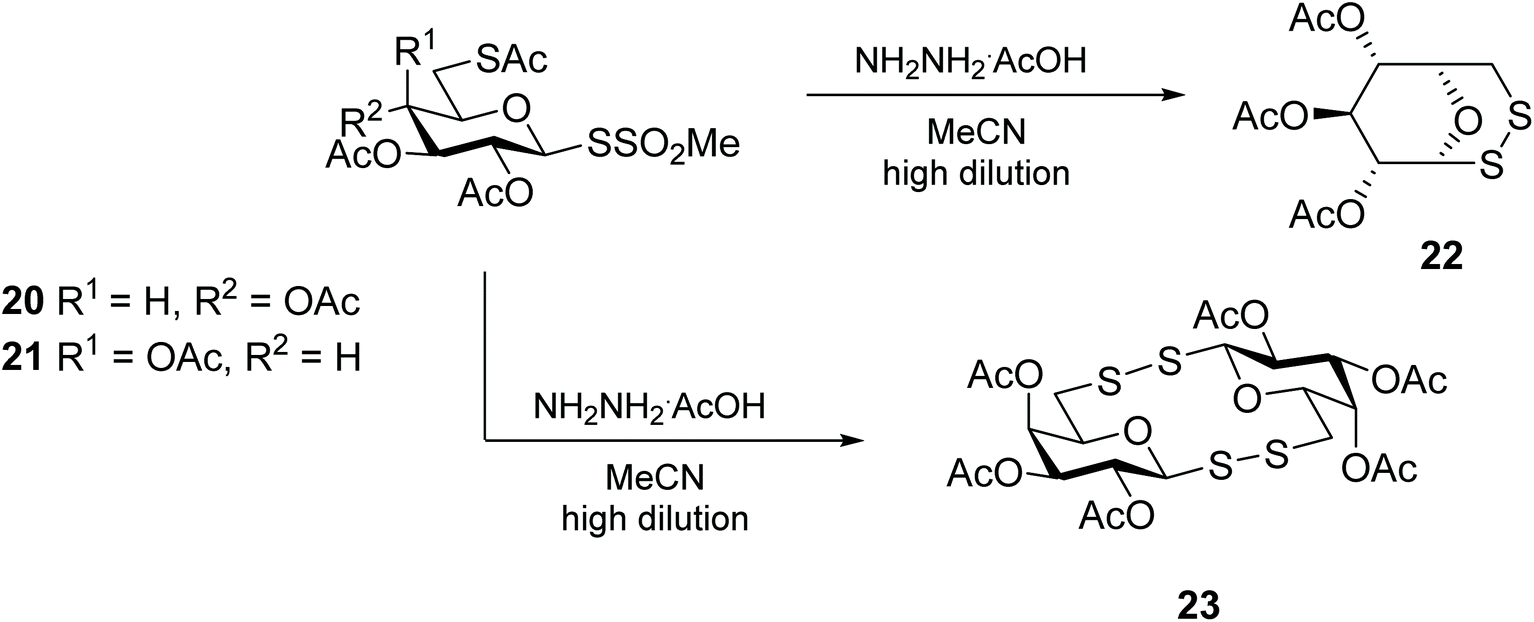 | ||
| Scheme 8 Synthesis of 1,6-disulfide bridged D-hexopyranoses. | ||
An alternative that exploited glycosulfenic acid as the electrophilic-thiol reactive synthon was developed,46 but its synthesis was less straightforward than that of the sulfur-transfer reagents described above. Interest in sulfenic acids stemmed from the knowledge that oxidation of the cysteine side chain into a disulfide bond was initiated by a cysteine sulfenic acid.47 Sulfenic acids are inherently unstable and tend to self-condense and convert into thiosulfonates48 and because of that, glycosulfenic acids could not be prepared and stored. Transient glycosyl sulfenics have been prepared via a β-syn-elimination of appropriate sulfoxides through thermolysis, and were condensed with various thiols to produce unsymmetrical glycosyl disulfides under mild conditions efficiently. Despite the need for more synthetic steps to prepare the glycosyl sulfenyl reagent, the extreme high reactivity of sulfenic acids allowed the construction of bi- and trivalent glycoconjugate disulfide systems based on benzene with very high yields.49,50 The authors claimed this approach to be suitable to glycosylate proteins, but the high temperatures used for the in situ generation of the transient glycosyl sulfenic acids might prove problematic in handling the proteins for glycosylation, and lead to denaturation instead.
Wang and co-workers established another less conventional approach to the preparation of unsymmetrical glucosyl disulfide 17,51 which was motivated by the previous finding that thiols could efficiently reduce 5-arylidene-1,3-dimethyllbarbituric acid,52 involving reduction of para-quinone methide analogues with a 1-thio-sugar (Scheme 9). In this work, addition of thioglucose 25 to the para-quinone methide derivative 24 generated the 1,6 conjugate adduct 26, which in turn was efficiently converted into unsymmetrical glycosyl disulfide 17, via an SN2 process, and reduced product 27 under basic conditions. While para-quinone methide oxindole 24 was reduced and produced an unsymmetrical glycosyl disulfide, other simpler systems like for example the para-quinone methide from an aryl aldehyde were not susceptible to nucleophilic attack by the second mercaptan, and in this example only the respective sulfide analogue was isolated. Moreover, if a chiral catalyst and methyl acrylate were added to the reaction, the synthesis of disulfide was accompanied by unsymmetric stereoselctive resolution of the sulfide 26 to yield chiral 3,3-disubstituted oxindoles in one-step. This strategy was generalised to different thiols and 3,3-disubstituted oxindoles.
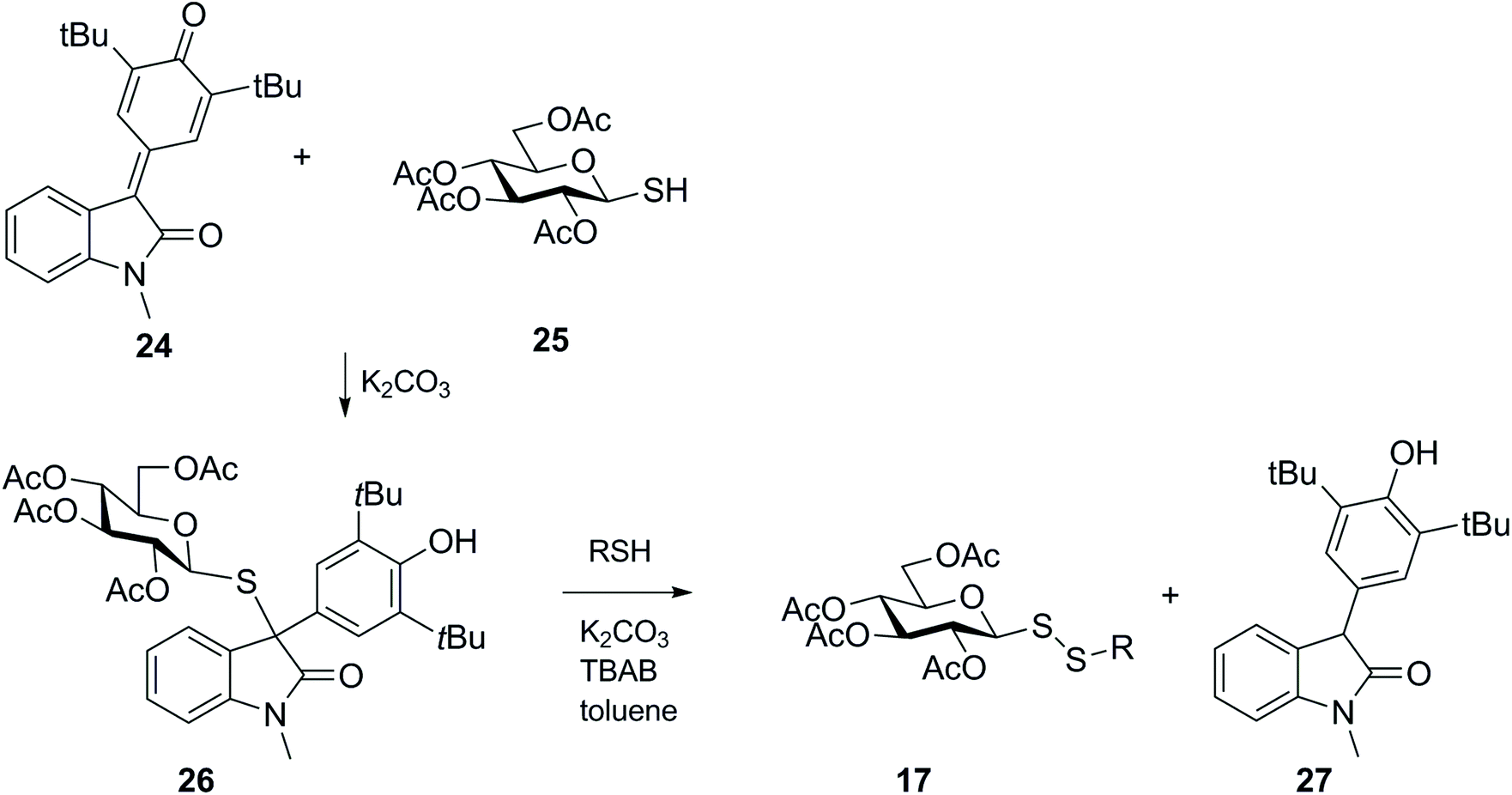 | ||
| Scheme 9 Synthesis of glycosyl disulfides via reduction of para-quinone methides with thiols (TBAB = tetrabutylammonium bromide). | ||
2.2. Synthesis via nucleophilic sulfur-based carbohydrates
An alternative approach to the introduction of a disulfide linkage at the anomeric centre is based on the nucleophilic attack by a glycosyl thiol (or thiolate) to a sulfenyl reagent. For example, reaction of a thiosugar with arylsulfenylchloride produced a series of unsymmetrical aryl glycosyl disulfides as potential solubilizing agents to isolate and purify membrane proteins.53 Dithiocarbamates have been another functionality used to introduce sulfur at the anomeric position. Treatment of a glycosyl dithiocarbamate with a strong base (e.g. NaOEt) gave the thiolate, which was highly reactive with dimethyl (methylthio)sulfonium tetrafluoroborate, with the corresponding S-methyl glycosyl disulfide being obtained in high (90%) yields. Despite these excellent yields, this approach is less appealing due to the need to use base-resistant protective groups (e.g. benzyl, acetonide), plus glycosyl disulfides were obtained as an undesirable mixture of anomers, as a result of partial anomerisation during the transesterification reaction.54Sialosyl disulfides also have potential to act as O-glycoside mimics or for application to the synthesis of oligosaccharides, and a few reports have described their synthesis. The first relies on the nucleophilic attack of a per-O-acetylated thiosialoside to a sulfenylhydrazine intermediate in a two-stage process.55 When pursuing thiosialosides as potential anticancer agents in our own research, we made the observation that during the synthesis of sialoside thioacetate 29 another product was apparent, depending on the level of oxidation of commercially-sourced reagent potassium thioacetate. Mechanistic studies uncovered that the unexpected product was in fact the respective dithioperoxyester analogue that resulted from the thiol-disulfide exchange reaction of sialoside thioacetate 29 with the oxidised acetyl disufide (AcSSAc).56 This observation motivated us to exploit this exchange reaction as an alternative approach to introduce a disulfide linkage into the sialoside scaffold. Sialosyl disulfides 30 were then prepared directly from the respective chlorosialoside 28, and the reaction proceeded through the thioacetate intermediate 29, which in turn then reacted with a symmetric disulfide carrying the desired functionality (Scheme 10). The efficiency of this thiol-disulfide exchange reaction was improved significantly (yields of up to 75%) when an organic base was added to the reaction.57 This exchange strategy was applicable to other carbohydrates, with glucosyl thioacetate 15 being converted equally efficiently into the respective unsymmetrical glucosyl disulfides 17 (Scheme 10). The thioacetate group of glucoside 15 was found to be unreactive with bases like diethylamine, diisopropylethylamine or morpholine. Hydrazine hydrate was required to promote S-deacetylation efficiently in this chemical setting.
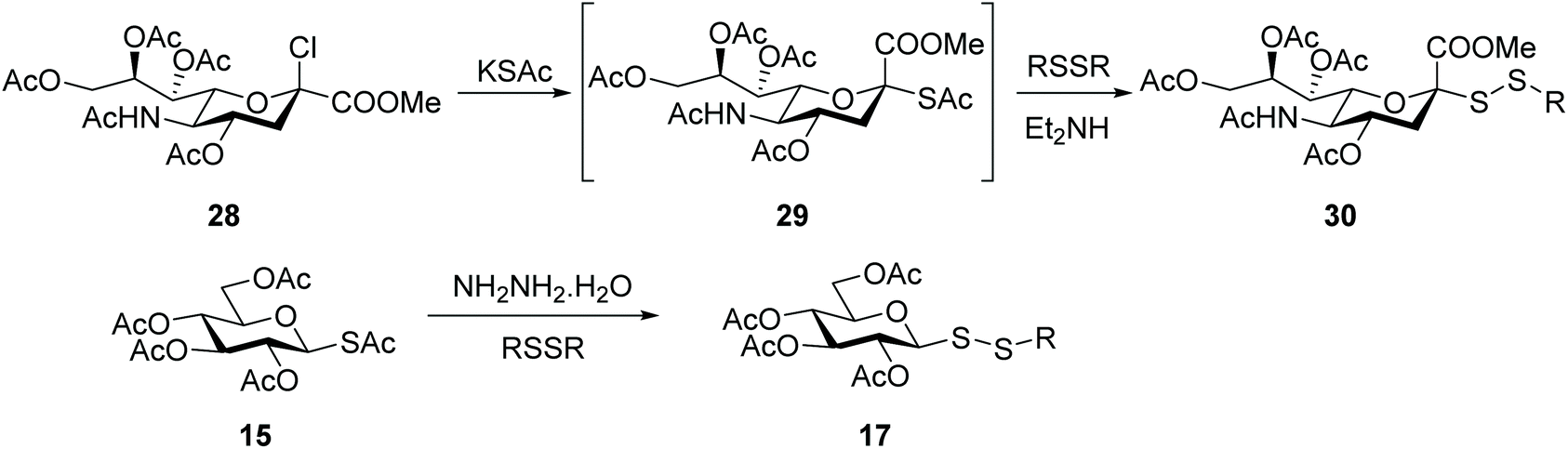 | ||
| Scheme 10 Synthesis of glycosyl disulfides via a thiol-exchange reaction. | ||
In addition, we investigated the utility of the identified acetyl disulfide derivative of sialic acid (represented by general structure 31, Scheme 11) for the preparation of disulfide-linked sialosides. In this case, two sulfur atoms were introduced in one step via an SN2 reaction, employing several alkyl halides. It was observed that sequential use of hydrazine acetate, to selectively cleave the C–S bond (instead of S–S bond cleavage), and triethylamine efficiently produced the unsymmetrical aryl sialosyl disulfide (Scheme 11A). In the presence of weak electrophiles, however, the generated dithiolate species rapidly decomposed, leading to several side reactions and poor yields. As a general method, this strategy was deemed sub-optimal.57 Around the same time, Jiang and co-workers also used the acetyl group to mask and stabilise hydrodisulfides (RSSH). The respective dithioperoxyesters were investigated as disulfurating reagents to access unsymmetrical disulfides by oxidative cross-coupling reaction (Scheme 11B). The efficiency of this approach was generalised with regards to aryl boronic acids and different disulfurating reagents, including use of galactose as substrate, and constituted the first examples of metal-catalysed reactions to synthesise unsymmetrical glycosyl disulfides.58 Furthermore, this copper-catalyzed cross coupling reaction could be tuned to different classes of products, and when a phenanthroline derivative was used as ligand and methanol as solvent, the same authors obtained the respective methoxy disulfide (SSOMe) instead. This umpolung strategy, in which the nucleophilic acetyl disulfide was converted into the electrophilic methoxy disulfide opened up a new avenue and enabled the reaction with a wide number of C-, N-, and S-nucleophiles to produce mixed glycosyl disulfides, aza-disulfides and trisulfides (Scheme 11C). Since sulfur and oxygen exhibit similar electronic effects, the challenge of this approach was to distinguish between the S–O bond cleavage over the S-S bond which in turn was overcome by employing a hard acid like tris(perfluorophenyl)borane directly coordinated to the oxygen.59 In summary, carbohydrate dithioperoxyesters (RSSAc) have been shown to be valuable disulfurating reagents to efficiently access unsymmetrical glycosyl disulfides.
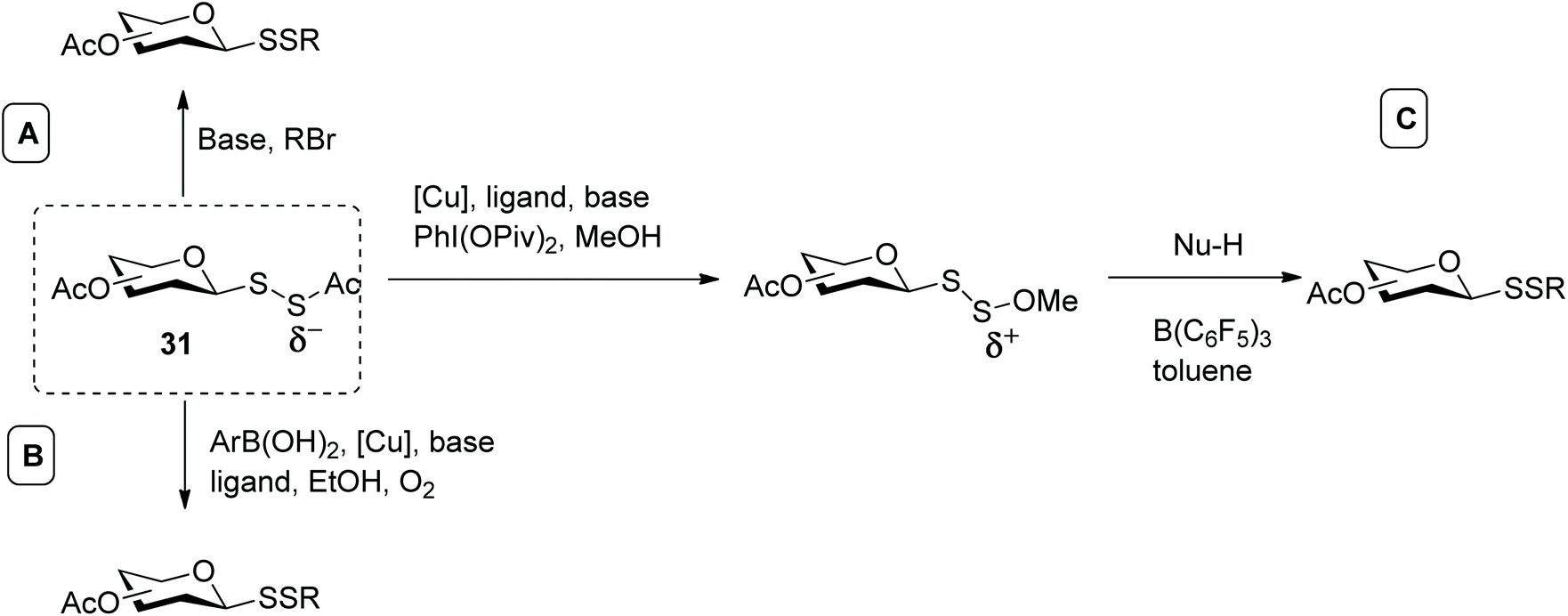 | ||
| Scheme 11 Different methods to prepare disulfide-linked glycosides from glycosyl dithioperoxyesther via: (A) A base-catalysed SN2 process; (B) a copper-catalysed oxidative cross-coupling reaction; (C) an umpolung strategy. | ||
Glycosyl disulfides have also formed key components of dynamic libraries through a combinatorial chemistry approach. Dynamic combinatorial chemistry (DCC) represents an evolutionary approach to molecular design and is a very efficient route to identify specific inhibitors, catalysts or receptor ligands. Fundamentally, the building blocks of the library exchange to form its constituents by reversible reactions, where the thermodynamic equilibrium shifts towards the best suitable ligands, upon introduction of the biological target. The amplification of the best ligands is accompanied by a decrease of the concentration of the products, that albeit containing a common building block, do not bind well to the target.60 Based on their interchangeable nature in the presence of thiolates, disulfides had been considered an attractive functionality to explore in DCC, and Ramoström and co-workers constructed carbohydrate libraries of glycosyl disulfides based on this principle.61,62 The dynamic carbohydrate libraries were generated from a pool of thiol components through a thiol-disulfide exchange (Fig. 2). This dynamic combinatorial strategy was also extended to prepare glycopeptide libraries, in which different thiosugars were mixed with cysteine-containing peptides.63,64
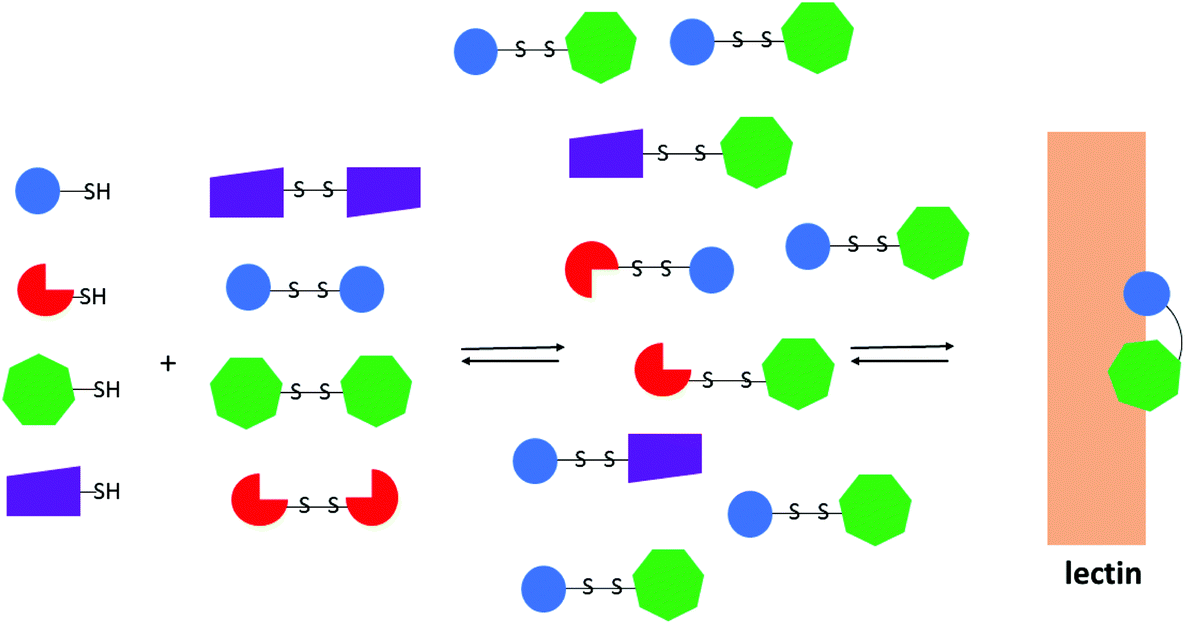 | ||
| Fig. 2 Representation of a dynamic combinatorial library using disulfide interchange in the presence of a lectin. The coloured geometrical forms are representative of different monosaccharides. | ||
2.3. Glycosylation of proteins via a disulfide linkage
Glycosylation is one of the most important post-translational modifications (PTMs) that is crucial in regulating protein expression, folding, activity, thermal and proteolytic stability.65 On the mammalian cell surface, glycosylated proteins play key roles in cellular communication, immune responses, and cell migration, all of which are important in multiple disease states, and in particular cancer.66,67 The crucial need for probes to aid structural and functional study of PTMs has led to significant efforts to chemically or enzymatically glycosylate those proteins. The uniqueness of the cysteine residue which allows for site-specific glycosylation of peptides/proteins has prompted the development of several elegant methods to produce S-linked glycoproteins, including through a disulfide linkage. As disulfides are flexible and can adopt conformations imposed by natural amide bonds at glycosylation sites,68 asymmetric glycosyl disulfides can be considered structural mimics of natural N-linked glycoproteins.Among the electrophilic thiol-specific carbohydrates created as cysteine-reactive reagents, the glycosyl dithiopyridine (33) developed by Boons and co-workers was the first.69 The 5-nitropyridine-2-sulfenyl (pNpys) group was introduced into thioglucosamine 32 in modest yield, while neoglycoconjugate 34 was obtained quantitatively with full control of anomeric stereochemistry, albeit employing a huge excess of 33 (50 molar equivalents) in an effort to maximise the reaction (Scheme 12). Moreover, this method was limited to the addition of a single GlcNAc monosaccharide unit, since attempts to prepare the corresponding sulfenyl transfer reagent from di- and trisaccharides failed, with mainly disulfide-linked homodimers obtained.70 As an alternative, glycosylation of peptides/proteins has been achieved through the reaction of the thiol group of a cysteine residue of proteins with the thiolate of thioaldoses, under buffered aqueous conditions. This oxidative coupling did indeed produce the respective disulfide-linked neoglycoconjugates, however homodimerisation of both peptide/protein and glycan was a significant undesirable side reaction.70 Both approaches are amenable to the use of fully deprotected glycans, but rely on the modification of an existing cysteine residue in the protein with no control of the glycosylation site. To solve this problem and allow glycosylation of proteins that have no natural cysteine in their sequence, Davis and co-workers established a combination of site-directed mutagenesis followed by chemical modification (Scheme 13).44 The inserted cysteine acts as chemoselective tag at the pre-determined position of the protein, which was then modified with a thiol-containing sugar. This elegant two-step strategy allowed for full control of both glycosylation site and sugar. Based on the use of methanethiosulfonates (MTS) in protein modification,71 the authors developed the corresponding carbohydrate 37 and explored it as a glycosyl sulfur transfer reagent that efficiently glycosylated proteins. This glycomethanethiosulfonate ester (glyco-MTS) was appropriate to different sugar units (e.g. glucose, glucosamine), but the efficiency of its preparation, which involved stereoselective displacement of the halide from the appropriately protected halo-sugar derivative 36 with sodium methanethiosulfonate, was dramatically affected by the synthetic conditions.72,73 Occasionally during the synthesis of glyco-MTS, the respective glycosyl methanedithiosulfonate (glyco-MDTS) was also formed, which then contaminated the glycosylation reactions.74 Since MTS reagents are relatively unstable under the basic conditions employed for the glycosylation, the same authors further modified the methyl group in the MTS aglycone to a phenyl group, and created a novel class of glycophenylthiosulfonates (glyco-PTS, 38).73 The authors reasoned that removal of the acidic α-methyl protons might enhance stability of the thiosulfonate during the preparation and subsequent reaction, and that introduction of a UV-active chromophore into the aglycone would facilitate monitoring of the reactions, both in the preparation of glyco-PTS and its use. The synthesis of glyco-PTS 38 was indeed more efficient than glyco-MTS 37, and glycosylation of mutant protein subtilisin from Bacillus lentus (SBL) was superior with the former (Scheme 13).73 While glycothiosulfonate esters were further used as sulfur transfer reagents to prepare other glycoproteins32,75 and glycodendriproteins,76 this approach introduced only protected glycans, and the desired free glycosides were only obtained after enzymatic deprotection on protein,77 which limited their utility.
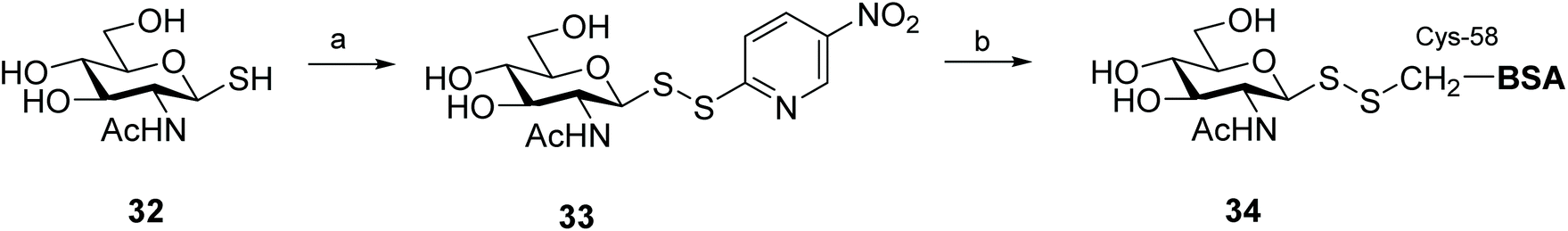 | ||
| Scheme 12 Synthesis of a disulfied-linked glycoprotein from a glycosyl dithiopyridine. Reaction conditions: (a) 2,2′-Dithiobis(5-nitropyridine), AcOH–water, 6 h, 58%; (b) bovine serum albumin (BSA), NH4OAc (1 M), pH 5, 15 min. | ||
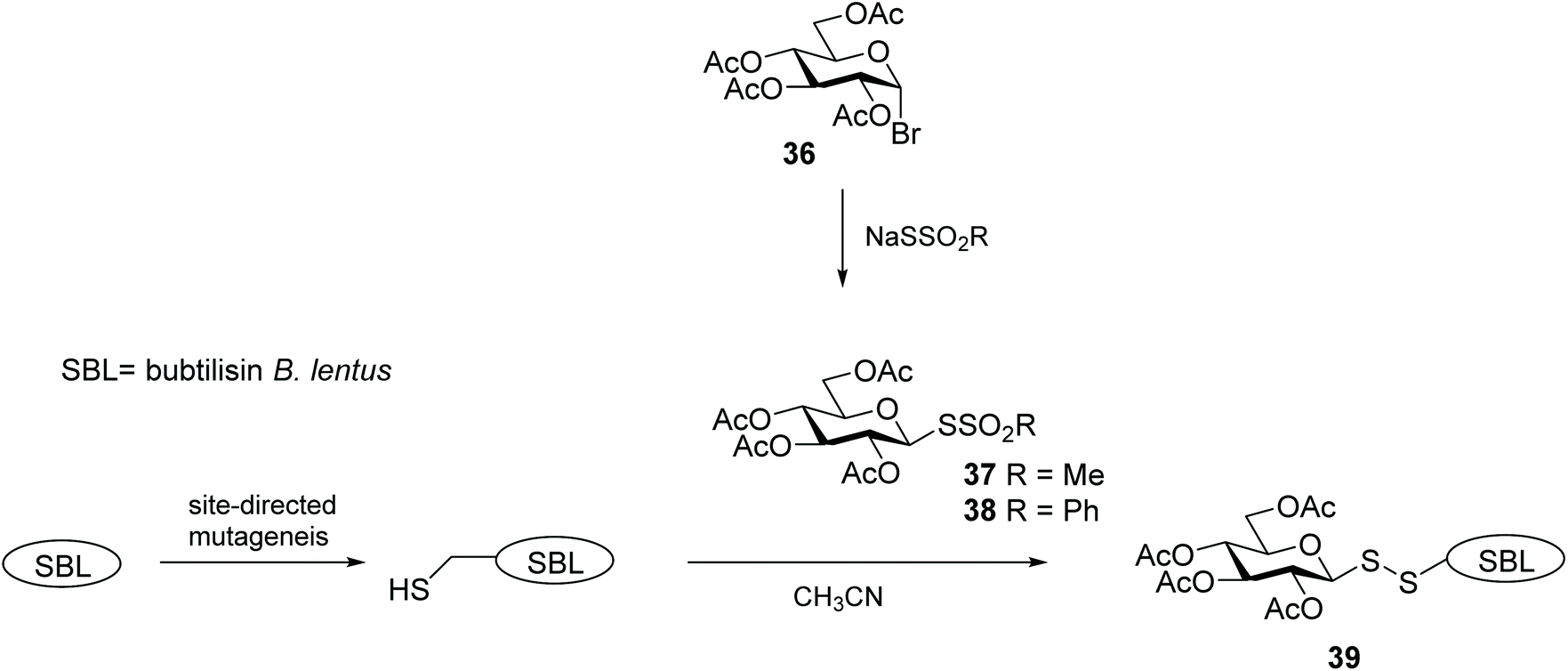 | ||
| Scheme 13 A combined site-directed mutagenesis and chemical modification approach to site-selective protein glycosylation. | ||
The knowledge that rare naturally-occurring selenenylsulfide proteins behaved as selective electrophiles78 motivated the same authors to explore (phenylselenenyl)sulfide-mediated protein glycoconjugation.79 In their investigations they explored cysteine either as a nucleophile to react with a glycoselenenylsulfide derivative, or as an electrophile, in the form of the corresponding selenenylsulfide analogue, that reacted with a deprotected 1-thiosugar (mono- or oligosaccharide). In both strategies the electrophilic character of the sulfur in the resulting S–Se bond made it susceptible to nucleophilic attack by the thiol and produced an interglycosidic disulfide bond. Glycoselenenylsulfides were prepared either from the corresponding protected or deprotected thiosugar with phenylselenenyl bromide. No homodimer formation was reported in this case, but attempts by Iadonisi and co-workers to prepare the galactosylselenenylsulfide reagent via a sulfur-selenium exchange reaction between the galactosylthiouronium salt and diphenyl diselenide produced symmetrical galactosyl disulfides as a side product to some extent (20–50%).23 The use of glycoselenylsulfides as sulfur transfer reagents allowed quantitative introduction of mono- and oligosaccharides of up to seven saccharide units in size at single and multiple sites in a variety of proteins. Moreover, the compatibility of this method with unprotected thiosugars is a significant advantage (i.e. removing the need for a post-modification de-protection step).79
3. Glycosyl disulfides as chemical intermediates
Glycosyl disulfides have also been employed successfully as useful intermediates in carbohydrate synthesis, to give access to a wide variety of analogues. For example, treatment of S-phenyl glucosyl disulfide (40) with a phosphine reagent led to the formation of the corresponding thioglycoside 43 plus the symmetrical diglucosyl sulfide (44). However, the mono-desulfurisation was only partial, and this was a suitable route to obtain the most synthetically challenging cis-related compounds at C1- and C-2. The inversion at the anomeric centre occurs following the generation of two thiolate anions, each of which attacks the C1 of the glycosylthiophosphonium salt 41 (Scheme 14).31 Later Davis and co-workers found that the nature of the aglycone subunit plays an important role in the efficiency of the desulfurisation reaction and the anomeric stereochemistry.32 Using tributylphosphine as reducing agent, they observed that the desulfurisation yields where significantly superior (6–7 fold) when p-nitrothiophenol was used as a component of the disulfide substrate, instead of an unsubstituted thiophenol or amino acid. The yields of thioglycoside 47 were increased by up to 75% when the desulfurisation was performed with the more electron rich tris(dimethylamino)phosphine. It was also observed that the desulfurised product of disulfide-linked glycosyl cysteine 45 had retained its anomeric configuration and it was obtained as a mixture cysteine diastereoisomers, which indicated that in this case the reaction mechanism was different from that originally reported,31 and subsequently described by themselves with aryl aglycones.32 NMR studies for mechanistic purposes indicated the formation of a dehydroalanine intermediate 46 as the result of the disulfide cleavage followed by β elimination of the cysteinyl phosphonium salt, which was slowly converted into thioether 47 (Scheme 15). This reaction was found to be applicable to other carbohydrates with different degrees of protection/deprotection, and to access the more enzymatically stable thioether-linked glycoproteins from disulfide-linked precursors.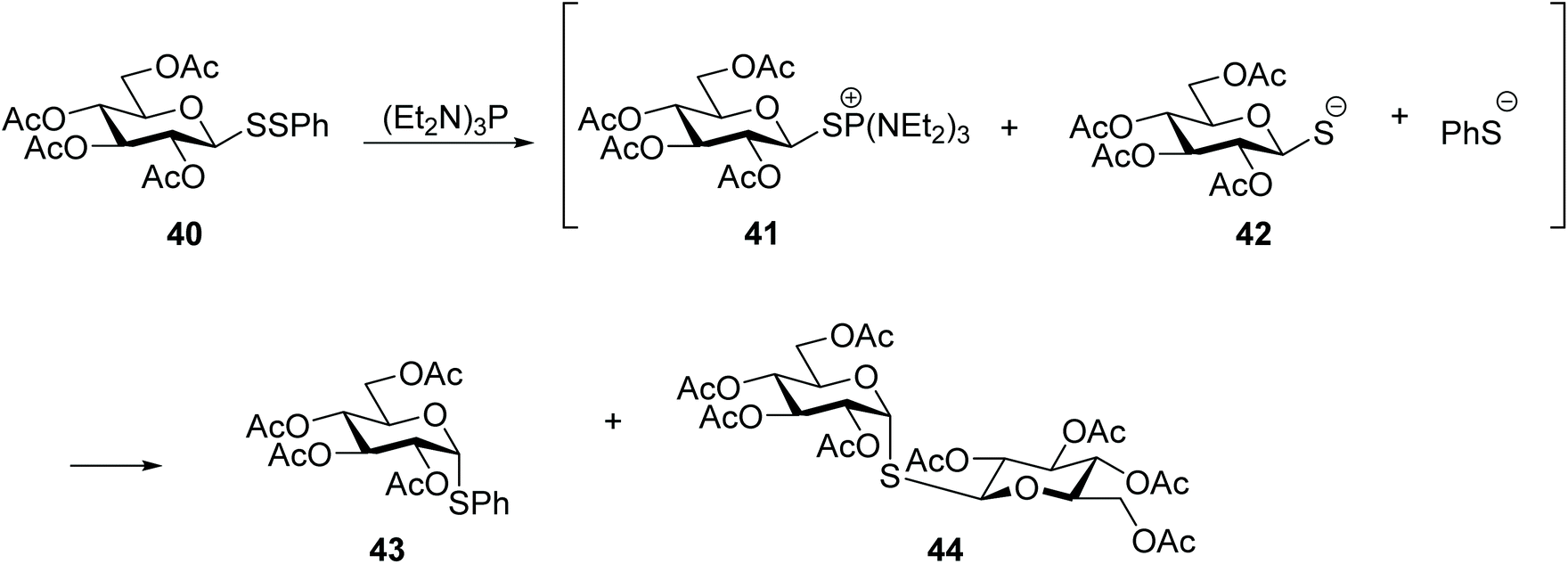 | ||
| Scheme 14 Desulfurisation of a glycosyl disulfide to obtain cis-thioglycosides. | ||
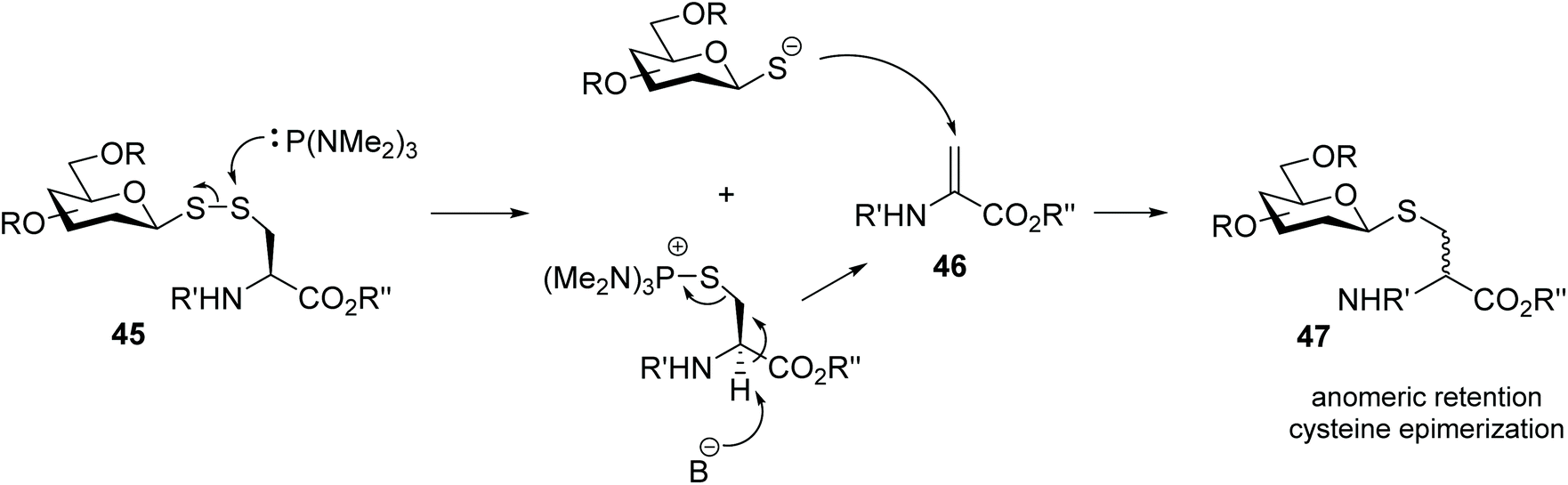 | ||
| Scheme 15 Proposed Sγ-attack followed by elimination-conjugate addition mechanism of desulfurisation of a disulfide-linked glycosyl cysteine. | ||
During the investigations into the thiomolybdate-catalysed synthesis of symmetrical disulfides, a different approach to the preparation of thioglycosides was uncovered, but in a related mechanism to that observed for the desulfurisation of glycosyl-cysteinyl disulfide. It involved the sequential addition of a Michael acceptor to the reaction after a glycosyl disulfide underwent reduction to the corresponding thiolate. This was achieved directly from a sugar bromide, a significant advantage for this method, and was extended to the synthesis of a variety of thiosugar analogues with excellent diastereoselectivity through inter- and intramolecular reactions.80
Von Itzstein and co-workers were pioneers in exploring disulfides as protecting groups for anomeric thiols, but the employed reductive conditions (Zn in Ac2O) were sub-optimal and disulfides were converted into the respective S-acetyl glycosides.12 This disadvantage was later solved by Hummel and Hindsgaul in their work on thio-oligosaccharides, where they established the utility of ethyl disulfide as an orthogonal protective group for the respective thiosugars. After immobilisation of mixed sialosyl- and glycosyl disulfides on a trityl chloride polystyrene resin, the anomeric thiol was successfully generated by the reductive deprotection of the disulfide with dithiothreitol (DTT). This strategy was explored for the synthesis of thio-oligosaccharides on solid phase, which were efficiently obtained after reaction of the free thiol 50 with a triflate-activated glycoside (Scheme 16).55 This protection of the anomeric thiol function as a tert-butyl disulfide was further utilised in the synthesis of a Man9GlnNAc2 oligosaccharide81 and S-linked oligosialic acid in solution.82,83 Alkyl disulfides as protective groups proved to be compatible with carbohydrate chemistry, while minimizing or preventing several of the side reactions associated with thiol-deprotection and oligomerisation (e.g. anomerisation, thiol-thiol exchange, elimination).
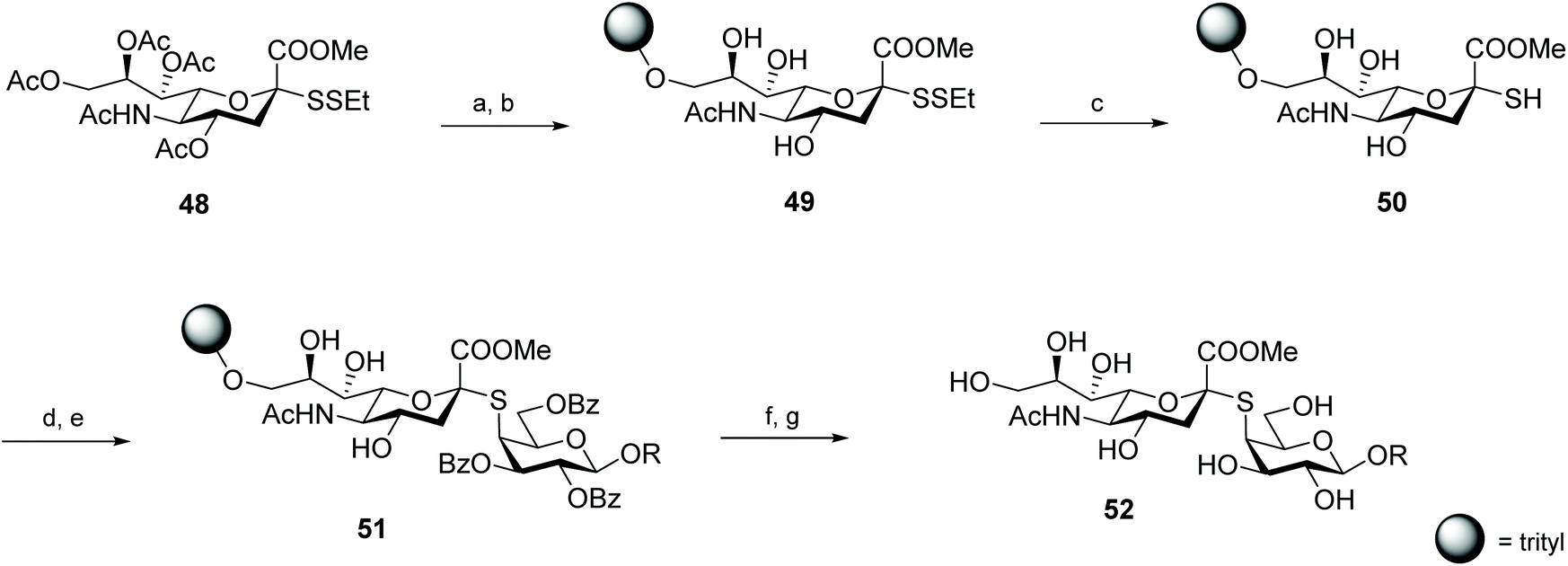 | ||
| Scheme 16 Synthesis of thio-oligosaccharides on solid phase via a glycosyl disulfide intermediate. Reaction conditions: (a) NaOMe, MeOH; (b) trityl chloride resin, pyridine, DMAP; (c) dithiothreitol, THF, MeOH; (d) NaOMe, THF; (e) O-triflate glycoside, [15]crown, THF; (f) NaOMe, THF; (g) TFA, CH2Cl2. | ||
Another example that represented the usefulness of disulfides as protective groups of the anomeric thiol was demonstrated in the assembly of micelles based on auranofin.84 Auranofin 53 (Scheme 17) is a gold complex with a sugar ligand that has been used extensively to treat rheumatoid arthritis for the past four decades, but recently it has been repurposed for use as a cancer chemotherapy agent, amongst other things.85 Its applicability as an in vivo anti-tumour agent is limited by its excessive reactivity with protein thiols. Incorporation of auranofin into the core of micellar systems is one approach to prevent interaction with serum proteins and enhance accumulation in tumour tissue. For this effect, the gold complex was coupled to a homopolymer containing pendant deacetylated thioglucose monomers, after reduction of the glycosyl dithiopyridine 55 (Scheme 17). While in this case the pyridinyl disulfide was explored as a protective group for the anomeric thiol,84 glycosyl dithiopyridines have also served as sulfur transfer agents for the glycosylation of proteins.69,86,87
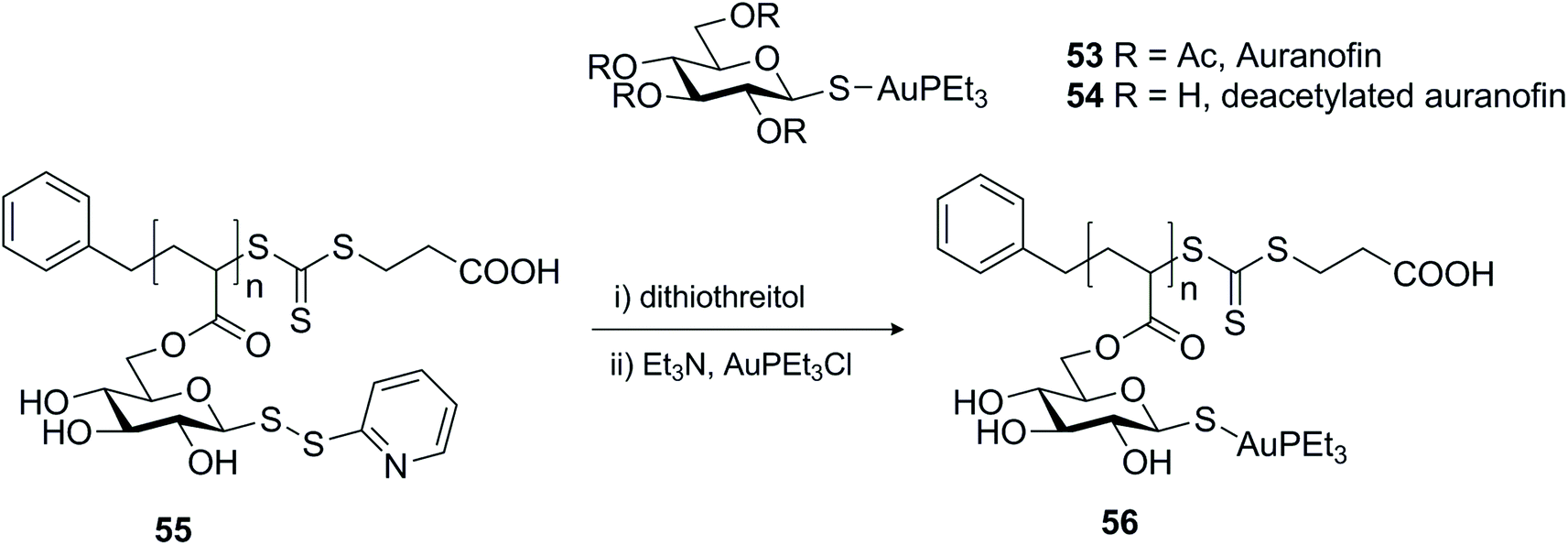 | ||
| Scheme 17 Synthesis of deacetylated auranofin micelles using dithiopyridine as a protective group for the anomeric thiol. | ||
Thioglycosides are versatile glycosyl donors, particularly for access to O-glycosides in oligosaccharide synthesis.88 Other glycosides with a sulfur atom at the anomeric centre have also been explored as glycosyl donors, like for example glycosyl sulfones,89 but also glycosyl disulfides, albeit to a much lesser extent. The first example of glycosyl disulfides being employed as glycosyl donors was demonstrated by Vasella and co-workers, where an activated methyl mannosyl disulfide was reacted with bromine, and the acceptor aniline to successfully produce an N-glycoside.22 Later, Davis went on to investigate the potential of several unsymmetrical glycosyl disulfides to prepare O-glycosides.90 Glycosyl disulfides offer several advantages as donors over traditional thioglycoside systems, including their higher chemical flexibility, and the ease of cleavage/exchange for adjustment of the aglycone, to fine-tune their reactivity as donors. In addition, since disulfides are better Lewis acids than their sulfide counterparts,91 they offer better coordination to thiophilic promoters, which in turn enhances reactivity towards glycosyl acceptors. Both ‘armed’ (ether protecting groups) and ‘disarmed’ (acyl protecting groups) protected mixed glycosyl disulfides behaved as glycosyl donors in the synthesis of O-glycosides (e.g. disaccharides, glycopeptides), but the former (ether protected) proved more efficient donors (yields up to 90%). Despite the lower yields, the reaction between ‘disarmed’ glycosyl disulfides 37 with glycosyl acceptors produced exclusively β-stereoselective O-glycosides 60, while the reaction with ‘armed’ glycosyl disulfides 57 lacked stereoselectivity (Scheme 18).90 A further study investigated the influence of the aglycone (SSEt vs. SSpNP) and the donor configuration (Glc vs. Gal)† on the donor properties of glycosyl disulfides. This study was extended to assess the effectiveness of different activators on the reactivity of glycosyl disulfides.72 Glycosyl disulfides have also been exploited as donors for the preparation of complex carbohydrates, while at the same time providing access to an anomeric thiol for site-specific attachment to a protein carrier through a thioether linkage, thereby producing glycoconjugates with specificity as immunogens.92 In summary, these reports represent a series of elegant methods for the preparation of O-glycosides, and provide evidence for the utility of glycosyl disulfides as efficient glycosyl donors in both solution- and solid-phase systems.
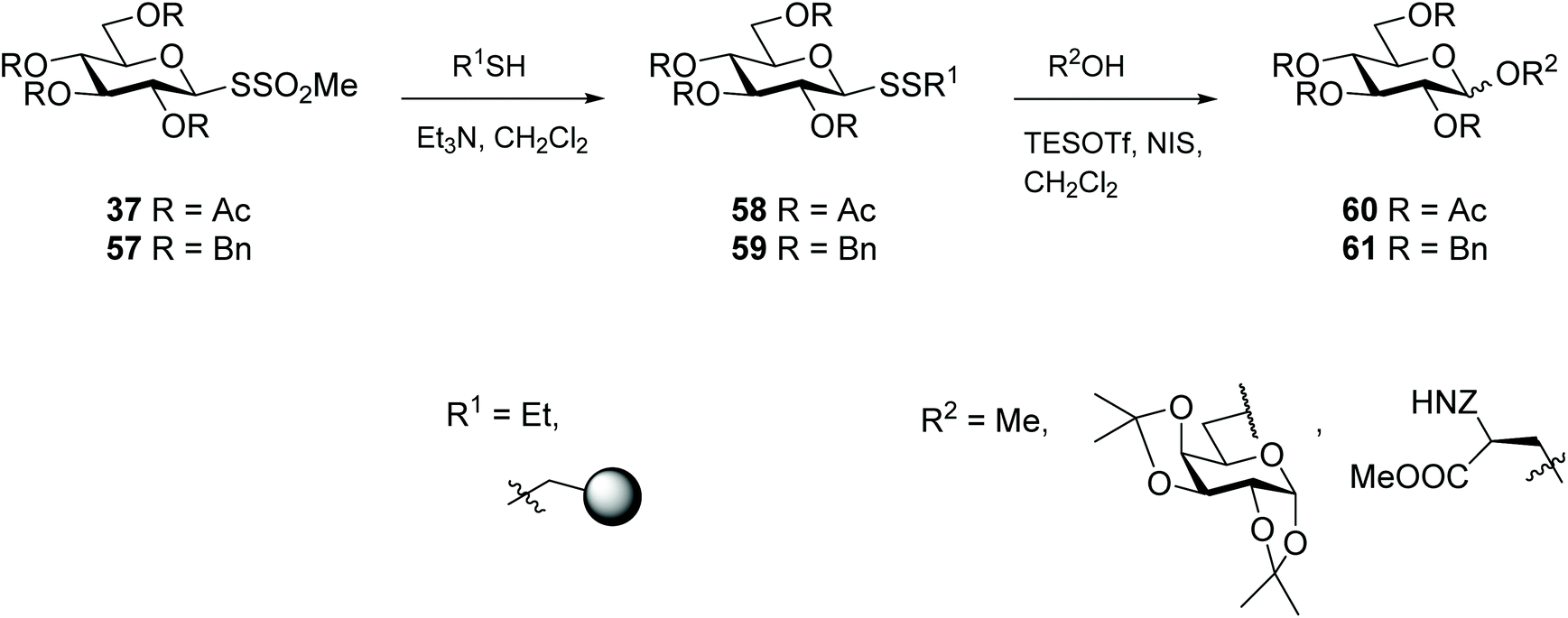 | ||
| Scheme 18 Unsymmetrical glycosyl disulfides as glycosyl donors. | ||
Thiocarbonyls are useful synthetic intermediates and are usually prepared from the respective O-lactones using Lawesson's reagent. Direct thionation of carbohydrate O-lactones gives only poor yields of the corresponding thionolactones, however. A more efficient approach was then established for the preparation of glyconothio-O-lactones 63 from glycosyl disulfides, either symmetrical or unsymmetrical. Oxidation of glycosyl disulfide 17 with 3-chloroperoxybenzoic acid produced the intermediate thiosulfinate 62, which upon thermolysis provided the desired glyconothio-O-lactone 63 (Scheme 19) in better yields and higher purity than direct thionation of the O-lactone employing Lawesson's reagent. Undesirable formation of glycosyl thiosulfonates, either by over-oxidation of the disulfide, or by disproportionation of the thiosulfinate under the reaction conditions were drawbacks of this methodology, however.54 Direct synthesis of the more stable glycosyl thiosulfinate from thiosugars, rather than oxidation of the glycosyl disulfide, partially overcomes this problem.93
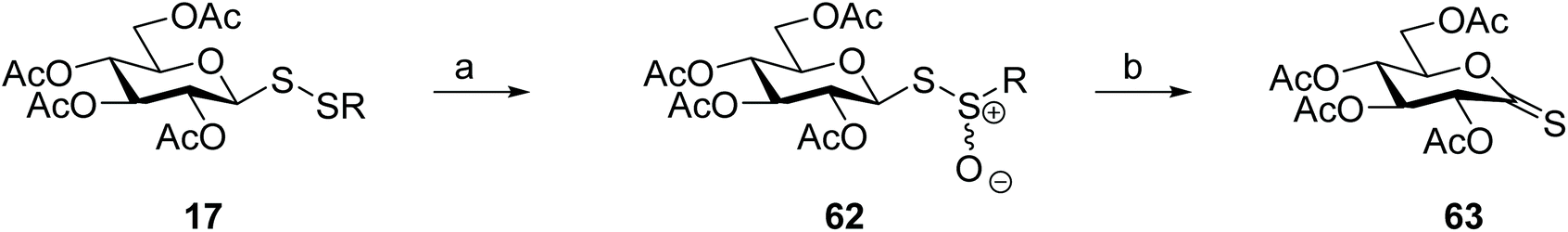 | ||
| Scheme 19 Use of glycosyl disulfides to prepare glyconothio-O-lactones. Reaction conditions: (a) 3-Chloroperoxybenzoic acid, CH2Cl2, 0 °C; (b) toluene, 110 °C. | ||
4. Biological applications
The development of synthetic methodologies associated with glycosyl disulfides has underpinned their importance and utility in biological systems. Glycosyl disulfides have applications as lectin ligands, as drugs (cytotoxic agents, and bioreductive prodrugs) and as detergents to name a few significant roles. These will be considered in the ensuing sections.4.1. Glycosyl disulfides as lectin ligands
Lectins are a large class of carbohydrate-binding proteins that are highly specific for sugar groups, and play key roles in recognition of events at both the cellular and molecular level.94 One of the best studied examples, Concanavalin A (Con A), is a plant lectin that binds to mannose residues of various glycoproteins, inducing those associated with acute hepatic inflammation.95 The crystal structure of ConA was solved with methyl α-D-mannopyranoside as a bound ligand.96 A mannosyl disulfide containing an aziridine aglycone was explored as a molecular tool to probe ConA. Upon binding of the sugar, the aglycone moiety covalently bound to ConA is sequentially converted into a fluorescent saccharide sensor. The mannose unit and disulfide bond were essential for lectin recognition and sugar release, respectively.87 Ramström and co-workers also explored lectins as targets for glycosides through dynamic combinatorial chemistry (DCC), in which synthesis of the ligand was amplified upon lectin binding (Fig. 2). They identified two active dimannosyl disulfides: 64 and 65 (Fig. 3), with binding affinities comparable to that of methyl α-D-mannopyranoside. The results also indicated that the hydroxyl group in the 6-position of the methyl α-D-mannopyranoside is crucial for the efficient binding to ConA, compared to the analogous sulfhydryl group.62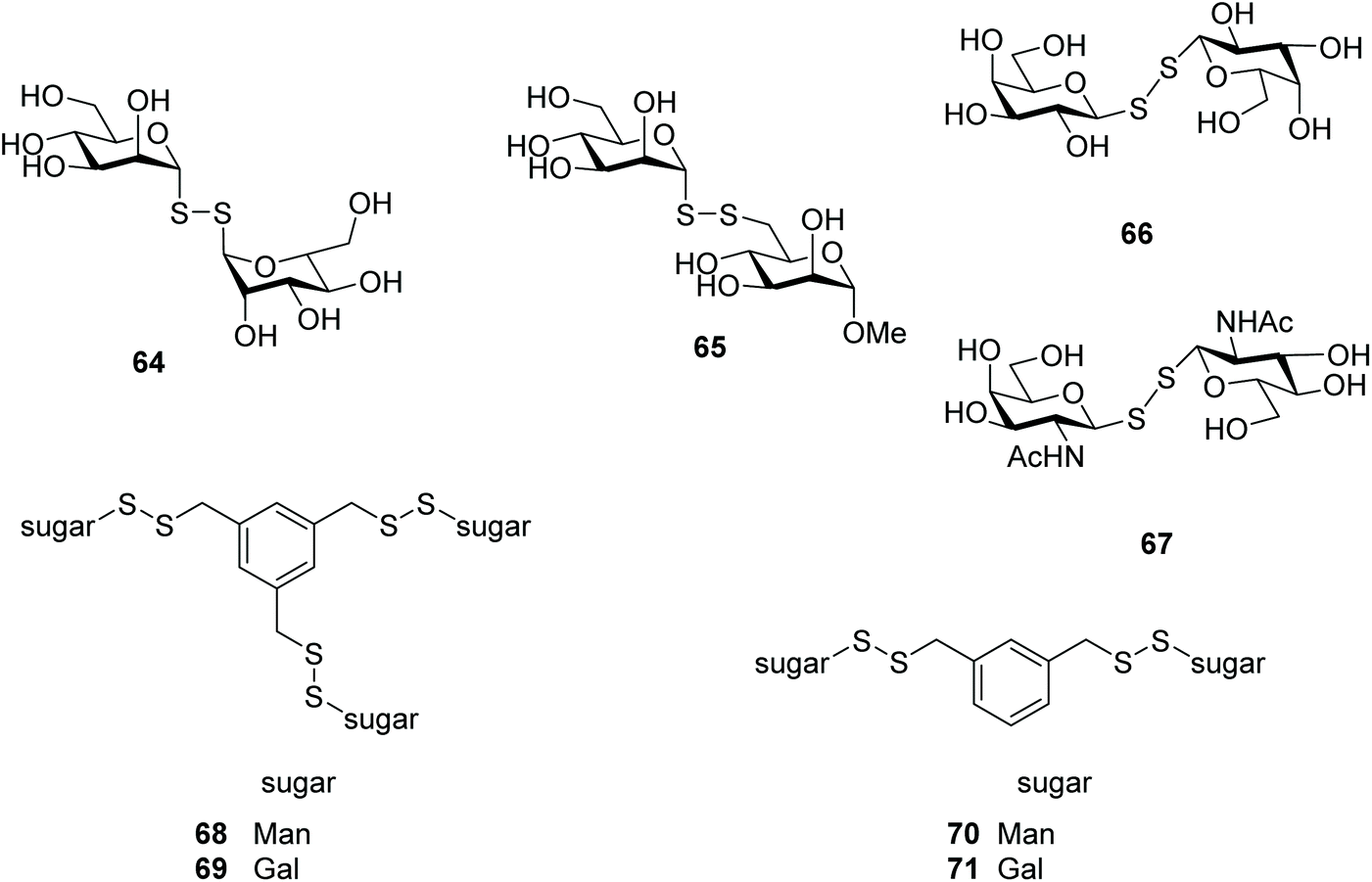 | ||
| Fig. 3 Glycosyl disulfides as lectin ligands. | ||
To study the impact of the differences in conformational space and electronic properties on carbohydrate recognition properties, the inhibitory effect of six thio-aldopyranoses (Man, Glc, Gal, GlcNAc, GalNAc, and methyl-6-thio-α-D-mannopyranoside)† was compared to that of corresponding symmetrical diglycosyl disulfides against ConA. With the exception of 1-thio-mannose, none of the thiosugar monomers demonstrated significant inhibition, while three of the glycosyl disulfides were clearly efficient in being able to bind and inhibit ConA. Amongst the diglycosyl disulfides, ManSSMan was the most effective inhibitor, followed by GlcSSGlc and GalSSGal. This activity was found to be cooperative, calcium-dependent and redox-responsive.17 Evaluation of a dithiodiglycosyl library by STD NMR studies, a spectroscopic technique for assessing ligand–protein interactions,97 confirmed that mannose derivatives (ManSSMan, ManSSGlu, ManSSGal) were indeed bound to ConA. GlcSSGln, GalSSGal, GlcSSGal were deemed inefficient in this investigation, however.29 A beneficial effect on the binding to ConA was observed when the number of mannosyl disulfides increased. For instance compounds 68 and 70 (Fig. 3) exhibited higher affinity than methyl α-D-mannopyranoside, and it was suggested that cross-linking was likely to occur in the interaction of these derivatives with ConA.43
Further combinatorial carbohydrate libraries were screened with lectins besides ConA, where dithiogalactose disulfide 66 (Fig. 3) was identified as being able to block mistletoe lectin (Viscum album agglutinin, VAA), a highly toxic plant lectin similar to the biohazard ricin, binding to human cell lines. An unsymmetrical disulfide of N-acetyl galactosamine and N-acetyl glucosamine 67 (Fig. 3) was also found to display recognition for galectin-3.14,61 Motivated by these findings, a library composed of mono-, di- (71) and trivalent galactosyl disulfides (69) attached to a phenyl group in the meta-position was assembled, with these disulfides also demonstrating inhibition of VAA. The inhibitory potency for the toxin increased with the valency (i.e. number of monosaccharide units) of the glycan. Inhibition of VAA by 71 was 18-fold and 6-fold higher than that observed for the dithiodigalactoside 66 and thiodigalactoside14 respectively, which confirmed that the inhibitory potency of the glycosyl disulfides was enhanced by glycocluster formation. Interaction of the galactosyl disulfides with human galectins was found to be insignificant.98 Likewise, di-valent galactosyl and lactosyl disulfides attached to a naphthalene core in the para-position exhibited little or no activity on human galectins.99
4.2. Glycosyl disulfides as cytototoxic agents
Several mono-, di- and trivalent glycosyl disulfides have been evaluated for their potential effects on cancer cell viability, but no significant antiproliferative effect was observed.100 The divalent benzene-based dithioglucoside disulfide also showed neither cytotoxic nor haemolytic effects when tested against human erythrocytes.50 Moreover, the lack of cytotoxicity of 64 and 66 (Fig. 3) was confirmed when tested against cancer A375 and HepG2 cell lines.25 These data confirm the potential utility of glycosyl disulfides described above as glycomimetics, as well as being biomedically safe.In view of the promising interaction with lectins demonstrated by some glycosyl disulfides, the Szilágyi group investigated their potential to treat infections caused by Trypanosoma cruzi, the parasite responsible for Chaga's disease.101 In that study, a series of mono and oligovalent disulfides based on galactosyl and mannosyl glycans were tested against different strains of T. cruzi with different levels of resistance to two drugs used in the treatment of Chagasic patients (namely nitrofuran and benznidazole). The bivalent galactosyl disulfide 71 (Fig. 3) was shown to be the most active compound against the three tested strains, with IC50 values in the range 4–10 μM. Remarkably, some of the unprotected glycosyl disulfides were cell-permeable and were effective against amastigotes, the intracellular stage of the parasite reproduction process.102
A series of glycosyl disulfides was introduced into the podophyllotoxin pharmacophore, and the respective conjugates 72 (Fig. 4) were evaluated for their cytotoxicity against human cancer cell lines, including KB (Mouth Epidermal Carcinoma Cells) and KB/VCR (Vincristine-resistant Mouth Epidermal Carcinoma Cells).103 The anticancer drug etoposide (Fig. 4) is a podophyllotoxin analogue widely used for treatment of numerous solid tumours,104 but like many cancer chemotherapeutics, is known to cause several undesirable side effects and drug-resistance problems in the clinic.105 With the aim of addressing these shortcomings, several etoposide derivatives were developed, including analogues with replacement of both the glycan by different aldopyranoses, or aryl/heteroaryl moieties and the interglycosidic oxygen atom by a disulfide bond. Though not the most active compounds of the derivatives synthesised, within the glycosyl analogues the galactosyl disulfide was also found to be very marginally more potent than the parent etoposide, not only for the sensitive cancer cell line KB (IC50 = 1.12 μM vs. 2.27 μM) but particularly in the multi-resistant KB/VR cancer cell line (IC50 = 9.96 μM vs. 16.8 μM).
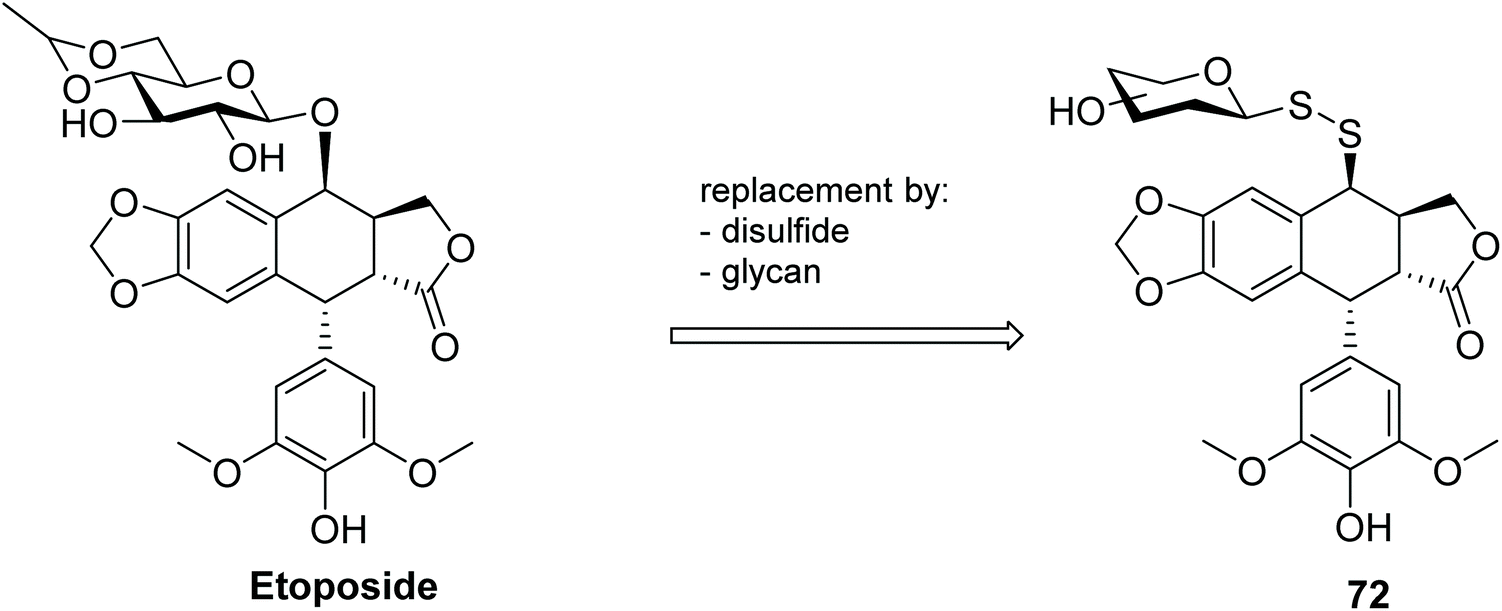 | ||
| Fig. 4 Glycosyl disulfide etoposide conjugates. | ||
This data indicated that introduction of the disulfide moiety was not detrimental to the cytotoxic activity of the analogues, and in some cases proved beneficial. Moreover, metabolic evaluation revealed that the disulfide bond was stable in human plasma for over 8 hours, further indicating the potential of these compounds (and glycosyl disulfides more generally) for in vivo use. The effect of the linker (i.e. disulfide and trisulfide) was also investigated, with all the glycosyl trisulfides exhibiting lower cytotoxic activity against the cancer cell lines than the corresponding disulfides.
4.3. Glycosyl disulfides as bioreductive prodrugs
Paclitaxel (PTX) is one of the most widely used chemotherapeutics globally, but it is another drug where dose-limiting systemic toxicity limits its efficacy. It furthermore suffers from poor aqueous solubility that limits its clinical use. On the other hand, the hypoxic tumour environment, which is a characteristic of many solid cancers, is an excellent target for the development of cancer therapeutics which elicit their cytotoxicity through bioreductive activation. Based on this principle, the glycosyl disulfide 73 (Fig. 5) was designed as an unsymmetrical polar disulfide prodrug of paclitaxel (PTX) to be activated in the reductive hypoxic environment of tumours. The highly polar sugar moiety was expected to increase the hydrophilicity of the conjugate and improve its water solubility, while the disulfide bond was anticipated to be cleaved in the tumour microenvironment via bioreduction. The enhanced hydrophilicity of glycosyl disulfide prodrug 73 was also expected to impair its ability to cross cell membranes, and to diminish its system cytotoxic activity. HPLC stability studies with human serum suggested that disulfide prodrug 73 was relatively stable, with only <10% paclitaxel released after 2 hours. Further antiproliferative assays using L2987 lung carcinoma cells showed that disulfide 73 was 30-fold less toxic than the parent PTX, while when 73 was incubated with DTT for reductive activation, the cytotoxicities of both were similar. No solubility studies were described.4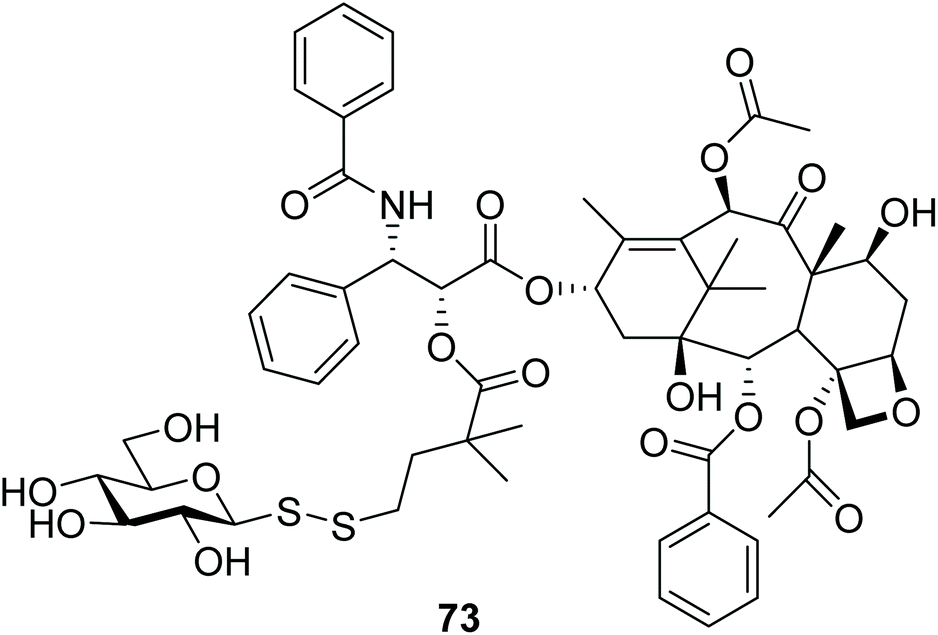 | ||
| Fig. 5 A reductively-activated glycosyl disulfide prodrug of paclitaxel. | ||
4.4. Glycosyl disulfides as chemically-cleavable detergents
Based on their important biological roles, membrane proteins (MPs) are attractive targets106 but their exploration and study has been difficult when compared to soluble proteins. One of the challenges is the extraction, purification, and consequent reconstitution of MPs. Extraction and purification of MPs from biological membranes is accomplished with detergents that must be removed and replaced, for example by phospholipids, to mimic the membrane systems and allow for biochemical and biophysical characterisation. However solubilizing detergents are difficult to remove completely, which complicates reconstitution of MPs and their biophysical characterisation. Conventional detergents can also cause denaturation and aggregation of MPs, which is another shortcoming of their use. Significant improvement was achieved by eliminating residual detergents through physical methodologies (e.g. Biobeads), however complete replacement was rarely achieved.107 As long ago as the 1980s, the use of unsymmetrical glycosyl disulfides as detergents was proposed to solve this problem.53 The idea behind this approach explored the amphiphilic character of glycosyl disulfides, and the cleavability of the disulfide linkage by mild protein-compatible agents, and that the thioglucose and apolar aglycone produced in the cleavage reaction should be readily removed by dialysis. Therefore the authors claimed that unprotected unsymmetrical disulfides could be considered as biologically compatible detergents and an alternative to ionic detergents for the isolation of MPs.53 However, no further studies have been described. Even so, Tao and co-workers revived this proof-of-concept, and last year reported the successful use of glycosyl disulfides with hydrophobic aglycones as chemically cleavable detergents (CCDs) to purify and reconstitute MPs.108 Essentially, they transformed the traditional detergent n-dodecyl-β-D-maltopyranoside (non-cleavable) into the respective disulfide as a CCD, which in turn behaved well in both solubilizing and refolding the protein A2R, also being readily degraded under mild conditions. Full cleavage and removal of CCD-2 allowed for the in situ reconstitution of the MPs into adequate commercial detergents for stabilisation screening, or into nanodiscs for electron microscopy analysis, under an approach that the authors termed sacrifice-replacement strategy (Fig. 6).108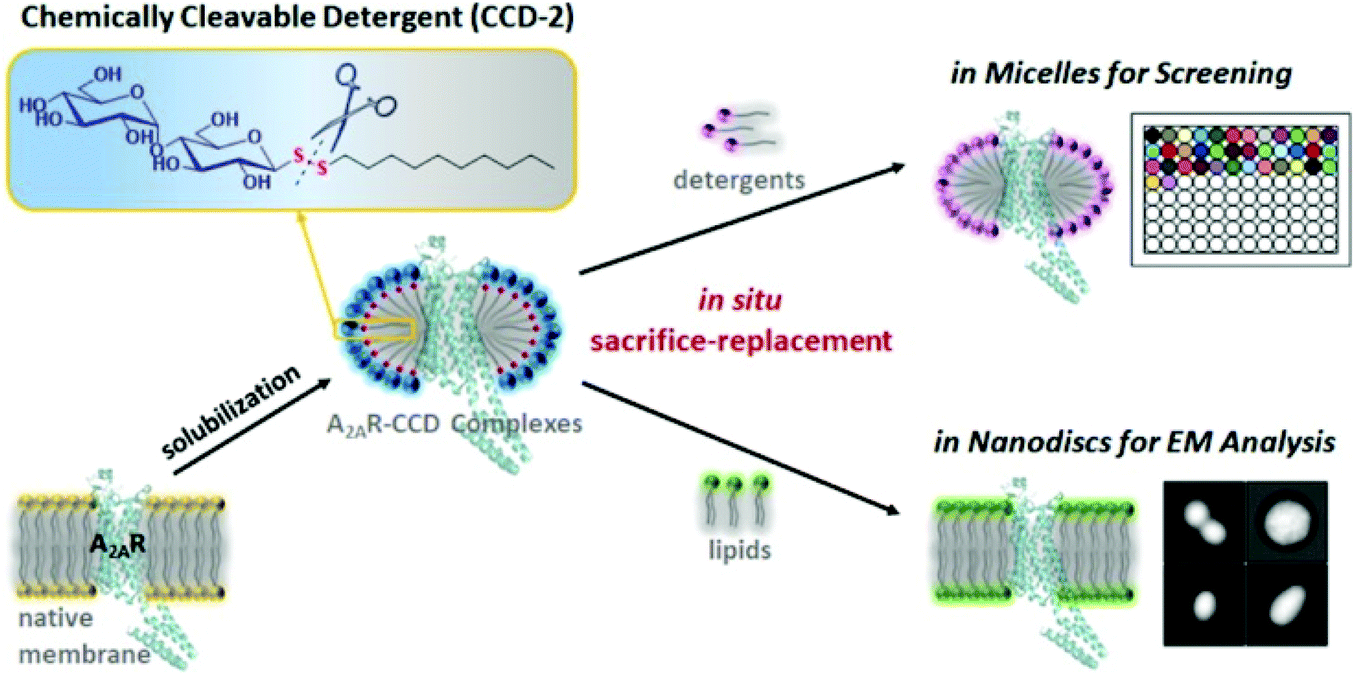 | ||
| Fig. 6 In situ sacrifice-replacement strategy for reconstitution of mebrane proteins mediated by chemically cleavable detergents.108 Reproduced by permission of ACS publications. | ||
5. Conclusions
The volume of methods to prepare glycosyl disulfides has increased significantly over recent decades, accompanied by enhanced chemical efficiency. The prior lack of efficient approaches to obtain glycosyl disulfides hampered their use, but nowadays the rapid access to this class of compounds has fuelled several applications either as chemical intermediates or biological agents. Of these, the exploration of glycosyl disulfides as glycosyl donors was highly significant and has been accompanied by an orthogonal protective group approach to access the anomeric thiol. In a biological context, a robust SAR has been generated around appropriate carbohydrate positioning to enhance lectin binding and a large body of evidence proved that glycosyldisulfides could interact efficiently with various lectins, and be useful glycomimetics to explore carbohydrate-binding targets. Clearly new glycosyl disulfides will continue to be developed and investigated as glycomimetics, and new potential biological applications will emerge, such as in the field of cancer drug discovery. In turn, the search for new biological applications will continue to prompt the development of innovative synthetic methods to introduce this most interesting of chemical linkages at the interglycosidic bond.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the Institute of Cancer Therapeutics (University of Bradford) Doctoral Training Centre for financial support.Notes and references
- M. Narayan, E. Welker, W. J. Wedemeyer and H. A. Scheraga, Acc. Chem. Res., 2000, 33, 805–812 CrossRef CAS
.
- L. R. Staben, S. G. Koenig, S. M. Lehar, R. Vandlen, D. Zhang, J. Chuh, S.-F. Yu, C. Ng, J. Guo, Y. Liu, A. Fourie-O'Donohue, M. Go, X. Linghu, N. L. Segraves, T. Wang, J. Chen, B. Wei, G. D. L. Phillips, K. Xu, K. R. Kozak, S. Mariathasan, J. A. Flygare and T. H. Pillow, Nat. Chem., 2016, 8, 1112–1119 CrossRef CAS
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS
- V. M. Vrudhula, J. F. MacMaster, Z. Li, D. E. Kerr and P. D. Senter, Bioorg. Med. Chem. Lett., 2002, 12, 3591–3594 CrossRef CAS
- C.-S. Jiang, W. E. G. Müller, H. C. Schröder and Y.-W. Guo, Chem. Rev., 2012, 112, 2179–2207 CrossRef CAS
- J. Chick, K. Gough, W. Falkowski, P. Kershaw, B. Hore, B. Mehta, B. Ritson, R. Ropner and D. Torley, Br. J. Psychiatry, 1992, 161, 84–89 CrossRef CAS
- E. Block, S. Ahmad, M. K. Jain, R. W. Crecely, R. Apitz-Castro and M. R. Cruz, J. Am. Chem. Soc., 1984, 106, 8295–8296 CrossRef CAS
- S.-J. Kim, S. Jin and G. Ishii, Biosci., Biotechnol., Biochem., 2004, 68, 2444–2450 CrossRef CAS
- S.-S. Wang, X. Gao, V. d. Solar, X. Yu, A. Antonopoulos, A. E. Friedman, E. K. Matich, G. E. Atilla-Gokcumen, M. Nasirikenari, J. T. Lau, A. Dell, S. M. Haslam, R. A. Laine, K. L. Matta and S. Neelamegham, Cell Chem. Biol., 2018, 25, 1519–1532 CrossRef CAS
- B. Mandal and B. Basu, RSC Adv., 2014, 4, 13854–13881 RSC
- M. Musiejuk and D. Witt, Org. Prep. Proced. Int., 2015, 47, 95–131 CrossRef CAS
- M. J. Kiefel, R. J. Thomson, M. Radovanovic and M. v. Itzstein, J. Carbohydr. Chem., 1999, 18, 937–959 CrossRef CAS
- R. Smith, X. Zeng, H. Müller-Bunz and X. Zhu, Tetrahedron Lett., 2013, 54, 5348–5350 CrossRef CAS
- S. Martín-Santamaría, S. André, E. Buzamet, R. Caraballo, G. Fernández-Cureses, M. Morando, J. P. Ribeiro, K. Ramírez-Gualito, B. de Pascual-Teresa, F. J. Cañada, M. Menéndez, O. Ramström, J. Jiménez-Barbero, D. Solís and H.-J. Gabius, Org. Biomol. Chem., 2011, 9, 5445–5455 RSC
- S. Knapp and D. S. Myers, J. Org. Chem., 2002, 67, 2995–2999 CrossRef CAS
- R. Zelli, S. Tommasone, P. Dumy, A. Marra and A. Dondoni, Eur. J. Org. Chem., 2016, 2016, 5102–5116 CrossRef CAS
- Z. Pei, T. Aastrup, H. Anderson and O. Ramström, Bioorg. Med. Chem. Lett., 2005, 15, 2707–2710 CrossRef CAS
- G. Ribeiro Morais and R. A. Falconer, Tetrahedron Lett., 2007, 48, 7637–7641 CrossRef CAS
- S. Knapp and E. Darout, Tetrahedron Lett., 2002, 43, 6075–6078 CrossRef CAS
- S. Knapp, E. Darout and B. Amorelli, J. Org. Chem., 2006, 71, 1380–1389 CrossRef CAS
- N. K. Richtmyer, C. J. Carr and C. S. Hudson, J. Am. Chem. Soc., 1943, 65, 1477–1478 CrossRef CAS
- M. Hürzeler, B. Bernet and A. Vasella, Helv. Chim. Acta, 1992, 75, 557–588 CrossRef
- M. Adinolfi, M. d'Ischia, A. Iadonisi, L. Leone, A. Pezzella and S. Valerio, Eur. J. Org. Chem., 2012, 2012, 4333–4338 CrossRef CAS
- D. Bhar and S. Chandrasekaran, Carbohydr. Res., 1997, 301, 221–224 CrossRef CAS
- M. Adinolfi, D. Capasso, S. Di Gaetano, A. Iadonisi, L. Leone and A. Pastore, Org. Biomol. Chem., 2011, 9, 6278–6283 RSC
- J.-T. Ge, L. Zhou, F.-L. Zhao and H. Dong, J. Org. Chem., 2017, 82, 12613–12623 CrossRef CAS
- T. Belz and S. J. Williams, Carbohydr. Res., 2016, 429, 38–47 CrossRef CAS
- B. D. Johnston and B. M. Pinto, J. Org. Chem., 2000, 65, 4607–4617 CrossRef CAS
- R. Caraballo, M. Sakulsombat and O. Ramström, Chem. Commun., 2010, 46, 8469–8471 RSC
- J. McNulty, V. Krishnamoorthy, D. Amoroso and M. Moser, Bioorg. Med. Chem. Lett., 2015, 25, 4114–4117 CrossRef CAS
- R. J. Ferrier, R. H. Furneaux and P. C. Tyler, Carbohydr. Res., 1977, 58, 397–404 CrossRef CAS
- G. J. L. Bernardes, E. J. Grayson, S. Thompson, J. M. Chalker, J. C. Errey, F. El Oualid, T. D. W. Claridge and B. G. Davis, Angew. Chem., Int. Ed., 2008, 47, 2244–2247 CrossRef CAS
- R. Caraballo, M. Rahm, P. Vongvilai, T. Brinck and O. Ramström, Chem. Commun., 2008, 6603–6605 RSC
- D. Horton and M. J. Miller, Carbohydr. Res., 1965, 1, 335–337 CrossRef CAS
- R. H. Bell and D. Horton, Carbohydr. Res., 1969, 9, 187–199 CrossRef CAS
- R. A. Falconer, I. Jablonkai and I. Toth, Tetrahedron Lett., 1999, 40, 8663–8666 CrossRef CAS
- J. P. Malkinson and R. A. Falconer, Tetrahedron Lett., 2002, 43, 9549–9552 CrossRef CAS
- K. Banisalman, F. M. Barnieh, P. Loadman, G. Ribeiro Morais and R. A. Falconer, Unpublished results.
- N. Stellenboom, R. Hunter, M. R. Caira and L. Szilágyi, Tetrahedron Lett., 2010, 51, 5309–5312 CrossRef CAS
- C. Venkateswarlu, V. Gautam and S. Chandrasekaran, Carbohydr. Res., 2015, 402, 200–207 CrossRef CAS
- T.-Z. Illyés, T. Szabó and L. Szilágyi, Carbohydr. Res., 2011, 346, 1622–1627 CrossRef
- L. Szilágyi, T.-Z. Illyés and P. Herczegh, Tetrahedron Lett., 2001, 42, 3901–3903 CrossRef
- B. N. Murthy, S. Sinha, A. Surolia, N. Jayaraman, L. Szilágyi, I. Szabó and K. E. Kövér, Carbohydr. Res., 2009, 344, 1758–1763 CrossRef CAS
- B. G. Davis, M. A. T. Maughan, M. P. Green, A. Ullman and J. B. Jones, Tetrahedron: Asymmetry, 2000, 11, 245–262 CrossRef CAS
- E. D. Goddard-Borger and R. V. Stick, Aust. J. Chem., 2005, 58, 188–198 CrossRef CAS
- M. C. Aversa, A. Barattucci and P. Bonaccorsi, Eur. J. Org. Chem., 2009, 36, 6355–6359 CrossRef
- M. D. Percival, M. Ouellet, C. Campagnolo, D. Claveau and C. Li, Biochemistry, 1999, 38, 13574–13583 CrossRef CAS
- M. C. Aversa, A. Barattucci, P. Bonaccorsi and P. Giannetto, Curr. Org. Chem., 2007, 11, 1034–1052 CrossRef CAS
- M. C. Aversa, A. Barattucci, G. Battaglia and P. Bonaccorsi, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 1220–1224 CrossRef CAS
- P. Bonaccorsi, M. C. Aversa, A. Barattucci, T. Papalia, A. Torre, F. Trischitta and C. Faggio, J. Sulfur Chem., 2013, 34, 684–691 CrossRef CAS
- K. Wang, Y. Xiang, Z. Shi, H. Wang, N. Li and B. Tang, Org. Biomol. Chem., 2019, 17, 6351–6354 RSC
- K. Tanaka, X. Chen and F. Yoneda, Tetrahedron, 1988, 44, 3241–3249 CrossRef CAS
- J. Cuomo, J. H. Merrifield and J. F. W. Keana, J. Org. Chem., 1980, 45, 4216–4219 CrossRef CAS
- M. Hürzeler, B. Bernet and A. Vasella, Helv. Chim. Acta, 1993, 76, 995–1012 CrossRef
- G. Hummel and O. Hindsgaul, Angew. Chem., Int. Ed., 1999, 38, 1782–1784 CrossRef CAS
- G. Ribeiro Morais, I. P. F. Oliveira, A. J. Humphrey and R. A. Falconer, Tetrahedron Lett., 2009, 50, 1642–1644 CrossRef
- G. Ribeiro Morais, B. R. Springett, M. Pauze, L. Schröder, M. Northrop and R. A. Falconer, Org. Biomol. Chem., 2016, 14, 2749–2754 RSC
- X. Xiao, M. Feng and X. Jiang, Angew. Chem., Int. Ed., 2016, 55, 14121–14125 CrossRef CAS
- X. Xiao, J. Xue and X. Jiang, Nat. Commun., 2018, 9, 2191–2199 CrossRef
- S. Otto, L. E. F. Ricardo and K. M. S. Jeremy, Science, 2002, 297, 590–593 CrossRef CAS
- S. André, Z. Pei, H.-C. Siebert, O. Ramström and H.-J. Gabius, Bioorg. Med. Chem., 2006, 14, 6314–6326 CrossRef
- Z. Pei, R. Larsson, T. Aastrup, H. Anderson, J.-M. Lehn and O. Ramström, Biosens. Bioelectron., 2006, 22, 42–48 CrossRef CAS
- S. Sando, A. Narita and Y. Aoyama, Bioorg. Med. Chem. Lett., 2004, 14, 2835–2838 CrossRef CAS
- T. Hotchkiss, H. B. Kramer, K. J. Doores, D. P. Gamblin, N. J. Oldham and B. G. Davis, Chem. Commun., 2005, 4264–4266 RSC
- R. A. Dwek, Chem. Rev., 1996, 96, 683–720 CrossRef CAS
- M. Ono and S. Hakomori, Glycoconjugate J., 2003, 20, 71–78 CrossRef
- K. Ohtsubo and J. D. Marth, Cell, 2006, 126, 855–867 CrossRef CAS
- A. Imberty and S. Pérez, Protein Eng., 1995, 8, 699–709 CrossRef CAS
- W. M. Macindoe, A. H. van Oijen and G.-J. Boons, Chem. Commun., 1998, 7, 847–848 RSC
- G. M. Watt and G.-J. Boons, Carbohydr. Res., 2004, 339, 181–193 CrossRef CAS
- P. Berglund, G. DeSantis, M. R. Stabile, X. Shang, M. Gold, R. R. Bott, T. P. Graycar, T. H. Lau, C. Mitchinson and J. B. Jones, J. Am. Chem. Soc., 1997, 119, 5265–5266 CrossRef CAS
- E. J. Grayson, S. J. Ward, A. L. Hall, P. M. Rendle, D. P. Gamblin, A. S. Batsanov and B. G. Davis, J. Org. Chem., 2005, 70, 9740–9754 CrossRef CAS
- D. P. Gamblin, P. Garnier, S. J. Ward, N. J. Oldham, A. J. Fairbanks and B. G. Davis, Org. Biomol. Chem., 2003, 1, 3642–3644 RSC
- G. J. L. Bernardes, J. P. Marston, A. S. Batsanov, J. A. K. Howard and B. G. Davis, Chem. Commun., 2007, 30, 3145–3147 RSC
- M. Salvadó, B. Amgarten, S. Castillón, G. J. L. Bernardes and O. Boutureira, Org. Lett., 2015, 17, 2836–2839 CrossRef
- B. G. Davis, Chem. Commun., 2001, 4, 351–352 RSC
- B. G. Davis, R. C. Lloyd and J. B. Jones, Bioorg. Med. Chem., 2000, 8, 1527–1535 CrossRef CAS
- T. Sandalova, L. Zhong, Y. Lindqvist, A. Holmgren and G. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9533–9538 CrossRef CAS
- D. P. Gamblin, P. Garnier, S. van Kasteren, N. J. Oldham, A. J. Fairbanks and B. G. Davis, Angew. Chem., Int. Ed., 2004, 43, 828–833 CrossRef CAS
- P. R. Sridhar, K. R. Prabhu and S. Chandrasekaran, Eur. J. Org. Chem., 2004, 23, 4809–4815 CrossRef
- J. J. Bailey and D. R. Bundle, Org. Biomol. Chem., 2014, 12, 2193–2213 RSC
- C.-F. Liang, M.-C. Yan, T.-C. Chang and C.-C. Lin, J. Am. Chem. Soc., 2009, 131, 3138–3139 CrossRef CAS
- C.-F. Liang, T.-C. Kuan, T.-C. Chang and C.-C. Lin, J. Am. Chem. Soc., 2012, 134, 16074–16079 CrossRef CAS
- S. Pearson, W. Scarano and M. H. Stenzel, Chem. Commun., 2012, 48, 4695–4697 RSC
- C. Roder and M. J. Thomson, Drugs R&D, 2015, 15, 13–20 CrossRef CAS
- M. Ishii, S. Matsumura and K. Toshima, Angew. Chem., Int. Ed., 2007, 46, 8396–8399 CrossRef CAS
- I. Hamachi, T. Nagase and S. Shinkai, J. Am. Chem. Soc., 2000, 122, 12065–12066 CrossRef CAS
- R. Das and B. Mukhopadhyay, ChemistryOpen, 2016, 5, 401–433 CrossRef CAS
- A. Xolin, R. Losa, A. Kaid, C. Tresse, J.-M. Beau, F.-D. Boyer and S. Norsikian, Org. Biomol. Chem., 2018, 16, 325–335 RSC
- B. G. Davis, S. J. Ward and P. M. Rendle, Chem. Commun., 2001, 189–190 RSC
- H. Böhme and H. Steudel, Justus Liebigs Ann. Chem., 1969, 730, 121–132 CrossRef
- E. J. Grayson, G. J. L. Bernardes, J. M. Chalker, O. Boutureira, J. R. Koeppe and B. G. Davis, Angew. Chem., Int. Ed., 2011, 50, 4127–4132 CrossRef CAS
- B. L. Wilkinson and A. J. Fairbanks, Tetrahedron Lett., 2008, 49, 4941–4943 CrossRef CAS
- S. André, H. Kaltner, J. C. Manning, P. V. Murphy and H.-J. Gabius, Molecules, 2015, 20, 1788–1823 CrossRef
- D. Gupta, S. Oscarson, T. S. Raju, P. Stanley, E. J. Toone and C. F. Brewer, Eur. J. Biochem., 1996, 242, 320–326 CrossRef CAS
- K. D. Hardman and C. F. Ainsworth, Biochemistry, 1976, 15, 1120–1128 CrossRef CAS
- S. Walpole, S. Monaco, R. Nepravishta and J. Angulo, Methods Enzymol., 2019, 615, 423–445 CAS
- S. André, K. E. Kövér, H.-J. Gabius and L. Szilágyi, Bioorg. Med. Chem. Lett., 2015, 25, 931–935 CrossRef
- H. Kaltner, T. Szabó, K. Fehér, S. André, S. Balla, J. C. Manning, L. Szilágyi and H.-J. Gabius, Bioorg. Med. Chem., 2017, 25, 3158–3170 CrossRef CAS
- P. Bonaccorsi, F. Marino-Merlo, A. Barattucci, G. Battaglia, E. Papaianni, T. Papalia, M. C. Aversa and A. Mastino, Bioorg. Med. Chem., 2012, 20, 3186–3195 CrossRef CAS
- A. D. Rassi, A. P. Rassi and J. A. P. Marin-Neto, Lancet, 2010, 375, 1388–1402 CrossRef
- B. Gutiérrez, C. Muñoz, L. Osorio, K. Fehér, T.-Z. Illyés, Z. Papp, A. A. Kumar, K. E. Kövér, H. Sagua, J. E. Araya, P. Morales, L. Szilágyi and J. González, Bioorg. Med. Chem. Lett., 2013, 23, 3576–3579 CrossRef
- S.-J. Zhu, H.-Z. Ying, Y. Wu, N. Qiu, T. Liu, B. Yang, X.-W. Dong and Y.-Z. Hu, RSC Adv., 2015, 5, 103172–103183 RSC
- P. Meresse, E. Dechaux, C. Monneret and E. Bertounesque, Curr. Med. Chem., 2004, 11, 2443–2466 CrossRef CAS
- J. G. Turner, J. L. Dawson, S. Grant, K. H. Shain, W. S. Dalton, Y. Dai, M. Meads, R. Baz, M. Kauffman, S. Shacham and D. M. Sullivan, J. Hematol. Oncol., 2016, 9, 73–11 CrossRef
- R. Santos, O. Ursu, A. Gaulton, A. P. Bento, R. S. Donadi, C. G. Bologa, A. Karlsson, B. Al-Lazikani, A. Hersey, T. I. Oprea and J. P. Overington, Nat. Rev. Drug Discovery, 2017, 16, 19–34 CrossRef CAS
- J.-L. Rigaud, G. Mosser, J.-J. Lacapere, A. Olofsson, D. Levy and J.-L. Ranck, J. Struct. Biol., 1997, 118, 226–235 CrossRef CAS
- D. Xue, J. Wang, X. Song, W. Wang, T. Hu, L. Ye, Y. Liu, Q. Zhou, F. Zhou, Z.-X. Jiang, Z.-J. Liu and H. Tao, Langmuir, 2019, 35, 4319–4327 CrossRef CAS
Footnote |
| † Mono- and disaccharides are represented as followed: D-Glucose (Glc), D-Galactose (Gal), D-Mannose (Man), L-Fucose (Fuc), N-Acetyl D-galactosamine (GalNAc), N-Acetyl D-glucosamine (GlcNAc) and Lactose (Lac). |
| This journal is © The Royal Society of Chemistry 2021 |