
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoosting hydrogen generation by anodic oxidation of iodide over Ni–Co(OH)2 nanosheet arrays†
Enlai
Hu
,
Yue
Yao
,
Yi
Chen
,
Yuanjing
Cui
 ,
Zhiyu
Wang
and
Guodong
Qian
*
,
Zhiyu
Wang
and
Guodong
Qian
*
State Key Laboratory of Silicon Materials, Cyrus Tang Center for Sensor Materials and Applications, School of Materials Science and Engineering, Zhejiang University, Hangzhou, 310027, China. E-mail: gdqian@zju.edu.cn
First published on 8th December 2020
Abstract
For overall water electrolysis, the hydrogen evolution reaction (HER) is severely limited by the sluggish kinetics of the anodic oxygen evolution reaction (OER). Therefore, replacing the OER with a more favorable anodic oxidation reaction with remarkable kinetics is of paramount significance, especially the one that can produce value-added chemicals. Moreover, time-saving and cost-effective strategies for the fabrication of electrodes are helpful for the wide application of electrolysis. Herein, thermodynamically more favorable iodide electrooxidation over Ni doped Co(OH)2 nanosheet arrays (NSAs) in alkaline solution is presented as the alternative to the OER to boost the HER. And the active species are determined to be the reverse redox of the Co(IV)/Co(III) couple. Remarkably, a negative shift of voltage of 320 mV is observed at a current density of 10 mA cm−2 after using iodide electrolysis to replace ordinary water splitting. The synthetic strategy and iodide oxidation in this work expand the application of Co-based materials in the field of energy-saving hydrogen production.
Introduction
Electrocatalytic water splitting has been widely explored for the purpose of sustainable hydrogen generation.1 However, the sluggish kinetics of the anodic oxygen evolution reaction (OER) always requires a high overpotential at high current density that severely inhibits the overall reaction rate.2 Although a variety of outstanding electrocatalysts with attractive OER performance have been fabricated, the overpotential for the OER is still too high to match with the rate of the hydrogen evolution reaction (HER), thus increasing energy consumption.3 In this regard, replacing the OER with more favorable oxidation reactions at the anode has triggered a lot of interest, such as tetrahydroisoquinoline oxidation, the urea oxidation reaction (UOR), amine oxidation, and N2 oxidation.4–15 Among them the UOR with a low equilibrium potential of 0.37 V has been regarded as a promising alternative to achieve energy-saving hydrogen generation and great process has been made.16–18 Unfortunately, the UOR also has sluggish kinetics due to its multi-electron process and requires highly active electrocatalysts to accelerate its reaction rate.19 Recently, nickel-based electrocatalysts have been demonstrated to have superior UOR activities with the oxidation of the Ni(III)/Ni(II) couple as active species.4 In this regard, the limitation of UOR performance has a strong relationship with the oxidation potential of Ni(II) species. Therefore, it is of great significance to explore other kinds of readily oxidized chemicals with a fast response from the redox of other metal species, such as Co(IV)/Co(III) species that are not as efficient as Ni(III)/Ni(II) species for the UOR.20,21 Moreover, the oxidation products (CO2 and N2) of the UOR are of low value and the generated gas may also peel off the active materials from the substrate, lowering the electrocatalytic activity. Thus, it is significant but still challenging to achieve the generation of both hydrogen and value-added oxidation products with low energy consumption. Iodide with a lower oxidation potential than water should be easily oxidized compared to water during the electrocatalysis process.22 The oxidation product of iodide in alkaline solution is always oxyiodide that has been widely regarded as an efficient additive to salt for the compensation of iodine deficiency.23–25 Consequently, it is possible and meaningful to achieve energy-saving hydrogen generation through iodide electrolysis in an alkaline medium.For the wide application of electrolysis for hydrogen production, another vital factor is to cut down the cost on the electrode fabrication process, which has long been ignored. Current strategies for the fabrication of electrodes can be classified into the following two categories: (1) direct coating of active materials onto current collectors,17,26–29 including glass carbon electrodes, metal foam, and carbon-based substrates; (2) in situ growth of active materials onto current collectors by different kinds of methods,30–34 such as hydrothermal/solvothermal methods, and electrodeposition. However, these two kinds of strategies also have some demerits. For example, polymer binders are inevitably involved in the coating strategy, which will inhibit the exposure of active sites. In addition, the massive gas evolution will also peel off the active materials from the current collector.35 For the second strategy, a sophisticated synthesis procedure, complex precursor solution, or/and heat treatment are always required, which will increase the time and cost of electrode fabrication, although the attachment between the active materials and substrates can be greatly enhanced by using this kind of strategy.36–39 Thus, developing time-saving and cost-effective methods for the simple fabrication of electrodes towards electrolysis is of crucial significance, yet remains a big challenge.
Herein, based on the redox reaction between Co(NO3)2 solution and a Ni foam substrate, the electrode of Ni doped Co(OH)2 nanosheet arrays on Ni foam (Ni–Co(OH)2 NSAs) can be simply fabricated at room temperature. Thanks to the easy oxidation feature of iodide, the anodic oxidation process can be efficiently triggered over the Ni–Co(OH)2 NSA electrode. The required potential is merely 1.30 V (versus the reversible hydrogen electrode, vs. RHE) to drive a current density of 20 mA cm−2, which is 30 and 240 mV lower than that of the UOR and OER, respectively. The similar performance of the Ni-free Co(OH)2 NSAs reveals that the reverse redox of the Co(IV)/Co(III) couple is the active species for the IOR. Furthermore, the overall iodide electrolyzer assembled from the as-prepared Ni–Co(OH)2 NSAs and a Ni–Mo electrode displays a low cell voltage of 1.34 V at 10 mA cm−2 and remarkable durability for over 12 h, further confirming the promising application of the IOR over Co-based materials for energy-saving hydrogen production.
Experimental
Synthetic procedures
Characterization
XRD patterns were obtained through a Shimadzu XRD7000 powder X-ray diffractometer at a scan rate of 0.15° min−1 at room temperature. The morphologies were elucidated by FESEM carried out on a Hitachi S-4800 field emission scanning electron microscope. TEM tests were conducted on a HT-7700 X-MAXn65T with an acceleration voltage of 120 kV. A Thermo Scientific K-Alpha+ X-ray photoelectron spectrometer was utilized to collect XPS spectra. Inductively coupled plasma (ICP) analyses were conducted on a PE Nexion 2000.Electrochemical measurements
Electrochemical measurements were conducted in a three-electrode cell through a 660E CH Instrument electrochemical workstation at room temperature with a carbon rod as the counter electrode and a saturated calomel electrode (SCE) as the reference electrode. The as-prepared electrode was directly utilized as the working electrode with a geometrical area of 1 cm−2. The electrolyte for the OER, IOR and UOR was 1 M KOH, 1 M KOH with 0.33 M KI and 1 M KOH with 0.33 M urea, respectively. All potentials were calibrated according to E(RHE) = E(SCE) + 0.059 pH + 0.242. The impedance data were collected through electrochemical impedance spectroscopy (EIS) measurements in the range of 0.01 to 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz with 5 mV sinusoidal perturbations. Polarization curves were obtained at a scan rate of 5 mV s−1. iR compensation was applied for all potentials. Tafel slopes were determined according to the following Tafel equation:
000 Hz with 5 mV sinusoidal perturbations. Polarization curves were obtained at a scan rate of 5 mV s−1. iR compensation was applied for all potentials. Tafel slopes were determined according to the following Tafel equation:| E = blogj + a |
The electrochemical active surface area (ECSA) of the electrocatalysts was estimated according to the following equation:
| ECSA = Cdl/Cs |
The Faraday efficiency of the IOR process was determined to be about 117.7%. The method to evaluate the Faraday efficiency of the IOR is described as follows and detailed in the Experimental section. The reason for the larger Faraday efficiency can be ascribed to the loss of AgI during the collection procedure.
Firstly, the IOR was conducted at a constant current of 50 mA for 2 h. Then, 35 mL of the electrolyte (total volume of the electrolyte at the anode is 35 mL) was obtained and 35 mL of Tollens was added (obtained by dissolving 14.0 g of AgNO3 in 35 mL of ammonia) into the above electrolyte. Finally, the precipitate (AgI) was collected, washed with water and dried at 60 °C for 2 h. AgI is determined to be about 2.54 g. Under these conditions the I− anion can be precipitated after adding Tollens, while the IO3− anion cannot be precipitated. As a result, the Faraday efficiency can be calculated.
Results and discussion
The Ni–Co(OH)2 NSAs were fabricated through a simple one-pot substrate oxidation strategy at room temperature, as schematically illustrated in Fig. 1. In the synthetic process, the hydrolysis of Co(NO3)2 will release H+ ions to build an acidic condition, which can promote the oxidation ability of NO3− ions. Then Ni foam can be oxidized by NO3− ions with the help of H+ ions to deliver Ni2+ species. Finally, with the consumption of H+ ions in the solution, pH near the Ni foam surface will increase thus generating Ni–Co(OH)2 NSAs. The crystallographic structure of the as-obtained electrocatalyst was first elucidated by the powder X-ray diffraction (XRD) test. As shown in Fig. S1,† except the three strong peaks of Ni foam, all of the rest of the peaks can be well indexed to Co(OH)2 (JCPDS card No. 51-1731). The morphology of the as-obtained electrocatalyst was examined by field emission scanning electron microscopy (FESEM). Panoramic FESEM images in Fig. 2A and B illustrate that the Ni foam surface is covered by a layer of interconnected nanosheets with vertical alignment. The distance between the adjacent nanosheets is several hundred nanometers, which is favorable for the penetration of electrolyte and transfer of reactants. In sharp contrast, only a flat surface is observed for a pristine Ni foam substrate (Fig. S2†). The nanosheet morphology of the Ni–Co(OH)2 NSAs is further confirmed by transition electron microscopy (TEM) images (Fig. 2C and D), in which a smooth surface with lateral size of hundreds of nanometers can be observed. Further, the spatial dispersion distribution of the Ni, Co and O elements in the nanosheet was elucidated by elemental mapping examination (Fig. 2E–G). As can be seen, the three elements homogeneously disperse in the whole nanosheet, indicating the uniform incorporation of Ni species. Raman scattering spectroscopy was conducted to further analyze the chemical structure of Ni–Co(OH)2 NSAs. Two main peaks can be observed at about 470 and 531 cm−1 in the Raman spectrum (Fig. S3†), corresponding to the characteristic peaks for Ni–O and Co–O vibrational modes.43 All of the above results imply the successful fabrication of Ni–Co(OH)2 NSAs with a homogeneous Ni distribution.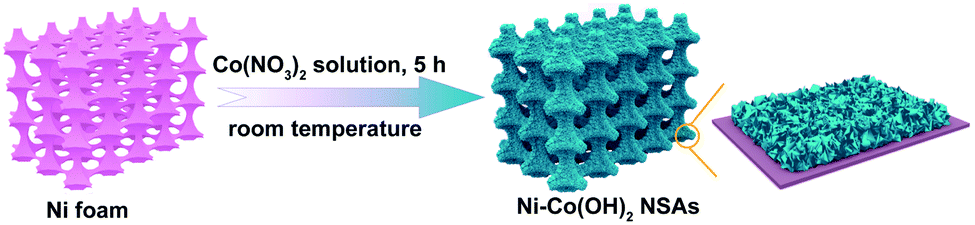 | ||
| Fig. 1 Schematic illustration of the fabrication of Ni–Co(OH)2 NSAs. | ||
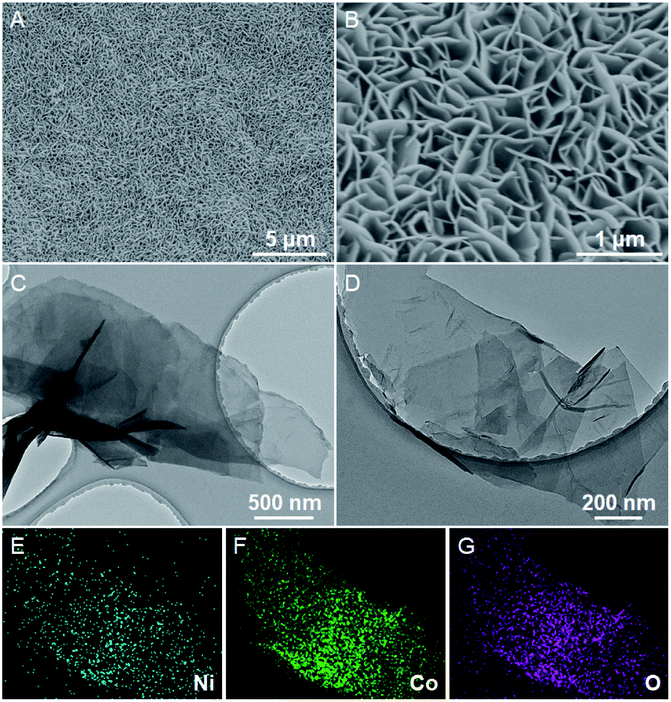 | ||
| Fig. 2 SEM (A and B) and TEM (C and D) images of Ni–Co(OH)2 NSAs. Mapping images of different elements for the nanosheets of Ni (E), Co (F) and O (G). | ||
X-ray photoelectron spectroscopy (XPS) analyses were performed to illustrate the surface chemical composition and oxidation state of Ni–Co(OH)2 NSAs. As shown in Fig. S4A,† the XPS spectrum of Ni–Co(OH)2 NSAs demonstrates the coexistence of Ni, Co, and O elements in the electrocatalyst. The spin–obit doublet in the high-resolution spectrum of Co 2p (Fig. S4B†) centered at 781.14 and 796.78 eV can be indexed to the Co(II) species with two satellite peaks at 785.67 and 802.85 eV. And the differential binding energy of 15.64 eV between these two peaks further prove the oxidation state of +2 for Co species in the electrocatalyst, consistent with previous literature.44 Similarly, there are also a spin–obit doublet and two satellite peaks in the high-resolution spectrum of Ni 2p (Fig. S4C†). The peaks at 855.87 and 873.46 eV correspond to the characteristic peaks of Ni(II) species in the electrocatalyst.45 And the peaks assigned to satellite peaks are observed at 861.54 and 879.26 eV. The high-resolution spectrum of O 1s uncovers the existence of three types of oxygen species, including metal–oxygen bonding at 530.70 eV, OH− species at 531.39 eV, and absorbed oxygen at 532.38 eV.46 The above results further confirm the successful incorporation of Ni species into Co(OH)2.
The electrocatalytic oxidation reaction of iodide (IOR) was performed in 1 M KOH solution and 0.33 M KI solution. Firstly, the optimal reaction time was determined through evaluating the IOR activity of the electrocatalysts synthesized with different times. The polarization curves clearly present that the highest activity can be achieved with a reaction time of 5 h (Fig. S5†). As can be seen in Fig. 3A, the potential to drive a current density of 20 mA cm−2 is determined to be 1.30 V (vs. RHE). To further demonstrate the superiority of the IOR on lowing the potential at the same current density, UOR (0.33 M urea) and OER measurements were also carried out. The required potentials at 20 mA cm−2 are as high as 1.33 and 1.54 V (vs. RHE) for the UOR and OER, respectively. Similar to the UOR process, the key to trigger the IOR process is the generation of high valence state metal species. As presented in the polarization curve of the OER (Fig. 3A), three obvious oxidation peaks can be found at about 1.09, 1.31 and 1.39 V (vs. RHE) that refer to the oxidation process of Co(II)/Co(III), Co(III)/Co(IV), and Ni(II)/Ni(III),47,48 respectively. After adding KI into electrolyte, only the oxidation peak of Co(II)/Co(III) can be observed in the polarization curve, implying that the oxidation of KI occurred at the potential where high-valence metal species (Co(IV) and Ni(III)) were generated. The faster increase of the anodic current density of the IOR than that of the UOR indicates a more favorable response of the IOR process than the UOR process driven from the redox of the Co(III)/Co(IV) couple.
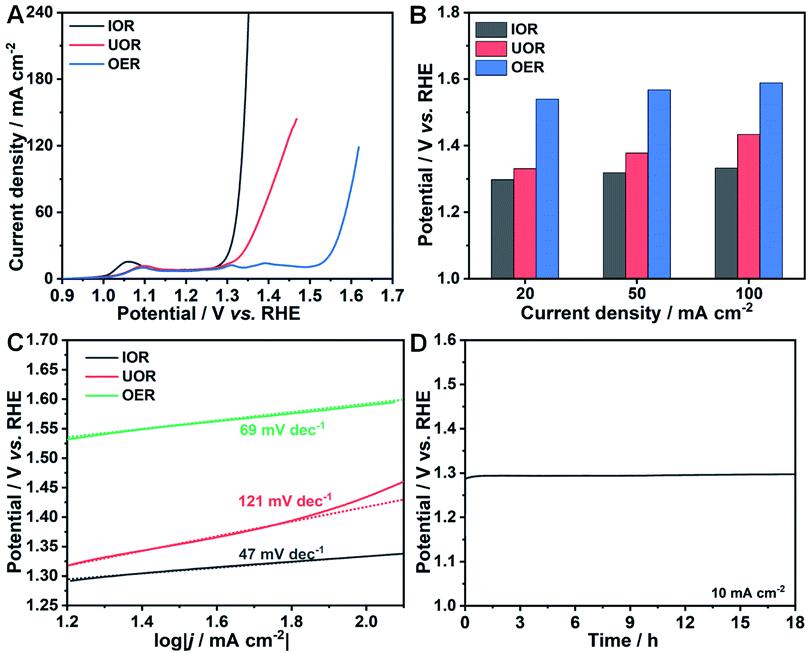 | ||
| Fig. 3 (A) Polarization curves of the IOR, UOR and OER over Ni–Co(OH)2 NSAs. (B) Comparison of potentials required to drive different current densities of the IOR, UOR and OER over Ni–Co(OH)2 NSAs. (C) Tafel plots of electrooxidation reactions from (A). (D) Chronopotentiometry curve of the IOR over Ni–Co(OH)2 NSAs. | ||
For the IOR, the potentials to reach current densities of 50 and 100 mA cm−2 are determined to be 1.32 and 1.33 V (vs. RHE), whereas they are much higher for UOR and OER processes (Fig. 3B). The highest activity for the IOR among the three electrocatalytic oxidation reactions well illustrate its advantage as a promising alternative to the OER to lower the energy consumption on hydrogen generation. The Tafel slopes of these three electrocatalytic oxidation reactions were calculated to evaluate their reaction kinetics. The Tafel slope for the IOR is only 47 mV dec−1, while it is 121 and 69 mV dec−1 for the UOR and OER, respectively (Fig. 3C). The smallest Tafel slope of the IOR points to its more favorable catalytic kinetics than the UOR and OER. Electrocatalytic durability, one of the most crucial evaluation criteria, is important to be measured. Accordingly, chronopotentiometry measurements at a constant current density of 10 mA cm−2 were carried out. Remarkably, the IOR process is quite durable with merely negligible changes in the potential over 18 h (Fig. 3D). And it also can smoothly work at 100 mA cm−2 for 5 h (Fig. S6A†). Moreover, the recorded multi-current curve (Fig. S6B†) displays the rapid response to the current change of Ni–Co(OH)2 NSAs towards the IOR. Once the current is changed, the required potential can immediately level off and be well preserved for 1000 s, suggesting the superior mass transfer properties and robust structure of Ni–Co(OH)2 NSAs towards the IOR. The ultrahigh activity and stability of the IOR over Ni–Co(OH)2 NSAs suggest that the IOR is a promising oxidation reaction to replace the OER for energy-saving hydrogen generation. Moreover, the application of Co-based electrocatalysts in the field of energy-saving hydrogen generation can be expanded at the same time through the IOR. The morphology of the Ni–Co(OH)2 NSAs after IOR durability measurements was measured. As shown in Fig. S7A–C,† the nanosheet arrays and microstructures are well preserved after the IOR durability test, suggesting the robust structure of Ni–Co(OH)2 NSAs. ICP analyses reveal that the atomic ratios of Co and Ni elements before and after the long-term reaction are evaluated to be 6.80 and 7.17, respectively. These results imply that some Co will be dissolved into the electrolyte during the IOR process, though the structure of Ni–Co(OH)2 NSAs can be well retained. In addition, XRD and XPS measurements were further conducted to elucidate the changes on Ni–Co(OH)2 NSAs after performing the IOR. As presented in the XRD pattern (Fig. S7D†), the peaks indexed to Co(OH)2 disappear, while some new weak peaks corresponding to CoOOH can be observed. XPS results also reveal the changes of the chemical oxidation state of elements. Although Co, Ni and O elements still exist in the electrocatalysts (Fig. S4A†), the oxidation states for these elements are obviously different, especially the Co and O. Two new peaks centered at 779.57 and 794.61 eV with an energy difference of 15.04 eV in the Co 2p spectrum after the IOR can be indexed to Co(III) species (Fig. S4C†).49 For the O 1 s spectrum, the peaks at 529.11 and 530.90 eV can be attributed to Co–O and Co–OH bonds, respectively (Fig. S4D†).50 And the typical peak for absorption of oxygen is also observed at a binding energy of 532.38 eV. The binding energies of Ni species only exhibit a slightly negative shift of about 0.23 and 0.25 eV for Ni 2p3/2 and Ni 2p1/2, respectively, with the chemical oxidation state unchanged. The phase transformation and chemical oxidation state change after the IOR process implies that the redox of Co(III)/Co(IV) and Ni(II)/Ni(III) couples is the key to the IOR, coincident with the electrochemical measurements. In the IOR process, Co(II) species are firstly oxidized to Co(III) species and Co(IV) at a high potential. Similarly, the Ni(II) species are also oxidized to Ni(III) at a high potential. Then, the I− ions can immediately transfer to IO3− ions once they attach the newly formed Co(IV) and Ni(III) species that will be reduced to Co(III) and Ni(II) species at the same time. As a result, the electrocatalyst after IOR tests is determined to be CoOOH with doped Ni(II).
To have a clear understanding of the crucial importance of Co species in lowering the IOR potential, Ni(OH)2 on Ni foam was also fabricated. The XRD pattern and FESEM images (Fig. S1 and S8†) well confirm the successful formation of Ni(OH)2 with the morphology of nanosheets on Ni foam. The activities towards the IOR of the as-obtained NSAs was recorded by the polarization curve. As shown in Fig. 4A, the IOR performance of Ni–Co(OH)2 NSAs far more outperforms that of Ni(OH)2 NSAs. The potential to reach a current density of 20 mA cm−2 is 1.34 V (vs. RHE) for Ni(OH)2 NSAs, which is almost 40 mV higher than that of Ni–Co(OH)2 NSAs. The IOR activity of the pristine Ni foam substrate was also evaluated and the required potential is 1.43 V (vs. RHE) at 20 mA cm−2. The Tafel plots of the recorded polarization curves are presented in Fig. 4B. Remarkably, the Tafel slopes of Ni–Co(OH)2 NSAs and Ni(OH)2 NSAs are 47 and 49 mV dec−1, while it is as high as 74 mV dec−1 for pristine Ni foam. The similar Tafel slopes of both NSAs suggest that these two electrocatalysts have similar reaction kinetics for the IOR. The ECSA of the Ni–Co(OH)2 NSAs and Ni(OH)2 NSAs was further measured to uncover their intrinsic activity towards the IOR. Since the ECSA is linearly proportional to the double-layer capacitances (Cdl), we first determined Cdl through cyclic voltammetry (CV) measurements at scan rates from 10 to 50 mV s−1. According to CV curves in Fig. S9,† the Cdl of Ni–Co(OH)2 NSAs and Ni(OH)2 NSAs was calculated to be 0.44 and 0.41 mF cm−2 (Fig. 4C), corresponding to the ECSA of 11 and 10.25 cm2, respectively. This result implies that the superior IOR performance for Ni–Co(OH)2 NSAs is independent of the ECSA, since both the electrocatalysts exhibit similar ECSAs with quite different current density at the same potential (Fig. 4D). The current density normalized to the ECSA of Ni–Co(OH)2 NSAs is higher than that of Ni(OH)2 NSAs, suggesting the remarkable intrinsic activity of Ni–Co(OH)2 NSAs towards the IOR, which can be attributed to its favorable response driven by the redox of Co(III)/Co(IV) species.
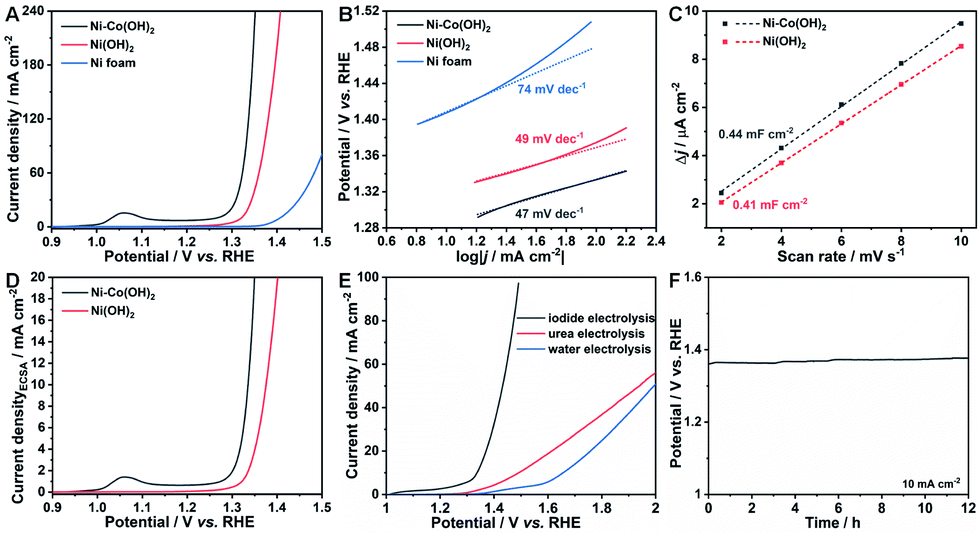 | ||
| Fig. 4 (A) Polarization curves of the IOR over Ni–Co(OH)2 NSAs, Ni(OH)2 NSAs and Ni foam. (B) The corresponding Tafel plots from (A). (C) Difference in current density plotted against the scan rate displaying the value of double-layer capacitances. (D) ECSA normalized polarization curves of Ni–Co(OH)2 NSAs, and Ni(OH)2 NSAs from (A). (E) Polarization curves of iodide electrolysis, urea electrolysis, and water electrolysis over the Ni–Co(OH)2 NSAs‖Ni–Mo electrode pair. (F) Chronopotentiometry curve of iodide electrolysis at a current density of 10 mA cm−2. | ||
Furthermore, the overall electrolysis of iodide, urea and water was carried out in a two electrode cell by using the as-obtained Ni–Co(OH)2 NSAs as the anode and the Ni–Mo electrode as the cathode. The Ni–Mo electrode was fabricated by a facile electrodeposition method according to the literature,41 and its morphology was characterized by FESEM (Fig. S10A and B†). The utilization of the Ni–Mo electrode as the cathode in this system can be ascribed to its low cost, convenient fabrication, and extremely superior HER activity with a low overpotential of 43 mV at 10 mA cm−2 (Fig. S10C†). As expected, the overall iodide electrolysis displays prominent activity by delivering a current density of 10 mA cm−2 at only 1.34 V, while it is as high as 1.49 and 1.66 V for urea and water electrolysis, respectively (Fig. 4E). Further, overall iodide electrolysis can operate smoothly for at least 12 h, confirmed by the chronopotentiometry test (Fig. 4F). These remarkable features imply that Ni–Co(OH)2 NSAs are an excellent electrocatalyst for the IOR and the IOR process can be one of the promising alternatives to the OER process to boost hydrogen generation efficiency with low energy consumption.
To exclude the effect of Ni ions on the IOR performance, a pure Co(OH)2-based electrocatalyst on Ni foam without Ni incorporation was synthesized with ZIF-67 as a precursor similar to the previous work of the Lou group.51 FESEM images (Fig. S11†) and XRD patterns (Fig. S12†) reveal the successful fabrication of ZIF-67 and Co(OH)2 on Ni foam with a triangular and nanosheet shape (denoted as Co(OH)2 NSAs), respectively. The IOR, UOR and OER performances of Co(OH)2 NSAs were also evaluated. As seen in polarization curves presented in Fig. 5A, the electrocatalytic performance for the IOR is significantly remarkable with a low potential of 1.31 V (vs. RHE) to drive a current density of 20 mA cm−2. However, the UOR and OER activities over Co(OH)2 NSAs are quite sluggish. The required potential at 20 mA cm−2 for the UOR is as high as 1.40 V (vs. RHE), and it is even higher than 1.5 V (vs. RHE) for the OER. The IOR performance over the as-prepared Co-based electrocatalysts is superior to that of other kinds of anodic oxidation reactions (Fig. 5B and Table S1†), such as the UOR over CoFeCr LDH/NF,52 S–MnO2,17 CoMn/CoMn2O4,53 and NF/NiMoO-Ar,16 and 5-hydroxymethylfurfural (HMF) oxidation over NixB,27 Ni2P NPA/NF,54 NiCo2O4,55 and Ni3S2/NF.56 Tafel slopes are calculated to be 52, 115 and 139 mV dec−1 for the IOR, UOR and OER, respectively, indicating more favorable catalytic kinetics of the IOR than the UOR and OER (Fig. 5C). The negligible change of potential over 18 h in chronopotentiometry measurements conducted at a constant current density of 20 mA cm−2 (Fig. 5D) suggests the prominent stability of the IOR over Co(OH)2 NSAs. All of the above results further imply that the Co-based electrocatalysts exhibit more efficient response to the IOR than to UOR and OER. And the Co(III)/Co(IV) couple is of key importance to lower the potential for iodide electrooxidation. It is worth noting that the application of Co-based electrocatalysts for energy-saving hydrogen generation can be expanded by the IOR process.
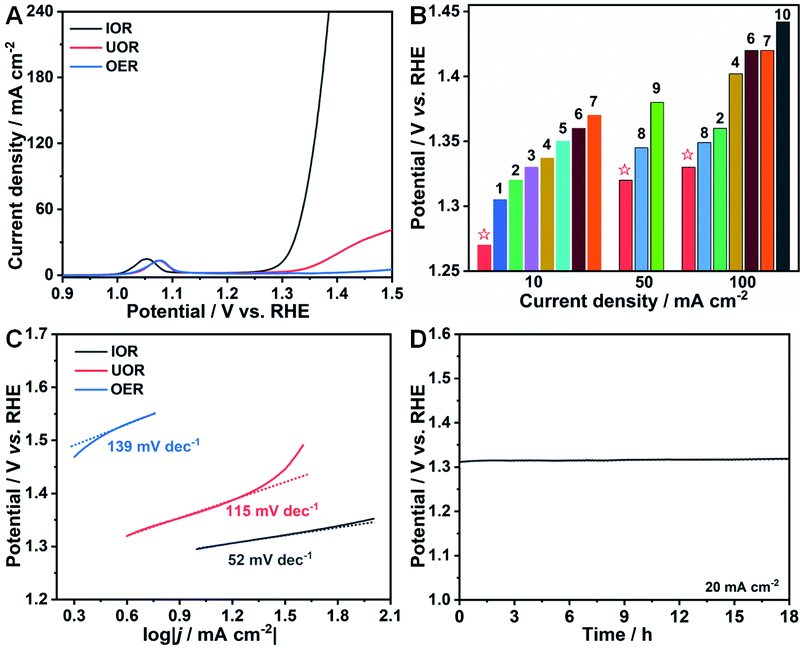 | ||
| Fig. 5 (A) Polarization curves of the IOR, OER, and UOR over Co(OH)2 NSAs. (B) Comparison of potentials at 10, 50, and 100 mA cm−2 of Ni–Co(OH)2 NSAs in this work with those of other representative electrocatalysts in references. (1): CoFeCr LDH/NF,52 (2): CoMn/CoMn2O4,53 (3): S–MnO2,17 (4): C-350,6 (5): Ni3N NA/CC,7 (6): FQD/CoNi-LDH/NF,8 (7): NF/NiMoO-Ar,16 (8): NiIr-MOF/NF,9 (9): MoS2/CoS/Co0.85Se HNT,34 and (10): MS-Ni2P/Ni0.96S/NF.10 (C) Corresponding Tafel plots from (A). (D) Chronopotentiometry curve of the IOR over Co(OH)2 NSAs at a current density of 20 mA cm−2. | ||
Conclusions
In summary, the highly active electrode of Ni–Co(OH)2 NSAs was successfully fabricated at room temperature by simply immersing Ni foam in Co(NO3)2 solution. And we also demonstrated that the electrooxidation of iodide can efficiently take place over Ni–Co(OH)2 NSAs with favorable kinetics and high durability, in which Co(IV)/Co(III) serves as the primary active species to accelerate the reaction rate. Thanks to the ready oxidation feature of iodide, integrating with the Ni–Mo electrode, overall iodide electrolysis exhibits an ultralow cell voltage of 1.34 V at 10 mA cm−2, which is almost 320 mV lower than that of water electrolysis. The outstanding stability also illustrates that the as-prepared Ni–Co(OH)2 NSAs are a promising electrocatalyst towards the IOR in an alkaline medium. We believe that this work can open a new avenue for the application of Co-based materials as anodic oxidation reaction electrocatalysts for energy-saving hydrogen generation with value-added oxidation products generated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (U1609219, 51632008, and 61721005).References
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS.
- J.-B. Tan and G.-R. Li, J. Mater. Chem. A, 2020, 8, 14326–14355 RSC.
- B. Zhu, Z. Liang and R. Zou, Small, 2020, 16, 1906133 CrossRef CAS.
- L. Chen and J. Shi, J. Mater. Chem. A, 2018, 6, 13538–13548 RSC.
- S. Hu, S. Wang, C. Feng, H. Wu, J. Zhang and H. Mei, ACS Sustainable Chem. Eng., 2020, 8, 7414–7422 CrossRef CAS.
- Q. Liu, L. Xie, F. Qu, Z. Liu, G. Du, A. M. Asiri and X. Sun, Inorg. Chem. Front., 2017, 4, 1120–1124 RSC.
- Y. Feng, X. Wang, J. Huang, P. Dong, J. Ji, J. Li, L. Cao, L. Feng, P. Jin and C. Wang, Chem. Eng. J., 2020, 390, 124525 CrossRef CAS.
- Y. Xu, X. Chai, T. Ren, S. Yu, H. Yu, Z. Wang, X. Li, L. Wang and H. Wang, Chem. Commun., 2020, 56, 2151–2154 RSC.
- M. He, C. Feng, T. Liao, S. Hu, H. Wu and Z. Sun, ACS Appl. Mater. Interfaces, 2020, 12, 2225–2233 CrossRef CAS.
- B. You, G. Han and Y. Sun, Chem. Commun., 2018, 54, 5943–5955 RSC.
- C. Huang, Y. Huang, C. Liu, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2019, 58, 12014–12017 CrossRef CAS.
- Y. Huang, X. Chong, C. Liu, Y. Liang and B. Zhang, Angew. Chem., Int. Ed., 2018, 57, 13163–13166 CrossRef CAS.
- X. Chong, C. Liu, Y. Huang, C. Huang and B. Zhang, Natl. Sci. Rev., 2019, 7, 285–295 CrossRef.
- Y. Wang, Y. Yu, R. Jia, C. Zhang and B. Zhang, Natl. Sci. Rev., 2019, 6, 730–738 CrossRef CAS.
- Z. Yu, C. Lang, M. Gao, Y. Chen, Q. Fu, Y. Duan and S. Yu, Energy Environ. Sci., 2018, 11, 1890–1897 RSC.
- S. Chen, J. Duan, A. Vasileff and S. Z. Qiao, Angew. Chem., Int. Ed., 2016, 55, 3804–3808 CrossRef CAS.
- D. Liu, T. Liu, L. Zhang, F. Qu, G. Du, A. M. Asiri and X. Sun, J. Mater. Chem. A, 2017, 5, 3208–3213 RSC.
- X. Hu, J. Zhu, J. Li and Q. Wu, ChemElectroChem, 2020, 7, 3211–3228 CrossRef CAS.
- L. Sha, K. Ye, G. Wang, J. Shao, K. Zhu, K. Cheng, J. Yan, G. Wang and D. Cao, J. Power Sources, 2019, 412, 265–271 CrossRef CAS.
- N. Senthilkumar, G. Gnana kumar and A. Manthiram, Adv. Energy Mater., 2018, 8, 1702207 CrossRef.
- A. J. Bard and L. R. Faulkner, ELECTROCHEMICAL METHODS-Fundamentals and Applications, JOHN WILEY & SONS, INC, 2001 Search PubMed.
- G. Arroyave, O. Pineda and N. S. Scrimshaw, Bull. W. H. O., 1956, 14, 183–185 CAS.
- X. Li, X. Cao, J. Li, J. Xu, W. Ma, H. Wang, J. Wang and Y. Zhang, J. Trace Elem. Med. Biol., 2020, 62, 126575 CrossRef CAS.
- H. de Santana and M. L. A. Temperini, J. Electroanal. Chem., 1991, 316, 93–105 CrossRef CAS.
- X. Zhu, X. Dou, J. Dai, X. An, Y. Guo, L. Zhang, S. Tao, J. Zhao, W. Chu, X. C. Zeng, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2016, 55, 12465–12469 CrossRef CAS.
- S. Barwe, J. Weidner, S. Cychy, D. M. Morales, S. Dieckhöfer, D. Hiltrop, J. Masa, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2018, 57, 11460–11464 CrossRef CAS.
- J. Wu, D. Wang, S. Wan, H. Liu, C. Wang and X. Wang, Small, 2020, 16, 1900550 CrossRef CAS.
- E. Hu, Y. Feng, J. Nai, D. Zhao, Y. Hu and X. W. Lou, Energy Environ. Sci., 2018, 11, 872–880 RSC.
- Y. Tong, P. Chen, M. Zhang, T. Zhou, L. Zhang, W. Chu, C. Wu and Y. Xie, ACS Catal., 2018, 8, 1–7 CrossRef.
- T. Liu, D. Liu, F. Qu, D. Wang, L. Zhang, R. Ge, S. Hao, Y. Ma, G. Du, A. M. Asiri, L. Chen and X. Sun, Adv. Energy Mater., 2017, 7, 1700020 CrossRef.
- M. Ranjani, N. Senthilkumar, G. Gnana kumar and A. Manthiram, J. Mater. Chem. A, 2018, 6, 23019–23027 RSC.
- J. Xie, W. Liu, F. Lei, X. Zhang, H. Qu, L. Gao, P. Hao, B. Tang and Y. Xie, Chem.–Eur. J., 2018, 24, 18408–18412 CrossRef CAS.
- Y. Sun, S. Wang, J. Ning, Z. Zhang, Y. Zhong and Y. Hu, Nanoscale, 2020, 12, 991–1001 RSC.
- K. Zeng and D. Zhang, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef CAS.
- X. Lu and C. Zhao, Nat. Commun., 2015, 6, 6616 CrossRef CAS.
- Y. Li, P. Hasin and Y. Wu, Adv. Mater., 2010, 22, 1926–1929 CrossRef CAS.
- L. Sha, K. Ye, J. Yin, K. Zhu, K. Cheng, J. Yan, G. Wang and D. Cao, Chem. Eng. J., 2020, 381, 122603 CrossRef CAS.
- E. Hu, J. Ning, D. Zhao, C. Xu, Y. Lin, Y. Zhong, Z. Zhang, Y. Wang and Y. Hu, Small, 2018, 14, 1704233 CrossRef.
- C. Guan, W. Zhao, Y. Hu, Z. Lai, X. Li, S. Sun, H. Zhang, A. K. Cheetham and J. Wang, Nanoscale Horiz., 2017, 2, 99–105 RSC.
- M. Xia, T. Lei, N. Lv and N. Li, Int. J. Hydrogen Energy, 2014, 39, 4794–4802 CrossRef CAS.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS.
- X. Cai, X. Shen, L. Ma, Z. Ji, C. Xu and A. Yuan, Chem. Eng. J., 2015, 268, 251–259 CrossRef CAS.
- Y. Luo, X. Li, X. Cai, X. Zou, F. Kang, H.-M. Cheng and B. Liu, ACS Nano, 2018, 12, 4565–4573 CrossRef CAS.
- F. Chen, C. Chen, Q. Hu, B. Xiang, T. Song, X. Zou, W. Li, B. Xiong and M. Deng, Chem. Eng. J., 2020, 401, 126145 CrossRef CAS.
- H. Liang, J. Lin, H. Jia, S. Chen, J. Qi, J. Cao, T. Lin, W. Fei and J. Feng, J. Mater. Chem. A, 2018, 6, 15040–15046 RSC.
- M. Yang, H. Cheng, Y. Gu, Z. Sun, J. Hu, L. Cao, F. Lv, M. Li, W. Wang, Z. Wang, S. Wu, H. Liu and Z. Lu, Nano Res., 2015, 8, 2744–2754 CrossRef CAS.
- L. Peng, L. Lv, H. Wan, Y. Ruan, X. Ji, J. Liu, L. Miao, C. Wang and J. Jiang, Materials Today Energy, 2017, 4, 122–131 CrossRef.
- J. Yang, H. Liu, W. N. Martens and R. L. Frost, J. Phys. Chem. C, 2010, 114, 111–119 CrossRef CAS.
- Z. Wang, L. Wang, S. Liu, G. Li and X.-P. Gao, Adv. Funct. Mater., 2019, 29, 1901051 CrossRef.
- H. Hu, B. Y. Guan and X. W. Lou, Chem, 2016, 1, 102–113 CAS.
- Z. Wang, W. Liu, Y. Hu, M. Guan, L. Xu, H. Li, J. Bao and H. Li, Appl. Catal., B, 2020, 272, 118959 CrossRef CAS.
- C. Wang, H. Lu, Z. Mao, C. Yan, G. Shen and X. Wang, Adv. Funct. Mater., 2020, 30, 2000556 CrossRef CAS.
- B. You, N. Jiang, X. Liu and Y. Sun, Angew. Chem., Int. Ed., 2016, 55, 9913–9917 CrossRef CAS.
- M. J. Kang, H. Park, J. Jegal, S. Y. Hwang, Y. S. Kang and H. G. Cha, Appl. Catal., B, 2019, 242, 85–91 CrossRef CAS.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00847h |
| This journal is © The Royal Society of Chemistry 2021 |