
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSwitching between TADF and RTP: anion-regulated photoluminescence in organic salts and co-crystals†
Zhen
Xu
 a,
Duane
Hean
a,
Clàudia
Climent
b,
David
Casanova
*cd and
Michael O.
Wolf
*a
a,
Duane
Hean
a,
Clàudia
Climent
b,
David
Casanova
*cd and
Michael O.
Wolf
*a
aDepartment of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, BC V6T 1Z1, Canada. E-mail: mwolf@chem.ubc.ca
bDepartamento de Física Teórica de la Materia Condensada, Universidad Autónoma de Madrid, E-28049 Madrid, Spain
cDonostia International Physics Center (DIPC), Paseo Manuel de Lardizabal 4, 20018 Donostia, Euskadi, Spain. E-mail: david.casanova@ehu.eus
dIKERBASQUE, Basque Foundation for Science, 48013 Bilbao, Euskadi, Spain
First published on 13th August 2021
Abstract
Thermally activated delayed fluorescence (TADF) and room temperature phosphorescence (RTP) are two photophysical phenomena which utilize triplet excitons. In this work, we demonstrate how variation of the anion in organic salts with carbazole and phenothiazine-5,5-dioxide donors and pyridinium and quinolinium acceptors may be used to switch between TADF and RTP. These compounds adopt similar molecular structures and packing modes with different anions and exhibit different types of photophysical behavior due to the electronic effects of the anions. With bromide anions, the salts exhibit TADF with some RTP. These compounds show fast reverse intersystem crossing and a short delayed lifetime, which is key to application in efficient and robust OLEDs. With BF4− and PF6− anions, RTP with long-lived lifetimes and afterglow are observed by eye. This behavior can be utilized for data encryption and anti-counterfeiting applications. Emission wavelengths and lifetimes are also anion-dependent. These results open up an avenue for developing novel luminescent materials through anion tuning and present a molecular model to understand the interplay of RTP and TADF.
Introduction
Organic luminescent materials that can effectively harvest triplet excitons are advantageous for numerous applications including organic light-emitting diode (OLED) displays, chemical sensors and as bioimaging agents.1–4 The two main strategies to utilize triplet excitons are thermally activated delayed fluorescence (TADF) and room temperature phosphorescence (RTP). TADF involves emission from a singlet excited state with a delayed component, and many highly efficient OLED devices have been fabricated using TADF emitters.5,6 In contrast, RTP arises from direct emission from the triplet excited state, and due to its ultralong lifetime, researchers have utilized the ‘afterglow’ feature in bioimaging, sensing, anti-counterfeiting and data encryption materials.7–10It is important to understand the relationship between TADF and RTP both to develop novel multifunctional luminescent materials and to shed light on triplet processes in organic molecules from a fundamental perspective.11,12 Many efforts have been made towards bridging these two phenomena. Through structural modifications, the singlet–triplet energy gap ΔEST or the triplet chromophore stacking can be tuned in favor of either TADF or RTP.13–17 Crystal polymorphs of the same molecule, on the other hand, may also exhibit different behaviors in triplet states.18,19 These strategies, however, often involve extensive synthesis or complex crystal engineering, which makes the process of understanding TADF vs. RTP difficult. Furthermore, it is also a challenge to study the electronic effects alone without altering the packing mode in the crystalline state at the same time.
In some cases, organic compounds have been found to exhibit both TADF and RTP concurrently.15,17,19–21 It has been reported that a hidden RTP state of a TADF molecule can potentially serve as a ‘springboard’ for long-lived triplet excitons and facilitate reverse intersystem crossing (RISC). This finding further demonstrates the importance and urgency of developing novel and facile strategies with versatile molecular models to understand the interplay of TADF and RTP.
Variation of the corresponding anions can be utilized to tune the photoluminescence properties of organic cations. Inorganic anions such as PF6− have been shown to enhance solid-state fluorescence in 1,2,3,4-tetraphenyloxazolium (TPO) molecular systems, which feature aggregation-induced emission (AIE),22,23 while phosphorescence efficiency can be improved by pairing anions such as bromide and iodide with the cations due to the heavy atom effect.20 In carbazole-containing phosphonium salts, the phosphorescent triplet state becomes accessible with halide anions, thereby reducing fluorescence and enhancing phosphorescence.24,25 Even though success has been found in achieving phosphorescence in organic crystals with inorganic anions such as BF4−,26 the utilization of anions to fine-tune RTP or TADF remains relatively unexplored.
We elected to use carbazole as the donor and N-heterocyclic cations including pyridinium and quinolinium27 as acceptors for RTP. Herein, we report the design, synthesis and characterization of a series of non-conjugated carbazole species containing pyridinium and quinolinium acceptor groups with different anions (Scheme 1). By varying the anion, switching between RTP and TADF can be realized. The origin of the differences in the photophysical properties of these compounds and the role of anions is rationalized by means of density functional theory (DFT) and time-dependent DFT (TDDFT) calculations, providing a perspective into anion regulated photoluminescence.
 | ||
| Scheme 1 Chemical structures of carbazole-containing pyridinium (Py) and quinolinium (Qui) compounds. | ||
Synthesis and structures
Compound Py-Br was synthesized via direct alkylation of pyridine by 9-(4-bromobutyl)-9H-carbazole. This reaction occurs under neat conditions, negating the need for any solvent. Further anion exchange by adding a saturated aqueous solution of NH4BF4 or NH4PF6 to an acetone solution of Py-Br resulted in precipitation of the desired product Py-BF4 and Py-PF6, which can then be isolated by filtration. Species containing the chloride and iodide counter anions (Py-Cl and Py-I) were obtained through reaction of Py-PF6 with either tetrabutylammonium chloride or iodide (NBu4Cl or NBu4I). Qui-Br and Qui-BF4 were synthesized in a similar fashion. These syntheses do not require any column chromatography, with purification occurring via a simple solvent wash and recrystallization, resulting in high product yields. All compounds were characterized using NMR spectroscopy and high-resolution mass spectrometry. 1H and 13C signals were assigned using COSY, HSQC and HMBC experiments.Solid-state structures of all the compounds were obtained by single crystal X-ray diffraction. Suitable single crystals of the species were grown by slow evaporation of acetone solutions (Py-BF4, Py-PF6 and Qui-Br) or CH2Cl2 solutions (Py-Cl, Py-Br, Py-I and Qui-BF4). Pyridinium salt (Py) crystals belong to the Pbca space group with similar unit cell dimensions whereas the two quinolinium salts adopted the P21/n space group. Similar molecular geometries with the anions positioned close to the cationic centers are found for the pyridinium (Py) and quinolinium (Qui) compounds, demonstrating that the specific anion present does not affect the individual molecular structures of the cations. Compounds with Cl− and Br− anions exhibit hydrated structures, due to the hydrogen-bonding tendency of these anions and the smaller anionic size.
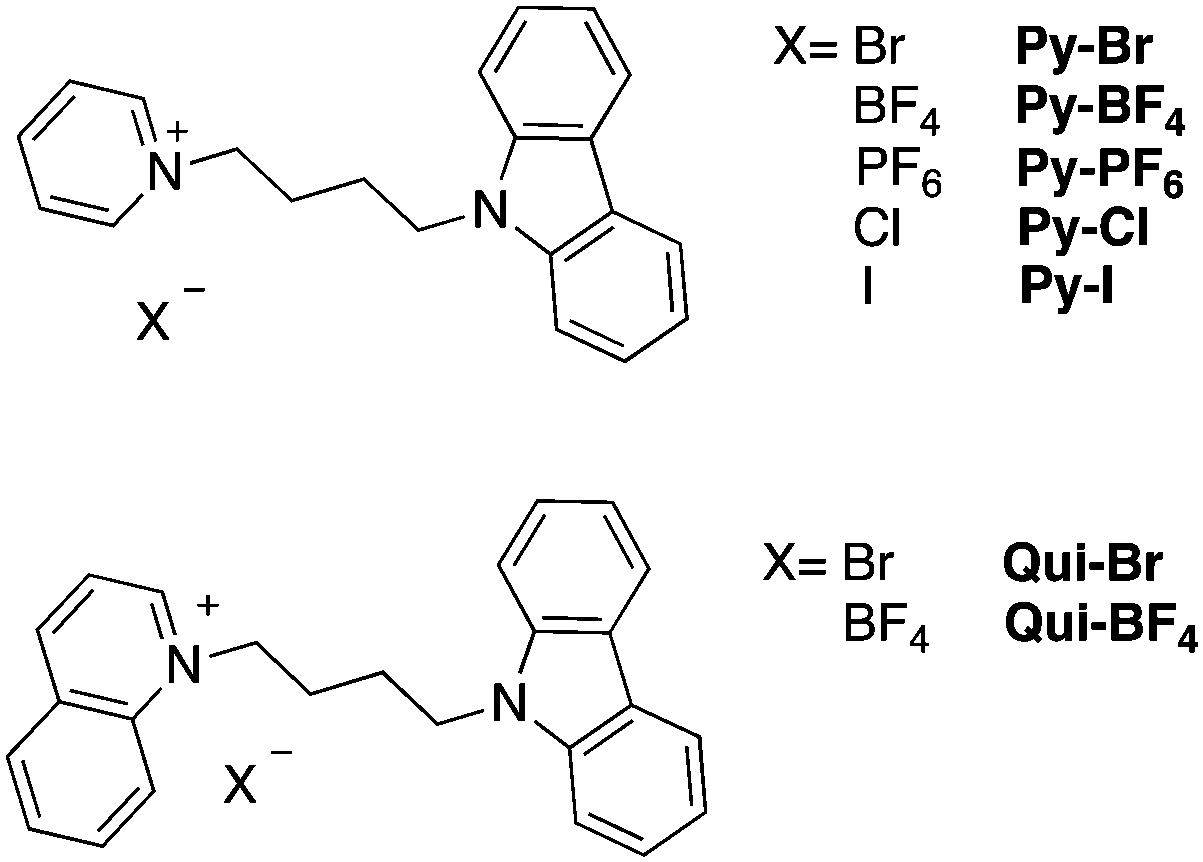
The intermolecular interactions in these salts have also been carefully examined by analyzing the packing diagrams (Fig. 1). These interactions are key to understanding the solid-state photoluminescence properties (see below). The pyridinium compounds all adopt similar packing modes in which two pyridinium rings are parallel in the center of the unit cell. The distances between the two pyridinium rings ranges between 3.468 Å in Py-I and 3.768 Å in Py-PF6, indicative of strong π+–π+ interactions in Py-I. Py-PF6 has the largest anion, which leads to the weakest π+–π+ interaction among the compounds.28 Three other types of intermolecular interactions including CH⋯N, CH⋯π+ and CH⋯CH are also observed in the structures. The hydrogen atoms on either the pyridinium ring or the carbazole ring can interact with another carbazole π-conjugated ring, with distances ranging from 2.418 to 2.900 Å. The hydrogen atom on the carbazole moiety can also have short contacts with the nitrogen atom on the carbazole ring to give CH⋯N interactions. CH⋯π+ and CH⋯CH interactions are also observed. It is worth noting that halogen-containing compounds such as Py-Br, Py-Cl and Py-I, exhibit halogen–hydrogen interactions with pyridinium, carbazole and with hydrogens at sp3 hybridized carbon atoms.
 | ||
| Fig. 1 Different crystal views of the intermolecular packing of Py-Br (a and b) and Qui-Br (e and f). (c and d) Molecular π+–π+ and Br–H interactions in Py-Br. (g) Molecular π–π+ and π+–π+ interactions in Qui-Br. | ||
The solid-state structures of the quinolinium salts Qui-Br and Qui-BF4 also contain abundant short contacts. Interestingly, they both show strong π–π+ and π+–π+ interactions. One quinolinium ring aligns in parallel with a carbazole unit and another quinolinium ring to form H-aggregates. The π–π+ distances in Qui-Br and Qui-BF4 are 3.439 and 3.355 Å, respectively. Similarly, CH⋯π interactions are also present in the crystal structures between the quinolinium hydrogens and carbazoles. However, there are no CH⋯π interaction between two carbazole moieties. In Qui-Br, Br⋯H halogen interactions also exist and are measured as ∼2.9 Å.
Photophysics
The absorption and photoluminescence (PL) spectra of all compounds in CH2Cl2 solutions are shown in Fig. S2 (ESI†). The absorption spectra of the pyridinium compounds appear virtually identical to those of the related 9-alkyl carbazole compounds, reported by Xue and co-workers.29 Compound Py-I, on the other hand, exhibits a broad feature between 360 nm and 420 nm, which is attributed to charge transfer between the pyridinium cation and the iodide anion. Electronic structure calculations confirm the presence of low-lying CT excitations of iodide 5p electrons to the pyridinium moiety (Fig. S27, ESI†). On the other hand, CT transitions are not observed with the other counter anions (Tables S3 and S4, ESI†). Quinolinium compounds (Qui-Br and Qui-BF4) exhibit slightly different absorption behavior between 300 nm to 350 nm, which result from contributions of the quinolinium moiety, as computationally characterized (Table S7, ESI†). All the species show structured emission peaks at 350 nm and 370 nm. These are attributed to the locally excited state on carbazole, although emission from a state localized on quinolinium cannot be discounted as the excitation energies of the two local states are rather close to each other (Table S7 and Fig. S30, ESI†).30 The localized nature of the low-lying excited singlets on either of the two moieties indicates that there is no strong interaction between the cationic heterocyclic units and carbazole in dilute CH2Cl2 solutions. The photoluminescence quantum yields (PLQYs) in CH2Cl2 solution are extremely low (<0.1%), consistent with the PLQYs reported for similar compounds in the literature.28Powder X-ray diffraction (pXRD) patterns of Py-Br, Py-Cl, Qui-Br and Qui-BF4 were also collected in order to determine the solid-state structures in the bulk phase (Fig. S1, ESI†). The experimental patterns match the simulated patterns generated from the single crystal structures well, indicating that the bulk powder samples of these compounds pack the same as in the single crystals. The single crystals of Py-BF4, Py-PF6 and Py-I were used for photophysical measurements.
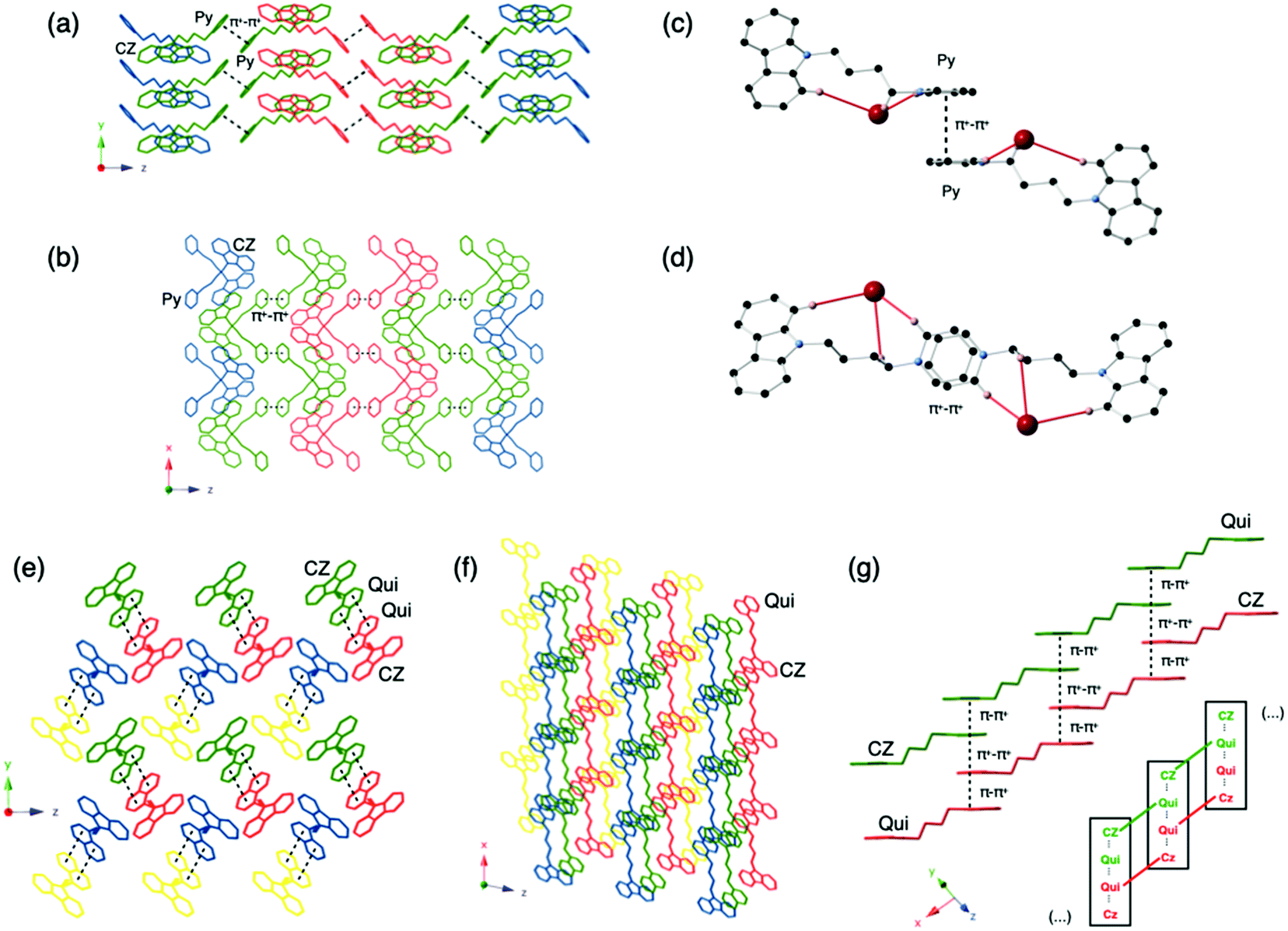
The solid-state photoluminescence of the pyridinium and quinolinium compounds (Py and Qui) exhibit entirely different behavior from solution. The luminescence intensity increases dramatically relative to in solution (Table 1) due to aggregation-induced emission (AIE).28 For the pyridinium compounds, Py-BF4, Py-PF6 and Py-Cl display similar PLQYs of ∼0.6, while Py-Br is slightly less emissive, possibly due to heavy-atom quenching31 triggered by the short contact between carbazole and the Br atom. The quinolinium compounds exhibit much weaker photoluminescence of 0.15 and 0.17 for Qui-Br and Qui-BF4, respectively. The strong π–π+ stacking may be the cause of the quenching.32 In accordance with this, electronic structure calculations show sizeable π–π+ overlaps around the Fermi level favored by the CBZ–Qui–Qui–CBZ coplanar arrangement in the crystal (Fig. 2), stabilizing spin singlet excitations with strong CT character between stacked carbazole and quinolinium cations (Table S7, ESI†). Moreover, the lowest singlet–singlet excitations have weak transition dipole moments, which may be related to the weak photoluminescence. These compounds are categorized into three types based on their photophysical behavior: those exhibiting thermally activated delayed fluorescence (TADF), room temperature phosphorescence (RTP) or low temperature phosphorescence (LTP).
| Fluorescence | Phosphorescence (air) | PLQY | Phosphorescence τ (ms) (77 K)b | |||
|---|---|---|---|---|---|---|
| λ em (nm) | τ (ns) | λ em (nm) | τ (ms) | |||
| a λ ex = 359 nm. b Relative amplitudes in parenthesis. | ||||||
| Py-Br | 488 | 43.2 (0.70), 354 (0.30) | 515 | 3.00 (0.08), 16.8 (0.30), 58.3 (0.62) | 0.47 | 39.9 (0.22), 169 (0.34), 698 (0.44) |
| Py-BF4 | 464 | 147 | 512 | 19.3 (0.25), 88.6 (0.544), 362 (0.21) | 0.67 | 154 (0.23), 538 (0.38), 1883 (0.39) |
| Py-PF6 | 442 | 125 | 500 | 9.03 (0.16), 47.2 (0.46), 131 (0.38) | 0.61 | 143 (0.26), 588 (0.43), 1913 (0.31) |
| Py-Cl | 490 | 117 | 524 | 2.87 (0.14), 14.0 (0.46), 56.1 (0.40) | 0.61 | 122 (0.39), 779 (0.61) |
| Qui-Br | 575 | 10.9 (0.39), 655 (0.61) | 575 | 1.51 (0.44), 8.0 (0.56) | 0.15 | 21.5 (0.54), 6.5 (0.29), 68.8 (0.17) |
| Qui-BF4 | 563 | 59.9 | 580 | 3.41 (0.29), 16.0 (0.38), 73.7 (0.33) | 0.17 | 41.6 (0.10), 210 (0.28), 804 (0.62) |
 | ||
| Fig. 2 Main orbital contribution (HOMO−2 → LUMO) to the lowest excited singlet computed for the Qui-BF4 crystal tetramer at the ωB97X-D/6-31+G(d) computational level. | ||
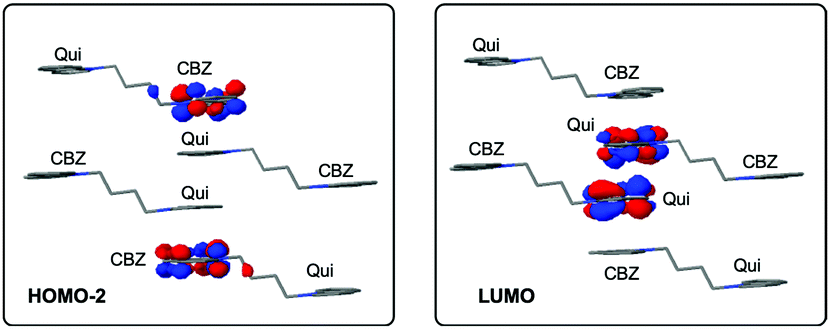
The steady-state emission spectrum of the crystalline powder of Py-Br shows a broad featureless peak with a maximum at 488 nm, red shifted compared to the emission in solution. This is attributed to through-space interactions between pyridinium and carbazole.28 The fluorescence decay profile measured by time-correlated single photon counting consists of two components, prompt fluorescence with a lifetime of 43 ns and a 354 ns delayed emission (Fig. 3(b)). The amplitudes of these two components are 70% and 30%, respectively.
 | ||
| Fig. 3 (a) Steady-state and time-resolved spectra of crystalline Py-Br at room temperature under air. Inset: Photograph of Py-Br after UV excitation is removed. (b) Fluorescence and (c) phosphorescence decay curve of crystalline Py-Br at room temperature under air. | ||
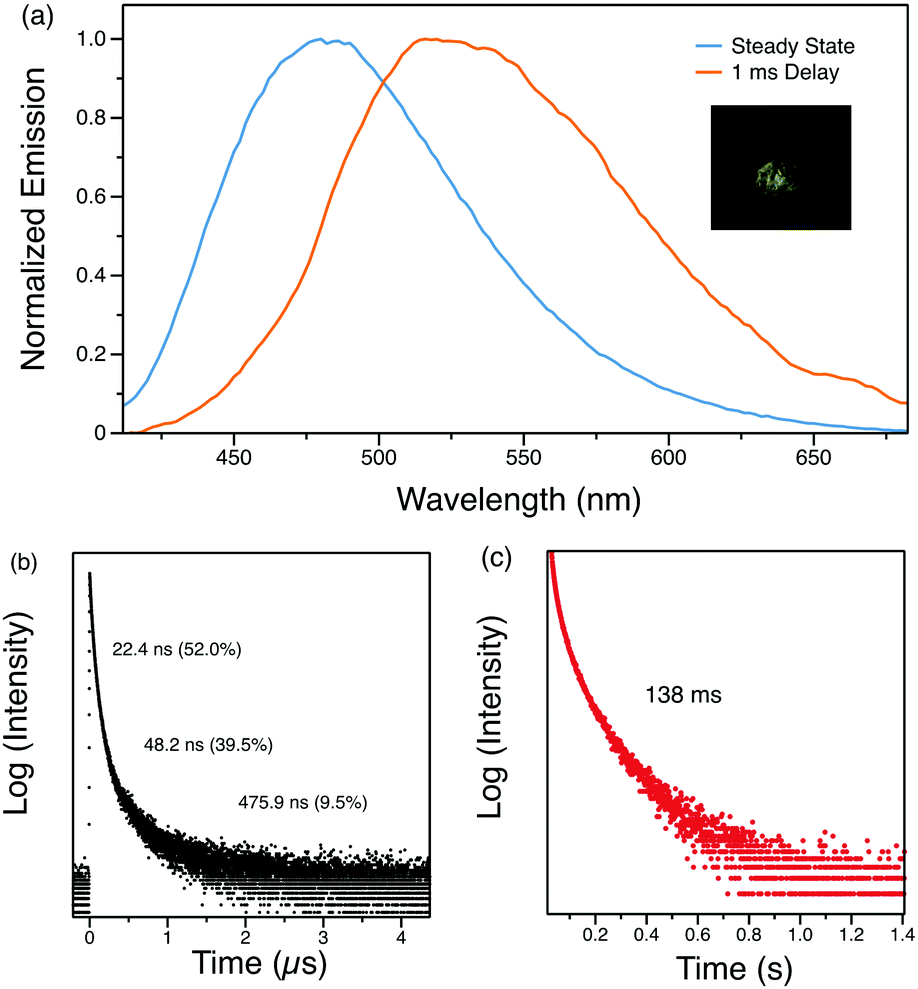
Time-resolved photoluminescence spectroscopy at room temperature was carried out to understand the emission features at different time ranges. The time-resolved spectra in the microsecond range are identical to the steady-state spectra, indicating that the delayed emission originates from the same singlet excited state as the prompt fluorescence. However, with a delay time of 1 ms, the luminescence spectrum of Py-Br shows a red-shift to 515 nm, indicative of a second radiative excited state (Fig. 3(a)). The photoluminescence decay of this long-lived component gives a long lifetime of 58 ms with a yellow afterglow, which is attributed to RTP from the lowest triplet state localized on the carbazole fragment (Table S4, ESI†). Therefore, Py-Br displays prompt fluorescence, delayed fluorescence (DF) and RTP concurrently.
The origin of this delayed feature was further investigated by probing the emission of Py-Br as a function of temperature. When cooled to 77 K, the 1 ms delayed spectrum shows well-structured emission with a lifetime of 544 ms that originates from the locally excited triplet states of carbazole (Fig. 4).30 Such long-lived phosphorescence gives an afterglow that lasts for seconds after the UV excitation source is removed. Since the delayed spectrum overlaps significantly with the prompt spectrum, it is difficult to extrapolate ΔEST from these measurements. Therefore, the fluorescence lifetimes were measured from 260 K to 310 K for every 10 K since kRISC is proportional to exp(ΔEST/kBT), where kB is the Boltzmann constant and T is the temperature.33 The resultant linear plot (R2 = 0.976) further proves the ‘thermally activated’ feature of the delayed emission and the fitted ΔEST is 178 meV, consistent with TDDFT computed gap of 0.15–0.17 eV between singlet and triplet states localized on carbazole (Tables S4 and S5, ESI†). The RTP lifetime is considerably longer than the DF, suggesting that they arise from two different triplets. This interpretation is in line with our calculations (Tables S4 and S5, ESI†), which locate a triplet state energetically close to the lowest singlet (related to DF) different than the lowest triplet (related to RTP). Alternatively, the DF could emerge from triplet–triplet annihilation, but this possibility can be eliminated since delayed emission intensity increases linearly with the excitation power (Fig. S4, ESI†). We therefore conclude that Py-Br is a TADF emitter that also exhibits RTP character.
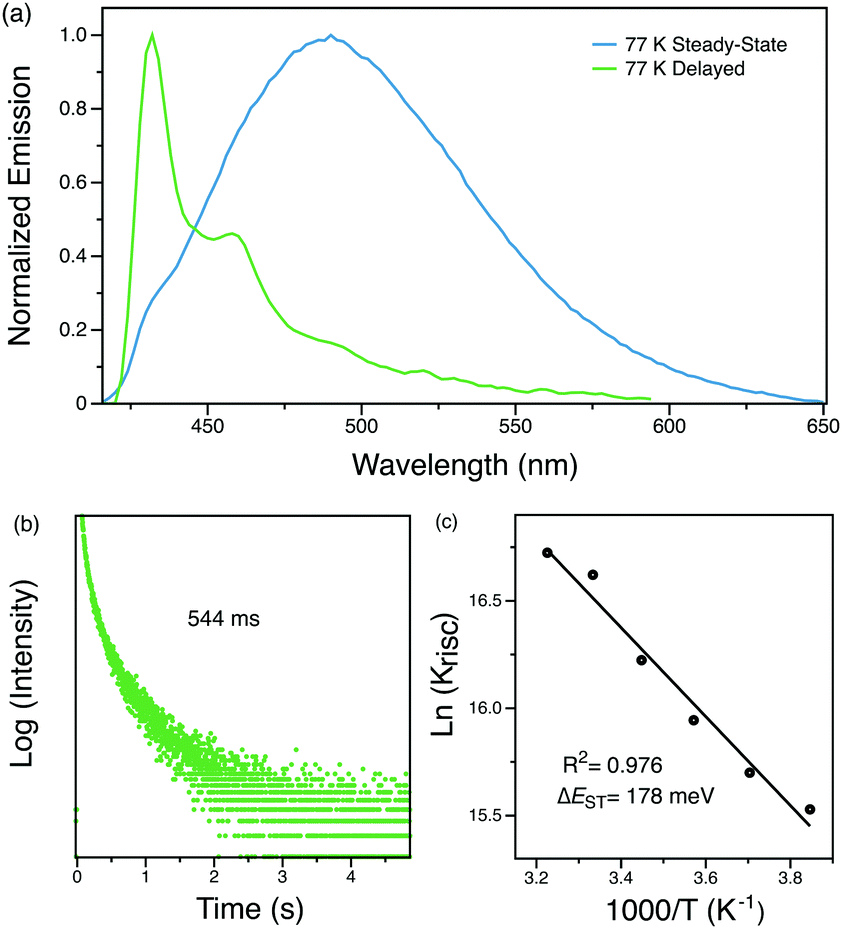 | ||
| Fig. 4 (a) Steady-state and time-resolved photoluminescence spectra of crystalline Py-Br at 77 K. (b) Phosphorescence decay curve of crystalline Py-Br at 77 K. (c) Arrhenius plot of ln(kRISC) versus temperature to calculate ΔEST. | ||
Replacing the pyridinium unit with a quinolinium unit results in an increase in π–π+ and π+–π+ interactions, as characterized by the orbital mixing between different units (Fig. S29, ESI†), and the Qui-Br crystalline powder is yellow in colour. Similar to Py-Br, Qui-Br also displays TADF (Fig. S3, ESI†). The steady-state photoluminescence is significantly red-shifted compared to Py-Br with a peak maximum at 575 nm. At room temperature, the emission decay curve contains two components with lifetimes of 11 ns and 657 ns. The time-resolved spectrum from 0 to 30 ns shows a blue-shift compared to the steady state spectrum, which can be attributed to a more localized excited state. The microsecond range emission, on the contrary, lines up well with steady-state fluorescence, indicating that both prompt and delayed emission are from the same singlet excited state. The time-resolved spectrum with a 1 ms delay shows a slight offset compared to the steady state spectrum, which results from the small ΔEST. This long-lived RTP has a lifetime of 8 ms. Temperature-dependent studies give a ΔEST of 50 meV from time-resolved spectra at 77 K and 62 meV from the Arrhenius plot, in agreement with the TDDFT singlet–triplet gap between the lowest singlet and the triplet state just below S1 computed at ∼40–85 meV (Table S7, ESI†). Meanwhile, the low temperature phosphorescence displays a 69 ms lifetime.
Pyridinium and quinolinium cations with a bromide anion (Py-Br and Qui-Br) exhibit simultaneous TADF and RTP in the crystalline state under ambient conditions. Moreover, the heavy-atom effect from the Br atom in these two compounds (Tables S9 and S10, ESI†) enables extremely fast RISC and thus short lifetimes of the delayed fluorescence, which is beneficial for prolonging OLED device lifetime and suppressing efficiency roll-off in devices.31
Compounds Py-BF4, Py-PF6, Py-Cl and Qui-BF4 were also studied by similar photoluminescence lifetime measurements as for Py-Br and Qui-Br. However, these compounds only exhibit mono-exponential fluorescence decays with no delayed component observed. Computed singlet–triplet gaps in Py-BF4, Py-PF6, Py-Cl and Qui-BF4 are very close to the one obtained for Py-Br regardless of the counter anion. Moreover, the nature of the electronic states does not change with different counter anions. Therefore, we hypothesize that the lack of delayed fluorescence with BF4−, Cl− and PF6− counter anions is due to weak RISC, which increases in Py-Br through the heavy atom effect. As shown in Fig. S6 (ESI†), the fluorescence lifetimes of these compounds in the crystalline state are all longer than 50 ns, that is, longer than conventional organic fluorophores. This can be attributed to the intermolecular and cation–anion interactions between the pyridinium group and the counter ions in the solid state. Steady-state photoluminescence reveals that anions can affect the emission wavelength of pyridinium compounds (Py-BF4, Py-PF6 and Py-Cl). Py-PF6 has an emission maximum at 442 nm while Py-Cl shows a red-shifted emission at 490 nm.
Time-resolved spectra of all four compounds (Py-BF4, Py-PF6, Py-Cl and Qui-BF4) display separation from the steady-state emission (Fig. 5a), indicative of a second emissive excited state. Py-BF4 has a peak maximum at 465 nm in the steady state spectrum. However, when a 1 ms delay is employed, a maximum at 512 nm is observed, with a shoulder at 540 nm. The delayed luminescence of the pyridinium compounds (Py-BF4, Py-PF6, Py-Cl) shares the same trend as seen in the steady-state emission, in which a red-shift from non-coordinating PF6− to coordinating Cl− is observed.
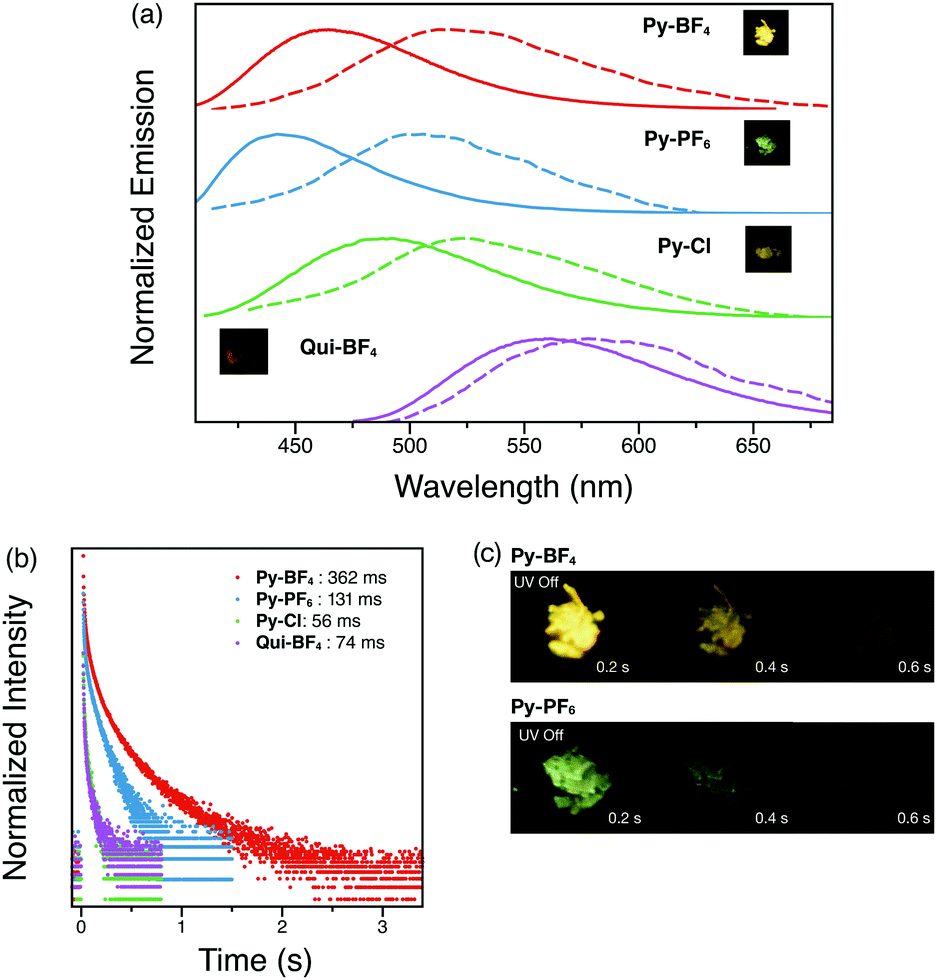 | ||
| Fig. 5 (a) Steady-state (solid lines) and time-resolved with 1 ms delay (dashed lines) photoluminescence spectra of Py-BF4, Py-PF6, Py-Cl and Qui-BF4 crystalline powders under ambient conditions. Inset: Photographs of Py-BF4, Py-PF6, Py-Cl and Qui-BF4 crystalline powders when UV excitation is removed. (b) Phosphorescence decay profiles of Py-BF4, Py-PF6, Py-Cl and Qui-BF4 crystalline powders under ambient conditions. (c) Photographs of Py-BF4 and Py-PF6 over 0.6 s when UV excitation is removed. | ||
All four compounds (Py-BF4, Py-PF6, Py-Cl and Qui-BF4) display long-lived lifetimes (longer than milliseconds)34–37 with visually observable afterglows that can be captured by a digital single-lens reflex (DSLR) camera, which is a characteristic feature of RTP (Fig. 5). Py-BF4 has the longest lifetime (362 ms), while Py-PF6 exhibits a shorter lifetime (131 ms). This can be rationalized by analyzing the crystal packing modes. The larger size of the PF6− counter anion prevents close interaction between triplet chromophores thus shortening the lifetime. Py-Br and Py-Cl have even shorter lifetimes, despite the fact that Py-Br and Py-Cl have shorter interaction distances than those of Py-PF6. This could be the result of water molecules in the crystal cavities. Qui-BF4, however, displays a 74 ms lifetime, which is a 5-fold decrease compared to its pyridinium counterpart (Py-BF4). Even though Qui-BF4 has more short contacts and π–π+ and π+–π+ interactions in the crystal structure, the triplet carbazole chromophores cannot effectively interact with each other, which might lead to the decrease in lifetime. More interestingly, Py-BF4 shows persistent yellow ‘afterglow’ for 1 s after UV excitation is removed, whereas Py-PF6 exhibits a green afterglow, both observable by the naked eye, which makes them potential candidates for anti-counterfeiting inks and for application in data encryption technology.
Low temperature studies in the crystalline state were also carried out (Fig. 6). The 1 ms delayed spectra exhibit similar phosphorescence at 500–650 nm as well as some new emission bands in the higher energy region possibly due to the more compacted packing. Measured lifetimes range from 779 ms to 1913 ms, demonstrating that at low temperature, molecular motions have been further suppressed. These findings provide a novel and facile toolkit to tune fluorescence and phosphorescence, in addition to RTP lifetimes through choice of anions.
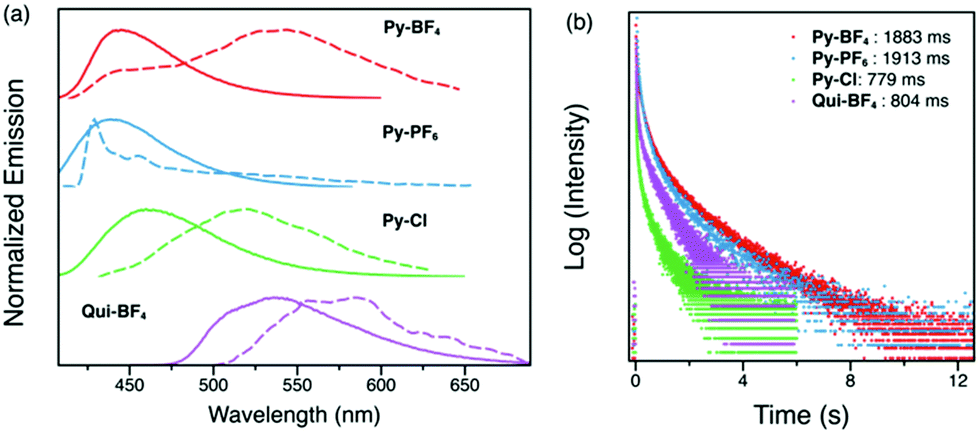 | ||
| Fig. 6 (a) Steady-state (solid lines) and time-resolved with 1 ms delay (dashed lines) photoluminescence spectra of Py-BF4, Py-PF6, Py-Cl and Qui-BF4 crystalline powders at 77 K. (b) Phosphorescence decay profiles of Py-BF4, Py-PF6, Py-Cl and Qui-BF4 crystalline powders at 77 K. | ||
Conversely, Py-I in the single crystalline state does not exhibit any photoluminescence at room temperature in air or under vacuum. Variable temperature photoluminescence studies were employed to understand the excited state of this compound. At temperatures above 150 K, the photoluminescence is extremely weak. However, when cooled to 100 K, Py-I exhibits green emission with a peak at 505 nm (Fig. 7). At 77 K, this broad emission peak grows drastically and gives relatively strong green luminescence. A time-resolved spectrum with 0.5 ms delay at 77 K is identical to the steady-state spectrum, indicating that emission originates from the long-lived triplet excited state. Therefore, by changing the anion to I−, room temperature luminescence is completely quenched and the phosphorescence pathway is only activated at low temperature.38 We tentatively hypothesize that the emission in Py-I is quenched by the low-energy CT excitations from the halogen to pyridinium (not present with the other anions).
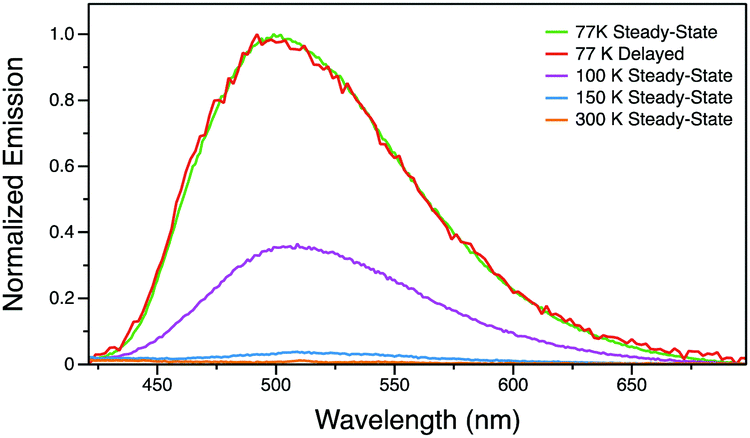 | ||
| Fig. 7 Variable temperature steady-state and time-resolved photoluminescence spectra of Py-I. | ||
To further expand the scope and examine the general applicability of this approach, a phenothiazine-5,5-dioxide moiety was used to replace the carbazole group as the triplet emitter (Scheme 2). Even though the RTP behavior of compounds containing this functional group has been thoroughly studied,39–41 reports of TADF from compounds containing phenothiazine-5,5-dioxide are scarce.17 Therefore, it is of interest to see whether incorporation of a bromide anion can enhance RISC in such systems as well. Unlike the Py-CBZ compounds, the structure of Py-Cs-Br is both water and CH2Cl2 solvated, and the crystal packing modes of Py-Cs-Br and Py-Cs-BF4 differ. In Py-Cs-BF4, the interaction between the phenothiazine-5,5-dioxide moiety and pyridinium group are stronger as they are close in space. However, the Py-Cs-Br single crystal sample still maintains a TADF feature with a 2.61 μs delayed component (Fig. S7(b), ESI†). The RTP lifetime of Py-Cs-Br is 125 ms, longer than for Py-Br. Py-Cs-BF4, on the other hand, exhibits a slight amount of TADF in the single crystal phase as shown in Fig. S8(b) (ESI†). Similarly, it has a longer RTP lifetime, compared to Py-Cs-Br. Even though crystal packing might play a role in the differing photophysical behavior between Py-Cs-Br and Py-Cs-BF4, it can be concluded that in general, the anion can regulate TADF and RTP behavior in such π–π+ building blocks.
 | ||
| Scheme 2 Chemical structures of pyridinium compounds with phenothiazine-5,5-dioxide group. | ||
To further demonstrate the influence anions can have on triplet luminescence processes, equimolar quantities of Py-BF4 and Py-Br were mixed and co-crystallized from an acetone solution to form an organic co-crystal (Py-A).42,43 The obtained single crystal contains BF4− and Br− anions in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 molar ratio. Interestingly, TADF and RTP properties originating from the parent compounds Py-BF4 and Py-Br are maintained in the alloy. The steady-state spectrum of Py-A displays a peak at 475 nm, intermediate in energy between the maxima of parent compounds Py-BF4 and Py-Br (Fig. 8a). Py-A exhibits a characteristic red-shifted phosphorescence spectrum when a 1 ms delay was employed with yellow afterglow observed. As a result, the singlet–triplet energy gap is larger than that of Py-Br but smaller than that of Py-BF4 demonstrating how TADF and RTP can be controlled. The amplitude of the delayed component of the TADF feature decreases and the RTP lifetime is prolonged compared to Py-Br (Fig. 8b and c). These results clearly show that manipulation of anions can effectively regulate the TADF and RTP features in an organic salt system.
:3 molar ratio. Interestingly, TADF and RTP properties originating from the parent compounds Py-BF4 and Py-Br are maintained in the alloy. The steady-state spectrum of Py-A displays a peak at 475 nm, intermediate in energy between the maxima of parent compounds Py-BF4 and Py-Br (Fig. 8a). Py-A exhibits a characteristic red-shifted phosphorescence spectrum when a 1 ms delay was employed with yellow afterglow observed. As a result, the singlet–triplet energy gap is larger than that of Py-Br but smaller than that of Py-BF4 demonstrating how TADF and RTP can be controlled. The amplitude of the delayed component of the TADF feature decreases and the RTP lifetime is prolonged compared to Py-Br (Fig. 8b and c). These results clearly show that manipulation of anions can effectively regulate the TADF and RTP features in an organic salt system.
 | ||
| Fig. 8 (a) Steady-state (blue) and time-resolved with 1 ms delay (orange) photoluminescence spectra of Py-A single crystal under ambient conditions. Inset: Photograph of Py-A single crystals when UV excitation is removed. (b) Fluorescence decay profile of Py-A single crystal under ambient conditions. (c) Phosphorescence decay profile of Py-A single crystal under ambient conditions. | ||
Conclusions
Seven carbazole-containing pyridinium and quinolinium compounds, two phenothiazine-containing pyridium compounds and one organic co-crystal are reported where the specific anion present plays a role in tuning the photophysical properties. These compounds adopt similar molecular structures and packing modes with different anions present and exhibit three types of photophysical behaviors due to the electronic effects from the anions. Py-Br, Qui-Br and Py-Cs-Br exhibit TADF with some RTP observed, these are the first examples to achieve these two interesting photophysical phenomena using a bromide anion. These compounds show fast reverse intersystem crossing and a short delayed lifetime, which is key to application in efficient and robust OLEDs. Py-BF4, Py-PF6, Py-Cl, Qui-BF4 and Py-Cs-BF4 display RTP with long-lived lifetimes and afterglow can be clearly observed by eye once the UV excitation is removed. This behavior can be further utilized for data encryption and anti-counterfeiting applications. Emission wavelengths and lifetimes are also dependent on the specific anions.Two examples of N-heterocyclic cations and triplet emitters demonstrate the general applicability of using anions to regulate photoluminescence. By altering the anions, reverse intersystem crossing rate can be tuned to tailor the desired photophysical behaviour. The showcasing of a co-crystal also expands the scope of this strategy and fine-tuning of TADF and RTP can be realized through co-crystallization. Previously reported cases utilized only the heavy-atom effect from halogen anions to achieve RTP. A key feature here is that the cation is involved in the low excited state transitions through interaction with the triplet emitters. These counter-intuitive results also open up an avenue for developing luminescent materials through anion tuning. Moreover, this approach enhances the design principles behind TADF and RTP materials and presents a molecular model to understand the interplay of RTP and TADF.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. O. W. acknowledges the Natural Sciences and Engineering Research Council of Canada for funding. D. C. acknowledges financial support from the Spanish Government MINECO/FEDER (project PID2019-109555GB-I00) and the Eusko Jaurlaritza (Basque Government, projects IT1254-19 and PIBA19-0004). C. C. is indebted to the European Research Council (ERC-2016-STG-714870) for a postdoctoral contract. We acknowledge the Laboratory for Advanced Spectroscopy and Imaging Research (LASIR) for facilities access and would like to thank Dr Saeid Kamal for assistance with spectroscopic studies and Anita Lam for pXRD measurements.Notes and references
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- Q. Li, Y. Tang, W. Hu and Z. Li, Small, 2018, 14, 1801560 CrossRef PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS PubMed.
- X. Yang and D. Yan, Chem. Sci., 2016, 7, 4519–4526 RSC.
- X. Yang and D. Yan, Adv. Opt. Mater., 2016, 4, 897–905 CrossRef CAS.
- R. Gao, D. Yan, D. G. Evans and X. Duan, Nano Res., 2017, 10, 3606–3617 CrossRef CAS.
- Z. Lin, R. Kabe, K. Wang and C. Adachi, Nat. Commun., 2020, 11, 191 CrossRef CAS PubMed.
- Z. Xu, C. Climent, C. M. Brown, D. Hean, C. J. Bardeen, D. Casanova and M. O. Wolf, Chem. Sci., 2021, 12, 188–195 RSC.
- J. S. Ward, R. S. Nobuyasu, A. S. Batsanov, P. Data, A. P. Monkman, F. B. Dias and M. R. Bryce, Chem. Commun., 2016, 52, 2612–2615 RSC.
- T. Serevičius, T. Bučiūnas, J. Bucevičius, J. Dodonova, S. Tumkevičius, K. Kazlauskas and S. Juršėnas, J. Mater. Chem. C, 2018, 6, 11128–11136 RSC.
- N. Gan, X. Wang, H. Ma, A. Lv, H. Wang, Q. Wang, M. Gu, S. Cai, Y. Zhang, L. Fu, M. Zhang, C. Dong, W. Yao, H. Shi, Z. An and W. Huang, Angew. Chem., Int. Ed., 2019, 58, 14140–14145 CrossRef CAS PubMed.
- W. Li, Q. Huang, Z. Mao, J. Zhao, H. Wu, J. Chen, Z. Yang, Y. Li, Z. Yang, Y. Zhang, M. P. Aldred and Z. Chi, Angew. Chem., Int. Ed., 2020, 59, 3739–3745 CrossRef CAS PubMed.
- Y. Wang, J. Yang, Y. Tian, M. Fang, Q. Liao, L. Wang, W. Hu, B. Z. Tang and Z. Li, Chem. Sci., 2020, 11, 833–838 RSC.
- C. Chen, R. Huang, A. S. Batsanov, P. Pander, Y.-T. Hsu, Z. Chi, F. B. Dias and M. R. Bryce, Angew. Chem., Int. Ed., 2018, 57, 16407–16411 CrossRef CAS PubMed.
- P. Data, M. Okazaki, S. Minakata and Y. Takeda, J. Mater. Chem. C, 2019, 7, 6616–6621 RSC.
- L. Zhan, C. Yang, Z. Chen, S. Gong, Y. Xiang, F. Ni, X. Zeng, G. Xie and C. Yang, Angew. Chem., Int. Ed., 2019, 58, 17651–17655 CrossRef CAS PubMed.
- X. Cai, Z. Qiao, M. Li, X. Wu, Y. He, X. Jiang, Y. Cao and S.-J. Su, Angew. Chem., Int. Ed., 2019, 58, 13522–13531 CrossRef CAS PubMed.
- F. Hu, G. Zhang, C. Zhan, W. Zhang, Y. Yan, Y. Zhao, H. Fu and D. Zhang, Small, 2015, 11, 1335–1344 CrossRef CAS PubMed.
- J. Wang, X. Gu, P. Zhang, X. Huang, X. Zheng, M. Chen, H. Feng, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 16974–16979 CrossRef CAS PubMed.
- J. Wang, X. Gu, H. Ma, Q. Peng, X. Huang, X. Zheng, S. H. P. Sung, G. Shan, J. W. Y. Lam, Z. Shuai and B. Z. Tang, Nat. Commun., 2018, 9, 2963 CrossRef PubMed.
- P. She, Y. Yu, Y. Qin, Y. Zhang, F. Li, Y. Ma, S. Liu, W. Huang and Q. Zhao, Adv. Opt. Mater., 2020, 8, 1901437 CrossRef CAS.
- G. Chen, S. Guo, H. Feng and Z. Qian, J. Mater. Chem. C, 2019, 7, 14535–14542 RSC.
- Z. Xu, Q. T. Liu, X. Wang, Q. Liu, D. Hean, K. C. Chou and M. O. Wolf, Chem. Sci., 2020, 11, 2729–2734 RSC.
- K. Leduskrasts and E. Suna, RSC Adv., 2019, 9, 460–465 RSC.
- Z. Zhang, L. Tang, X. Fan, Y. Wang, K. Zhang, Q. Sun, H. Zhang, S. Xue and W. Yang, J. Mater. Chem. C, 2018, 6, 8984–8989 RSC.
- X. Wang, H. Ma, M. Gu, C. Lin, N. Gan, Z. Xie, H. Wang, L. Bian, L. Fu, S. Cai, Z. Chi, W. Yao, Z. An, H. Shi and W. Huang, Chem. Mater., 2019, 31, 5584–5591 CrossRef CAS.
- A. Kretzschmar, C. Patze, S. T. Schwaebel and U. H. F. Bunz, J. Org. Chem., 2015, 80, 9126–9131 CrossRef CAS PubMed.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- Q. Zhang, H. Kuwabara, J. William, J. Potscavage, S. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070–18081 CrossRef CAS PubMed.
- S. Cai, H. Shi, Z. Zhang, X. Wang, H. Ma, N. Gan, Q. Wu, Z. Cheng, K. Ling, M. Gu, C. Ma, L. Gu, Z. An and W. Huang, Angew. Chem., Int. Ed., 2018, 130, 4069–4073 CrossRef.
- Y. Tao, R. Chen, H. Li, J. Yuan, Y. Wan, H. Jiang, C. Chen, Y. Si, C. Zheng, B. Yang, G. Xing and W. Huang, Adv. Mater., 2018, 30, 1803856 CrossRef PubMed.
- L. Zhang, M. Li, Q.-Y. Gao and C.-F. Chen, Chem. Commun., 2020, 56, 4296–4299 RSC.
- L. Xu, K. Zhou, H. Ma, A. Lv, D. Pei, G. Li, Y. Zhang, Z. An, A. Li and G. He, ACS Appl. Mater. Interfaces, 2020, 12, 18385–18394 CrossRef CAS PubMed.
- R. Giri, Spectrochim. Acta, Part A, 2004, 60, 757–763 CrossRef.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- J. Yang, H. Gao, Y. Wang, Y. Yu, Y. Gong, M. Fang, D. Ding, W. Hu, B. Z. Tang and Z. Li, Mater. Chem. Front., 2019, 3, 1391–1397 RSC.
- S. Tian, H. Ma, J. Li, X. Wang, A. Lv, H. Shi, Y. Geng, F. Liang, Z.-M. Su, Z. An and W. Huang, Angew. Chem., Int. Ed., 2019, 58, 6645–6649 CrossRef CAS PubMed.
- D. Yan and D. G. Evans, Mater. Horiz., 2014, 1, 46–57 RSC.
- B. Zhou and D. Yan, Adv. Funct. Mater., 2019, 29, 1807599 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, electronic structure calculations details. CCDC 2024261–2024267, 2024270 and 2052371. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ma00314c |
| This journal is © The Royal Society of Chemistry 2021 |