
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Near-canopy horizontal concentration heterogeneity of semivolatile oxygenated organic compounds and implications for 2-methyltetrols primary emissions†
Jianhuai
Ye
 *a,
Carla E.
Batista
bc,
Patricia C.
Guimarães
bc,
Igor O.
Ribeiro
bc,
Charles
Vidoudez
d,
Rafael G.
Barbosa
c,
Rafael L.
Oliveira
c,
Yongjing
Ma
ae,
Kolby J.
Jardine
f,
Jason D.
Surratt
g,
Alex B.
Guenther
h,
Rodrigo A. F.
Souza
bc and
Scot T.
Martin
*ai
*a,
Carla E.
Batista
bc,
Patricia C.
Guimarães
bc,
Igor O.
Ribeiro
bc,
Charles
Vidoudez
d,
Rafael G.
Barbosa
c,
Rafael L.
Oliveira
c,
Yongjing
Ma
ae,
Kolby J.
Jardine
f,
Jason D.
Surratt
g,
Alex B.
Guenther
h,
Rodrigo A. F.
Souza
bc and
Scot T.
Martin
*ai
aSchool of Engineering and Applied Sciences, Harvard University, Cambridge, Massachusetts 02138, USA. E-mail: scot_martin@harvard.edu; jye@seas.harvard.edu
bPost-graduate Program in Climate and Environment, National Institute of Amazonian Research, Manaus, Amazonas 69060-001, Brazil
cSchool of Technology, Amazonas State University, Manaus, Amazonas 69065-020, Brazil
dHarvard Center for Mass Spectrometry, Harvard University, Cambridge, Massachusetts 02138, USA
eCollege of Atmospheric Sciences, Lanzhou University, Lanzhou, 730000, China
fClimate and Ecosystem Sciences Division, Earth Science Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
gDepartment of Environmental Sciences and Engineering, Gillings School of Global Public Health, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA
hDepartment of Earth System Science, University of California, Irvine, California 92697, USA
iDepartment of Earth and Planetary Sciences, Harvard University, Cambridge, Massachusetts 02138, USA
First published on 21st December 2020
Abstract
Semivolatile oxygenated organic compounds (SV-OVOCs) are important atmospheric species, in particular for the production and chemistry of atmospheric particulate matter and related impacts on air quality and climate. In this study, SV-OVOCs were collected in the horizontal plane of the roughness layer over the tropical forest in the central Amazon during the wet season of 2018. A sampler mounted to a copter-type, hovering unmanned aerial vehicle was used. Underlying the collection region, a plateau forest transitioned into a slope forest across several hundred meters. The concentrations of pinonic and pinic acids, which are monoterpene oxidation products, had no statistical difference over the two forests. By comparison, across the study period, differences in the concentration of 2-methyltetrols, which are products of isoprene oxidation, ranged from −70% to +480% over the two forests. The chemical lifetime of 2-methyltetrols in the atmosphere is sufficiently long that heterogeneity in the isoprene emission rate from the two forests followed by atmospheric oxidation does not explain the concentration heterogeneity of 2-methyltetrols. Standing waves and local meteorology also cannot account for the heterogeneity. Of the possibilities considered, the most plausible explanation is the direct emission from the forest of 2-methyltetrols produced through biological processes within the plants. Under this explanation, the rate of direct SV-OVOC emissions should be modulated by forest type and related environmental stressors. Direct emissions of SV-OVOCs should be more broadly considered for constraining and improving models of atmospheric composition, transport, and chemistry over tropical forests.
Environmental significanceSemivolatile oxygenated organic compounds (SV-OVOCs) are key precursors of atmospheric particulate matter and play crucial roles in regional and global climate. Over forests, SV-OVOCs are largely considered as produced through in situ atmospheric oxidation and functionalization of volatile organic compounds emitted from forests. The evolution of SV-OVOCs in the atmosphere is, therefore, treated in that way in models of chemical transport and climate. Based on field observations and model simulations, the results herein highlight the possibility of direct emission from the forests of 2-methyltetrols, which is a family of SV-OVOC products from isoprene oxidation. The results also demonstrate how mapping out near-canopy SV-OVOC heterogeneity can be an effective strategy for isolating atmospheric SV-OVOC secondary production from direct primary forest emissions. |
1. Introduction
Oxygenated volatile organic compounds (OVOCs) emitted from vegetation are important components in forest ecosystem functioning, atmospheric chemistry, and climate.1–4 They have an annual estimated emission of around 200 Tg and contribute a significant part of the organic carbon flux into the atmosphere.5,6 Among biogenic OVOCs, methanol is the most emitted, estimated as 100 Tg per year globally. Some other important OVOCs include acetone, ethanol, short-chain aldehydes and acids, and long-chain terpenoid compounds.5These biogenic OVOCs are typically metabolic by-products or intermediates of biochemical processes in plants.1–3 As such, the emission rates are variable, both with plant and forest type as well as environmental stress (e.g., pathogen attack, wounding, exposure to high ozone concentrations, flooding, and so on).1–4 As an example of the latter, ethanol emissions from plant roots greatly increase when flooding induces anoxic conditions and fermentation.4 As another example, emissions of C6- and C9-aldehydes and alcohols such as hexenal and nonadienol, which are produced by lipoxygenase action in plants, increase during times of plant stress.2 Emission of a suite of OVOCs (i.e., 3-methyl furan and 2-methyl-3-buten-2-ol) was also observed from plants, such as mango branches, in response to environmental stress, such as high temperature or high light intensity.7
More recently, direct emissions of OVOCs of isoprene oxidation products and related organic species have been reported.7–10 Direct emissions are also called primary emissions. After methane, isoprene is the most abundant volatile organic compound (VOC) emitted by vegetation globally.5 A link between the rate of isoprene emission and the rate of photosynthesis is a common feature of many different tree genera in tropical forests, although not all.11,12 Isoprene is important both for anabolism in plants and for emission and subsequent chemistry in the atmosphere.11,12 Isoprene also has an important role in plant thermotolerance. Several different antioxidant pathways are associated with isoprene, such as the consumption of the excess products of photosynthesis (i.e., ATP and NADPH) through a tight coupling of isoprene biosynthesis with photochemical electron transport, direct antioxidant reactions between isoprene and reactive oxygen species, and the enhancement of the physical stability of membranes.8,13,14 Pyruvate-2-13C leaf and branch feeding experiments show that methyl vinyl ketone (MVK) and methacrolein (MACR) can be emitted from plants to the atmosphere.8 These species had previously been considered as produced through the atmospheric oxidation of isoprene. This mechanism is referred to as a secondary atmospheric source, as distinguished from the primary emissions from the forest to the atmosphere. Reactions between ozone and diterpenes stored in glandular trichomes of plants (e.g., tobacco) can also lead to direct emissions of MVK.9 Cappellin et al.10 likewise showed that methyl ethyl ketone (MEK), which had been considered mostly an anthropogenic species, can also be produced via in-plant transformation of MVK and then released from the plant into the atmosphere.
Of OVOCs, semivolatile oxygenated organic compounds (SV-OVOCs) are a further sub-classification. SV-OVOCs are defined as OVOC species that have a vapor pressure between 10 and 10−10 Pa.15 Over forests, SV-OVOCs are largely regarded as the secondary products of atmospheric oxidation. More specifically, they are produced through multi-generation oxidation by ozone and hydroxyl radical in the atmosphere of VOCs previously emitted from the forest.16 The SV-OVOCs in turn are key precursors of atmospheric particulate matter (PM).17 Submicron PM can regulate cloud properties over forests, among other features, and play crucial roles in regional and global climate systems.18 The atmospheric reactions to produce SV-OVOCs can take place in the gas phase and in the condensed phase. The latter is represented in the atmosphere by humid aerosol particles and cloud droplets.
Although SV-OVOCs are widely regarded as produced in situ in the atmosphere and simulated in that way in chemical transport models of atmospheric chemistry, increasing evidence shows that SV-OVOCs can also be biologically produced. Biological emissions may thus be another important category of direct emissions of SV-OVOCs. For example, semivolatile fatty acids, fatty alcohols, and long-chain n-alkanals (e.g., C12 to C26 aldehydes) are produced in plants and subsequently released in some fraction to the atmosphere, both as vegetative detritus in the form of coarse particles and as evaporated species from plant waxes if sufficiently volatile.19–22
For isoprene, the 2-methyltetrols family is an important class of SV-OVOCs. 2-Methyl-D-erythritol, which is one of the diastereomers of 2-methyltetrols, is an isoprene metabolism byproduct in plants of deoxyxylulose phosphate and methylerythritol phosphate pathways.23–27 This compound and 2-methylthreitol are diastereomers that together make up the 2-methyltetrols family. The 2-methyltetrols family has four members, including the two aforementioned diastereomers, each of which also has two enantiomers (i.e., 2-methyl-D-threitol, 2-methyl-L-threitol, 2-methyl-D-erythritol, and 2-methyl-L-erythritol). This family is identified as produced by isoprene oxidation in the atmosphere.28,29 Specifically, the heterogeneous chemical reactions of isoprene epoxydiols (IEPOX, first-generation oxidation products of isoprene) in acidic sulfate aerosol particles can produce 2-methyltetrols and the 2-methyltetrols organosulfates.28,30,31 González et al.32 performed chiral analysis of the 2-methyltetrols family for atmospheric PM collected on filters during the wet and dry seasons of the central Amazon. Non-biological atmospheric oxidation of isoprene mainly produces racemic mixtures whereas biological metabolism produces non-racemic mixtures. This principle allowed the authors to conclude that direct emissions of 2-methyltetrols from the forest accounted under some conditions for more than half of the atmospheric PM concentrations of this species.33 Cahill et al.34 observed a strong correlation between the concentrations of 2-methyltetrols and sugars, such as glucose and fructose, over a temperate coniferous forest, also suggesting that 2-methyltetrols, to some extent, may arise from similar biological sources as the sugars, which are biomarkers for airborne fungal spores.35,36
Assessment of the relative importance of primary SV-OVOC emissions compared to secondary production in the atmosphere is technically and scientifically challenging. Leaf-level measurements have demonstrated emission and quantified the rate for specific plants across a range of environmental conditions.7,9 Despite this success, scaling from leaf, to plant, and further to forest has large uncertainties because of immense forest biodiversity, especially in tropical regions. Shifting environmental stressors with seasons is a further complicating factor.
An alternative observational strategy for isolating the contribution of in situ atmospheric production of SV-OVOCs from direct emissions of the underlying forest could be to map out the near-canopy horizontal heterogeneity in the concentration of SV-OVOCs. The assumption for this strategy is that uniform atmospheric production should not contribute to near-canopy heterogeneity of SV-OVOC concentrations because SV-OVOCs have atmospheric lifetimes of several hours to days. By comparison, different emission rates with changes in underlying forest composition and environmental stressors can induce near-canopy heterogeneity of SV-OVOC concentrations. Even so, this observational strategy, including its possible limitations, remains not fully tested and evaluated in real-world practice, largely because of the absence of appropriate platforms to investigate the spatiotemporal variations of SV-OVOCs at intermediate scales over forests. For example, traditional tower measurements are excellent for long-term observations at fixed forest locations (e.g., usually over plateau forests characterized by firm soils),37 yet by being in one location towers do not capture the variability inherent over the surrounding heterogeneous forests. Aircraft measurements obtain valuable chemistry data sets, but in a heterogeneous landscape these data sets necessarily average across different types of landcover given the height and the speed of the aircraft.38 Satellite-based sensors have unprecedented views across 10's of km, yet they are limited to just a few detectable organic species.39 The emerging technology of hovering unmanned aerial vehicles (UAVs) has the capability to collect data sets at intermediate scales and thus to map out the heterogeneity of SV-OVOC concentrations in the roughness layer just over the forest canopy.40,41 Those data in turn should lead to insights into direct emissions of SV-OVOCs. This strategy, when developed, well tested, and understood, could evolve into a good complement to the scaling-up approach of leaf-level emission rates.
To these ends, in this study a hovering UAV was equipped with sorbent cartridges for SV-OVOCs and flown in the roughness layer over a forest. The objective of this study is to investigate and quantify the concentration distribution of SV-OVOCs and provide insights into the relative importance of primary SV-OVOC emissions compared to their secondary production in the atmosphere. In this regard, near-canopy concentrations of 2-methyltetrols (isoprene oxidation products) and pinonic and pinic acids (monoterpene oxidation products) were examined. The research was conducted in the central Amazon across four weeks during the wet season from 20 February to 15 March 2018. Sampling was performed over two different forests (namely, “plateau, P” and “slope, S” forests) at the Adolfo Ducke Forest Reserve, Manaus, Brazil. The collected SV-OVOCs were analyzed off-line by thermal-desorption gas chromatography coupled with on-line derivatization. VOCs were also analyzed, and this complementary work was previously reported in Batista et al.40 Herein, the SV-OVOC analysis is presented. The atmospheric observations are employed to probe heterogeneity in SV-OVOC concentration in the near-canopy atmosphere and, in combination with a two-dimensional gradient transport model of SV-OVOC production and transport, to probe the contribution of direct emissions.
2. Methods
2.1 Flight platform and sampling locations
A sampler for SV-OVOCs was mounted to a hexacopter hovering-type UAV (DJI Matrice 600). The custom-built sampler used adsorbent cartridges, and further details can be found in McKinney et al.41 Analysis of the disturbance of the local concentration field by the UAV propellers was also discussed therein, and this disturbance was not significant for the conditions of the present study. The UAV flights took place at the Adolfo Ducke Forest Reserve located on the northern outskirts of Manaus city (Brazil; Fig. 1). Two sampling locations, shown as points P and S in Fig. 1 and separated by 711 m, corresponded to two different underlying forests.40 The forest under location P resided in a plateau region with well-drained soils. The forest under location S populated a slope between plateau and valley regions. The plants making up the plateau, slope, and valley forests differed considerably (Section 3.2.3). Canopy height varied from 25 to 35 m for both locations P and S. The UAV was launched and recovered atop a tower at location P at the Manaus Botanical Gardens within the reserve. To maintain visual contact between the UAV operator and the UAV, sampling at both locations was conducted at the same absolute altitude, which was 15 m above local canopy at location P and 47 m above it at location S. For the four weeks of measurements, the variability in the meteorological parameters, such as temperature, relative humidity, wind speed, and wind direction, was small during the time periods that samples were obtained (Table S1†). The implication is that meteorology did not significantly contribute to the variability of relative SV-OVOC concentrations between locations P and S. | ||
| Fig. 1 Sampling site at the Adolfo Ducke Forest Reserve. The left panel shows the location of the reserve on the northern outskirts of Manaus city, Amazonas, Brazil. The right panel shows the local topography in the region of the two sampling sites. Location P is over a plateau forest. Location S is over a slope forest. | ||
2.2 Sampling methods
SV-OVOC samples were collected by drawing atmospheric air through adsorbent cartridges (Markes International, Inc. C2-AXXX-5149). The cartridges were packed with a combination of Tenax TA and Carbograph 5TD. These adsorbents were hydrophobic and suitable for atmospheric sampling at a wide range of relative humidity. They collected a wide range of organic compounds from carbon numbers of C3 through C30 (ref. 42) based on species affinity to the adsorbents. Prior to use, the cartridges were pre-conditioned at 320 °C by passing 0.1 L min−1 ultra-pure nitrogen (Airgas) for 8 h. During use on the hovering UAV, the sampling rate into a cartridge was 0.15 to 0.20 L min−1. This sampling rate allowed efficient collection of SV-OVOCs while maximizing the total sampling volume and thereby the total collected SV-OVOC mass. No filter was installed prior to the sampling inlet. Therefore, both gas- and particle-phase SV-OVOCs were collected by the cartridges.UAV-facilitated samples were collected by hovering at location S in four sequential time periods of a day, as follows: 09:00–10:00, 10:10–11:10, 11:20–12:20, and 12:30–13:30, all in local time (4 h earlier than UTC). Two flights were carried out in each time period. Within a flight, a sampling duration of 2.5 min was used for VOCs for one set of cartridges (work reported in Batista et al.40) and 17.5 min for SV-OVOCs for another set of cartridges. Specific sampling information, including time period of collection for each sample, appeared in Batista et al.,40 which focused on VOC analysis from the first set of cartridges. SV-OVOC collection was simultaneously performed at location P by an operator using an air sampling pump (GilAir Plus, Gilian; 0.20 L min−1) at the top of the tower. For both locations, the same set of SV-OVOC cartridges employed in the first day in a specific time window was used repeatedly in the same time window for other days of the week until a total sample collection of >100 min (i.e., >15 L) was achieved for each cartridge. This approach ensured that sufficient SV-OVOC mass was collected on a cartridge so that off-line chromatographic analysis was possible. For comparison, multi-day filter collection of samples for organic analysis is a common approach for the Amazon forest because of the low atmospheric concentrations of many species.32,43 After sample collection, the cartridges were sealed with Swagelok fittings and kept at room temperature before chromatographic analysis in the laboratory. Although some reactive SV-OVOCs can decompose at least in part during storage and prior to analysis, the highly oxidized SV-OVOC compounds targeted in the present study were stable, as confirmed by laboratory standards (Section 2.3). Moreover, the analysis herein is based on the relative concentrations at locations S and P, and any decomposition would be similar for the samples collected at both locations and thus canceling out in the relative analysis.
2.3 Analysis by thermal desorption gas chromatography mass spectrometry (TD-GC-MS)
In the laboratory, for TD-GC-MS analysis the air samples collected on the sorbent cartridges were held at 320 °C for 20 min for thermal desorption. The desorbed SV-OVOCs were chemically derivatized by reaction with N-methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA; ≥98.5%, Sigma Aldrich) in a custom-built system. MSTFA derivatization improved the volatility and thermal stability of the targeted compounds, allowing efficient separation along the GC column of the analytes. For derivatization, helium gas flowed over the headspace of MSTFA, and a dilution flow with pure helium was added to adjust the MSTFA concentration injected into the system. During desorption and derivatization, the SV-OVOC derivatives were re-concentrated at 0 °C in a cold trap.The custom-built system was coupled to chemical analysis by gas chromatography for analyte separation and electron-ionization mass spectrometry for quantification (Quattro Micro GC-MS; Waters). After collection in the trap at 0 °C, the SV-OVOC derivatives were subsequently transferred to the GC by heating the trap to 320 °C. This step also converted 2-methyltetrols organosulfates into 2-methyltetrols,30 meaning that the 2-methyltetrols family reported herein is inclusive of both atmospheric 2-methyltetrols and atmospheric 2-methyltetrols organosulfates. The GC inlet was operated at a split-mode ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 during the analysis. The column flow was 2 mL min−1. The temperature profile for the GC column was as follows: 3 min at 40 °C, 4 °C min−1 to 170 °C, 25 °C min−1 from 170 °C to 300 °C, and 300 °C for 5 min. The mass spectrometer began sampling 16 min after the start of the GC program to avoid contamination from high concentrations of MSTFA (i.e., which elutes early). The sampling delay limited the GC-MS analysis to chemical species having retention times greater than 16 min, corresponding to a retention index greater than that of a C14n-alkane.
:1 during the analysis. The column flow was 2 mL min−1. The temperature profile for the GC column was as follows: 3 min at 40 °C, 4 °C min−1 to 170 °C, 25 °C min−1 from 170 °C to 300 °C, and 300 °C for 5 min. The mass spectrometer began sampling 16 min after the start of the GC program to avoid contamination from high concentrations of MSTFA (i.e., which elutes early). The sampling delay limited the GC-MS analysis to chemical species having retention times greater than 16 min, corresponding to a retention index greater than that of a C14n-alkane.
Authentic standards were used for identification and calibration of 2-methyltetrols, cis-pinonic acid (98%, Sigma Aldrich), and pinic acid (99%, Sigma Aldrich). A 50:50 mix of 2-methylthreitol and 2-methylerythritol, prepared by the method of Bondy et al.,44 was used for the 2-methyltetrols calibration. Standards solutions (1 μL) were prepared in acetonitrile (HPLC grade, Sigma Aldrich) and injected into a flow of helium at 100 mL min−1. This flow was collected on a cartridge for 5 min. The helium flow carried away the acetonitrile, which was not retained by the sorbent in the cartridge.
Internal standards of lauric-D23-acid (≥98%, Sigma Aldrich) and 13C6-adipic acid (99%, Cambridge Isotope Laboratories, Inc.) were used to correct for any loss of analyte during the analysis sequence of thermal desorption, derivatization, sample transfer, and ionization. This approach improved the precision of quantitative analysis for the TD-GC-MS system. These compounds have different volatilities and different numbers of carboxyl functional groups. As a result, their co-use tested the analysis sequence for consistency across many compounds. Fig. S3† shows a 1:1 line of integrated peak areas of authentic analytes when adjusted for each of the two different internal standards for concentrations of 5 to 20 μM. The correlation coefficient of 0.99 confirms the effectiveness of the analysis sequence across the range of tested compounds. Based on the observed reproducibility in laboratory controls using the authentic standards and the internal standards, as well as taking into account the flow accuracy of the UAV-based cartridge sampler, the overall propagated uncertainty for two-sigma accuracy was better than 30% for the analyte concentrations, starting from SV-OVOC sampling over the forest through TD-GC-MS analysis in the laboratory.
3. Results and discussion
3.1 SV-OVOC concentrations at locations P and S
Targeted semivolatile organic compounds included 2-methylthreitol, 2-methylerythritol, pinonic acid, and pinic acid. These various SV-OVOCs were quantified by selecting the characteristic ion fragments of their trimethylsilyl derivatives in the GC mass spectra. Specifically, signal intensities at m/z 129, m/z 171, and m/z 219 were used to quantify pinic acid, pinonic acid, and 2-methyltetrols, respectively (Fig. S1†). Although m/z 219 was used for both 2-methylthreitol and 2-methylerythritol, their trimethylsilyl derivatives had different retention times on the GC column. Moreover, 2-methyltetrols organosulfates also contributed to the 2-methyltetrols signals from the decomposition of organosulfates during the high-temperature desorption step.30 The 2-methyltetrols family is thus operationally defined herein as the combined group of 2-methyltetrols and 2-methyltetrols organosulfates. In the atmosphere, pinonic and pinic acids are typically associated with monoterpene oxidation.45 In addition to the targeted SV-OVOCs, peaks for other derivatized oxygenated organic compounds also appeared in the chromatograms, such as for long-chain fatty acids (e.g., tetradecanoic acid), dicarboxyl acids (e.g., succinic acid), and polyols (e.g., levoglucosan). These compounds were previously observed for the Amazon forest,43 but quantification with authentic standards was beyond the scope of the current study.The SV-OVOC concentrations at locations P and S are plotted in Fig. 2A by hour for each week of measurements (Table S2†). Between the two locations, the concentrations of pinonic and pinic acids had no difference within the experimental uncertainty of 30% for the entire data set (Table S2†). In contrast, the concentration of the 2-methyltetrols family at location P differed beyond the experimental uncertainty with that of location S for 8 of the 15 samples. Across the data set, the concentrations at location S differed by −70% to +480% from those at location P (Fig. 2A).
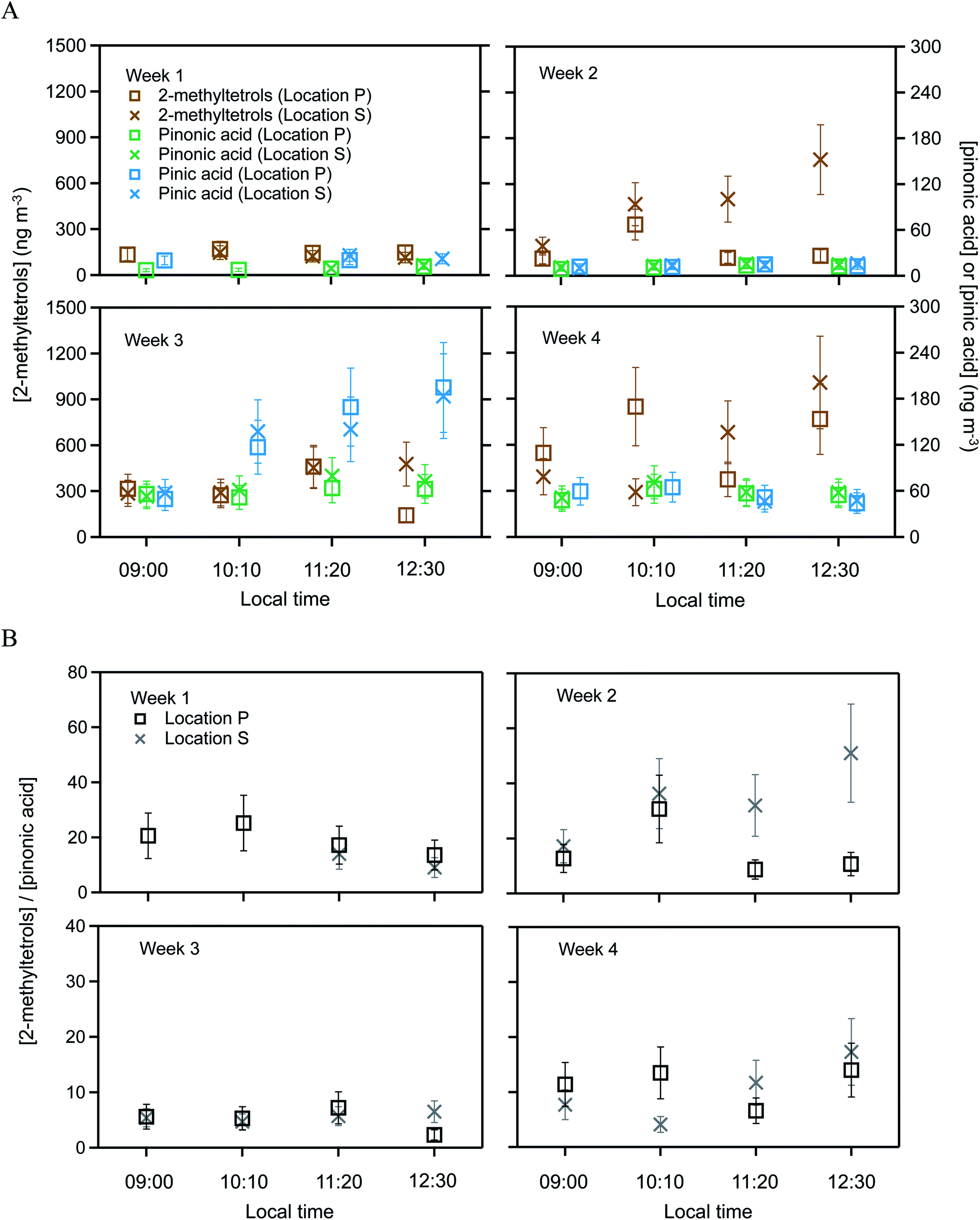 | ||
| Fig. 2 SV-OVOC concentrations across the study period. (A) Concentrations of 2-methyltetrols, pinonic acid, and pinic acid over the plateau forest at location P and the slope forest at location S. (B) Ratios of the concentrations of 2-methyltetrols and pinonic acid. Error bars on the data points in Panel A represent 30% experimental uncertainty. Error bars on the points in Panel B are the propagated uncertainties from concentration measurements of 2-methyltetrols and pinonic acid. Data are plotted for each hourly sampling period of each week. Local time was 4 h earlier than UTC. Plotted concentrations are listed in Table S2.† The 2-methyltetrols concentration is the sum of 2-methylthreitol concentration and 2-methylerythritol concentration. For clarity of presentation, the data markers for 2-methyltetrols and pinic acid are offset along the abscissa to the left and to the right in Panel A, respectively. Within experimental uncertainty (i.e., <30%), the concentrations of pinonic and pinic acids had no difference between the two locations over the four weeks. By comparison, the concentrations of the 2-methyltetrols family at location P differed beyond experimental uncertainty (i.e., >30%) with that of location S for 8 of the 15 samples. | ||
The concentration ratio of 2-methyltetrols to pinonic acid is plotted in Fig. 2B. Similar results are obtained for the concentration ratio of 2-methyltetrols to pinic acid, and the plot is omitted herein. In an ideal scenario of constant chemical conditions and emission rates in constant proportion between the chemical species, this ratio should remain unchanged over time and have the same value at both locations P and S. The value of the denominator (i.e., the concentration of pinonic acid) is observed as the same at both locations even as the absolute concentration varies over time (Fig. 2A), and it can thus serve as a normalization factor for effects of shifting meteorology (i.e., dilution) on the absolute concentrations. At a single location, such as location P, the ratio varied from 2 to 51 across the different weeks. Given that this ratio acts as a normalization factor for dilution, precursor emissions, and the strength of atmospheric oxidation, the implication to account for this variability is that the value of the numerator has a component of direct emissions (Section 3.2). Furthermore, the trends in Fig. 2A and B are similar. The ratios at location S were −70% to +380% compared to those at location P across the four weeks of measurements (Fig. 2B). Error propagation of 30% uncertainty in the numerator and in the denominator suggests that this ratio should be banded by −40% to +40%. Given that the observations exceed this range in uncertainty, the implication is that the direct emissions differ between the two locations.
3.2 Possible explanations of SV-OVOC 2-methyltetrols heterogeneity
Several factors could contribute to the heterogeneity of 2-methyltetrols concentrations between locations P and S. Different emission rates of precursor VOC compounds (i.e., isoprene) from the different forests underlying locations P and S, distinct deposition rates to the forests underlying the two locations due to differences in turbulent mixing over each canopy, and different direct emission rates from the two underlying forests are all possible contributing factors.A two-dimensional gradient transport model (eqn (1)) is used to aid in the examination of these possible factors. The model resolves atmospheric processes by taking into consideration emission and deposition rates, longitudinal advection, vertical convection, and chemical reaction rates of VOCs (e.g., isoprene) and corresponding SV-OVOCs (e.g., 2-methyltetrols). The governing equation is as follows:
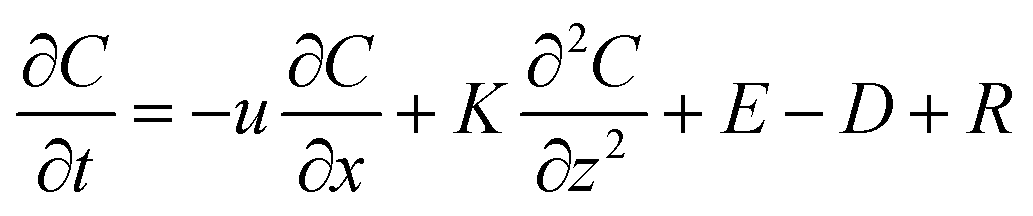 | (1) |
3.2.1 Non-uniform VOC emission rates
Concurrent VOC measurements and analysis by Batista et al.40 observed weekly-average differences of −64% to −31% in isoprene concentrations between locations S and P. For an overall monthly-average difference of −44%, a difference in isoprene emission rates of 220% to 330% between the two underlying forests was implied to fully explain this concentration difference. The hypothesis explored in this section is that the uneven distribution of isoprene concentration could lead to differences in the production and hence the concentration of 2-methyltetrols over the two forests.Simulations 1 and 2 in Table 1 consider this possibility. Simulation 1 is performed for a difference in emission rate of 200% between the two forests (i.e., EP = 200% ES) and simulation 2 for a difference of 300%. The baseline emission rate of isoprene is from the Model of Emissions of Gases and Aerosols from Nature (MEGAN, version 2.1) for tropical forests.5 The isoprene concentrations in simulations 1 and 2 at locations P and S differ by −34% and −36% (Table 1 and Fig. 3A), respectively, and the concentrations and the differences are within the range of concurrent observations at these locations (i.e., 4.4 ± 1.9 ppb at location P and 2.4 ± 2.0 ppb at location S, as presented in Batista et al.40). By comparison, the simulated 2-methyltetrols concentrations are 157 ng m−3 at location P and 153 ng m−3 at location S for both simulations 1 and 2. These concentrations are within the range of the observations (Table S2†).
| Isoprene emission rate (ppb m s−1) | 2-Methyltetrols emission rate (ppb m s−1) | 2-Methyltetrols deposition rate (m s−1) | Eddy diffusivity (m2 s−1) | Isoprene concentration (ppb) | 2-Methyltetrols concentration (ng m−3) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P | S | U | P | S | U | P | S | U | P | S | Δ ¶ (%) | P | S | Δ ¶ (%) | f pri ‖ (%) | ||
| 1 | 1* | 0.5 | 0.5 | 0 | 0 | 0 | 0.03‡ | 0.03‡ | 0.03‡ | 30 to 10; 10§ | 3.98 | 2.61 | −34 | 157 | 153 | −3 | 0 |
| 2 | 1.5† | 0.5 | 0.5 | 0 | 0 | 0 | 0.03 | 0.03 | 0.03 | 30 to 10; 10 | 4.05 | 2.61 | −36 | 157 | 153 | −3 | 0 |
| 3 | 1 | 0.5 | 0.5 | 0 | 0 | 0 | 0.01 | 0.01 | 0.01 | 30 to 10; 10 | 3.98 | 2.61 | −34 | 193 | 189 | −2 | 0 |
| 4 | 1 | 0.5 | 0.5 | 0 | 0 | 0 | 0.03 | 0.03 | 0.03 | 15 to 5; 5 | 5.86 | 3.84 | −34 | 200 | 194 | −3 | 0 |
| 5 | 1 | 0.5 | 0.5 | 0 | 0 | 0 | 0.03 | 0.10 | 0.03 | 30 to 10; 10 | 3.98 | 2.61 | −34 | 157 | 128 | −18 | 0 |
| 6 | 1 | 0.5 | 0.5 | 0 | 0 | 0 | 0.01 | 0.10 | 0.01 | 30 to 10; 10 | 3.98 | 2.61 | −34 | 193 | 149 | −23 | 0 |
| 7 | 1 | 0.5 | 0.5 | 0 | 0.001 | 0 | 0.03 | 0.03 | 0.03 | 30 to 10; 10 | 3.98 | 2.61 | −34 | 157 | 170 | +8 | 10 |
| 8 | 1 | 0.5 | 0.5 | 0 | 0.005 | 0 | 0.03 | 0.03 | 0.03 | 30 to 10; 10 | 3.98 | 2.61 | −34 | 157 | 237 | +51 | 35 |
| 9 | 1 | 0.5 | 0.5 | 0 | 0.015 | 0 | 0.03 | 0.03 | 0.03 | 30 to 10; 10 | 3.98 | 2.61 | −34 | 157 | 404 | +157 | 62 |
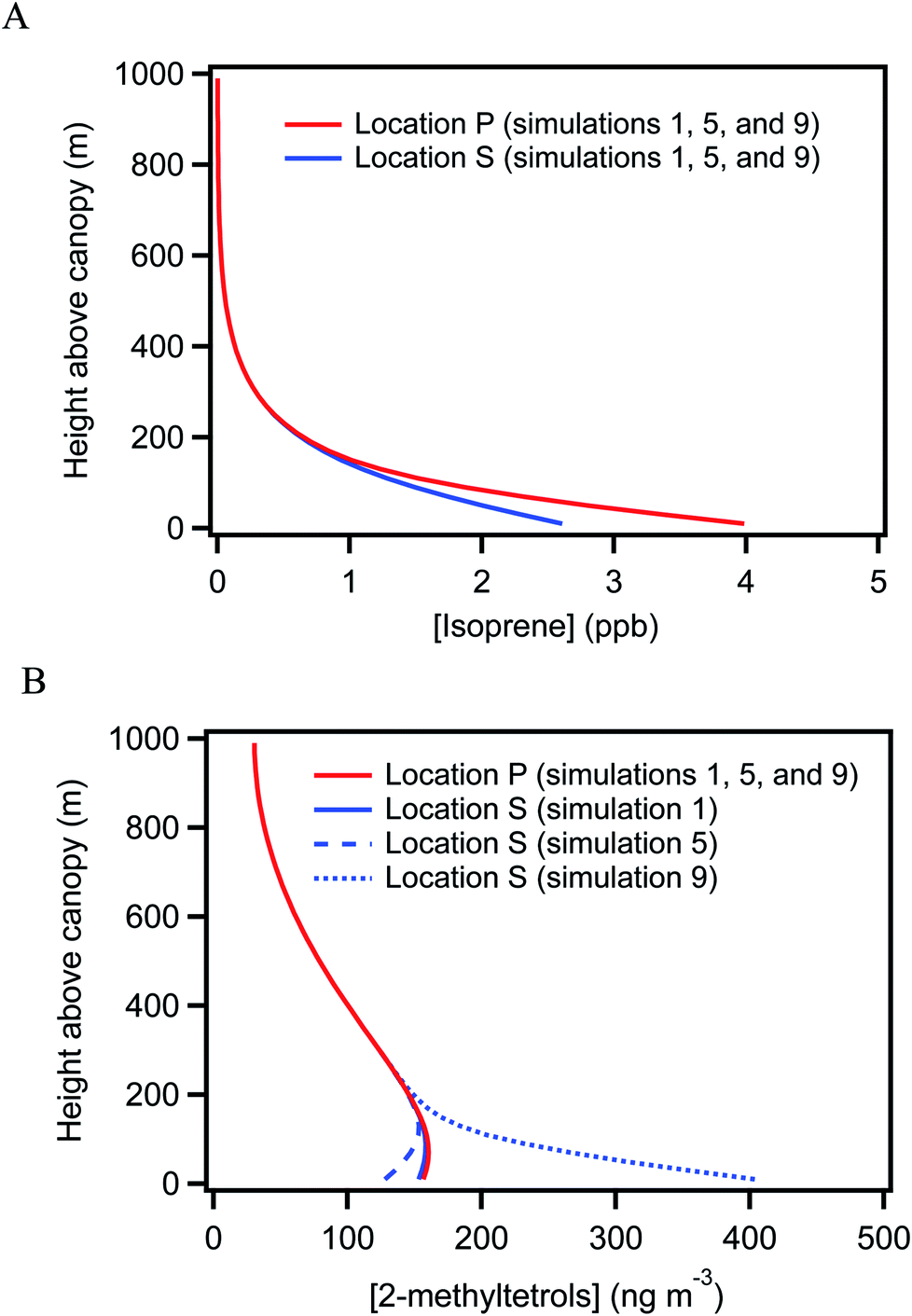 | ||
| Fig. 3 Simulated vertical concentration profiles. Vertical concentration distribution for simulations 1, 5, and 9 at locations P and S for (A) isoprene (B) 2-methyltetrols. | ||
For both simulations 1 and 2, the relative difference in 2-methyltetrols concentration over the two locations is negligible. The simulation results thus do not agree with the observations. The conclusion is that the atmospheric observations are not explained by a non-uniform emission rate of isoprene. The result can be understood by consideration of the atmospheric lifetime of 2-methyltetrols. The lifetime is estimated as 4 to 230 days for OH concentrations ranging from 0.2 to 11 × 1012 mol m−3 in the central Amazon (Table S3†). This lifetime of several days or more exceeds the atmospheric residence time in the region, and the lifetime thus anticipates a well-mixed background concentration across the central Amazon, at least in the absence of any strong local sources or sinks. Neither a decrease in the deposition rate of 2-methyltetrols by three-fold (simulation 3) nor a reduction in vertical convection by decreasing the eddy diffusivity by two-fold (simulation 4) in both locations changes the conclusion. The difference between sampling heights at 15 m above local canopy for location P and at 47 m above local canopy for location S also cannot explain the observed heterogeneity in 2-methyltetrols concentration (Fig. 3B).
A separate possibility, but one that is mathematically related and therefore also explored in this section, is that a heterogeneous distribution of oxidant concentration (e.g., OH concentration) leads to an uneven distribution of reaction rates and hence 2-methyltetrols concentration in the atmosphere (see Text S2 in ESI†). The simulations for the effects of non-uniform isoprene emission rates are mathematically similar to the effects of the possible spatial heterogeneity in oxidation rates. As such, the same conclusion is reached that the observed heterogeneity in 2-methyltetrols concentration cannot be explained in this way. In both cases, the multi-day lifetime of 2-methyltetrols implies that atmospheric turbulence evens out the 2-methyltetrols concentration across a spatial scale that is much larger than the observed heterogeneity of <1000 m. For this same reason of spatial averaging across a long lifetime, the possible presence of 2-methyltetrols organosulfates, which have atmospheric lifetimes of several days,46 does not affect the conclusion of this study (Text S3†).
In short, the various possibilities considered and ruled out in this section all come to the same conclusion that significant differences in some combination of localized sinks or sources (i.e., deposition or emission) related to the two different forests are needed to explain the observed localized heterogeneity in the 2-methyltetrols concentration.
3.2.2 Heterogeneous SV-OVOC deposition
The hypothesis explored in this section is that differential deposition of 2-methyltetrols from the gas and particle phases to the two different forest canopies might lead to significant heterogeneity in the overlying atmospheric SV-OVOC concentrations. Strong coherent eddies in the turbulent mixing within the roughness sublayer over the forest can develop at the canopy edge when air sweeps into the forest, thereby promoting the exchange of air between the forest and the overlying atmosphere.47 These eddies can increase the depositional loss of atmospheric species. The typical overlying eddies can differ with forest type because the air flow can respond to local topography over the rolling hills of the plateau, slope, and valley regions.48 Forest growth and plant structure likewise responds to this topography because of soil type and drainage, among other factors.49 In this way, heterogeneity in eddies over different forests can contribute to the heterogeneity in SV-OVOC concentrations over those forests. Another mechanism for heterogeneous deposition could be active uptake of an SV-OVOC by plants, and a dependence of uptake rate on plant type, number, and activity, which differ with forest type. Horstmann and McLachlan,49 for example, showed that lipophilic SV-OVOCs such as dibenzo-p-dioxins and dibenzofurans can be taken up from the atmosphere by plant stomata, as well as the leaf and needle cuticle, and the observed uptake rates differed between coniferous and deciduous forests.The possibility of heterogenous SV-OVOC deposition was simulated by adjusting the deposition rate (i.e., D of eqn (1)) at location S to mimic the processes of turbulent mixing, coherent eddies, and enhanced species deposition. For location P, the tested deposition rates of 2-methyltetrols over the two forests included 0.03 m s−1 (estimated for IEPOX; simulation 5) and 0.01 m s−1 (estimated for isoprene hydroxy hydroperoxides, ISOPOOHs; simulation 6).50 IEPOX and ISOPOOHs have chemical structures that are similar to those of the 2-methyltetrols family. For location S, a deposition rate of 0.10 m s−1, which is the upper limit reported in the literature for measurements over the forest canopy, was used in both simulations 5 and 6 to test for the effects of differential deposition because of strong coherent eddies.51 The obtained heterogeneity in the SV-OVOC concentration was −18% and −23% in simulations 5 and 6, respectively (Table 1). This simulated heterogeneity is not sufficient to explain the observed concentration difference of 2-methyltetrols of −70% to +480% between the two locations. Moreover, if differential deposition were the explanation, then similar percent differences would be expected in the concentrations of pinonic and pinic acids between the two locations. Such differences, however, were not observed for these acids. The conclusion is that the atmospheric observations of heterogeneity are not explained by heterogeneity in the deposition rate of 2-methyltetrols across the two forests.
3.2.3 Direct SV-OVOC emissions
The hypothesis explored in this section for explaining the observed heterogeneity in 2-methyltetrols concentration between locations P and S is two-fold: (i) forests can serve as strong local emission sources of 2-methyltetrols and (ii) the 2-methyltetrols emission rate differs among forests. The vegetation cover differed for the forests underlying locations P and S.40,52 The forest underlying location P consisted of several plant families, including Arecaceae, Caryocaraceae, Lecythidaceae, Fabaceae, Mimosaceae, and Solanaceae. The forest for location S mainly had Arecaceae, Fabaceae, and Lecythidaceae. Although there was overlap in the family supersets between the two locations, there was limited overlap in the family subsets of the actual growing plant species at each location.40,52 Biological production of OVOCs differs among plant species as well as local environmental conditions (e.g., water table). These several different factors can lead to differing 2-methyltetrols emission rates for the forests underlying locations P and S.This possibility was further evaluated by carrying out several different simulations to test if this mechanism can quantitatively account for the observed differences in 2-methyltetrols concentration. The introduction of direct emissions of 2-methyltetrols in the simulation significantly enhances the heterogeneity (Table 1 and Fig. 3). The concentration of 2-methyltetrols is +8%, to +51%, to +157% for location S compared to location P, for respective direct emission rates of 0.001, 0.005, and 0.015 ppb m s−1 underlying location S while maintaining no emission underlying location P (simulations 7, 8, and 9). The simulated heterogeneity (e.g., +157% in simulation 9) is in good agreement with the atmospheric observations (e.g., weekly-average differences of up to +176% in Table S2†). Furthermore, for these direct emission rates the modeled fraction of 2-methyltetrols contributed by direct emission to the total 2-methyltetrols concentration at location S ranges from 10% to 62% (Table 1). This result is consistent with the report by González et al.32 These several lines of evidence all support that direct emissions can explain the observed horizontal heterogeneity of 2-methyltetrols concentration in the near canopy over the forest.
3.3 Additional evidence: isomeric ratio and apparent yield of 2-methyltetrols
The methylthreitol-to-methylerythritol concentration ratio for the particle phase has helped to identify the production pathways of 2-methyltetrols in the atmosphere.53,54 For a deciduous broadleaf forest, Miyazaki et al.53 found that diastereomeric ratio was sensitive to sulfate and hydronium concentrations in atmospheric PM. The implication was that production pathways in humid aerosol particles and cloud droplets in the atmosphere must be important, at least in part, relative to reactions in the gas phase as well as direct biological emissions. In a chamber study of isoprene oxidation, under HO2-dominant conditions the ratio was sensitive to the relative concentrations of OH and ozone.54Herein, the diastereomeric ratio of the total concentration, meaning without resolving into gas-phase and particle-phase concentrations, was 0.60 ± 0.10 at location P and 0.57 ± 0.10 at location S during the study period (mean ± one-sigma variation; Table S2 and Fig. S2†). Isaacman-VanWertz et al.16 separately measured the concentrations in both phases in the wet season of the central Amazon. 2-Methylthreitol partitioned as 69% in the gas phase and 31% in the particle phase, and 2-methylerythritol partitioned as 45% in the gas phase and 55% in the particle phase. Applying these fractions to the results of the present study leads to estimated particle-phase diastereomeric ratios of 0.34 ± 0.06 at location P and 0.32 ± 0.06 at location S. These ratios are in good agreement with the results of four other studies for the central Amazon during the wet season (Table S4†). The present study was also carried out in the wet season.
For comparison, the ratio is reported to shift significantly for the dry season in the central Amazon. It ranges from 0.07 ± 0.06 for background conditions to 0.37 ± 0.06 for polluted conditions (Table S4†). The seasonal differences might be explained by shifts in the atmospheric chemistry as a result of higher concentrations of pollutants such as ozone and sulfate as well as other factors in the dry season. The results of the present study, however, suggest that another contributing factor could be that the relative direct emission rates of the 2-methylthreitol and 2-methylerythritol from forests differ between the seasons, possibly because of differing influences of environmental stress or possibly because of competing sources of direct emissions. In regard to the latter, for China Fu et al.55 observed a high correlation between the diastereomeric ratio and the concentration of levoglucosan, which is an important tracer of biomass burning. This result could suggest that biomass burning can also serve as a source of direct emissions that favors 2-methylthreitol over 2-methylerythritol.
A yield of particle-phase 2-methyltetrols from isoprene, meaning [2-methyltetrols]particle/[isoprene]reacted, can be defined. Laboratory gas-phase experiments for HO2-dominant conditions like the wet season of the Amazon estimate a yield of 0.6%.29 The measured 2-methyltetrols concentrations of this study imply apparent yields of 0.4 ± 0.3% at location P and 1.2 ± 0.8% at location S (Table S5†) based on the concurrent isoprene concentrations reported in Batista et al.40 and the gas/particle partitioning fractions of Isaacman-VanWertz et al.16 Within experimental uncertainty, the apparent yield at location P is in agreement with the laboratory-measured yield. The apparent yield at location S is higher, however. The difference in apparent yields at locations P and S is robust within the measurement uncertainty. An apparent yield can be explained as higher than the intrinsic yield of gas-phase chemistry in the vicinity of direct local emissions of 2-methyltetrols from the forest.
4. Conclusion
The emission, deposition, and chemistry of SV-OVOCs are dynamic over the heterogenous landscape of the Amazon tropical forest. In this study, the concentration and distribution of SV-OVOCs from isoprene and monoterpene oxidation were investigated in the wet season of the central Amazon with the aid of a hovering unmanned aerial vehicle. There was heterogeneity in the concentration of 2-methyltetrols over two forests that were several hundred meters apart from each other. By comparison, the concentrations of pinonic and pinic acids had no statistical difference.Results from a gradient transport model suggest that non-uniform isoprene emission over the two forests does not explain the heterogeneity of 2-methyltetrols concentration. Chemical yields are too low and atmospheric lifetimes are too long. The lifetime of 2-methyltetrols against chemical loss is on the order of a week for typical OH concentrations. Differences in deposition rates because of distinct local forest micrometeorology and associated coherent eddies can contribute to but not fully explain the magnitude of the heterogeneity in 2-methyltetrols concentration. Furthermore, other species such as pinonic and pinic acids would be expected to have differences in concentrations, too, if heterogeneity in deposition were a dominant mechanism, whereas the observations show homogeneity in the concentrations of these two acids. Simulations show that heterogeneity in the emission rate depending on the underlying forest can quantitatively account for the differences observed in the overlying atmospheric concentrations.
More generally, these results demonstrate how mapping out the near-canopy horizontal heterogeneity in the concentration of SV-OVOCs can be an effective strategy for isolating the contribution of in situ atmospheric production of SV-OVOCs from direct emissions of the underlying forest. This strategy can serve as a complementary approach for traditional bottom-up leaf-level emission measurements. In a diverse forest such as the Amazon tropical forest where direct SV-OVOC emissions may vary among plant species and families and under different environmental conditions, a comprehensive program of leaf-level measurements for accurately scaling up can become impractical. Near-canopy measurements can represent a complementary alternative approach that captures the combined effects of local forest emissions, albeit without the detailed view of leaf-level measurements.
Further development, improvement, and quantification of this strategy to infer heterogeneity in emissions based on near-canopy horizontal concentration heterogeneity is thus well motivated, and several recommendations can be considered. In particular, a complementary focus on vertical profiles of SV-OVOC concentrations at each horizontal sampling point might prove quite valuable. Simulation 1, representing an absence of emissions, shows that the 2-methyltetrols concentration changes little in the lower part of the atmospheric boundary layer of a few hundred meters, mostly because of the long lifetime of 2-methyltetrols. Many other SV-OVOCs of common interest have similarly long lifetimes. Strong deposition to the forest or strong direct emission from the forest can change the shape of the concentration profile for these SV-OVOCs (simulations 5 and 9). Collection of vertical profiles can thus be complementary in a scientific sense to horizontal mapping of SV-OVOC concentrations.
Another recommendation for future efforts is that the concentrations of strong biomarkers of direct forest emissions only, such as sugars and sugar alcohols,34,35 and the concentrations of strong chemical tracers of atmospheric secondary processes, such as inorganic nitrate and sulfate salts,56 could be simultaneously measured. In a combined data set, a correlation analysis between the SV-OVOC concentrations produced by the two mechanisms and the concentrations of species produced dominantly by one mechanism (i.e., primary or secondary) might provide additional robustness and quantification with respect to atmospheric SV-OVOCs sources. For the specific case of 2-methyltetrols, incorporation of 2-methyltetrols organosulfates in the measurement protocols could be fruitful as a strong chemical tracer of secondary processes.
Building upon earlier work for VOCs, ozone, and black carbon,40,41,57,58 the SV-OVOC findings of the present study demonstrate the capability of using UAVs as valuable mobile sampling platforms for atmospheric chemical sensing and addressing scientific unknowns in atmospheric chemistry. The heterogeneity in 2-methyltetrols concentration observed in this study in the horizontal plane of the atmospheric boundary layer of the central Amazon highlights the more general need for understanding and quantifying emissions and atmospheric process of SV-OVOCs, especially at intermediate scales.
Data availability
Results for simulations 1 to 9 of Table 1 are available at DOI: 10.7910/DVN/RROEDO. Concentrations of SV-OVOCs measured by the TD-GC-MS system are summarized in Table S2.†Author contribution
J. Y. and S. T. M. designed the research; J. Y., C. E. B., P. C. G., I. O. R., R. G. B., and R. L. O. conducted the research; J. Y. and C. V. performed the chemical analysis; J. Y. and Y. M. carried out simulations using the gradient transport model; J. Y., K. J. J., J. D. S., A. B. G., R. A. F. S., and S. T. M. analyzed the data; and J. Y. and S. T. M. wrote the paper.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was funded by the Division of Atmospheric and Geospace Sciences (AGS) of the USA National Science Foundation (AGS-1829025 and AGS-1829074). The Brazilian Federal Agency for Support and Evaluation of Graduate Education (CAPES), the Brazilian National Council for Scientific and Technological Development (CNPq), a Senior Visitor Research Grant of the Amazonas State Research Foundation (FAPEAM) (062.00568/2014 and 062.00491/2016), and the Harvard Climate Change Solutions Fund are acknowledged. Jianhuai Ye acknowledges support from a postdoctoral fellowship from the Natural Sciences and Engineering Research Council of Canada and a fellowship in Environmental Chemistry from the Dreyfus Foundation. The synthesis of 2-methyltetrols was supported through a grant from the USA National Science Foundation (AGS-1703535).References
- R. Seco, J. Peñuelas and I. Filella, Short-chain oxygenated VOCs: emission and uptake by plants and atmospheric sources, sinks, and concentrations, Atmos. Environ., 2007, 41, 2477–2499 CrossRef CAS.
- A. C. Heiden, K. Kobel, C. Langebartels, G. Schuh-Thomas and J. Wildt, Emissions of oxygenated volatile organic compounds from plants Part I: emissions from lipoxygenase activity, J. Atmos. Chem., 2003, 45, 143–172 CrossRef CAS.
- J. Kesselmeier, Exchange of short-chain oxygenated volatile organic compounds (VOCs) between plants and the atmosphere: a compilation of field and laboratory studies, J. Atmos. Chem., 2001, 39, 219–233 CrossRef CAS.
- P. Perata and A. Alpi, Plant responses to anaerobiosis, Plant Sci., 1993, 93, 1–17 CrossRef CAS.
- A. B. Guenther, X. Jiang, C. L. Heald, T. Sakulyanontvittaya, T. Duhl, L. K. Emmons and X. Wang, The model of emissions of gases and aerosols from nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 2012, 5, 1471–1492 CrossRef.
- H. B. Singh, L. J. Salas, R. B. Chatfield, E. Czech, A. Fried, J. Walega, M. J. Evans, B. D. Field, D. J. Jacob, D. Blake, B. Heikes, R. Talbot, G. Sachse, J. H. Crawford, M. A. Avery, S. Sandholm and H. Fuelberg, Analysis of the atmospheric distribution, sources, and sinks of oxygenated volatile organic chemicals based on measurements over the Pacific during TRACE-P, J. Geophys. Res.: Atmos., 2004, 109, D15S07 Search PubMed.
- K. J. Jardine, K. Meyers, L. Abrell, E. G. Alves, A. M. Yanez Serrano, J. Kesselmeier, T. Karl, A. Guenther, J. Q. Chambers and C. Vickers, Emissions of putative isoprene oxidation products from mango branches under abiotic stress, J. Exp. Bot., 2013, 64, 3697–3708 CrossRef.
- K. J. Jardine, R. K. Monson, L. Abrell, S. R. Saleska, A. Arneth, A. Jardine, F. Y. Ishida, A. M. Y. Serrano, P. Artaxo, T. Karl, S. Fares, A. Goldstein, F. Loreto and T. Huxman, Within-plant isoprene oxidation confirmed by direct emissions of oxidation products methyl vinyl ketone and methacrolein, Glob. Change Biol., 2012, 18, 973–984 CrossRef.
- W. Jud, L. Fischer, E. Canaval, G. Wohlfahrt, A. Tissier and A. Hansel, Plant surface reactions: an opportunistic ozone defence mechanism impacting atmospheric chemistry, Atmos. Chem. Phys., 2016, 16, 277–292 CrossRef CAS.
- L. Cappellin, F. Loreto, F. Biasioli, P. Pastore and K. McKinney, A mechanism for biogenic production and emission of MEK from MVK decoupled from isoprene biosynthesis, Atmos. Chem. Phys., 2019, 19, 3125–3135 CrossRef CAS.
- K. J. Jardine, R. F. Zorzanelli, B. O. Gimenez, L. R. d. Oliveira Piva, A. Teixeira, C. G. Fontes, E. Robles, N. Higuchi, J. Q. Chambers and S. T. Martin, Leaf isoprene and monoterpene emission distribution across hyperdominant tree genera in the Amazon basin, Phytochemistry, 2020, 175, 112366 CrossRef CAS PubMed.
- A. M. Yáñez-Serrano, E. Bourtsoukidis, E. G. Alves, M. Bauwens, T. Stavrakou, J. Llusià, I. Filella, A. Guenther, J. Williams, P. Artaxo, K. Sindelarova, J. Doubalova, J. Kesselmeier and J. Peñuelas, Amazonian biogenic volatile organic compounds under global change, Glob. Change Biol., 2020, 1–30 Search PubMed.
- T. B. Rodrigues, C. R. Baker, A. P. Walker, N. McDowell, A. Rogers, N. Higuchi, J. Q. Chambers and K. J. Jardine, Stimulation of isoprene emissions and electron transport rates are a key mechanism of thermal tolerance in the tropical species Vismia guianensis, Glob. Change Biol., 2020, 1–14 Search PubMed.
- E. L. Singsaas, M. Lerdau, K. Winter and T. D. Sharkey, Isoprene increases thermotolerance of isoprene-emitting species, Plant Physiol., 1997, 115, 1413–1420 CrossRef CAS PubMed.
- R. Harkov, in Air Pollution. The Handbook of Environmental Chemistry, ed. H. Brauer, J. S. Gaffney, R. Harkov, M. A. K. Khalil, F. W. Lipfert, N. A. Marley, E. W. Prestbo and G. E. Shaw, Springer Berlin Heidelberg, Berlin, Heidelberg, 1989, vol. 4/4B, pp. 39–68 Search PubMed.
- G. Isaacman-VanWertz, L. D. Yee, N. M. Kreisberg, R. Wernis, J. A. Moss, S. V. Hering, S. S. De Sá, S. T. Martin, M. L. Alexander, B. B. Palm, W. Hu, P. Campuzano-Jost, D. A. Day, J. L. Jimenez, M. Riva, J. D. Surratt, J. Viegas, A. Manzi, E. Edgerton, K. Baumann, R. Souza, P. Artaxo and A. H. Goldstein, Ambient gas-particle partitioning of tracers for biogenic oxidation, Environ. Sci. Technol., 2016, 50, 9952–9962 CrossRef CAS PubMed.
- J. H. Kroll and J. H. Seinfeld, Chemistry of secondary organic aerosol: formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 2008, 42, 3593–3624 CrossRef CAS.
- U. Pöschl, S. T. Martin, B. Sinha, Q. Chen, S. S. Gunthe, J. A. Huffman, S. Borrmann, D. K. Farmer, R. M. Garland, G. Helas, J. L. Jimenez, S. M. King, A. Manzi, E. Mikhailov, T. Pauliquevis, M. D. Petters, A. J. Prenni, P. Roldin, D. Rose, J. Schneider, H. Su, S. R. Zorn, P. Artaxo and M. O. Andreae, Rainforest aerosols as biogenic nuclei of clouds and precipitation in the Amazon, Science, 2010, 329, 1513–1516 CrossRef PubMed.
- I. G. Kavouras, N. Mihalopoulos and E. G. Stephanou, Secondary organic aerosol formation vs. primary organic aerosol emission: in situ evidence for the chemical coupling between monoterpene acidic photooxidation products and new particle formation over forests, Environ. Sci. Technol., 1999, 33, 1028–1037 CrossRef CAS.
- L. L. D. Cooper, J. E. Oliver, E. D. De Vilbiss and R. P. Doss, Lipid composition of the extracellular matrix of Botrytis cinerea germlings, Phytochemistry, 2000, 53, 293–298 CrossRef CAS.
- E. G. Stephanou and N. Stratigakis, Oxocarboxylic and α,ω-dicarboxylic acids: photooxidation products of biogenic unsaturated fatty acids present in urban aerosols, Environ. Sci. Technol., 1993, 27, 1403–1407 CrossRef CAS.
- A. Kubátová, R. Vermeylen, M. Claeys, J. Cafmeyer, W. Maenhaut, G. Roberts and P. Artaxo, Carbonaceous aerosol characterization in the Amazon basin, Brazil: novel dicarboxylic acids and related compounds, Atmos. Environ., 2000, 34, 5037–5051 CrossRef.
- T. Duvold, J.-M. Bravo, C. Pale-Grosdemange and M. Rohmer, Biosynthesis of 2-C-methyl-D-erythritol, a putative C5 intermediate in the mevalonate independent pathway for isoprenoid biosynthesis, Tetrahedron Lett., 1997, 38, 4769–4772 CrossRef CAS.
- S. Sagner, W. Eisenreich, M. Fellermeier, C. Latzel, A. Bacher and M. H. Zenk, Biosynthesis of 2-C-methyl-D-erythritol in plants by rearrangement of the terpenoid precursor, 1-deoxy-D-xylulose 5-phosphate, Tetrahedron Lett., 1998, 39, 2091–2094 CrossRef CAS.
- J. Kitajima, T. Ishikawa, E. Fujimatu, K. Kondho and T. Takayanagi, Glycosides of 2-C-methyl-D-erythritol from the fruits of anise, coriander and cumin, Phytochemistry, 2003, 62, 115–120 CrossRef CAS PubMed.
- E. E. Jacobsen and T. Anthonsen, 2-C-Methyl-D-erythritol. Produced in plants, forms aerosols in the atmosphere. An alternative pathway in isoprenoid biosynthesis, Biocatal. Biotransform., 2015, 33, 191–196 CrossRef CAS.
- H. Enomoto, K. Kohata, M. Nakayama, Y. Yamaguchi and K. Ichimura, 2-C-methyl-D-erythritol is a major carbohydrate in petals of Phlox subulata possibly involved in flower development, J. Plant Physiol., 2004, 161, 977–980 CrossRef CAS PubMed.
- J. D. Surratt, A. W. H. Chan, N. C. Eddingsaas, M. Chan, C. L. Loza, A. J. Kwan, S. P. Hersey, R. C. Flagan, P. O. Wennberg and J. H. Seinfeld, Reactive intermediates revealed in secondary organic aerosol formation from isoprene, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6640–6645 CrossRef CAS PubMed.
- M. Claeys, B. Graham, G. Vas, W. Wang, R. Vermeylen, V. Pashynska, J. Cafmeyer, P. Guyon, M. O. Andreae, P. Artaxo and W. Maenhaut, Formation of secondary organic aerosols through photooxidation of isoprene, Science, 2004, 303, 1173–1176 CrossRef CAS PubMed.
- T. Cui, Z. Zeng, E. O. dos Santos, Z. Zhang, Y. Chen, Y. Zhang, C. A. Rose, S. H. Budisulistiorini, L. B. Collins, W. M. Bodnar, R. A. F. de Souza, S. T. Martin, C. M. D. Machado, B. J. Turpin, A. Gold, A. P. Ault and J. D. Surratt, Development of a hydrophilic interaction liquid chromatography (HILIC) method for the chemical characterization of water-soluble isoprene epoxydiol (IEPOX)-derived secondary organic aerosol, Environ. Sci.: Processes Impacts, 2018, 20, 1524–1536 RSC.
- M. Glasius, M. S. Bering, L. D. Yee, S. S. de Sá, G. Isaacman-VanWertz, R. A. Wernis, H. M. J. Barbosa, M. L. Alexander, B. B. Palm, W. Hu, P. Campuzano-Jost, D. A. Day, J. L. Jimenez, M. Shrivastava, S. T. Martin and A. H. Goldstein, Organosulfates in aerosols downwind of an urban region in central Amazon, Environ. Sci.: Processes Impacts, 2018, 20, 1546–1558 RSC.
- N. J. D. González, A. K. Borg-Karlson, P. Artaxo, A. Guenther, R. Krejci, B. Nozière and K. Noone, Primary and secondary organics in the tropical Amazonian rainforest aerosols: chiral analysis of 2-methyltetraols, Environ. Sci.: Processes Impacts, 2014, 16, 1413–1421 RSC.
- B. Nozière, N. J. D. González, A.-K. Borg-Karlson, Y. Pei, J. P. Redeby, R. Krejci, J. Dommen, A. S. H. Prevot and T. Anthonsen, Atmospheric chemistry in stereo: a new look at secondary organic aerosols from isoprene, Geophys. Res. Lett., 2011, 38, L11807 CrossRef.
- T. M. Cahill, V. Y. Seaman, M. J. Charles, R. Holzinger and A. H. Goldstein, Secondary organic aerosols formed from oxidation of biogenic volatile organic compounds in the Sierra Nevada Mountains of California, J. Geophys. Res.: Atmos., 2006, 111, D16312 CrossRef.
- A. P. M. Emygdio, M. d. F. Andrade, F. L. T. Gonçalves, G. Engling, R. H. d. S. Zanetti and P. Kumar, Biomarkers as indicators of fungal biomass in the atmosphere of São Paulo, Brazil, Sci. Total Environ., 2018, 612, 809–821 CrossRef CAS PubMed.
- D. H. Lewis and D. C. Smith, Sugar alcohols (polyols) in fungi and green plants, New Phytol., 1967, 66, 143–184 CrossRef CAS.
- S. Pressley, B. Lamb, H. Westberg, J. Flaherty, J. Chen and C. Vogel, Long-term isoprene flux measurements above a northern hardwood forest, J. Geophys. Res.: Atmos., 2005, 110, D07301 CrossRef.
- T. Karl, A. Guenther, R. J. Yokelson, J. Greenberg, M. Potosnak, D. R. Blake and P. Artaxo, The tropical forest and fire emissions experiment: emission, chemistry, and transport of biogenic volatile organic compounds in the lower atmosphere over Amazonia, J. Geophys. Res.: Atmos., 2007, 112, D18302 CrossRef.
- M. P. Barkley, T. P. Kurosu, K. Chance, I. De Smedt, M. Van Roozendael, A. Arneth, D. Hagberg and A. Guenther, Assessing sources of uncertainty in formaldehyde air mass factors over tropical South America: implications for top-down isoprene emission estimates, J. Geophys. Res.: Atmos., 2012, 117, D13304 CrossRef.
- C. E. Batista, J. Ye, I. O. Ribeiro, P. C. Guimarães, A. S. S. Medeiros, R. G. Barbosa, R. L. Oliveira, S. Duvoisin, K. J. Jardine, D. Gu, A. B. Guenther, K. A. McKinney, L. D. Martins, R. A. F. F. Souza, S. T. Martin and S. Duvoisin Jr, Intermediate-scale horizontal isoprene concentrations in the near-canopy forest atmosphere and implications for emission heterogeneity, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19318–19323 CrossRef CAS.
- K. A. McKinney, D. Wang, J. Ye, J. B. D. Fouchier, P. C. Guimarães, C. E. Batista, R. A. F. Souza, E. G. Alves, D. Gu, A. B. Guenther and S. T. Martin, A sampler for atmospheric volatile organic compounds by copter unmanned aerial vehicles, Atmos. Meas. Tech., 2019, 12, 3123–3135 CrossRef CAS.
- N. Watson, S. Davies and D. Wevill, Air monitoring: new advances in sampling and detection, Sci. World J., 2011, 11, 2582–2598 CrossRef PubMed.
- B. Graham, P. Guyon, P. E. Taylor, P. Artaxo, W. Maenhaut, M. M. Glovsky, R. C. Flagan and M. O. Andreae, Organic compounds present in the natural Amazonian aerosol: characterization by gas chromatography–mass spectrometry, J. Geophys. Res.: Atmos., 2003, 108, D24 Search PubMed.
- A. L. Bondy, R. L. Craig, Z. Zhang, A. Gold, J. D. Surratt and A. P. Ault, Isoprene-derived organosulfates: vibrational mode analysis by raman spectroscopy, acidity-dependent spectral modes, and observation in individual atmospheric particles, J. Phys. Chem. A, 2018, 122, 303–315 CrossRef CAS PubMed.
- J. Yu, D. R. Cocker, R. J. Griffin, R. C. Flagan and J. H. Seinfeld, Gas-phase ozone oxidation of monoterpenes: gaseous and particulate products, J. Atmos. Chem., 1999, 34, 207–258 CrossRef CAS.
- H. K. Lam, K. C. Kwong, H. Y. Poon, J. F. Davies, Z. Zhang, A. Gold, J. D. Surratt and M. N. Chan, Heterogeneous OH oxidation of isoprene-epoxydiol-derived organosulfates: kinetics, chemistry and formation of inorganic sulfate, Atmos. Chem. Phys., 2019, 19, 2433–2440 CrossRef CAS.
- J. Finnigan, Turbulence in plant canopies, Annu. Rev. Fluid Mech., 2000, 32, 519–571 CrossRef.
- S. E. Belcher and J. C. R. Hunt, Turbulent flow over hills and waves, Annu. Rev. Fluid Mech., 1998, 30, 507–538 CrossRef.
- M. Horstmann and M. S. McLachlan, Atmospheric deposition of semivolatile organic compounds to two forest canopies, Atmos. Environ., 1998, 32, 1799–1809 CrossRef CAS.
- T. B. Nguyen, J. D. Crounse, A. P. Teng, J. M. S. Clair, F. Paulot, G. M. Wolfe and P. O. Wennberg, Rapid deposition of oxidized biogenic compounds to a temperate forest, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E392–E401 CrossRef CAS PubMed.
- B. B. Hicks, R. D. Saylor and B. D. Baker, Dry deposition of particles to canopies—A look back and the road forward, J. Geophys. Res.: Atmos., 2016, 121, 14691–14707 Search PubMed.
- J. E. L. S. Ribeiro, B. W. Nelson, M. F. d. Silva, L. S. S. Martins and M. Hopkins, Reserva florestal ducke: diversidade e composição da flora vascular, Acta Amazonica, 1994, 24, 19–30 CrossRef.
- Y. Miyazaki, P. Fu, K. Ono, E. Tachibana and K. Kawamura, Seasonal cycles of water-soluble organic nitrogen aerosols in a deciduous broadleaf forest in northern Japan, J. Geophys. Res.: Atmos., 2014, 119, 1440–1454 CAS.
- W. Wang, Y. Iinuma, A. Kahnt, O. Ryabtsova, A. Mutzel, R. Vermeylen, P. Van der Veken, W. Maenhaut, H. Herrmann and M. Claeys, Formation of secondary organic aerosol marker compounds from the photooxidation of isoprene and isoprene-derived alkene diols under low-NOx conditions, Faraday Discuss., 2013, 165, 261–272 RSC.
- P. Q. Fu, K. Kawamura, J. Chen, J. Li, Y. L. Sun, Y. Liu, E. Tachibana, S. G. Aggarwal, K. Okuzawa, H. Tanimoto, Y. Kanaya and Z. F. Wang, Diurnal variations of organic molecular tracers and stable carbon isotopic composition in atmospheric aerosols over Mt. Tai in the North China Plain: an influence of biomass burning, Atmos. Chem. Phys., 2012, 12, 8359–8375 CrossRef CAS.
- S. S. de Sá, B. B. Palm, P. Campuzano-Jost, D. A. Day, M. K. Newburn, W. Hu, G. Isaacman-VanWertz, L. D. Yee, R. Thalman, J. Brito, S. Carbone, P. Artaxo, A. H. Goldstein, A. O. Manzi, R. A. F. Souza, F. Mei, J. E. Shilling, S. R. Springston, J. Wang, J. D. Surratt, M. L. Alexander, J. L. Jimenez and S. T. Martin, Influence of urban pollution on the production of organic particulate matter from isoprene epoxydiols in central Amazonia, Atmos. Chem. Phys., 2017, 17, 6611–6629 CrossRef.
- P. Guimarães, J. Ye, C. Batista, R. Barbosa, I. Ribeiro, A. Medeiros, R. Souza and S. T. Martin, Vertical profiles of ozone concentration collected by an unmanned aerial vehicle and the mixing of the nighttime boundary layer over an amazonian urban area, Atmosphere, 2019, 10, 599 CrossRef.
- B. Liu, C. Wu, N. Ma, Q. Chen, Y. Li, J. Ye, S. T. Martin and Y. J. Li, Vertical profiling of fine particulate matter and black carbon by using unmanned aerial vehicle in Macau, China, Sci. Total Environ., 2020, 709, 136109 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ea00006j |
| This journal is © The Royal Society of Chemistry 2021 |