
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigating the evolution of water-soluble organic carbon in evaporating cloud water†
Vikram
Pratap
 a,
Amy E.
Christiansen
b,
Annmarie G.
Carlton
b,
Sara
Lance
c,
Paul
Casson
c,
Jed
Dukett
c,
Hesham
Hassan
c,
James J.
Schwab
c and
Christopher J.
Hennigan
*a
a,
Amy E.
Christiansen
b,
Annmarie G.
Carlton
b,
Sara
Lance
c,
Paul
Casson
c,
Jed
Dukett
c,
Hesham
Hassan
c,
James J.
Schwab
c and
Christopher J.
Hennigan
*a
aDepartment of Chemical, Biochemical and Environmental Engineering, University of Maryland, Baltimore County, Baltimore, MD 21250, USA. E-mail: hennigan@umbc.edu; Tel: +1-410-455-3515
bDepartment of Chemistry, University of California, Irvine, CA 92697, USA
cAtmospheric Sciences Research Center, University at Albany, State University of New York, Albany, NY 12203, USA
First published on 22nd December 2020
Abstract
Cloud cycling plays a key role in the evolution of atmospheric particles and gases, producing secondary aerosol mass and transforming the optical properties and impacts of aerosols globally. In this study, bulk cloud water samples collected at Whiteface Mountain (Wilmington, NY) in the summer of 2017 were aerosolized, dried to 50% RH, and analyzed for the evaporative loss of water soluble organic carbon (WSOC) and for brown carbon (BrC) formation. Systematic WSOC evaporation occurred in all cloud water samples, while no evidence for drying induced BrC formation was observed. On average, 11% (±3%) of WSOC evaporated when the aerosolized cloud droplets were dried to 50% RH, though this represents a lower bound on the WSOC reversibly partitioned to clouds due to experimental constraints. To our knowledge, this represents the first direct measurements of organic evaporation from actual cloud water undergoing drying. Formate and acetate contributed 19%, on average, to the evaporated WSOC, while no oxalate evaporation occurred. GECKO-A model simulations were carried out to predict the production of WSOC compounds that reversibly partition to cloud water from photooxidation of an array of VOCs. The model results suggest that precursor VOC identity and oxidation regime (VOC:NOx) have a dramatic effect on the reversible partitioning of WSOC to cloud water and the abundance of aqSOA precursors, though the higher abundance of reversibly partitioned WSOC predicted by the model may be due to aqueous production of low-volatility material in the actual cloud samples. This study underscores the importance of the large fraction of unidentified compounds that contribute to WSOC in cloud water and their aqueous processing.
Environmental significanceClouds serve as important reservoirs for the transformation of organics in Earth's atmosphere. Water-soluble gases can partition to cloud droplets and react to form lower-volatility compounds that increase the SOA burden. Such aqueous reactions can also form light-absorbing brown carbon (BrC), a process accelerated when droplets evaporate. Despite their potential importance for global aerosol concentrations and radiative effects, there are immense challenges in directly measuring in-cloud transformations of organics in situ. Herein, we aerosolized and dried cloud water samples collected at Whiteface Mountain (NY) to mimic a cloud cycle. All samples showed WSOC evaporation with drying but negligible BrC production. The results advance our understanding of in-cloud organic chemistry and provide a critical link between prior laboratory and field observations. |
1. Introduction
Low-level clouds are abundant in Earth's atmosphere and interact with boundary layer gases and particles to affect the fate and transport of trace species. Cloud processing alters the size distribution and chemical composition of ambient aerosol.1 Photooxidation of anthropogenic and biogenic volatile organic compounds (VOCs) forms water-soluble organic compounds (WSOC) commonly observed in clouds and fogs.2 Partitioned species undergo chemical transformation in the aqueous phase that is often different from gas-phase oxidation pathways, and can favor the formation of low volatility compounds that contribute to secondary organic aerosol (aqSOA) mass.1 Cloud radiative impacts are also substantial and include potential production of brown carbon (BrC), a short-lived climate forcer, aloft.3 While ubiquitous and important, large uncertainties in cloud physical predictions and aqueous-phase chemical mechanisms hinder accurate quantitative understanding of aqSOA contributions to tropospheric aerosol burdens, though it is expected to be substantial.4,5Ambient cloud physics and cloud microphysics are routinely studied in field and laboratory experiments,6 but the investigation of cloud chemistry is not as widespread. The majority of liquid water droplets (>90%) evaporate after air parcels pass through clouds.7 The fundamental chemistry and potential WSOC evaporative loss during drying of ambient cloud droplets remain poorly understood, as does BrC formation during cloud droplet drying. Laboratory studies of organic partitioning during droplet evaporation investigate individual compounds8–11 and find that observed gas- and condensed-phase concentrations can deviate from thermodynamic partitioning predictions up to several orders of magnitude, especially for more polar species.12 This inhibits the development of chemical mechanisms to describe cloud processing of complex organic mixtures given that single component systems are difficult to predict. Evaporative formation of BrC is observed in laboratory experiments with simplified droplet and particle compositions, but not in the drying of ambient aerosols sampled in the eastern US.11,13,14 Such conflicting results challenge efforts to develop parameterizations that can be used in models to evaluate this process on larger scales. During a comprehensive and nearly ideal Lagrangian study of cloud chemistry at Schmücke Mountain, it was difficult to constrain physical losses (droplet impaction on vegetation), and chemical transformations from concurrent upwind, in-cloud, and downwind measurements, precluding quantitative assessment of gas-aqueous partitioning for most organic compounds.15 Further, the majority of WSOC in clouds and fogs are unidentified on a molecular basis,2 so the source of these compounds (whether present in the activated CCN or partitioned from the gas phase upon cloud droplet formation) and their subsequent fate under cloud droplet evaporation remain uncharacterized.16
In this study, we evaluate the cloud processing of WSOC by analyzing cloud water samples collected at Whiteface Mountain, Wilmington, NY, a remote forested site influenced by airmasses from the Central Midwest, Great Lakes and Canada.17–19 We characterize the evaporative loss of WSOC and formation of BrC in aerosolized cloud water under conditions of drying. We also model the equilibrium partitioning of WSOC formed from the photooxidation of various precursor VOCs under high- and low-NOx conditions with the GECKO-A box model.
2. Methods
2.1 Cloud water sampling
Whiteface Mountain (WFM) is a peak in the Adirondack Mountain Range in northern New York. The summit (latitude N 44°21′58′′ and longitude W 73°54′10′′) is situated above tree line at 1483 m a.s.l., and is isolated from other peaks.17 Clouds are regularly intercepted (25–60%) at the summit in summertime.16 WFM is mostly affected by airmasses transported from the western and central Unites States, Great Lakes region and Canada, suggesting the organics (gas and aerosol) are aged and often dominated by contributions derived from precursor biogenic species.17–20 Aged biomass burning smoke intercepts the summit of WFM and episodically influences cloud water composition.20,21 The Atmospheric Sciences Research Center (ASRC) at the University at Albany, Albany, NY, operates a research observatory at the summit, where cloud water sampling has been conducted regularly since 1994, historically focusing on acid deposition.17 The cloud water sampling is usually conducted in summertime (June–September) due to prohibitive wintertime weather, while gas phase measurements are performed year round.Cloud water samples were collected during August 2017 as a part of the Cloud Processing of Organics within Clouds (CPOC) pilot study.20 Cloud water was collected using a Möhnen omni-directional passive cloud water collector,22 which was deployed when the following conditions were met: (1) liquid water content measured by the Gerber Particle Volume Meter (Gerber PVM-100)23 exceeded 0.05 g m−3, (2) wind speed exceeded 2 m s−1, (3) temperature exceeded 2 °C (to avoid freezing), and (4) no rain was detected by a CAPMoN (Canadian Air and Precipitation Monitoring Network) resistive heating element rain sensor. The collected samples were frozen and stored in a deep freezer (−20 °C), and thawed immediately before analysis. The cloud water samples analyzed in this study were collected on August 12 (06:00–09:00), August 13 (06:00–09:00), August 13 (10:00–11:00), August 18 (06:00–09:00) and August 18 (11:00–12:00), where the time specified is local time (UTC – 4 h). In situ measurements of below-cloud aerosol composition on August 18 were concluded to have been urban-influenced, with an organic mass fraction of 75%, which was slightly lower than average for the CPOC pilot study.20 On these 3 days (August 12, 13 and 18), cloud water samples are estimated to have been collected at 300, 490 and 400 m above cloud base, respectively, corresponding to 2.5–4 minutes of time in cloud assuming 2 m s−1 updraft velocity.
2.2 Cloud water experiments and analysis
Cloud water samples were analyzed for WSOC, BrC via absorbance at 365 nm (Abs365), and other inorganic and organic ions. The aim of the analyses was to (1) measure the fraction of WSOC that partitions to the gas phase upon drying, mimicking the cloud cycling effect and cloud processing, and (2) evaluate drying-induced BrC formation through the Abs365 measurement. The experimental setup was adapted from a previous study from our group (Fig. 1).14 Cloud water was atomized (TSI, model 9302) and alternated between a dry channel, which contained a silica-gel diffusion-dryer, and a bypass channel every 15 minutes using an automated 3-way valve (Brechtel manufacturing). Note here, the cloud water samples were not exposed to any oxidants in the lab (in either the bulk or as aerosolized droplets), and no UV light source was used to initiate aqueous reactions. The drying induced BrC formation chemistry takes place due to reactants already present in the cloud water. The particles were dried to ∼50% RH (not fully dried) in the dry channel, while particles in the bypass-channel experienced an RH of ∼85% due to the low humidity in the makeup air. Each cloud water sample measurement was repeated 2–4 times through each channel. Particle losses in the experimental setup were investigated with a mixed sucrose/NaCl solution. We observed no difference in particle concentrations between the two channels during the control experiments, consistent with the QA/QC results of our prior studies.14,24 The samples from either channel passed through an activated carbon denuder to remove VOCs prior to collection in a particle-into-liquid sampler (PILS). The PILS samples were analyzed on-line for WSOC using a total organic carbon analyzer (Sievers 900 portable, GE analytical instruments) in turbo mode (4 s resolution) and UV-absorbance using a liquid waveguide capillary cell (LWCC, model 3050, World Precision Instrument) coupled to a monochromatic 365 nm light source (Oceanoptics, LLS-LED) and a spectrometer (Oceanoptics, FLAME-S, 200–800 nm). The effective path length of the LWCC was 50 cm. The difference in WSOC and Abs365 from the two channels provided the measurement of drying-induced evaporation of WSOC and BrC formation, respectively. Additional samples from the PILS were analyzed off-line for inorganic (SO42−) and organic (formate, acetate, oxalate) ions via ion chromatography (Metrohm 850 dual IC, Metrohm). The cation and anion columns used were Metrosep C4 – 150/4.0 and Metrosep A Supp 5 – 150/4.0, respectively. Since SO42− is non-volatile, the SO42− measurement through each channel was used to normalize the measurements to account for any minor differences in atomizer output during an experiment, assuming that changes in the atomizer output do not affect the relative mass concentrations of solute within the droplets produced. Since formate and acetate were not chromatographically resolved, we describe their collective behavior and concentration (together F + A) using their individual IC calibrations and the average F/A molar ratio measured in a previous study at WFM (1.9) to interpret the present results.25 The uncertainty in formate, acetate and oxalate measurements were 0.15 ppbC, 0.23 ppbC and 0.1 ppbC, respectively.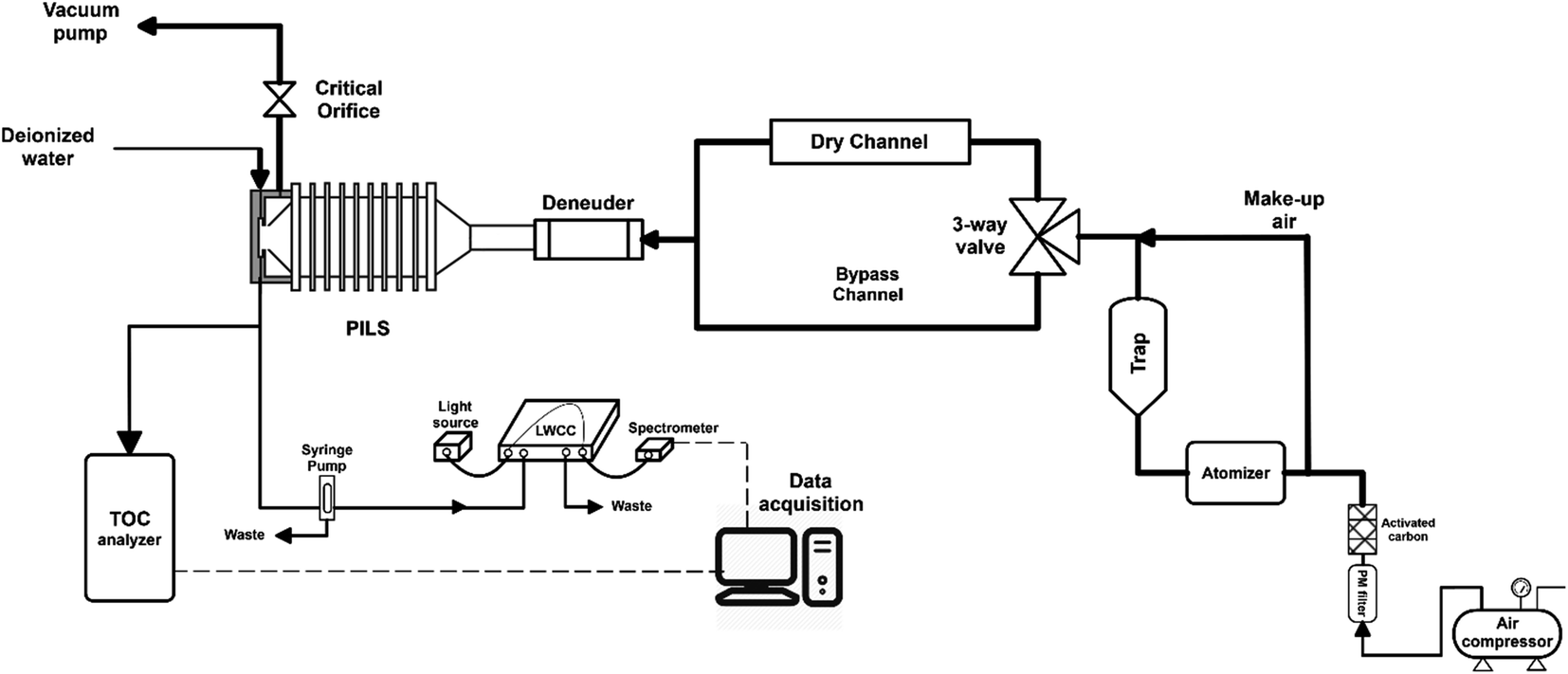 | ||
| Fig. 1 Schematic of the experimental setup for laboratory analysis of the cloud water. | ||
Note that the RH through the bypass channel was not 100%, thus some WSOC may have evaporated from the aerosolized droplets relative to the bulk cloud water. However, the 35% RH reduction (from 85% to 50%) appears to be a reasonable drying based on RH measurements at the WFM summit and downwind lodge site. The median RH at the summit was 98% during August 2017, consistent with the frequent presence of clouds, while the median RH at the downwind lodge site was 80% for the same period (Fig. S1†). It should be noted that the airmasses may experience both an increase in RH or further drying downwind of the lodge, so a range of dry channel RH levels should be investigated. However, limited sample volumes constrained the extent of drying RH values in this study. The drying time of ∼7 s is sufficient for water equilibration with particles, however, organics may take longer to fully evaporate.8,24,26,27 In addition, further evaporation of WSOC is expected at longer drying times and lower RHs. Together, this indicates that our measurements of WSOC evaporation from the cloud water represent a lower bound on the amount of organics that may evaporate in the atmosphere during an actual cloud cycle.
2.3 Model
The photooxidation of ten atmospherically relevant organic compounds under high- and low-NOx conditions was simulated by the Generator of Explicit Chemistry and Kinetics of Organics in the Atmosphere (GECKO-A) box model. GECKO-A models the reaction of atmospheric organic gases using laboratory-generated kinetic data or through estimates using structure–activity relationships.28 The model also estimates the water solubility (Henry's constant, KH) and vapor pressure (volatility, C*) of thousands of oxidation products through structure–activity relationships.29 For compounds whose physical constants have not been determined experimentally, the vapor pressure was estimated using the methodology in Nannoolal et al. (2008)30 while the Henry's constant was estimated using the group contribution method for Henry's law estimate (GROMHE).12The specific simulations used for this study have been described in detail before.29,31 Briefly, the gas-phase photooxidation of the following ten VOCs was simulated under mid-latitude tropospheric conditions: C8,12,18,22-alkanes, benzene, toluene, m-, o-, p-xylenes (xylenes considered together), C12-alkenes (with internal and terminal double bonds, considered separately), and α-pinene. The relative abundance, KH, and C* values of each oxidation product were determined after photooxidation equivalent to ∼2 days of atmospheric aging, an appropriate value for airmasses impacting WFM.19,32 The simulations predict hundreds or thousands of oxidation products with a wide distribution of KH and C* values for each VOCs considered.29 Here, we extend our prior work by calculating the partitioning of each oxidation product after 2 days of gas-phase photooxidation, with no treatment of condensed-phase reactions in the aerosols or cloud water. The fraction (molar basis) of oxidation products dissolved in cloud water, εCW, was calculated according to Seinfeld and Pandis (2006) assuming cloud liquid water content of 1 g m−3, very close to the median value for the WFM samples analyzed (1.12 g m−3, Table S1†).33 In this work, we assume liquid clouds only, to avoid complexities in partitioning that arise with ice or mixed-phase clouds.34 This is consistent with summertime conditions at WFM (Table S1†). We assume equilibrium partitioning, an approach taken by most regional and global climate models, neglecting deviations that can occur due to mass transfer limitations.35,36
3. Results and discussion
3.1 Experimental results and discussion
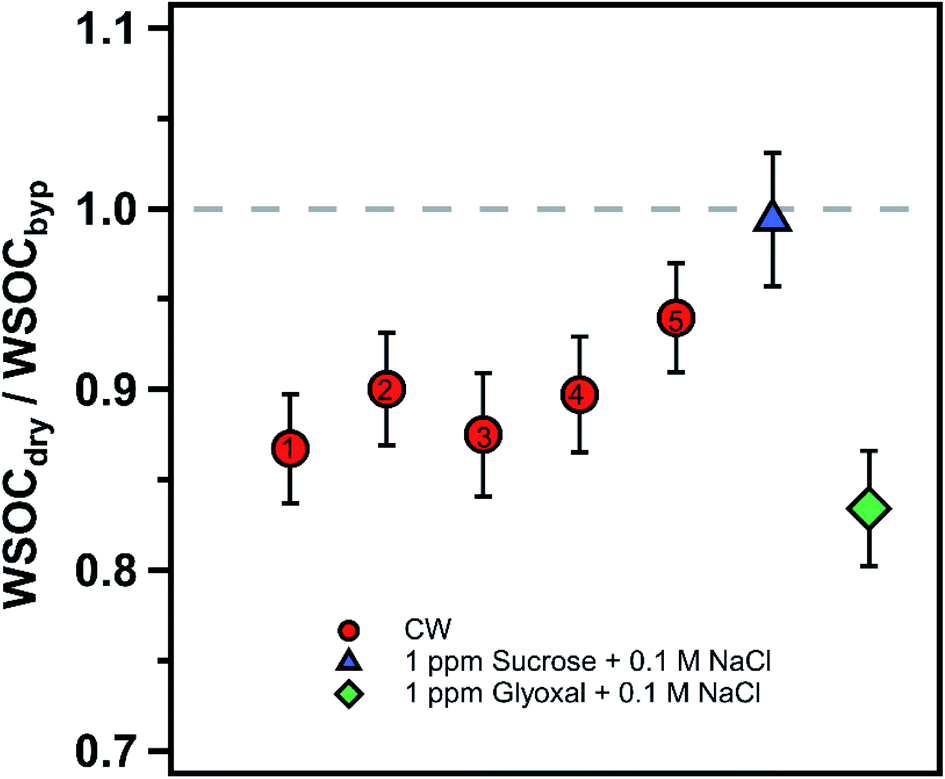 | ||
| Fig. 2 Ratio of WSOC measured in the dry and bypass channels for the cloud water (CW) samples (red circles). The numbers inside the red circles correspond to the cloud water sample index (i.e., 1 denotes CW1). The error bars represent the measurement uncertainty, propagated from the accuracy of WSOC measurements. The cloud water samples are contrasted with experiments performed with non-volatile (sucrose, blue triangle) and volatile (glyoxal, green diamond) species. The dashed line at unity represents no organic evaporation due to drying, shown for visual reference. | ||
To our knowledge, this represents the first direct measurements of organic evaporation from cloud water undergoing drying. In contrast, previous studies conducted on ambient aerosols in the eastern United States during summer have shown 10–30% evaporation of particulate WSOC when dried.14,37 The dry channel RH in the prior studies was lower (41% and 35%, respectively) than the one in the current study (∼50%), but was subject to the same residence time of 7 s in the drying system.14,37 In the atmosphere, cloud droplets may undergo a greater extent of drying than aerosol depending upon the cloud properties and evolution of meteorology that may lead to additional evaporation of WSOC.33 Future work should investigate variable drying configurations and temperatures to characterize a wider range of atmospheric conditions.
The combined contribution of formate, acetate, and oxalate to CW WSOC decreased with increasing WSOC (Fig. 3a), from a high of ∼35% at the lowest CW WSOC (CW3, ∼50 μg-C L−1) to a low of only 7% at the highest CW WSOC (CW5, ∼167 μg-C L−1). Formic, acetic, and oxalic acids are generally the most abundant carboxylic acids found in cloud water, and the relatively high vapor pressure of formic and acetic acid indicate their likely contribution to the evaporated WSOC.25,38–42 Our observations suggest an array of individual compounds contributed to the evaporated WSOC, as formate and acetate (F + A) together accounted for an average of only 19% of the evaporated WSOC (C mass basis). The evaporation of F + A was highly variable across the five CW samples; we found that 0–30% of F + A was lost due to evaporation (Fig. 3b). Notably, we observed no F + A loss due to drying in CW5, the sample with the strongest signature of photochemical and aqueous-phase processing (ESI†). Multiple studies have shown that the gas-particle partitioning of formic and acetic acid do not typically agree with thermodynamic predictions, as the condensed-phase fraction is often higher by orders of magnitude.43,44 This may explain the relatively high fraction of F + A remaining after drying. We observed no oxalate evaporation in any of the cloud water samples analyzed. Fig. 3b also shows that the evaporative loss of F + A was linearly proportional to that of WSOC. While the majority of evaporated WSOC was not identified, our correlation is consistent with prior observations that F + A share similar sources and/or condensed-phase processes with this broader pool of WSOC compounds.45–49 For example, Millet et al., (2015) showed a large concentration of formic acid in the southeastern United States, which is dominated by biogenic emissions in summertime.46 Similarly, Paulot et al., (2011) reported biogenic emissions as a major source of global formic and acetic acid concentrations.47 As discussed previously, airmasses at WFM are expected to be predominantly influenced by biogenic emissions.20 In addition, a number of studies have also established that WSOC is efficiently produced from biogenic sources, at least in summertime in the eastern United States, when the cloud water samples were collected.50–52
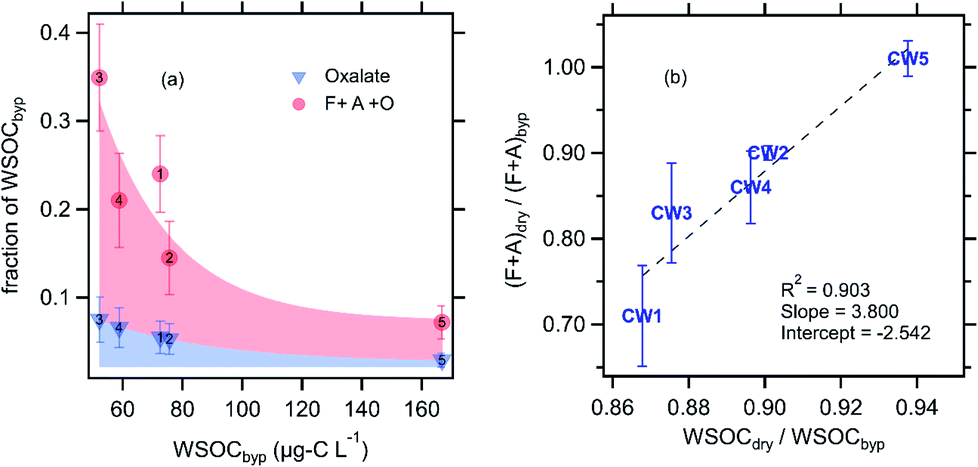 | ||
| Fig. 3 (a) Contribution of oxalate (blue triangles) and the sum of oxalate, formate and acetate (red circles) to the total WSOC as a function of the WSOC concentration, all measured in the bypass channel (not dried, see Fig. 1). (b) Fraction of WSOC and F + A that evaporated with drying. Note that no oxalate evaporation was observed with drying. Error bars in each panel represent the estimated uncertainty derived from error propagation of the individual measurements. The numbers in panel ‘a’ correspond to the CW sample number. | ||
These results demonstrate that a small but significant fraction of WSOC in clouds derives from organic gases reversibly partitioned to water. Our findings complement measurements of gas scavenging by clouds,34 and direct Lagrangian sampling of gas uptake in clouds.15 These results have importance for the budget of reactive carbon in the atmosphere, and for the associated effects on air quality and climate. The reversible partitioning of WSOC to cloud water impacts the lifetime and transport of reactive organics in the atmosphere since the removal of oxygenated (and water-soluble) gases via dry deposition and photolysis can be substantially reduced through transfer to the aqueous phase.53 The uptake of small, volatile WSOC compounds can also alter the evolution of cloud droplets and their physical properties, with implications for the climate system.54 Volatile WSOC compounds have long been known to have the potential to form SOA through aqueous chemistry in clouds,2,4 and many of these compounds are semi-volatile and reversibly partition to water (e.g., glyoxal, methylglyoxal).10,11,55 Our results are qualitatively consistent with observations from the WACS-2010 study, which suggested the formation of SOA in cloud water from dissolved organics that were too volatile to exist in the aerosol phase.56 Although the contribution of aqueous reactions in clouds to ambient OA concentrations remains very difficult to measure or constrain, our results add to the body of work suggesting the importance of this phenomenon.1
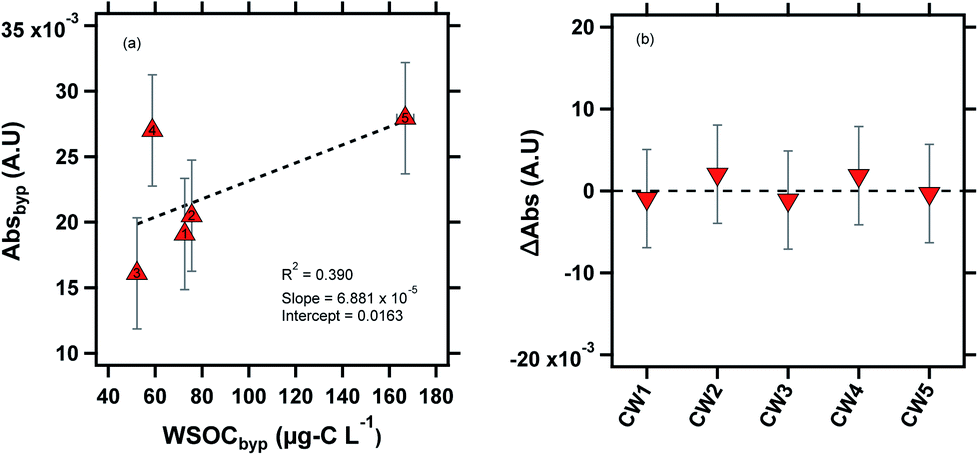 | ||
| Fig. 4 Absorbance measurements of the cloud water samples at 365 nm. CW1, CW2 and CW5 were likely influenced by biomass burning, as indicated by the potassium ion concentrations (Table S2†). (a) Linear correlation between Absbyp and WSOCbyp, (b) ΔAbs (ΔAbs = Absdry − Absbyp) is plotted for the cloud water samples. The subscript “byp” represents measurements through the bypass channel, and the dashed line at ΔAbs = 0 line is shown for visual reference. | ||
Previous laboratory studies have confirmed BrC formation by aqueous reactions between dicarbonyls (glyoxal, methylglyoxal) and reduced nitrogen species (ammonium sulfate, ammonia, glycine).57,58 In drying particles, these reactions accelerate by several orders of magnitude due to the concentration of reactants, suggesting that secondary BrC formation in evaporating cloud droplets may contribute significantly to the aerosol direct effect.8,11,13 Our previous study did not find evidence of drying-induced BrC formation in ambient particles,14 most likely due to the low concentration of dicarbonyls in ambient particles relative to laboratory studies that have observed such a phenomenon.25 However, in clouds, the partitioning of BrC precursors is expected to be several orders of magnitude higher, due to the high LWC.59,60 For example, Gly + MeGly can constitute up to ∼10% of WSOC in clouds and fogs,2,61 demonstrating the greater potential for drying-induced BrC formation compared to aqueous particles. Fig. 4b shows the ΔAbs value (ΔAbs = Absdry − Absbyp) for all five cloud water samples. Similar to our prior results for the eastern US aerosol, we did not observe evidence of BrC formation in the aerosolized cloud water samples as a result of drying within our method detection limit of 1 × 10−3 A.U.14 There are a few possible explanations for this observation. As mentioned earlier, air-masses reaching WFM were likely aged, suggesting that any secondary BrC production may have occurred during transport to WFM, largely depleting the airmass of BrC precursors.18,25,62 Photobleaching (decreasing absorbance) of secondary BrC occurs on time scales of minutes to days,63–65 so any BrC formation upwind of WFM would likely have been affected by this process.65,66 Another possible explanation for the lack of BrC formation in our experiments is that the drying RH of 50% may not be sufficient to produce substantial BrC. In a previous study, we observed BrC formation in aqueous glyoxal/(NH4)2SO4 particles when dried to ∼30% RH, but not when the particles were dried to ∼50% RH.14 In the same study, we observed BrC formation from MeGly/(NH4)2SO4 when aqueous particles were dried to both 50% and 30% RH, but a significant enhancement at the lower RH level. A drying time of ∼7 s was sufficient to produce BrC formation in aqueous particles undergoing drying in previous laboratory studies.11,14 Notably, in our previous study conducted with a nearly identical experimental setup, we observed BrC formation in aqueous glyoxal and methylglyoxal droplets with only ∼7 s of drying.14 However, the effect of variable drying times on BrC formation has not been systematically studied.
3.2 Model results
Simulations from GECKO-A were used to predict the reversible partitioning of VOC oxidation products to cloud water after the equivalent of two days of gas-phase photooxidation. There is a wide distribution of WSOCdry/WSOCcw predicted for the VOC oxidation products, with precursor molecule identity and oxidation regime (i.e., VOC/NOx) both contributing to this variability (Fig. 5). While the simulations are simplistic in that they do not account for reactions of the organics in CW, which can shift the partitioning of compounds produced or consumed in the aqueous phase, they are instructive in a number of ways. For most of the simulated VOCs, photooxidation under high-NOx conditions leads to a decrease in the predicted WSOCdry/WSOCcw due to an increase in the relative abundance of volatile oxidation products (compounds like Gly and MeGly). This is most evident in the single ring aromatic simulations, which all show <2% of the WSOC that partitions to CW remains in the condensed phase after drying (mole fraction basis). The simulations also indicate far greater production of higher volatility products from these compounds compared to the species with much higher predicted WSOCdry/WSOCcw, for example, C22- and C18-alkanes.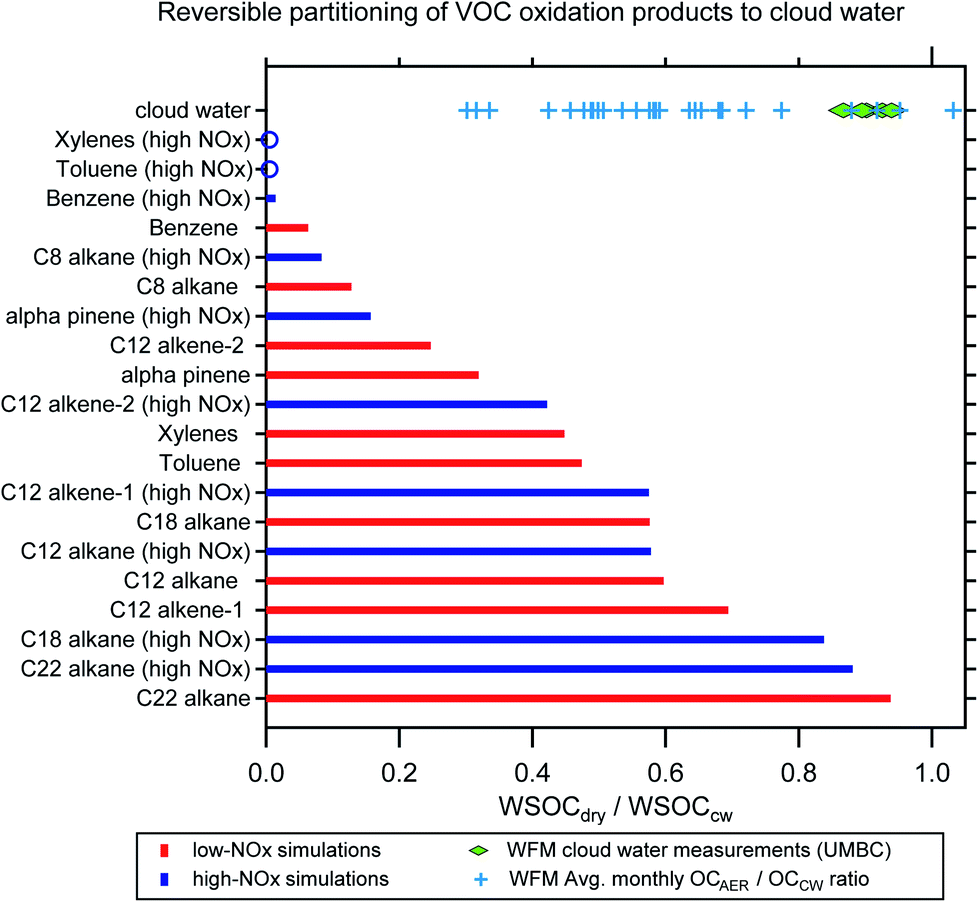 | ||
| Fig. 5 GECKO model predictions of WSOCdry/WSOCCW for the VOC photooxidation products (red and blue bars). The model results were compared with the laboratory cloud water measurements (green diamonds) and with the ratio of organic aerosol to cloud water organics measured at WFM (summertime monthly averages from 2009–2018, blue crosses). | ||
The simulations are interesting to contrast with the lab measurements of WSOCdry/WSOCcw in the WFM samples (green diamonds in Fig. 5). The measured WSOCdry/WSOCcw shows a relatively narrow distribution, which is perhaps not surprising given that the CW samples were collected over such a short period of time (12-Aug-2017 to 18-Aug-2017, Table S1†). While we observed systematic evaporation of some WSOC in all five CW samples (11% on average), the majority of the WSOC compounds had sufficiently low volatility to remain in the condensed phase upon drying.
Broader analysis of WFM data suggests a wider distribution of volatilities in the CW than our five samples showed (blue crosses in Fig. 5). The data presented are summertime monthly averages (June–September, 2009–2018) of the ratio of organic aerosol concentrations (OAAER) measured downwind of the WFM summit relative to CW organics (both concentrations expressed per m3 of air for direct comparison).67 While the summit and downwind sites are not always sampling the same airmass, simultaneous O3 measurements suggest that the prevailing conditions at the summit where CW collection occurs and the aerosol sampling site are representative (Fig. S3, ESI†). The monthly average OAAER/OACW ratio (blue crosses in Fig. 5) has a wide range of values, from a low of 0.3 to a high of ∼1, with an average value of 0.6. This reflects influence from the oxidation of numerous VOCs, wide-ranging atmospheric conditions, and variable aging times during transport to WFM. Even when the site conducting aerosol sampling is downwind of the summit, there can be difficulties directly connecting the CW and the aerosol composition measurements due to droplet deposition.15,20 Thus, the values of the blue crosses in Fig. 5 have a high level of uncertainty and would be biased low if deposition removed organics during transport from the summit to the downwind site. Nevertheless, the lower OA concentrations in nearly all months suggests the evaporation of organics from clouds at this site is a systematic occurrence during the summertime. The modeling and long-term WFM results in Fig. 5 suggest that a much broader range of WSOCdry/WSOCcw values would be observed if the laboratory measurements were expanded to include cloud water samples influenced by a broader range of source regions and atmospheric ageing conditions.
These results reveal the importance of clouds in the uptake of oxidized VOCs, which demonstrates significant potential to produce aqSOA. A lower predicted WSOCdry/WSOCcw indicates a more volatile product distribution and thus a greater potential to form aqSOA in cloud water. Note that the GECKO modeling results include vastly more oxygenated organics than have been identified through molecular measurements, in agreement with the finding that the majority of WSOC in cloud water is unidentified on a molecular level.2 Together, this suggests that aqSOA formation may be even more substantial than estimates using known precursors.
4. Conclusions
Cloud cycling is an important process that leads to physical and chemical transformations of dissolved organic carbon. We found the drying (to ∼50% RH) of cloud water droplets resulted in ∼11% evaporative loss of WSOC. This observation confirms that organics in cloud water are reversibly partitioned to the aqueous phase, and likely represents a lower bound on the evaporation of organics during an actual cloud cycle (due to experimental measurement constraints). The relatively low evaporative loss of WSOC (∼11%) compared to the GECKO simulations also suggests the occurrence of aqueous processing of organics in cloud water during transport to WFM. Formic and acetic acid account for a small fraction, 19% on average, of this reversibly partitioned organic material. This may be an important process that affects the distribution and lifetime of reactive carbon in the atmosphere. The lifetime of semi-volatile organics is lower in the gas phase than in the condensed phase, so reversible partitioning to cloud water may reduce deposition losses and enhance the transport of these compounds.53 Although our laboratory analysis included a relatively small sample size (n = 5), the results from long-term observations at WFM show systematically lower organic aerosol concentrations downwind of the summit than in cloud water. This supports our laboratory measurements and suggests the nearly constant presence of volatile WSOC reversibly partitioned to cloud water. GECKO box model simulations predict abundant formation of compounds that reversibly partition to cloud water from the photooxidation of ten different VOCs under high- and low-NOx conditions. However, the relatively low evaporative loss of WSOC (∼11%) compared to the GECKO simulations also suggests the occurrence of aqueous processing in cloud water during transport to WFM.No evidence of drying-induced BrC formation was observed in the cloud water samples. The most likely reason appears to be the airmass reaching WFM may have been depleted of BrC precursors.18,25 Many studies have reported the loss of absorbance of BrC due to photooxidative aging, which could also contribute to the depletion of BrC and BrC precursors during transport.62 Studies have confirmed the need to account for photobleaching of BrC in order to accurately constrain BrC and global radiative balance, so this may have had an impact on the observations.66
The work highlights the potential importance of the large unidentified fraction of WSOC in cloud and fog water, as well, suggesting that current detailed molecular measurements of cloud water will likely be incomplete and may not capture broader trends in the behavior of organics.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
CH and AC were supported by the National Science Foundation through awards AGS-1719252 and AGS-1719245. The CPOC pilot study was funded by NSF RAPID grant: AGS-1753278. WFM trace gas and meteorological measurements were supported by the New York State Energy Research and Development Authority (NYSERDA) Contract 48971. Cloud water measurements during the time period discussed in this paper were supported by NYSERDA under the Adirondack Long Term Monitoring project.References
- B. Ervens, Modeling the Processing of Aerosol and Trace Gases in Clouds and Fogs, Chem. Rev., 2015, 115, 4157–4198 CrossRef CAS.
- P. Herckes, K. T. Valsaraj and J. L. Collett, A review of observations of organic matter in fogs and clouds: origin, processing and fate, Atmos. Res., 2013, 132–133, 434–449 CrossRef CAS.
- Y. Zhang, H. Forrister, J. Liu, J. Dibb, B. Anderson, J. P. Schwarz, A. E. Perring, J. L. Jimenez, P. Campuzano-Jost, Y. Wang, A. Nenes and R. J. Weber, Top-of-atmosphere radiative forcing affected by brown carbon in the upper troposphere, Nat. Geosci., 2017, 10, 486 CrossRef CAS.
- B. Ervens, B. J. Turpin and R. J. Weber, Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies, Atmos. Chem. Phys., 2011, 11, 11069–11102 CrossRef CAS.
- E. A. Marais, D. J. Jacob, J. L. Jimenez, P. Campuzano-Jost, D. A. Day, W. Hu, J. Krechmer, L. Zhu, P. S. Kim, C. C. Miller, J. A. Fisher, K. Travis, K. Yu, T. F. Hanisco, G. M. Wolfe, H. L. Arkinson, H. O. T. Pye, K. D. Froyd, J. Liao and V. F. McNeill, Aqueous-phase mechanism for secondary organic aerosol formation from isoprene: application to the southeast United States and co-benefit of SO2 emission controls, Atmos. Chem. Phys., 2016, 16, 1603–1618 CrossRef CAS.
- J. Fan, Y. Wang, D. Rosenfeld and X. Liu, Review of aerosol-cloud interactions: mechanisms, significance, and challenges, J. Atmos. Sci., 2016, 73, 4221–4252 CrossRef.
- H. R. Pruppacher, in Chemistry of Multiphase Atmospheric Systems, ed. W. Jaeschke, Springer Berlin Heidelberg, Berlin, Heidelberg, 1986, pp. 133–190 Search PubMed.
- M. M. Galloway, M. H. Powelson, N. Sedehi, S. E. Wood, K. D. Millage, J. A. Kononenko, A. D. Rynaski and D. O. De Haan, Secondary organic aerosol formation during evaporation of droplets containing atmospheric aldehydes, amines, and ammonium sulfate, Environ. Sci. Technol., 2014, 48, 14417–14425 CrossRef CAS.
- L. N. Hawkins, M. J. Baril, N. Sedehi, M. M. Galloway, D. O. De Haan, G. P. Schill and M. A. Tolbert, Formation of semisolid, oligomerized aqueous SOA: lab simulations of cloud processing, Environ. Sci. Technol., 2014, 48, 2273–2280 CrossRef CAS.
- D. O. De Haan, A. L. Corrigan, M. A. Tolbert, J. L. Jimenez, S. E. Wood and J. J. Turley, Secondary organic aerosol formation by self-reactions of methylglyoxal and glyoxal in evaporating droplets, Environ. Sci. Technol., 2009, 43, 8184–8190 CrossRef CAS.
- A. K. Y. Lee, R. Zhao, R. Li, J. Liggio, S.-M. Li and J. P. D. Abbatt, Formation of Light Absorbing Organo-Nitrogen Species from Evaporation of Droplets Containing Glyoxal and Ammonium Sulfate, Environ. Sci. Technol., 2013, 47, 12819–12826 CrossRef CAS.
- T. Raventos-Duran, M. Camredon, R. Valorso, C. Mouchel-Vallon and B. Aumont, Structure-activity relationships to estimate the effective Henry's law constants of organics of atmospheric interest, Atmos. Chem. Phys., 2010, 10, 7643–7654 CrossRef CAS.
- T. B. Nguyen, P. B. Lee, K. M. Updyke, D. L. Bones, J. Laskin, A. Laskin and S. A. Nizkorodov, Formation of nitrogen- and sulfur-containing light-absorbing compounds accelerated by evaporation of water from secondary organic aerosols, J. Geophys. Res.: Atmos., 2012, 117(D1), 1–14 CrossRef.
- V. Pratap, M. A. Battaglia, A. G. Carlton and C. J. Hennigan, No evidence for brown carbon formation in ambient particles undergoing atmospherically relevant drying, Environ. Sci.: Processes Impacts, 2020, 22, 442–450 RSC.
- D. Van Pinxteren, A. Plewka, D. Hofmann, K. Müller, H. Kramberger, B. Svrcina, K. Bächmann, W. Jaeschke, S. Mertes, J. L. Collett and H. Herrmann, Schmücke hill cap cloud and valley stations aerosol characterisation during FEBUKO (II): organic compounds, Atmos. Environ., 2005, 39, 4305–4320 CrossRef.
- S. Lance, M. Barth and A. Carlton, Multiphase chemistry: experimental design for coordinated measurement and modeling studies of cloud processing at a mountaintop, Bull. Am. Meteorol. Soc., 2017, 98, ES163–ES167 CrossRef.
- J. J. Schwab, D. Wolfe, P. Casson, R. Brandt, K. L. Demerjian, L. Husain, V. A. Dutkiewicz, K. L. Civerolo and O. V. Rattigan, Atmospheric science research at Whiteface Mountain, NY: site description and history, Aerosol Air Qual. Res., 2016, 16, 827–840 CrossRef CAS.
- J. J. Schwab, P. Casson, R. Brandt, L. Husain, V. Dutkewicz, D. Wolfe, K. L. Demerjian, K. L. Civerolo, O. V. Rattigan, H. D. Felton and J. E. Dukett, Atmospheric Chemistry measurements at Whiteface Mountain, NY: cloud water chemistry, precipitation chemistry, and particulate matter, Aerosol Air Qual. Res., 2016, 16, 841–854 CrossRef CAS.
- X. Zhou, G. Huang, K. Civerolo, U. Roychowdhury and K. L. Demerjian, Summertime observations of HONO, HCHO, and O3 at the summit of Whiteface Mountain, New York, J. Geophys. Res., 2007, 112, D08311 Search PubMed.
- S. Lance, J. Zhang, J. J. Schwab, P. Casson, R. E. Brandt, D. R. Fitzjarrald, M. J. Schwab, J. Sicker, C.-H. Lu, S.-P. Chen, J. Yun, J. M. Freedman, B. Shrestha, Q. Min, M. Beauharnois, B. Crandall, E. Joseph, M. J. Brewer, J. R. Minder, D. Orlowski, A. Christiansen, A. G. Carlton and M. C. Barth, Overview of the CPOC Pilot Study at Whiteface Mountain, NY: Cloud Processing of Organics within Clouds (CPOC), Bull. Am. Meteorol. Soc., 2020, E1820–E1841 Search PubMed.
- R. D. Cook, Y.-H. Lin, Z. Peng, E. Boone, R. K. Chu, J. E. Dukett, M. J. Gunsch, W. Zhang, N. Tolic, A. Laskin and K. A. Pratt, Biogenic, urban, and wildfire influences on the molecular composition of dissolved organic compounds in cloud water, Atmos. Chem. Phys., 2017, 17, 15167–15180 CrossRef CAS.
- V. A. Mohnen and J. A. Kadlecek, Cloud chemistry research at Whiteface Mountain, Tellus B, 1989, 41 B, 79–91 CrossRef.
- H. Gerber, Direct measurement of suspended particulate volume concentration and far-infrared extinction coefficient with a laser-diffraction instrument, Appl. Opt., 1991, 30, 4824–4831 CrossRef CAS.
- M. M. H. El-Sayed, Y. Wang and C. J. Hennigan, Direct atmospheric evidence for the irreversible formation of aqueous secondary organic aerosol, Geophys. Res. Lett., 2015, 42, 5577–5586 CrossRef.
- H. A. Khwaja, S. Brudnoy and L. Husain, Chemical characterization of three summer cloud episodes at whiteface mountain, Chemosphere, 1995, 31, 3357–3381 CrossRef CAS.
- W. C. Hinds, Aerosol technology: properties, behavior, and measurement of airborne particles, 1999 Search PubMed.
- T. B. Nguyen, M. M. Coggon, K. H. Bates, X. Zhang, R. H. Schwantes, K. A. Schilling, C. L. Loza, R. C. Flagan, P. O. Wennberg and J. H. Seinfeld, Organic aerosol formation from the reactive uptake of isoprene epoxydiols (IEPOX) onto non-acidified inorganic seeds, Atmos. Chem. Phys., 2014, 14, 3497–3510 CrossRef.
- B. Aumont, S. Szopa and S. Madronich, Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach, Atmos. Chem. Phys., 2005, 5, 2497–2517 CrossRef CAS.
- A. Hodzic, B. Aumont, C. Knote, J. Lee-Taylor, S. Madronich and G. Tyndall, Volatility dependence of Henry's law constants of condensable organics: application to estimate depositional loss of secondary organic aerosols, Geophys. Res. Lett., 2014, 41, 4795–4804 CrossRef CAS.
- Y. Nannoolal, J. Rarey and D. Ramjugernath, Estimation of pure component properties: Part 3. Estimation of the vapor pressure of non-electrolyte organic compounds via group contributions and group interactions, Fluid Phase Equilib., 2008, 269, 117–133 CrossRef CAS.
- C. J. Hennigan, M. M. H. El-Sayed and A. Hodzic, Detailed characterization of a mist chamber for the collection of water-soluble organic gases, Atmos. Environ., 2018, 188, 12–17 CrossRef CAS.
- H. A. Khwaja and A. Narang, Carbonyls and non-methane hydrocarbons at a rural mountain site in northeastern United States, Chemosphere, 2008, 71, 2030–2043 CrossRef CAS.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 2006 Search PubMed.
- M. M. Bela, M. C. Barth, O. B. Toon, A. Fried, C. Ziegler, K. A. Cummings, Y. Li, K. E. Pickering, C. R. Homeyer, H. Morrison, Q. Yang, R. M. Mecikalski, L. Carey, M. I. Biggerstaff, D. P. Betten and A. A. Alford, Effects of Scavenging, Entrainment, and Aqueous Chemistry on Peroxides and Formaldehyde in Deep Convective Outflow Over the Central and Southeast United States, J. Geophys. Res.: Atmos., 2018, 123, 7594–7614 CAS.
- C. Rose, N. Chaumerliac, L. Deguillaume, C. Mouchel-Vallon, M. Leriche, L. Patryl and P. Armand, Modeling the partitioning of organic chemical species in cloud phases with CLEPS (1.1), Atmos. Chem. Phys., 2018, 18, 2225–2242 CrossRef CAS.
- Y. Yin, D. J. Parker and K. S. Carslaw, Simulation of trace gas redistribution by corrective clouds - Liquid phase processes, Atmos. Chem. Phys., 2001, 1, 19–36 CrossRef CAS.
- M. M. H. El-Sayed, D. Amenumey and C. J. Hennigan, Drying-Induced Evaporation of Secondary Organic Aerosol during Summer, Environ. Sci. Technol., 2016, 50, 3626–3633 CrossRef CAS.
- A. Marinoni, P. Laj, K. Sellegri and G. Mailhot, Cloud chemistry at the Puy de Dôme: variability and relationships with environmental factors, Atmos. Chem. Phys., 2004, 4, 715–728 CrossRef CAS.
- J. Guo, Y. Wang, X. Shen, Z. Wang, T. Lee, X. Wang, P. Li, M. Sun, J. L. Collett, W. Wang and T. Wang, Characterization of cloud water chemistry at Mount Tai, China: seasonal variation, anthropogenic impact, and cloud processing, Atmos. Environ., 2012, 60, 467–476 CrossRef CAS.
- M. Löflund, A. Kasper-Giebl, B. Schuster, H. Giebl, R. Hitzenberger and H. Puxbaum, Formic, acetic, oxalic, malonic and succinic acid concentrations and their contribution to organic carbon in cloud water, Atmos. Environ., 2002, 36, 1553–1558 CrossRef.
- W. Winiwarter, H. Fierlinger, H. Puxbaum, M. C. Facchini, B. G. Arends, S. Fuzzi, D. Schell, U. Kaminski, S. Pahl, T. Schneider, A. Berner, I. Solly and C. Kruisz, Henry's law and the behavior of weak acids and bases in fog and cloud, J. Atmos. Chem., 1994, 19, 173–188 CrossRef CAS.
- Y. Tao and J. G. Murphy, Evidence for the Importance of Semivolatile Organic Ammonium Salts in Ambient Particulate Matter, Environ. Sci. Technol., 2019, 53, 108–116 CrossRef CAS.
- W. C. Keene, A. A. P. Pszenny, J. R. Maben, E. Stevenson and A. Wall, Closure evaluation of size-resolved aerosol pH in the New England coastal atmosphere during summer, J. Geophys. Res.: Atmos., 2004, 109, 1–16 CrossRef.
- J. Liu, X. Zhang, E. T. Parker, P. R. Veres, J. M. Roberts, J. A. de Gouw, P. L. Hayes, J. L. Jimenez, J. G. Murphy, R. A. Ellis, L. G. Huey and R. J. Weber, On the gas-particle partitioning of soluble organic aerosol in two urban atmospheres with contrasting emissions: 2. Gas and particle phase formic acid, J. Geophys. Res.: Atmos., 2012, 117(D21), 1–15 CrossRef.
- T. Stavrakou, J.-F. Müller, J. Peeters, A. Razavi, L. Clarisse, C. Clerbaux, P.-F. Coheur, D. Hurtmans, M. De Mazière, C. Vigouroux, N. M. Deutscher, D. W. T. Griffith, N. Jones and C. Paton-Walsh, Satellite evidence for a large source of formic acid from boreal and tropical forests, Nat. Geosci., 2012, 5, 26–30 CrossRef CAS.
- D. B. Millet, M. Baasandorj, D. K. Farmer, J. A. Thornton, K. Baumann, P. Brophy, S. Chaliyakunnel, J. A. De Gouw, M. Graus, L. Hu, A. Koss, B. H. Lee, F. D. Lopez-Hilfiker, J. A. Neuman, F. Paulot, J. Peischl, I. B. Pollack, T. B. Ryerson, C. Warneke, B. J. Williams and J. Xu, A large and ubiquitous source of atmospheric formic acid, Atmos. Chem. Phys., 2015, 15, 6283–6304 CrossRef CAS.
- F. Paulot, D. Wunch, J. D. Crounse, G. C. Toon, D. B. Millet, P. F. Decarlo, C. Vigouroux, N. M. Deutscher, G. G. Abad, J. Notholt, T. Warneke, J. W. Hannigan, C. Warneke, J. A. De Gouw, E. J. Dunlea, M. De Mazière, D. W. T. Griffith, P. Bernath, J. L. Jimenez and P. O. Wennberg, Importance of secondary sources in the atmospheric budgets of formic and acetic acids, Atmos. Chem. Phys., 2011, 11, 1989–2013 CrossRef CAS.
- H. D. Alwe, D. B. Millet, X. Chen, J. D. Raff, Z. C. Payne and K. Fledderman, Oxidation of Volatile Organic Compounds as the Major Source of Formic Acid in a Mixed Forest Canopy, Geophys. Res. Lett., 2019, 46, 2940–2948 CrossRef CAS.
- P. Khare, N. Kumar, K. M. Kumari and S. S. Srivastava, Atmospheric formic and acetic acids: an overview, Rev. Geophys., 1999, 37, 227–248 CrossRef CAS.
- M. M. H. El-Sayed, D. L. Ortiz-montalvo and C. J. Hennigan, The effects of isoprene and NOx on secondary organic aerosols formed through reversible and irreversible uptake to aerosol water, Atmos. Chem. Phys., 2018, 1171–1184 CrossRef CAS.
- C. J. Hennigan, M. H. Bergin, J. E. Dibb and R. J. Weber, Enhanced secondary organic aerosol formation due to water uptake by fine particles, Geophys. Res. Lett., 2008, 35, 1–5 CrossRef.
- R. J. Weber, A. P. Sullivan, R. E. Peltier, A. Russell, B. Yan, M. Zheng, J. de Grouw, C. Warneke, C. Brock, J. S. Holloway, E. L. Atlas and E. Edgerton, A study of secondary organic aerosol formation in the anthropogenic-influenced southeastern United States, J. Geophys. Res.: Atmos., 2007, 112, 1–13 CrossRef.
- T. B. Nguyen, J. D. Crounse, A. P. Teng, J. M. St. Clair, F. Paulot, G. M. Wolfe and P. O. Wennberg, Rapid deposition of oxidized biogenic compounds to a temperate forest, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E392–E401 CrossRef CAS.
- N. Sareen, A. N. Schwier, T. L. Lathem, A. Nenes and V. F. McNeill, Surfactants from the gas phase may promote cloud droplet formation, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2723–2728 CrossRef CAS.
- A. A. Rodriguez, A. De Loera, M. H. Powelson, M. M. Galloway and D. O. De Haan, Formaldehyde and Acetaldehyde Increase Aqueous-Phase Production of Imidazoles in Methylglyoxal/Amine Mixtures: Quantifying a Secondary Organic Aerosol Formation Mechanism, Environ. Sci. Technol. Lett., 2017, 4, 234–239 CrossRef CAS.
- A. K. Y. Lee, K. L. Hayden, P. Herckes, W. R. Leaitch, J. Liggio, A. M. Macdonald and J. P. D. Abbatt, Characterization of aerosol and cloud water at a mountain site during WACS 2010: secondary organic aerosol formation through oxidative cloud processing, Atmos. Chem. Phys., 2012, 12, 7103–7116 CrossRef CAS.
- M. H. Powelson, B. M. Espelien, L. N. Hawkins, M. M. Galloway and D. O. De Haan, Brown Carbon Formation by Aqueous-Phase Carbonyl Compound Reactions with Amines and Ammonium Sulfate, Environ. Sci. Technol., 2014, 48, 985–993 CrossRef CAS.
- K. M. Updyke, T. B. Nguyen and S. A. Nizkorodov, Formation of brown carbon via reactions of ammonia with secondary organic aerosols from biogenic and anthropogenic precursors, Atmos. Environ., 2012, 63, 22–31 CrossRef CAS.
- E. A. Betterton and M. R. Hoffmann, Henry's Law Constants of Some Environmentally Important Aldehydes, Environ. Sci. Technol., 1988, 22, 1415–1418 CrossRef CAS.
- H. J. Lim, A. G. Carlton and B. J. Turpin, Isoprene forms secondary organic aerosol through cloud processing: model simulations, Environ. Sci. Technol., 2005, 39, 4441–4446 CrossRef CAS.
- B. Ervens, Y. Wang, J. Eagar, W. R. Leaitch, A. M. Macdonald, K. T. Valsaraj and P. Herckes, Dissolved organic carbon (DOC) and select aldehydes in cloud and fog water: the role of the aqueous phase in impacting trace gas budgets, Atmos. Chem. Phys., 2013, 13, 5117–5135 CrossRef.
- R. F. Hems and J. P. D. Abbatt, Aqueous Phase Photo-oxidation of Brown Carbon Nitrophenols: Reaction Kinetics, Mechanism, and Evolution of Light Absorption, ACS Earth Space Chem., 2018, 2, 225–234 CrossRef CAS.
- A. Laskin, J. Laskin and S. A. Nizkorodov, Chemistry of Atmospheric Brown Carbon, Chem. Rev., 2015, 115, 4335–4382 CrossRef CAS.
- P. K. Aiona, J. L. Luek, S. A. Timko, L. C. Powers, M. Gonsior and S. A. Nizkorodov, Effect of Photolysis on Absorption and Fluorescence Spectra of Light-Absorbing Secondary Organic Aerosols, ACS Earth Space Chem., 2018, 2, 235–245 CrossRef CAS.
- J. P. S. Wong, A. Nenes and R. J. Weber, Changes in Light Absorptivity of Molecular Weight Separated Brown Carbon Due to Photolytic Aging, Environ. Sci. Technol., 2017, 51, 8414–8421 CrossRef CAS.
- J. P. S. Wong, M. Tsagaraki, I. Tsiodra, N. Mihalopoulos, K. Violaki, M. Kanakidou, J. Sciare, A. Nenes and R. J. Weber, Atmospheric Evolution of Molecular Weight Separated Brown Carbon from Biomass Burning, Atmos. Chem. Phys., 2019, 19, 7319–7334 CrossRef CAS.
- J. Zhang, S. Lance, R. Brandt, J. Marto, M. Ninneman and J. Schwab, Observed below-Cloud and Cloud Interstitial Submicron Aerosol Chemical and Physical Properties at Whiteface Mountain, New York, during August 2017, ACS Earth Space Chem., 2019, 3, 1438–1450 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Discussion on the analytical results of cloud water samples. See DOI: 10.1039/d0ea00005a |
| This journal is © The Royal Society of Chemistry 2021 |