
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of main and side chain chlorination on the photovoltaic properties of benzodithiophene-alt-benzotriazole polymers†
Haijun
Bin
a,
Indunil
Angunawela
b,
Ruijie
Ma
 c,
Asritha
Nallapaneni
de,
Chenhui
Zhu
d,
Pieter J.
Leenaers
a,
Bart W. H.
Saes
a,
Martijn M.
Wienk
a,
He
Yan
c,
Harald
Ade
*b and
René A. J.
Janssen
*af
c,
Asritha
Nallapaneni
de,
Chenhui
Zhu
d,
Pieter J.
Leenaers
a,
Bart W. H.
Saes
a,
Martijn M.
Wienk
a,
He
Yan
c,
Harald
Ade
*b and
René A. J.
Janssen
*af
aMolecular Materials and Nanosystems & Institute for Complex Molecular Systems, Eindhoven University of Technology, P. O. Box 513, 5600 MB, Eindhoven, The Netherlands. E-mail: r.a.j.janssen@tue.nl
bDepartment of Physics and Organic and Carbon Electronics Laboratories (ORaCEL), North Carolina State University, Raleigh, NC 27695, USA. E-mail: hwade@ncsu.edu
cDepartment of Chemistry, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong
dAdvanced Light Source Lawrence Berkeley National Laboratory Berkeley, CA 94720, USA
eDepartment of Polymer Engineering, University of Akron, Akron, Ohio 44325, USA
fDutch Institute for Fundamental Energy Research, De Zaale 20, 5612 AJ, Eindhoven, The Netherlands
First published on 22nd September 2020
Abstract
In developing organic semiconductor polymers for photovoltaic applications, chlorine substitution has become an effective strategy in replacing fluorine substitution to overcome the drawbacks of low yield and high cost, commonly associated with fluorination. In general, several molecular positions are available for chlorination. To obtain a clear understanding of the impact of chlorine substitution on the intrinsic polymer properties, an investigation of structure–property relationships is necessary. Herein, four donor–acceptor type polymers with the same conjugated backbone and flexible alkyl chains, but with chlorine atoms in different positions, are employed to systematically investigate the effect of the site of chlorination on the optoelectronic properties and photovoltaic performance. Substitution of fluorine by chlorine in the backbone slightly increases open circuit voltage (Voc) and fill factor (FF) of the solar cells but causes a loss of short-circuit current density (Jsc). The introduction of chlorine in the conjugated side chains, however, significantly improves Voc, FF, and power conversion efficiency, benefiting from a lower HOMO energy level, efficient and well-balanced transport properties, and superior nanoscale morphology.
Introduction
Organic solar cells (OSCs) have made rapid progress recently. The low power conversion efficiency (PCE) faced in the past has been overcome and PCE now exceeds 17%.1–3 Coupled with its low cost, lightweight, promise for flexible roll-to-roll processing, and semi-transparent characteristics,4 the gap to successful commercialization is decreasing.5–7 The breakthrough in efficiency was mainly due to the discovery and development of new non-fullerene acceptors (NFAs) in the active layer,8,9 such as ITIC10 and Y6 and their derivatives.11 Meanwhile, the donor materials that match the acceptors in the photoactive blend, play an equally important role.12 To enable practical applications, researchers have developed efficient, lower cost donor materials, such as the polymers PM6 and PTQ10.6,13–17 Preferably, electron donor and acceptor materials possess complementary optical absorption spectra to capture photons over a large part of the solar spectrum and have appropriate energy levels to promote exciton dissociation and charge separation. Furthermore molecular orientation,18 molecular packing, and domain purity that are closely related to nanoscale morphology and charge carrier mobility,19–22 strongly affect the photovoltaic performance. These properties find their origin in the molecular structure, which can be tailored with subtle structural changes.Over the years, many molecular design strategies have been adopted to alter optoelectronic properties and morphology of photovoltaic materials, including main-chain engineering (donor–acceptor units selection),23,24 side-chain engineering,25–30 and atomic or functional group substitution.7,31–33 Among the common strategies, fluorination is a widely studied and a well-established approach for designing efficient photovoltaic materials.31,34–36 Attributed to its small van der Waals radius of 1.35 Å and high electronegativity, fluorine (F) effectively modifies the energy levels and enhances optical absorption, without a negative influence on molecular packing. Moreover, benefitting from non-covalent intermolecular interactions, fluorine-containing materials generally feature enhanced coplanarity, molecular packing, and charge carrier mobility. Hence, fluorination has provided numerous excellent donors and acceptors, delivering outstanding PCEs in OSCs.5,6,35 However, some intrinsic demerits of fluorination exist. The selective formation of aromatic carbon–fluorine bonds is synthetically challenging.37 Hou et al. reported e.g. that the synthesis of 2-ethylhexyl-3-fluorothiophene requires four steps from 3-bromothiophene with tedious purification of intermediates giving an overall yield of 15%.31,38 In another example Leclerc et al. reported the synthesis of fluorinated dithienyldiketopyrrolopyrroles in six steps,39 which can be compared to a one-step procedure for the nonfluorinated analog. The multi-step synthesis and complex purification of the intermediates lead to higher costs of fluorinated conjugated materials and may hamper large-scale synthesis of these materials towards their commercial application.39–41
To work around this issue, researchers are now devoting considerable attention to chlorination. The synthesis of chlorine-containing materials is simpler, its reaction steps provide higher yields, and the raw materials are cheaper, resulting in lower costs.40,41 More importantly, chlorine (Cl) substituted organic semiconductors have similar or even superior optoelectronic properties compared to fluorine substituted analogs. In conjugated polymers for OSCs the incorporation of chlorine was found to enhance the stability compared to non-chlorinated and fluorinated analogs.40 In acceptor–donor–acceptor (A–D–A) type NFAs, the F or Cl atoms are usually on the terminal acceptor “A”. The higher dipole moment of the carbon–chlorine bond enhances intramolecular charge transfer, such that the optical band gap of chlorine-containing NFAs is often narrower compared to their fluorinated analogs,42,43e.g. in IT4Cl versus IT4F,44,45 and in IEICO-4Cl versus IEICO-4F.35,46 Also, the planarity, crystallinity, and charge mobility of some chlorine-containing NFAs have been reported to increase.40,47 These characteristics may enhance the short-circuit current density (Jsc) and fill factor (FF) in OSCs. Recently, Cui et al. demonstrated that BTP-4Cl, a chlorinated NFA, exhibits less non-radiative energy loss than its fluorinated analog BTP-4F (Y6) in combination with PM6 as donor. As a result, a higher open-circuit voltage (Voc) is achieved with BTP-4Cl, despite its lower lowest unoccupied molecular orbital (LUMO) energy level.48 For the donor–acceptor (D–A) type conjugated polymers, the availability of empty d-orbitals on chlorine enhances the delocalization of π-electrons. Hence, chlorine-containing materials often offer deeper energy levels, even though the electronegativity is less than that of fluorine.40,41,49,50 This is a favorable characteristic for donor materials because the lower highest occupied molecular orbital (HOMO) energy level can afford a higher Voc in OSCs. Unlike the NFAs, most D–A polymers offer a range of positions for chlorination, either in the main chain or in a conjugated side chain. Some groups have reported main chain and side chain chlorination, obtaining impressive results, but a systematic study, in particular, on the synergistic effect of bi-chlorination in the side and main chain is lacking at present.
To fill this gap and better understand the effect of the chlorination position on material properties, we synthesized and studied four benzodithiophene (BDT)-alt-benzotriazole (BTz) based polymers with different motifs of chlorine substitution (Scheme 1). Compared to the reference material, J71 with two F's on the main chain, J71–Cl has two extra Cl's in the side chains. J100 is similar to J71, but has two Cl's instead of two F's per repeating unit, while J101 has four Cl's; two in the main and two in the side chains per repeating unit. The halogens on the thiophenes adjacent to the BTz can planarize polymers via through-space noncovalent conformational locks, a topic recently discussed in detail by Marks et al.51 We apply these polymers as donors in OSCs by blending with IT4F as acceptor. Introduction of Cl in the side chains has a positive effect on the energy levels, transport properties, molecular packing, and photovoltaic performance. Using diphenyl ether (DPE) as processing additive, J71–Cl-based and J101-based devices, with chlorine in the conjugated side chains, afford PCEs over 11.0%, featuring high Voc, Jsc, and FF. Although introduction of chlorine in the backbone reduces the HOMO energy level of J100 to obtain a higher Voc, suboptimal morphologies and transport properties result in a low Jsc and FF and cause the optimized PCE of OSCs based on J100 to be less than those based on the J71 reference.
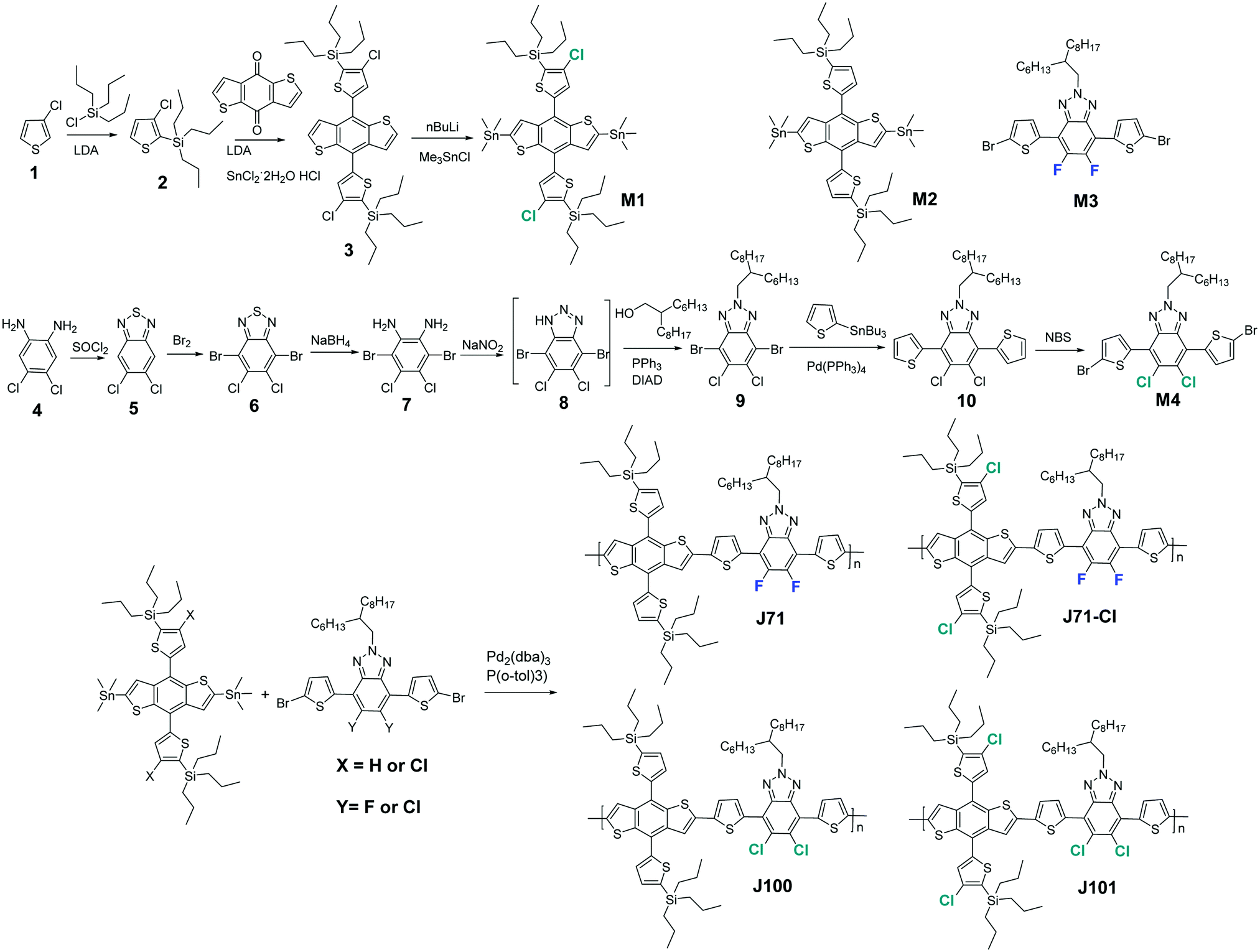 | ||
| Scheme 1 Synthetic routes of the monomers and polymers. | ||
Results and discussion
The synthesis routes to prepare the monomers and polymers are shown in Scheme 1. Monomer M1 and M4 were synthesized according to previous reported procedures.15,28,49,52–55 Although the synthetic route to M4 is long, isolation of intermediates is not required until intermediate 9 and the synthesis of M4 is therefore straightforward and has high overall yield. The four polymers are prepared by palladium-catalyzed Stille polymerization. The synthetic procedures are described in the Experimental section of the ESI.† These four polymers are well soluble in chloroform, which enables their solution processing for thin-film devices. The number-average molecular weights (Mn) of J71, J71–Cl, J100, and J101 were 28.2, 18.9, 35.5, and 24.6 kDa with a polydispersity index (PDI) of 2.16, 1.94, 2.71, and 2.31, respectively, as listed in Table 1.| Polymer | M n (kDa) | PDI | λ onset (nm) | E optg (eV) | HOMO (eV) | LUMO (eV) | μ h (cm2 V−1 s−1) | d (010) (Å) | L c (010) (Å) |
|---|---|---|---|---|---|---|---|---|---|
| J71 | 28.2 | 2.16 | 632 | 1.96 | −5.31 | −2.94 | 2.04 × 10−6 | 3.79 | 25 |
| J71–Cl | 18.9 | 1.94 | 632 | 1.96 | −5.33 | −2.98 | 1.96 × 10−6 | 3.79 | 25 |
| J100 | 35.5 | 2.71 | 636 | 1.95 | −5.35 | −3.00 | 3.65 × 10−6 | 3.88 | 24 |
| J101 | 24.6 | 2.31 | 632 | 1.96 | −5.47 | −2.97 | 3.49 × 10−6 | 3.83 | 27 |
The normalized UV-vis-NIR spectra of thin films of the donor polymers and the IT4F acceptor are shown in Fig. 1a, and the corresponding optical data are summarized in Table 1. The four polymers share an absorption edge at ∼632 nm and have virtually identical optical bandgaps of 1.95–1.96 eV (Table 1). J71 and J71–Cl exhibit two distinct absorption maxima, but the corresponding peaks are less defined for J100 and, especially, for J101. The relative intensities of the first and second peaks in the absorption spectra are higher for the fluorinated polymers J71 and J71–Cl than for J100 and J101. This suggests that fluorine in the main chain promotes the π–π stacking and intermolecular aggregation of the molecules, while introduction of chlorine in the side chain has little effect on the molecular packing. For J100 and J101, containing chlorine in the backbone, the π–π stacking seems to be less strong. Apparently, fluorine provides a stronger intermolecular interaction than chlorine of which the larger Van de Waals radius (1.80 Å) may reduce π–π stacking in the backbone. The thin film absorption spectra of J71 and J71–Cl are only slightly redshifted compared to that of diluted solutions (Fig. S1, ESI†). In contrast, J100 and J101 films show a significantly redshift compared to the absorption in solution (Fig. S1, ESI†). This implies a higher tendency for J71 and J71–Cl to aggregate in solution as a consequence of stronger π–π stacking. The absorption spectrum of the four donor polymers is complementary to that of the IT4F acceptor (Fig. S2, ESI†).
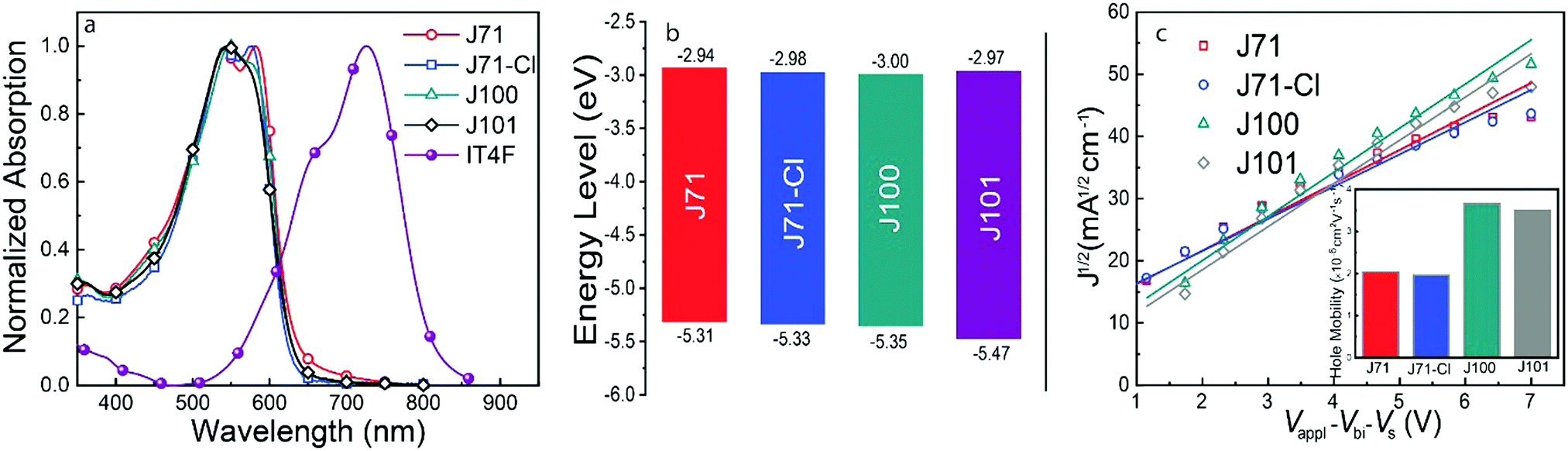 | ||
| Fig. 1 Optoelectronic properties of the polymers. (a) Normalized UV-vis-NIR absorption spectra of polymers and IT4F in thin film. (b) Electronic energy level diagrams of donors. (c) Hole mobility of the pure polymer films. | ||
The polymer energy levels were measured by square-wave voltammetry on thin films (Fig. S3, ESI†) and are depicted in Fig. 1b and summarized in Table 1. The energy levels were determined from the onset potentials of the redox waves (φox/red), referenced against the redox potential of ferrocene/ferrocenium (Fc/Fc+) versus Ag/AgCl in the measurement system (φ1/2(Fc/Fc+) = 0.37 eV), and converted to absolute energies by setting E(Fc/Fc+) vs. vacuum to −4.8 eV using EHOMO/LUMO = −q(φox/red − φ1/2(Fc/Fc+) + 4.8) [eV]. Apart from an unknown uncertainty in the energy of E(Fc/Fc+) vs. vacuum, the accuracy of the values is estimated to be ±0.05 eV. As expected, the Cl-containing polymer J100 has a lower HOMO level than J71. Two additional Cl atoms introduced into the conjugated side chains of J71–Cl and J101 further reduce the HOMO energy levels, compared to J71 and J100, which is expected to give a higher Voc in OSCs.
The hole mobility of the polymers was determined from space-charge-limited current (SCLC) measurements on pure polymer films in hole-only devices (ITO/PEDOT:PSS/active layer/MoO3/Ag) (Fig. 1c). The hole mobilities of J71 and J71–Cl are approximately 2.0 × 10−6 cm2 V−1 s−1, while those of J100 and J101 are about 3.5 × 10−6 cm2 V−1 s−1 (Table 1). Hence, replacing fluorine by chlorine on the backbone improves the hole mobility, while introducing chlorine in the side-chain has virtually no effect on the hole mobility (Table 1).40 The surface morphologies of the neat films were investigated by atomic force microscopy (AFM) in tapping mode. As shown in Fig. S4 (ESI†), AFM indicates that J71 and J71–Cl thin films have a somewhat higher surface roughness (Rq ≈ 0.43 nm) than J100 (Rq = 0.39 nm) and J101 (Rq = 0.29 nm). A higher surface roughness can be expected when the polymers have stronger tendency to aggregate and along this line it suggests that double chlorination causes less aggregation.
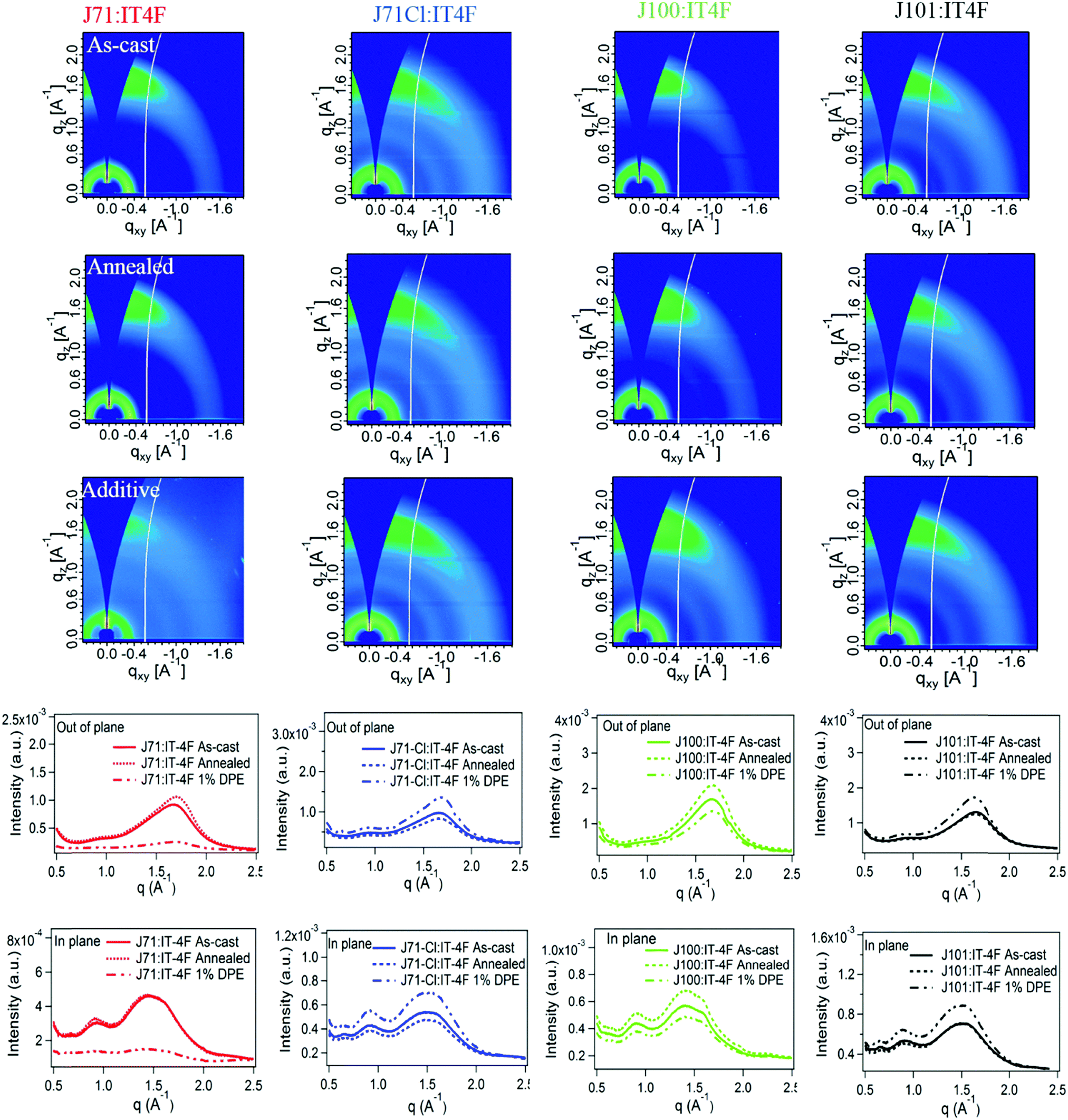
The effect of chlorination on molecular packing in the polymer films was studied by grazing-incidence wide-angle X-ray scattering (GIWAXS).56 As shown in Fig. 2 and Table 1, all polymers show a strong π–π stacking (010) reflection at around 1.64 Å−1 in the out-of-plane (OOP) direction. The OOP π–π stacking diffraction peaks reveal that J71 and J71–Cl have a somewhat closer π–π staking distance (d) than J100 and J101 (Table 1), consistent with information extracted from the optical absorption spectra. The coherence length (Lc) derived from the (010) reflection does not change much with different Cl substitutions (Table 1), suggesting that the spatial extent of ordered aggregates of the four polymers is similar along the π–π stacking direction in pristine films. Analyzing the texture of (100) and (010) peaks qualitatively, we note an apparent high intensity of the (010) peak in the OOP direction. This is often interpreted to indicate preferred face-on orientation. However, we note that the diffraction conditions (Ewald sphere) creates an apparent signal for the (010) peak in the OOP direction even for a random 2D isotropic materials (with the backbone in-plane) (see Note S1 in the ESI†).57,58 The weak enhancement in the IP direction of the complementary (100) peak suggests that these materials have a large isotropic population. Quantitative and even further qualitative analysis would require sophisticated pole-figure analysis that properly takes the isotropic population into account and furthermore considers that the π–π stacking and lamellar stacking intensities and coherence length can be independent of each other.59,60 Such analysis is outside the current scope.
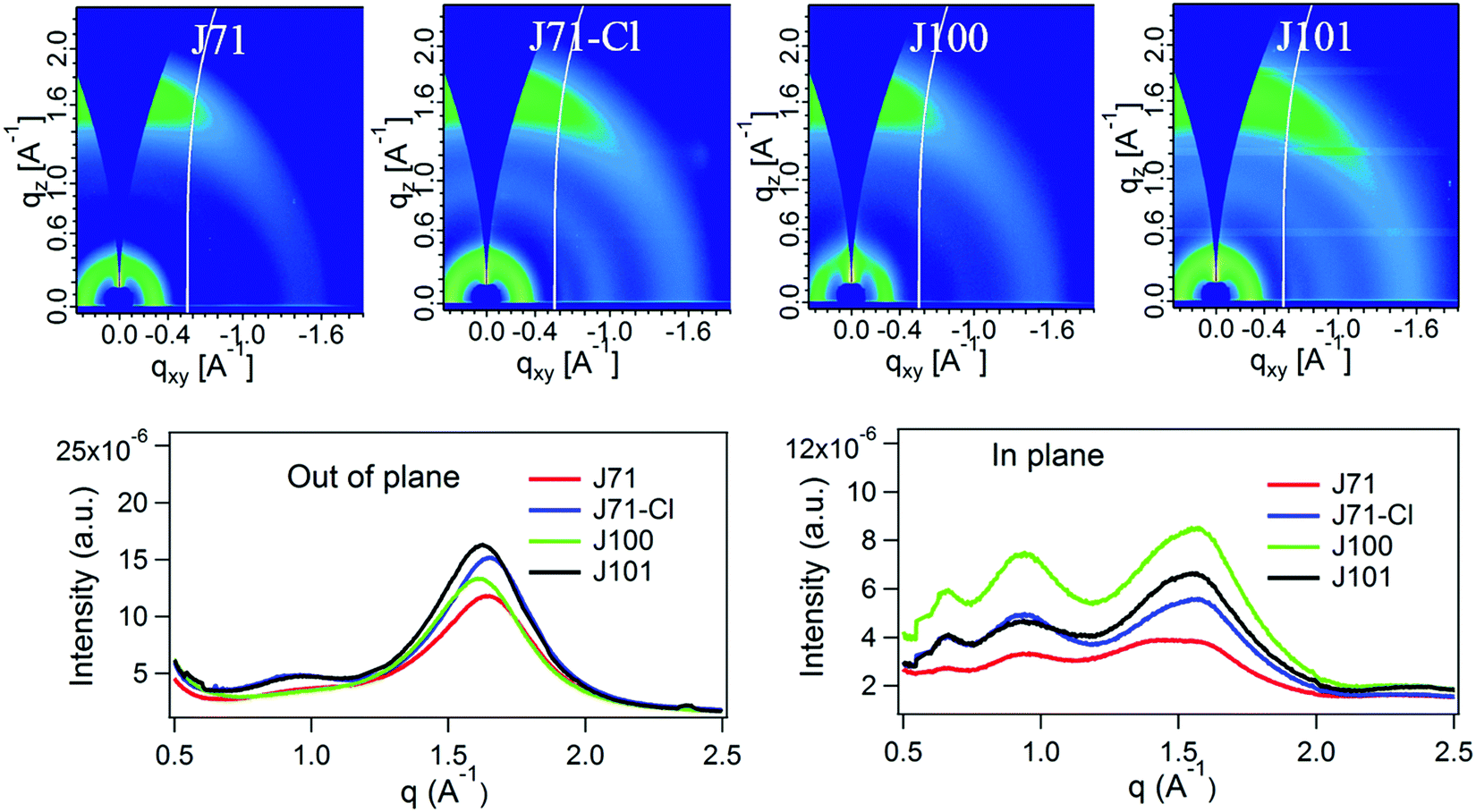 | ||
| Fig. 2 2D GIWAXS patterns, out-of-plane and in-plane scattering profiles of the four polymers. | ||
The combined results from optical absorption, π–π stacking, and AFM demonstrate that if chlorine is located on the main chain, it has a tendency to increase the π–π stacking distance and decrease aggregation, but that it has little structural effect when it is on the conjugated side chain.
To investigate the photovoltaic properties of the four polymers, OSCs with a conventional ITO/PEDOT:PSS/active layer (polymer:IT4F)/PDINO/Al device structure and active area of 0.09 cm2 were fabricated. The optimized polymer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IT4F weight ratio was 1.2:1. Active layers with a thickness of about 120 nm were prepared by spin coating blend solutions with a total concentration of 20 mg mL−1 in chloroform at 2000 rpm for 60 s (because of its higher Mn, the concentration for J100:IT4F was 12 mg mL−1). The active layers were processed under three different conditions: without any extra treatment (as-cast), with thermal annealing at 120 °C for 10 min. (annealed), and using 1% DPE as processing additive combined with annealing at 100 °C for 10 min. (additive). The photovoltaic performance is shown in Fig. 3 and the data are presented in Table 2. Compared to as-cast devices, thermal annealing slightly improves the Jsc and FF, whilst largely retaining the original Voc such that the PCE of the devices increases after annealing. Compared to J71, introduction of Cl in the backbone enhanced the Voc from 0.80 V for J71 to 0.86 V for J100. The increase in Voc is even more pronounced when Cl is in the side chain, resulting in a high Voc of 0.92 V for J71–Cl and of 0.96 V for J101. These changes are consistent with the differences in the HOMO energy levels. However, due to a limiting FF, which may result from suboptimal morphologies or transport properties, the device efficiencies of the four polymers are lower. Only for the J71–Cl based device, the PCE exceeds 10%. To improve device performance, 1% (v/v) DPE was added to the blend solution as processing additive to optimize the active layer morphology. DPE has a higher boiling point (b.p. 258.55 °C) and lower vapor pressure (p = 2.7 × 10−3 kPa) than chloroform (b.p. 61.15 °C, p = 25.9 kPa) and is thus expected to evaporate slowly which causes DPE enrichment in the solvent mixture as drying progresses. Using the additive, OSCs based on J101:IT4F give the best PCE of 11.34%, with the highest Voc of 0.93 V, a high Jsc and FF of 18.20 mA cm−2 and 0.67, respectively. Using DPE, also the PCE of J71–Cl:IT4F based OSCs increased, to 11.1%, with a high Voc of 0.87 V, a high Jsc of 18.41 mA cm−2 and FF of 0.69. However, the performance of devices based on J71 and J100 could not be improved with the additive. Thermal annealing provided the J71:IT4F based OSC an optimized PCE of 8.16%, with a low Voc and FF of 0.79 V and 0.52, respectively. Here, the additive hardly improves Jsc and FF, but reduces Voc to 0.72 V, leading to a decrease in PCE. The optimal J100:IT4F based OSC only shows a PCE of 7.7%, due to a low Jsc of 17.50 mA cm−2 and low FF of 0.52. Although the additive improves the FF to 0.57, the Voc decreases concomitantly from 0.86 to 0.76 V, and the PCE is not improved. The external quantum efficiencies (EQE) of the optimized devices were measured (Fig. S5, ESI†) and the Jsc values calculated from the EQE curves (Table 2) match well with those obtained from the current density–voltage (J–V) measurements.
:IT4F weight ratio was 1.2:1. Active layers with a thickness of about 120 nm were prepared by spin coating blend solutions with a total concentration of 20 mg mL−1 in chloroform at 2000 rpm for 60 s (because of its higher Mn, the concentration for J100:IT4F was 12 mg mL−1). The active layers were processed under three different conditions: without any extra treatment (as-cast), with thermal annealing at 120 °C for 10 min. (annealed), and using 1% DPE as processing additive combined with annealing at 100 °C for 10 min. (additive). The photovoltaic performance is shown in Fig. 3 and the data are presented in Table 2. Compared to as-cast devices, thermal annealing slightly improves the Jsc and FF, whilst largely retaining the original Voc such that the PCE of the devices increases after annealing. Compared to J71, introduction of Cl in the backbone enhanced the Voc from 0.80 V for J71 to 0.86 V for J100. The increase in Voc is even more pronounced when Cl is in the side chain, resulting in a high Voc of 0.92 V for J71–Cl and of 0.96 V for J101. These changes are consistent with the differences in the HOMO energy levels. However, due to a limiting FF, which may result from suboptimal morphologies or transport properties, the device efficiencies of the four polymers are lower. Only for the J71–Cl based device, the PCE exceeds 10%. To improve device performance, 1% (v/v) DPE was added to the blend solution as processing additive to optimize the active layer morphology. DPE has a higher boiling point (b.p. 258.55 °C) and lower vapor pressure (p = 2.7 × 10−3 kPa) than chloroform (b.p. 61.15 °C, p = 25.9 kPa) and is thus expected to evaporate slowly which causes DPE enrichment in the solvent mixture as drying progresses. Using the additive, OSCs based on J101:IT4F give the best PCE of 11.34%, with the highest Voc of 0.93 V, a high Jsc and FF of 18.20 mA cm−2 and 0.67, respectively. Using DPE, also the PCE of J71–Cl:IT4F based OSCs increased, to 11.1%, with a high Voc of 0.87 V, a high Jsc of 18.41 mA cm−2 and FF of 0.69. However, the performance of devices based on J71 and J100 could not be improved with the additive. Thermal annealing provided the J71:IT4F based OSC an optimized PCE of 8.16%, with a low Voc and FF of 0.79 V and 0.52, respectively. Here, the additive hardly improves Jsc and FF, but reduces Voc to 0.72 V, leading to a decrease in PCE. The optimal J100:IT4F based OSC only shows a PCE of 7.7%, due to a low Jsc of 17.50 mA cm−2 and low FF of 0.52. Although the additive improves the FF to 0.57, the Voc decreases concomitantly from 0.86 to 0.76 V, and the PCE is not improved. The external quantum efficiencies (EQE) of the optimized devices were measured (Fig. S5, ESI†) and the Jsc values calculated from the EQE curves (Table 2) match well with those obtained from the current density–voltage (J–V) measurements.
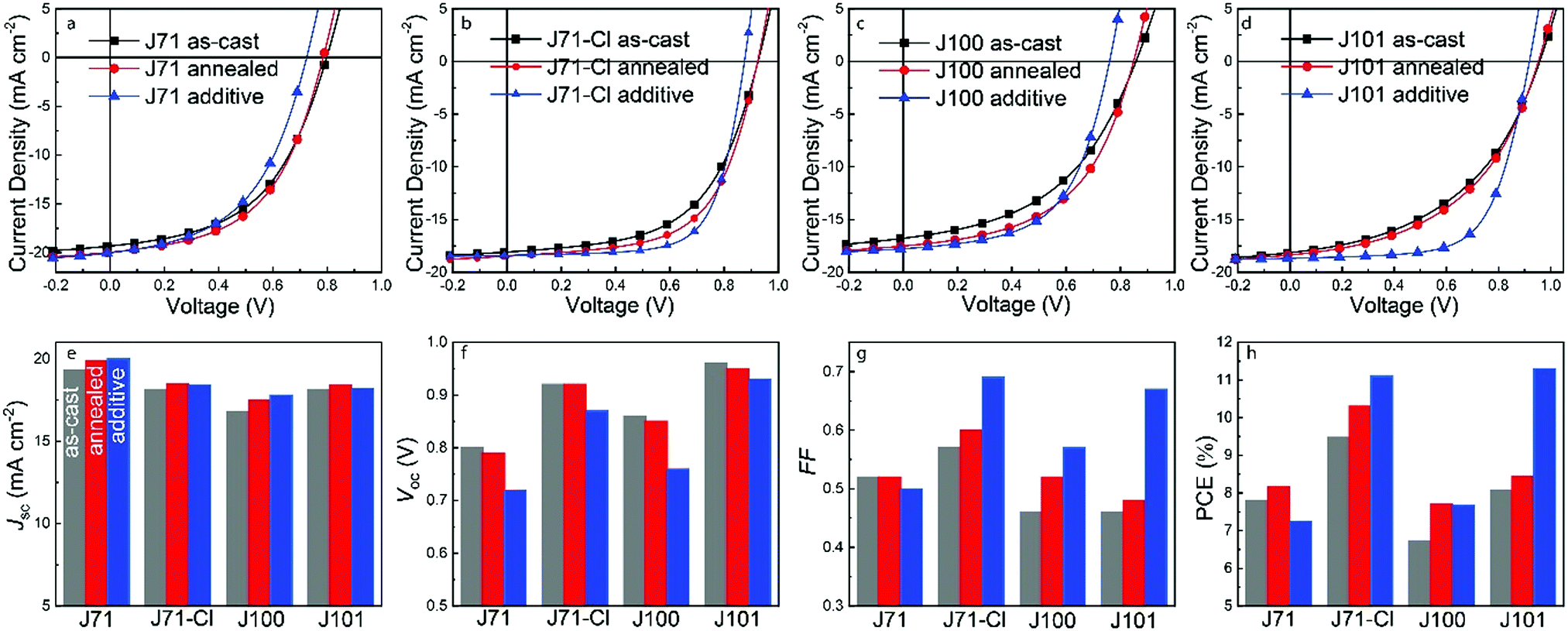 | ||
| Fig. 3 (a–d) J–V characteristics of as-cast, annealed and additive OSCs based on J71, J71–Cl, J100, and J101 as donor and IT4F as acceptor. (e–h) Histograms of the Jsc, Voc, FF and PCE values for the four OSCs under different processing conditions. | ||
| Device | Processing | J sc (mA cm−2) | V oc (V) | FF (%) | PCE (%) | μ h (cm2 V −1 s−1) | μ e (cm2 V−1 s−1) |
|---|---|---|---|---|---|---|---|
| a Values are calculated from EQE. | |||||||
| J71:IT4F | As-cast | 19.30 | 0.80 | 0.50 | 7.79 | 3.05 × 10−5 | 3.32 × 10−5 |
| Annealed | 19.90 (19.57)a | 0.79 | 0.52 | 8.16 | 3.06 × 10−5 | 4.60 × 10−5 | |
| Additive | 20.00 | 0.72 | 0.50 | 7.25 | 4.06 × 10−5 | 4.16 × 10−5 | |
| J71–Cl:IT4F | As-cast | 18.10 | 0.92 | 0.57 | 9.47 | 2.97 × 10−5 | 4.28 × 10−5 |
| Annealed | 18.50 | 0.92 | 0.60 | 10.30 | 4.26 × 10−5 | 4.35 × 10−5 | |
| Additive | 18.41 (18.40)a | 0.87 | 0.69 | 11.10 | 4.98 × 10−5 | 5.26 × 10−5 | |
| J100:IT4F | As-cast | 16.80 | 0.86 | 0.46 | 6.71 | 1.48 × 10−5 | 0.45 × 10−5 |
| Annealed | 17.50 | 0.85 | 0.52 | 7.70 | 1.72 × 10−5 | 0.41 × 10−5 | |
| Additive | 17.80 (17.58)a | 0.76 | 0.57 | 7.67 | 4.10 × 10−5 | 0.85 × 10−5 | |
| J101:IT4F | As-cast | 18.10 | 0.96 | 0.46 | 8.07 | 2.23 × 10−5 | 2.81 × 10−5 |
| Annealed | 18.40 | 0.95 | 0.48 | 8.43 | 2.30 × 10−5 | 2.86 × 10−5 | |
| Additive | 18.20 (18.13)a | 0.93 | 0.67 | 11.30 | 4.64 × 10−5 | 4.71 × 10−5 | |
As can be seen from Fig. 3e–h, the Jsc of all devices is not very sensitive to the different processing conditions. Although the DPE additive causes a decrease in Voc for all four polymers, the loss is comparatively less for side-chain Cl-containing polymers J71–Cl and J101 where Voc remains at a high level. Especially for J101, the Voc loss is small. In other words, the introduction of chlorine in the side chain not only successfully improved Voc, but also reduced Voc loss caused by device processing conditions (or morphology change). The main factor in enhancing the PCE using DPE as an additive is the FF. For the Cl-containing polymers J71–Cl and J101 the increase is very outspoken (Table 2). Interestingly, when we used PNDIT-F3N and Ag to replace PDINO and Al as the electrode (Fig. S6 and Table S1, ESI†), we found that the PCE of J71–Cl can be further increased to 12.4%, with Voc of 0.90 V, Jsc of 18.59 mA cm−2 and FF of 0.74. But the PCE of J101:IT4F remained 11.3%, with Voc of 0.92 V, Jsc of 18.6 mA cm−2 and FF of 0.66. The increased FF (from 0.69 to 0.74) for J71–Cl:IT4F indicates an improved contact with the PNDIT-F3N/Ag electrode, but this improvement does not occur for J101 (FF goes from 0.67 to 0.66), demonstrating that the effects of interlayers in modifying the contact are rather delicate and difficult to predict. Overall, the results demonstrate that introducing chlorine in the side chains improves the photovoltaic performance, while introducing chlorine into the main chain of BTD-alt-BTz polymers does not give a clear advantage. Of course the four polymers have somewhat different molecular weights (Table 1) which may affect the results. A higher Mn often improves device performance.61 In that respect, we note that the two polymers with chlorinated side chains (J71–Cl and J101) have somewhat lower Mn (Table 1) than the other two (J71 and J100) but still give rise to higher PCEs. This supports our conclusion that chlorination of the side chains causes the beneficial effect.
To investigate the charge transport properties of these four bulk heterojunction blend films, hole and electron mobilities (μh and μe) were determined from SCLC measurements (Fig. S6, ESI†). The resulting μh and μe values are listed in Table 2. Unlike in the pure polymer films, the blend films of F-containing polymers (J71 and J71–Cl) have higher mobilities than the blend films of Cl-containing polymers (J100 and J101), and the mobilities of polymers with chlorine in the side-chain are higher than those without, implying that in the blend films, the crystallinity of the donor and acceptor affect each other. The mobilities of J71:IT4F blends are relatively high and vary little with processing conditions. This is consistent with the small difference in Jsc and FF under different conditions. The mobilities of J71–Cl:IT4F increase with thermal annealing and use of the DPE additive, its optimal μh and μe are up to 4.98 × 10−5 and 5.26 × 10−5 cm2 V−1 s−1, these mobilities are the highest in the whole series and give a high FF of 0.67. For J100:IT4F and J101:IT4F, the mobilities significantly improved by using the DPE additive. The optimal μh and μe were determined to be 4.10 × 10−5 and 0.85 × 10−5 cm2 V−1 s−1 for J100:IT4F, and 4.64 × 10−5 and 4.71 × 10−5 cm2 V−1 s−1 for J101:IT4F. Clearly, the J100:IT4F blend shows unbalanced mobilities due to the low μe. This may contribute to the low Jsc of its OSCs. J101 is similar to J71–Cl, with high and balanced mobilities when using the processing additive, which suppresses charge recombination and improves charge extraction and collection, leading to a high Jsc and FF in OSCs. The UV-vis-NIR absorption spectra of the blend films measured with the three different processing conditions (Fig. S2, ESI†), reveal only minor differences between the as-cast films and the thermally annealed films. In contrast, when using the additive, the absorption intensity corresponding to the acceptor region (630–800 nm) decreased for the four blends. Concurrently, the additive slightly increased the absorption intensities of the donor for the J71–Cl and J101 based films, but not for the J71 and J100 based blends. These results indicate that DPE can inhibit the strong crystallization and self-aggregation behavior of IT4F, and at the same time seems to promote aggregation of J71–Cl and J101, allowing the donor and acceptor to achieve synergistic molecular packing in blend films,62 which may result in better nanoscale phase separation and more appropriate morphology.
The surface morphology of all active layers was studied by tapping-mode AFM (Fig. 4). The height and phase images reveal that the blend films with the F-containing polymers J71 and J71–Cl have similar surface features, and that their roughness is relatively large under the same processing conditions. The surface features of blends with the Cl-containing polymers J100 and J101 are also very similar, but their surfaces are smoother and the roughness is less than for the blends with F-containing polymers, consistent with the AFM results of neat polymer films. Compared to J71 and J71–Cl, the size of the surface features in the lateral dimensions is larger for blends with J100 and J101. Blend films of the polymers J71 and J100, which do not contain chlorine in the side-chains, have significantly increased roughness when using the additive (Rq from 0.74 to 1.44 nm for J71; Rq from 0.42 to 1.01 nm for J100). For blends of the polymers with Cl in the side-chain J71–Cl and J101, the roughness of the blend film increased less (Rq from 0.99 to 1.06 nm for J71–Cl; Rq from 0.46 to 0.81 nm for J101). Changes in surface morphology as a result of processing conditions often coincide with a loss of Voc,63 presumably because surface roughness is a sign of increased aggregation which often causes a reduction of Voc, via a shift of energy levels and reduction of the optical band gap.
 | ||
| Fig. 4 AFM height and phase images of the four blend films for different processing conditions. | ||
The domain spacing distribution and average purity of the mixed domains of the blends was characterized by resonant soft X-ray scattering (R-SoXS).64,65 In multiphase systems, the square root of the integrated scattering intensity (ISI) and the component scattering intensity (CSI) of organic blend films (that track the standard deviation of the composition) correlate in a mostly linear relation with the device FF.21,22,66,67 The higher the ISI and CSI, the higher the domain purity of the related domain populations and the higher the FF. Fig. S7 and Table S2 (ESI†) show that for each blend system both the normalized total ISI and CSI is highest when the FF is highest (annealed for J71 based blends, and additive for the other three blends). Overall, the blends with the highest FF, shows the highest domain purity. The R-SoXS data also shows differences in domain spacing, with J71–Cl exhibiting the largest spacing. Furthermore, the scattering intensity increases with the addition of the additive DPE for all but J71. The annealing had a disparate effect, with J71–Cl having unchanged intensity, but decreased intensity for J71 and increased intensity for J100 and J101. This indicates that the thermodynamics (miscibility between the polymers and the acceptor, solubility) might be different, or that the aggregation kinetics during casting and film formation is impacted due to differences in packing or molecular weight dependences impacting solidification.68
To obtain more insight into the morphology of the blends, GIWAXS has been carried out to investigate the molecular packing and orientation/texture in the four blend films without and with different post-treatments. Fig. 5 shows the 2D GIWAXS patterns and the corresponding line-cut profiles in the OOP and IP directions of each series blends. The values of π–π stacking distance, π–π stacking coherence length, and the integrated intensity of π–π peak in OOP directions are summarized in Table S3 (ESI†). All blends show apparent, but very weak face-on orientation. Further texture analysis requires pole figure analysis that takes into account the isotropic population properly. The calculated g parameters show that all the blends are amorphous in the π–π direction with only subtle differences in molecular packing, and the different processing conditions do not have any significant impact on this parameter (Table S3, ESI†). However, the diffraction intensities are clearly different with the use of 1% DPE. For J71, the molecular packing is suppressed, whereas it is enhanced for the other three polymers. Furthermore, the intensities of J71 and J100 based blends are highest in the annealed blends, which agrees with the device PCE of the blends. The GIWAXS intensities mirror and correlate with the observations regarding the relative soft X-ray scattering intensities and emphasize the same conclusions here: Kinetics (solubility, Mw-dependence)68 and possibly thermodynamics properties (solubility, miscibility)21,69 are impacted by the chlorine substitution and even the particular substitution locations. Yet, these changes might be beneficial or at least not detrimental as excellent device performance could be achieved.
 | ||
| Fig. 5 2D GIWAXS patterns and in-plane and out-of-plane scattering profiles of the blend films without and with post-treatments. | ||
Conclusions
By employing four BDT-alt-BTz polymers with the same conjugated main chain and alkyl side chains, but different chlorine substitution motifs, we systematically studied the effect of the site-specific chlorine substitution on the optical and electronic properties and on the photovoltaic performance in combination with IT4F as a common acceptor. Compared to the chlorine-free polymer J71, replacing the main chain fluorine by chlorine reduces the HOMO energy level of J100, increases the π–π stacking distance, and suppresses the tendency to aggregate. However, in OSCs, J100:IT4F blends have a suboptimal morphology and transport properties, leading to reduced photovoltaic performance (PCE = 7.70%) compared to J71-IT4F (8.16%). On the other hand, introducing chlorine into the conjugated side chain of J71 does not affect the π–π stacking, hole mobility, and aggregation of the polymer, but significantly reduces the HOMO energy level. In OSCs, with the aid of additives, the molecular packing, phase separation, and charge transport properties of J71–Cl:IT4F blend are improved, thus contributing to a much better photovoltaic performance (PCE = 11.10%) than for J71:ITF4 under optimized conditions. For J101, in which both the main chain and the conjugated side-chain have chlorine substitution, the π–π stacking and aggregation are further weakened, but the HOMO energy level is more reduced. With the help of additives, J101:IT4F provides an efficient solar cell (PCE = 11.30%) with well-balanced transport properties. The blend morphology was similar to J71–Cl:IT4F, yet with more pure and more ordered/aggregated domains. The results demonstrate that optimization of polymer photovoltaic performance by introducing chlorine into the conjugated side chains can yield more efficient OSCs. Incorporation of chlorine in the side chains reduces the HOMO more and affects the π–π stacking less than introduction of chlorines in the main chain and so enables reaching more optimal blend morphologies. We think this concept provides useful guidance for the future design of new materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research has received funding from the Netherlands Organisation for Scientific Research via the NWO Spinoza grant awarded to R. A. J. Janssen. We further acknowledge funding from the Ministry of Education, Culture and Science (Gravity program 024.001.035). The work was further supported by the Ministry of Education, Culture and Science (Gravity program 024.001.035). NCSU gratefully acknowledges the support of ONR grants N000141712204 and N000142012155. X-ray data were acquired at beamlines 11.0.1.2 and 7.3.3 at the Advanced Light Source, which is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.References
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma, C. An, C. He, Z. Wei, F. Gao and J. Hou, Adv. Mater., 2020, 1908205 CrossRef CAS.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H. L. Yip, Y. Cao and Y. Chen, Science, 2018, 361, 1094–1098 CrossRef CAS.
- R. Ma, T. Liu, Z. Luo, Q. Guo, Y. Xiao, Y. Chen, X. Li, S. Luo, X. Lu, M. Zhang, Y. Li and H. Yan, Sci. China: Chem., 2020, 63, 325–330 CrossRef CAS.
- E. Ravishankar, R. E. Booth, C. Saravitz, H. Sederoff, H. W. Ade and B. T. O’Connor, Joule, 2020, 4, 490–506 CrossRef CAS.
- X. Li, F. Pan, C. Sun, M. Zhang, Z. Wang, J. Du, J. Wang, M. Xiao, L. Xue, Z. G. Zhang, C. Zhang, F. Liu and Y. Li, Nat. Commun., 2019, 10, 519 CrossRef CAS.
- C. Sun, F. Pan, H. Bin, J. Zhang, L. Xue, B. Qiu, Z. Wei, Z. G. Zhang and Y. Li, Nat. Commun., 2018, 9, 743 CrossRef.
- C. Sun, S. Qin, R. Wang, S. Chen, F. Pan, B. Qiu, Z. Shang, L. Meng, C. Zhang, M. Xiao, C. Yang and Y. Li, J. Am. Chem. Soc., 2020, 142, 1465–1474 CrossRef CAS.
- X. Wan, C. Li, M. Zhang and Y. Chen, Chem. Soc. Rev., 2020, 49, 2828–2842 RSC.
- J. Hou, O. Inganas, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS.
- Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H. L. Yip, T. K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- C. Cui and Y. Li, Energy Environ. Sci., 2019, 12, 3225–3246 RSC.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS.
- Q. Fan, Y. Wang, M. Zhang, B. Wu, X. Guo, Y. Jiang, W. Li, B. Guo, C. Ye, W. Su, J. Fang, X. Ou, F. Liu, Z. Wei, T. C. Sum, T. P. Russell and Y. Li, Adv. Mater., 2018, 30, 1704546 CrossRef.
- H. Bin, L. Gao, Z. G. Zhang, Y. Yang, Y. Zhang, C. Zhang, S. Chen, L. Xue, C. Yang, M. Xiao and Y. Li, Nat. Commun., 2016, 7, 13651 CrossRef CAS.
- Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174–179 CrossRef CAS.
- L.-W. Feng, J. Chen, S. Mukherjee, V. K. Sangwan, W. Huang, Y. Chen, D. Zheng, J. W. Strzalka, G. Wang, M. C. Hersam, D. DeLongchamp, A. Facchetti and T. J. Marks, ACS Energy Lett., 2020, 5, 1780–1787 CrossRef CAS.
- J. R. Tumbleston, B. A. Collins, L. Yang, A. C. Stuart, E. Gann, W. Ma, W. You and H. Ade, Nat. Photonics, 2014, 8, 385–391 CrossRef CAS.
- Y. F. Li, Acc. Chem. Res., 2012, 45, 723 CrossRef CAS.
- H. Hu, K. Jiang, P. C. Y. Chow, L. Ye, G. Zhang, Z. Li, J. H. Carpenter, H. Ade and H. Yan, Adv. Energy Mater., 2018, 8, 1701674 CrossRef.
- L. Ye, H. Hu, M. Ghasemi, T. Wang, B. A. Collins, J. H. Kim, K. Jiang, J. H. Carpenter, H. Li, Z. Li, T. McAfee, J. Zhao, X. Chen, J. L. Y. Lai, T. Ma, J. L. Bredas, H. Yan and H. Ade, Nat. Mater., 2018, 17, 253–260 CrossRef CAS.
- S. Mukherjee, C. M. Proctor, J. R. Tumbleston, G. C. Bazan, T. Q. Nguyen and H. Ade, Adv. Mater., 2015, 27, 1105–1111 CrossRef CAS.
- L. Ye, S. Zhang, L. Huo, M. Zhang and J. Hou, Acc. Chem. Res., 2014, 47, 1595–1603 CrossRef CAS.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS.
- H. Huang, H. Bin, Z. Peng, B. Qiu, C. Sun, A. Liebman-Pelaez, Z. G. Zhang, C. Zhu, H. Ade, Z. Zhang and Y. Li, Macromolecules, 2018, 51, 6028–6036 CrossRef CAS.
- Q. Fan, W. Su, X. Guo, Y. Wang, J. Chen, C. Ye, M. Zhang and Y. Li, J. Mater. Chem. A, 2017, 5, 9204–9209 RSC.
- Z. G. Zhang, J. Min, S. Zhang, J. Zhang, M. Zhang and Y. Li, Chem. Commun., 2011, 47, 9474–9476 RSC.
- H. Bin, Y. Yang, Z. Peng, L. Ye, J. Yao, L. Zhong, C. Sun, L. Gao, H. Huang, X. Li, B. Qiu, L. Xue, Z. G. Zhang, H. Ade and Y. Li, Adv. Energy Mater., 2018, 8, 1702324 CrossRef.
- D. Deng, Y. Zhang, L. Yuan, C. He, K. Lu and Z. Wei, Adv. Energy Mater., 2014, 4, 1400538 CrossRef.
- C. Cui, Z. He, Y. Wu, X. Cheng, H. Wu, Y. Li, Y. Cao and W. Y. Wong, Energy Environ. Sci., 2016, 9, 885–891 RSC.
- Q. Zhang, M. A. Kelly, N. Bauer and W. You, Acc. Chem. Res., 2017, 50, 2401–2409 CrossRef CAS.
- A. Casey, S. D. Dimitrov, P. Shakya-Tuladhar, Z. Fei, M. Nguyen, Y. Han, T. D. Anthopoulos, J. R. Durrant and M. Heeney, Chem. Mater., 2016, 28, 5110–5120 CrossRef CAS.
- G. E. Park, S. Choi, S. Y. Park, D. H. Lee, M. J. Cho and D. H. Choi, Adv. Energy Mater., 2017, 7, 1700566 CrossRef.
- T. J. Aldrich, M. Matta, W. Zhu, S. M. Swick, C. L. Stern, G. C. Schatz, A. Facchetti, F. S. Melkonyan and T. J. Marks, J. Am. Chem. Soc., 2019, 141, 3274–3287 CrossRef CAS.
- H. Yao, Y. Cui, R. Yu, B. Gao, H. Zhang and J. Hou, Angew. Chem., 2017, 129, 3091–3095 CrossRef.
- A. C. Stuart, J. R. Tumbleston, H. Zhou, W. Li, S. Liu, H. Ade and W. You, J. Am. Chem. Soc., 2013, 135, 1806–1815 CrossRef CAS.
- S. Yamada, A. Gavryushin and P. Knochel, Angew. Chem., Int. Ed., 2010, 49, 2215–2218 CrossRef CAS.
- T. Bura, S. Beaupré, O. A. Ibraikulov, M. A. Légaré, J. Quinn, P. Lévêque, T. Heiser, Y. Li, N. Leclerc and M. Leclerc, Macromolecules, 2017, 50, 7080–7090 CrossRef CAS.
- S. Zhang, Y. Qin, J. Zhu and J. Hou, Adv. Mater., 2018, 30, 1800868 CrossRef.
- G. P. Kini, S. J. Jeon and D. K. Moon, Adv. Mater., 2020, 32, 1906175 CrossRef CAS.
- B. Qiu, S. Chen, H. Li, Z. Luo, J. Yao, C. Sun, X. Li, L. Xue, Z. G. Zhang, C. Yang and Y. Li, Chem. Mater., 2019, 31, 6558–6567 CrossRef CAS.
- Y. Li, J. D. Lin, X. Che, Y. Qu, F. Liu, L. S. Liao and S. R. Forrest, J. Am. Chem. Soc., 2017, 139, 17114–17119 CrossRef CAS.
- B. Kan, H. Feng, H. Yao, M. Chang, X. Wan, C. Li, J. Hou and Y. Chen, Sci. China: Chem., 2018, 61, 1307–1313 CrossRef CAS.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS.
- Y. Zhang, H. Yao, S. Zhang, Y. Qin, J. Zhang, L. Yang, W. Li, Z. Wei, F. Gao and J. Hou, Sci. China: Chem., 2018, 61, 1328–1337 CrossRef CAS.
- Y. Cui, C. Yang, H. Yao, J. Zhu, Y. Wang, G. Jia, F. Gao and J. Hou, Adv. Mater., 2017, 29, 1703080 CrossRef.
- F. Yang, C. Li, W. Lai, A. Zhang, H. Huang and W. Li, Mater. Chem. Front., 2017, 1, 1389–1395 RSC.
- Y. Cui, H. Yao, J. Zhang, T. Zhang, Y. Wang, L. Hong, K. Xian, B. Xu, S. Zhang, J. Peng, Z. Wei, F. Gao and J. Hou, Nat. Commun., 2019, 10, 2515 CrossRef.
- W. Su, G. Li, Q. Fan, Q. Zhu, X. Guo, J. Chen, J. Wu, W. Ma, M. Zhang and Y. Li, J. Mater. Chem. A, 2019, 7, 2351–2359 RSC.
- T. Wang, R. Sun, S. Xu, J. Guo, W. Wang, J. Guo, X. Jiao, J. Wang, S. Jia, X. Zhu, Y. Li and J. Min, J. Mater. Chem. A, 2019, 7, 14070–14078 RSC.
- H. Huang, L. Yang, A. Facchetti and T. J. Marks, Chem. Rev., 2017, 117, 10291–10318 CrossRef CAS.
- H. Bin, Z. G. Zhang, L. Gao, S. Chen, L. Zhong, L. Xue, C. Yang and Y. Li, J. Am. Chem. Soc., 2016, 138, 4657–4664 CrossRef CAS.
- K. Sun, Z. Xiao, S. Lu, W. Zajaczkowski, W. Pisula, E. Hanssen, J. M. White, R. M. Williamson, J. Subbiah, J. Ouyang, A. B. Holmes, W. W. H. Wong and D. J. Jones, Nat. Commun., 2015, 6, 6013 CrossRef CAS.
- X. Liu, P. Cai, Z. Chen, L. Zhang, X. Zhang, J. Sun, H. Wang, J. Chen, J. Peng, H. Chen and Y. Cao, Polymer, 2014, 55, 1707–1715 CrossRef CAS.
- H. Bin, Y. Yang, Z. G. Zhang, L. Ye, M. Ghasemi, S. Chen, Y. Zhang, C. Zhang, C. Sun, L. Xue, C. Yang, H. Ade and Y. Li, J. Am. Chem. Soc., 2017, 139, 5085–5094 CrossRef CAS.
- A. Hexemer, W. Bras, J. Glossinger, E. Schaible, E. Gann, R. Kirian, A. MacDowell, M. Church, B. Rude and H. Padmore, J. Phys.: Conf. Ser., 2010, 247, 012007 CrossRef.
- D. E. Johnston, K. G. Yager, H. Hlaing, X. Lu, B. M. Ocko and C. T. Black, ACS Nano, 2014, 8, 243–249 CrossRef CAS.
- L. Ye, X. Jiao, M. Zhou, S. Zhang, H. Yao, W. Zhao, A. Xia, H. Ade and J. Hou, Adv. Mater., 2015, 27, 6046–6054 CrossRef CAS.
- K. A. Page, A. Kusoglu, C. M. Stafford, S. Kim, R. J. Kline and A. Z. Weber, Nano Lett., 2014, 14, 2299–2304 CrossRef CAS.
- J. H. Carpenter, M. Ghasemi, E. Gann, I. Angunawela, S. J. Stuard, J. J. Rech, E. Ritchie, B. T. O’Connor, J. Atkin, W. You, D. M. DeLongchamp and H. Ade, Adv. Funct. Mater., 2019, 29, 1806977 Search PubMed.
- Z. B. Henson, K. Müllen and G. C. Bazan, Nat. Chem., 2012, 4, 699–704 CrossRef CAS.
- C. Li, X. Wu, X. Sui, H. Wu, C. Wang, G. Feng, Y. Wu, F. Liu, X. Liu, Z. Tang and W. Li, Angew. Chem., Int. Ed., 2019, 131, 15678–15686 CrossRef.
- S. M. Menke, N. A. Ran, G. C. Bazan and R. H. Friend, Joule, 2018, 2, 25–35 CrossRef CAS.
- E. Gann, A. T. Young, B. A. Collins, H. Yan, J. Nasiatka, H. A. Padmore, H. Ade, A. Hexemer and C. Wang, Rev. Sci. Instrum., 2012, 83, 045110 CrossRef CAS.
- B. A. Collins, Z. Li, J. R. Tumbleston, E. Gann, C. R. Mcneill and H. Ade, Adv. Energy Mater., 2013, 3, 65–74 CrossRef CAS.
- X. Jiao, L. Ye and H. Ade, Adv. Energy Mater., 2017, 7, 1700084 CrossRef.
- I. Angunawela, L. Ye, H. Bin, Z. G. Zhang, A. Gadisa, Y. Li and H. Ade, Mater. Chem. Front., 2019, 3, 137–144 RSC.
- L. Ye, S. Li, X. Liu, S. Zhang, M. Ghasemi, Y. Xiong, J. Hou and H. Ade, Joule, 2019, 3, 443–458 CrossRef CAS.
- Z. Liang, M. Li, Q. Wang, Y. Qin, S. J. Stuard, Z. Peng, Y. Deng, H. Ade, L. Ye and Y. Geng, Joule, 2020, 4, 1–18 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc03095c |
| This journal is © The Royal Society of Chemistry 2020 |