
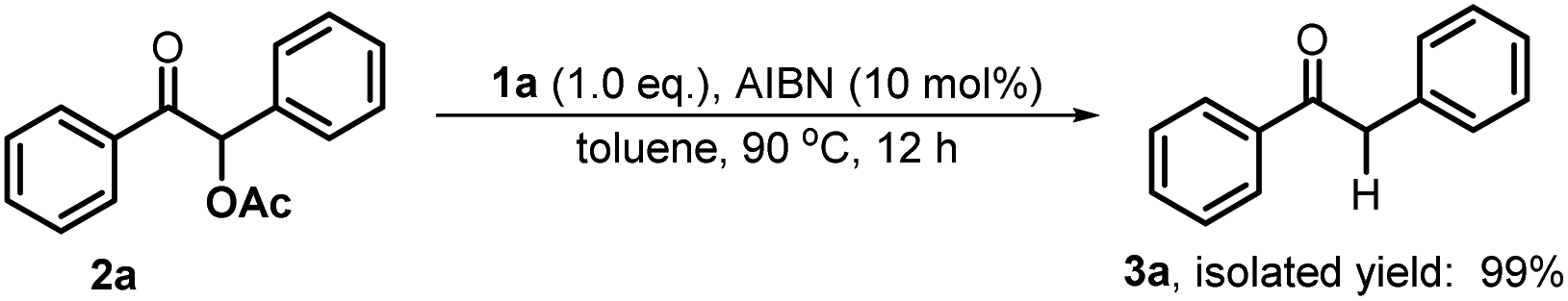
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiazaphosphinyl radical-catalyzed deoxygenation of α-carboxy ketones: a new protocol for chemo-selective C–O bond scission via mechanism regulation†
Jingjing
Zhang
a,
Jin-Dong
Yang
 *a and
Jin-Pei
Cheng
*ab
*a and
Jin-Pei
Cheng
*ab
aDepartment of Chemistry, Center of Basic Molecular Science, Tsinghua University, Beijing 100084, China. E-mail: jdyang@mail.tsinghua.edu.cn; jinpei_cheng@mail.tsinghua.edu.cn
bState Key Laboratory of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China
First published on 27th July 2020
Abstract
C–O bond cleavage is often a key process in defunctionalization of organic compounds as well as in degradation of natural polymers. However, it seldom occurs regioselectively for different types of C–O bonds under metal-free mild conditions. Here we report a facile chemo-selective cleavage of the α-C–O bonds in α-carboxy ketones by commercially available pinacolborane under the catalysis of diazaphosphinane based on a mechanism switch strategy. This new reaction features high efficiency, low cost and good group-tolerance, and is also amenable to catalytic deprotection of desyl-protected carboxylic acids and amino acids. Mechanistic studies indicated an electron-transfer-initiated radical process, underlining two crucial steps: (1) the initiator azodiisobutyronitrile switches originally hydridic reduction to kinetically more accessible electron reduction; and (2) the catalytic phosphorus species upconverts weakly reducing pinacolborane into strongly reducing diazaphosphinane.
The importance of reductive deoxygenation can be gauged by the wide use of Barton–McCombie deoxygenation in organic syntheses.1 Such C–O bond cleavage is also a crucial step in the degradation of natural polymers (e.g., sugars and lignins) to recycle sustainable resources.2 Consequently, a great variety of methodologies were explored for activation of these strong C–O bonds.3 Among them, deoxygenation of α-acyloxy ketones3b,c,4 (represented by benzoin derivatives, stemming from simple aldehydes via benzoin condensation5) has attracted considerable attention, because it may provide a facile way for accessing commonly useful building blocks (aryl ketones).6 As known, benzoin derivatives bear two types of C–O bonds—the carbonyl π-C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and the benzyl σ-C–O bond (Scheme 1). While reduction of the carbonyl π-CO bonds has been well established through transition metal-7 or Lewis acid-mediated8 hydride transfers, chemo-selective cleavage of the benzyl σ-C–O bonds is challenging and has seldom been achieved.9 The later process is occasionally seen, however, in some radical or electron reductions, but toxic tin hydrides1c or aggressive metal reagents (like Raney nickel,10 zinc dust,11etc.) are inevitably employed.
O bond and the benzyl σ-C–O bond (Scheme 1). While reduction of the carbonyl π-CO bonds has been well established through transition metal-7 or Lewis acid-mediated8 hydride transfers, chemo-selective cleavage of the benzyl σ-C–O bonds is challenging and has seldom been achieved.9 The later process is occasionally seen, however, in some radical or electron reductions, but toxic tin hydrides1c or aggressive metal reagents (like Raney nickel,10 zinc dust,11etc.) are inevitably employed.
 | ||
| Scheme 1 Possible reactive sites for benzoin reduction. | ||
The recent successful development of super electron donors (SEDs),4,12 which are defined as ground-state organic electron-donors capable of reducing aryl halides to aryl radicals or aryl anions,13 and photocatalytic systems3b,c may provide alternative protocols for reductive cleavage of the σ-C–O bonds in O-acetylated benzoin. However, these electron transfer-initiated reductions also suffer from some drawbacks, such as, excessive use of SEDs and their tedious synthetic procedures, expensive photoredox catalysts and ligands, and group-tolerance issues. In fact, there have been few reports to date on metal-free systems for efficiently catalytic deoxygenation with commercially available inexpensive reductants.3h Given the ubiquity of C–O bonds in nature, it is still an unmet need for development of efficient and economical methods for their degradation.
N-Heterocyclic phosphines (NHPs)8c,14 have recently found plentiful applications in hydridic reductions8b,15 owing to their outstanding hydricity.16 However, this seems to blind one to search for their other promising reaction patterns, like radical and electron transfer reactivities. Up to now, the catalytic potential of NHPs in radical or electron reductions has never been explored. Given the logical understanding that a deliberately manipulated mechanism variation usually leads to diverse reactivity and selectivity, we anticipate that an intended mechanism switch for NHP-based reactions from the conventional hydride transfer to an alternative electron transfer might provide a chance for originally inaccessible chemo-selectivity in the reduction of the substrates bearing multiple reactive sites. As known from previous studies, NHPs could transfer a hydride ion to carbonyl CO bonds to deliver the corresponding alcohol counterparts (Scheme 2a).8b This is indeed what we have seen. When NHPs are mixed with O-acetylated benzoins, an exclusive hydridic reduction of the carbonyl π-CO bonds is observed, leaving the benzyl σ-C–O bonds intact. How could we make the propensity of NHP reduction to switch from the original hydridic path to a radical one? Inspired by our recent findings that NHPs are also capable of serving as good hydrogen-atom donors (by P–H bond homolysis) and their corresponding phosphinyl radicals are excellent electron donors17 (Scheme 2b, bottom), we envisioned that if phosphinyl radicals can be in situ generated, their super electron-donicity may promote the initial electron transfer to benzoin, and trigger the subsequent benzyl σ-C–O bond scission. If this is realizable, chemo-selective deoxygenation of benzoin derivatives with NHPs may be achieved via such a mechanism switch.
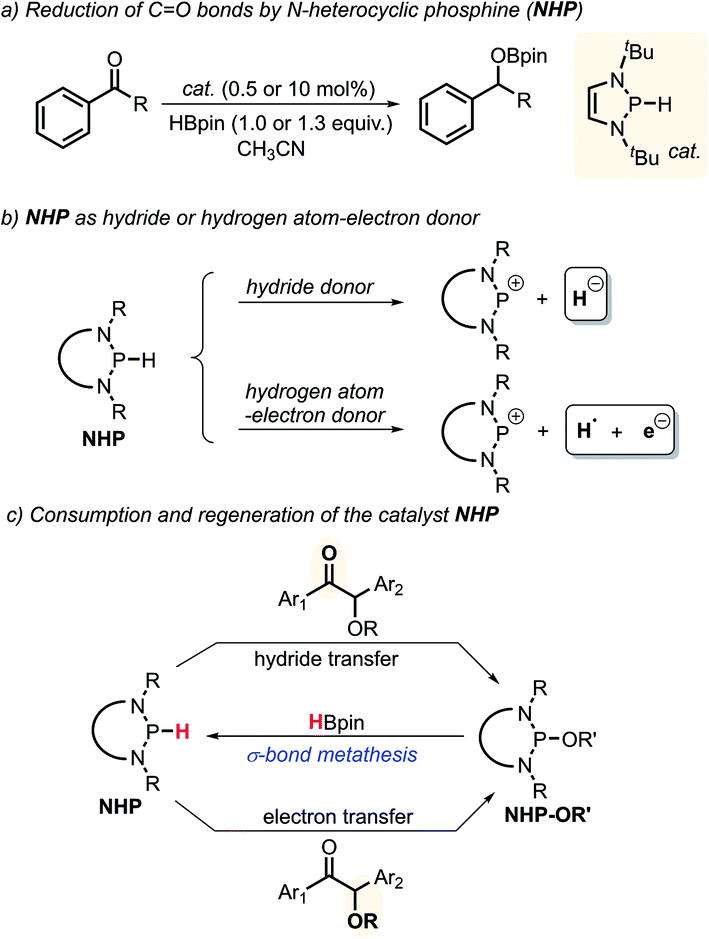 | ||
| Scheme 2 Chemical transformations of N-heterocyclic phosphines NHPs in reduction reactions. | ||
It is noted that the phosphorus species NHP-OR′ is produced in either the hydride or electron reduction of benzoin derivatives (Scheme 2c). Based on the previous knowledge that NHP-OR′ can be recycled back to NHP through a σ-bond metathesis between its exocyclic P–O bond and the B–H bond of pinacolborane (HBpin),8b we envisioned that the present deoxygenation may operate in a catalytic fashion with readily available HBpin as the terminal reductant to avoid the use of stoichiometric NHP. To verify this plot, we chose dimethyl 4,4′-(1-acetoxy-2-oxoethane-1,2-diyl)dibenzoate X as the testing substrate, and 1,3-di-tert-butyl-1,3,2-diazaphosphinane 1a as the catalyst based on its compatible reducing capacity (Scheme 3, for structure of 1a, cf.Table 1).17a It is observed that, under the previously established catalytic conditions for carbonyl reduction (20 mol% of 1a and 1.2 equiv. HBpin),8b the product X1 of hydridic reduction was obtained in 86% yield and 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.69 of diastereomer ratio in 12 h. On the other hand, when 10% azodiisobutyronitrile (AIBN) was added as a radical initiator, the σ-C–O bonds were, indeed, selectively cleaved to give the anticipated product X2 in 87% yield. This distinct chemo-selectivity did echo our proposed mechanism switch from the direct hydridic pathway to an electron reduction. In the following, we report this catalytic transformation in a more inclusive fashion. To our best knowledge, this is the first example of catalytic electron reduction mediated by NHPs.
:0.69 of diastereomer ratio in 12 h. On the other hand, when 10% azodiisobutyronitrile (AIBN) was added as a radical initiator, the σ-C–O bonds were, indeed, selectively cleaved to give the anticipated product X2 in 87% yield. This distinct chemo-selectivity did echo our proposed mechanism switch from the direct hydridic pathway to an electron reduction. In the following, we report this catalytic transformation in a more inclusive fashion. To our best knowledge, this is the first example of catalytic electron reduction mediated by NHPs.
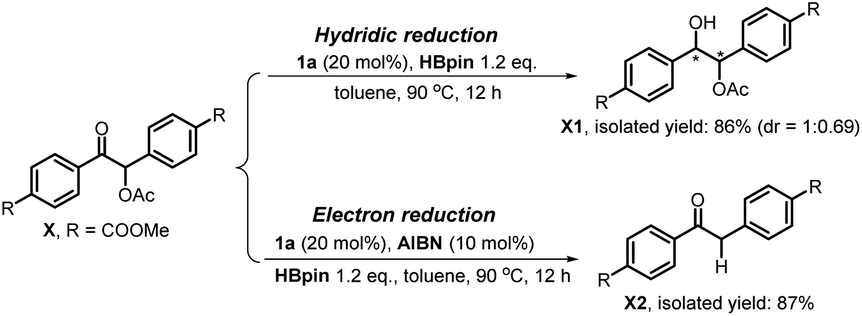 | ||
| Scheme 3 Chemo-selectively reductive cleavage of C–O bonds in O-acetylated benzoin X by diazaphosphinane 1a. | ||
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Conditiona | Yieldb |
| a Conditions for C–O bond activation: 2 (0.4 mmol), AIBN (0.04 mmol), 1a (0.08 mmol), HBpin (0.48 mmol) in toluene (1.0 mL). b Isolated yields. c NMR yields using 1,3,5-trimethoxybenzene as the internal standard. | |||
| 1 | 1a | Standard condition | 92% |
| 2 | 1a | 10 mol% 1a | 62% |
| 3 | 1b | Standard condition | <10%c |
| 4 | 1c | Standard condition | <5%c |
| 5 | 1d | Standard condition | <5%c |
| 6 | 1e | Standard condition | 46% |
| 7 | 1a | NH3BH3 as reductant | <5%c |
| 8 | 1a | No AIBN | <5%c |
| 9 | 1a | No heat | <5%c |
| 10 | — | Standard condition | <5%c |

|
|||
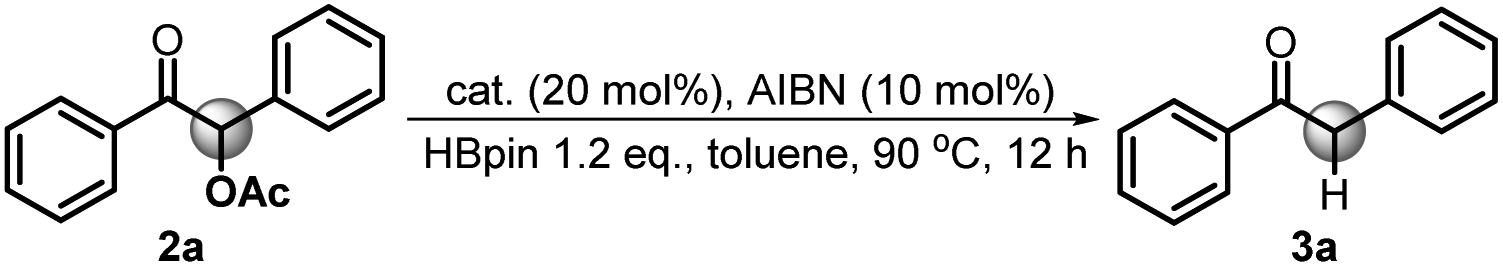
To verify the necessity of each component in the above catalytic system, a series of comparative experiments were conducted. We commenced the condition optimization with simple O-acetylated benzoin 2a as the standard substrate – an attractive precursor for accessing α-aryl ketone which is a common pharmacophore and also present in numerous biologically active natural products.18 As shown in Table 1, treatment of 2a with 20 mol% catalyst 1a, 10 mol% initiator AIBN and 1.2 equiv. HBpin in toluene solution harvested the product 1,2-diphenylethanone 3a in 92% isolated yield (entry 1). Decreasing the catalyst loading led to an inferior result (62%, entry 2). Replacement of 1a with structurally similar 1b gave a much lower yield (<10%, entry 3), which is primarily because the weak reducing capacity of 1b-derived phosphinyl radical (Eox = −1.94 V vs. Fc in MeCN)17a prevents its electron transfer to 2a. The same reason can be applied to account for the poor results of 1c and 1d catalysts (<5%, entry 4 and 5). When a stronger hydride donor 1e was employed, a moderate yield (46%, entry 6) was obtained along with 40% byproduct of direct hydride transfer. This may be because enhancing the reducing ability of 1e can simultaneously accelerate its hydride transfer to carbonyl groups, which competes with the electron transfer between its derived phosphinyl radical and benzoin. Commercially available borane ammonia (NH3·BH3) was also examined, furnishing no desired product (<5%, entry 7). In addition, the absence of AIBN, heating or catalyst 1a cannot render efficient C–O bond cleavage (entry 8–10). Therefore, 20 mol% 1a, 10 mol% AIBN and 1.2 equiv. HBpin in toluene solution were eventually used as the standard conditions.
Next, we explored the substrate scope starting with different benzoin derivatives 2 (Scheme 4). Besides the acetate, the reaction presented here also worked very well for other leaving groups, such as pivalate 2b, benzoate 2c and 4-cyanophenolate 2d, affording the product 1,2-diphenylethanone 3a in good to excellent yields (72–99%). Then, a series of benzoin derivatives with diverse substituents (2e–i) were synthesized to examine the functional group tolerance. As seen, the substrates with electron-withdrawing F (2e) and Cl (2f) groups gave almost quantitative yields (99%). Noteworthily, in contrast to the previously reported Ru-based photocatalytic deoxygenation,3b the reaction presented here could tolerate the ester group well and gave 3g in 87% yield. As for electron-donating substituents, such as methyl (2h) and methoxy groups (2i), the reaction yields were slightly reduced (72% and 75%), which may be ascribed to their lower reduction potentials. Replacement of the phenyl group with naphthyl (2j) afforded the product 3j in a good yield (80%). Furthermore, some cross-benzoin analogues were also investigated. The unsymmetrical counterpart 2k gave 3k in a moderate yield (62%). Similarly, heteroaromatic substrates (2l and 2m) generated corresponding products in 65% and 55% yields, respectively. Additionally, we examined the acyloin derivative 2n which was previously reported to give a base promoted aldol-type cyclization byproduct in the SED system.4 Notably, our conditions are mild enough for selective cleavage of its C–O bond in a moderate yield (52%), although 1a was necessarily employed as a stoichiometric reductant. However, the analogs 2o and 2p gave poor yields, possibly due to the less stability of their corresponding radical intermediates.
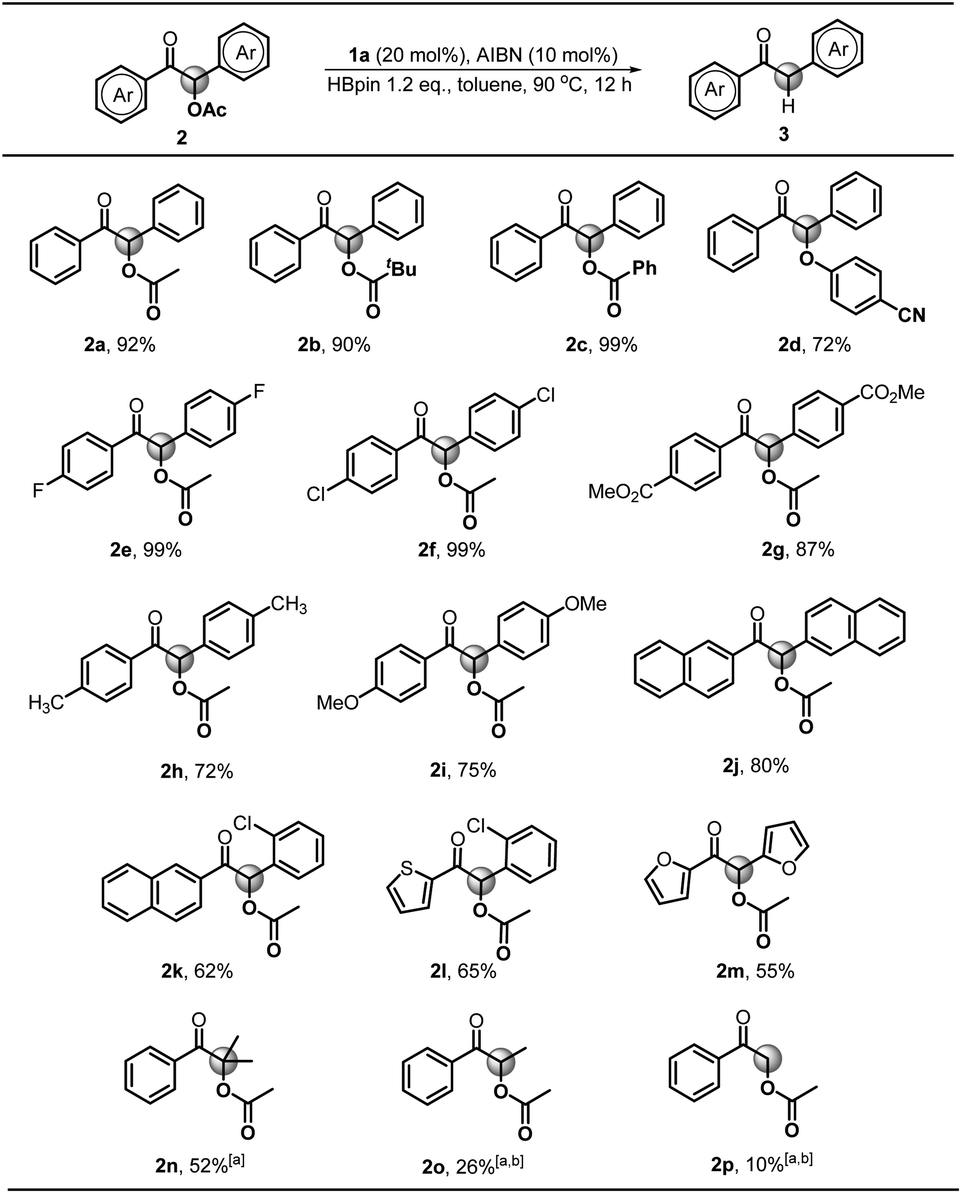 | ||
| Scheme 4 Substrate scope for C–O bond activation. Conditions unless otherwise specified: 2 (0.4 mmol), AIBN (0.04 mmol), 1a (0.08 mmol), HBpin (0.48 mmol) in toluene (1.0 mL). Isolated yields were given. [a] 0.4 mmol of 1a was used. [b] NMR yields using 1,3,5-trimethoxybenzene as the internal standard. | ||
Desyl is a classical protection group in organic chemistry and biology.3c,19 We wondered whether the same reaction could serve as a practical strategy to realize catalytic deprotection of various desyl-protected carboxylic acids under metal-free conditions. To assess its feasibility, we tested some carboxylic acids, including aromatic, aliphatic, and amino acids. The results revealed a good tolerance for the present method. As shown in Scheme 5, the substrate 4a gave benzoic acid 5a in a quantitative yield (99%) under the standard conditions. And, the reaction was compatible well with the susceptive acetal moiety and furnished 5b in a good yield (88%). This result indicated the high selectivity of our system to the targeted C–O bond. Substrate 4c with electron-donating groups was also found feasible, and afforded the deprotected product 5c in a slightly lower yield (74%). Interestingly, for the isophthalic acid system whose two carboxylic groups were both protected by desyl groups, the deprotection was proved to be highly reactive, and afforded the fully-deprotected product 5d in an excellent yield (92%). 1-Naphthoic acid 5e could be obtained in a good yield of 85% after deprotection. Furthermore, the deprotection of aliphatic acids 4f furnished 5f in an almost quantitative yield (99%). However, similar 5g with an additional conjugated double bond was obtained in a diminished yield (71%). More importantly, our protocol is also applicable in amino acid systems. As seen, 5h was obtained in 90% yield with conformational retention, and the deprotection of 4i was not affected by other commonly-used protecting group Boc, giving the product 5i in 91% yield.
 | ||
| Scheme 5 Substrate scope for catalytic deprotection with desyl as the protecting group. Conditions: 4 (0.4 mmol), AIBN (0.04 mmol), 1a (0.08 mmol), HBpin (0.48 mmol) in toluene (1.0 mL). Isolated yields were given. [a] 0.96 mmol of HBpin was used. | ||
Furthermore, we investigated the reaction mechanism by taking substrate 2a as the template compound. As previously established in SED systems, benzoin derivates were deemed to be reduced via a successive double-electron transfer mechanism, affording enolates as the intermediates which eventually captured a proton from the solvent.4 Different from this double-electron transfer pathway, our system would operate in a single-electron reduction mechanism, however. This was deduced from the fact that one equivalent reductant 1a could afford almost quantitative product 3a (eqn (1)). Consequently, the present process clearly displays a superiority in atom economy over the previous SED systems. With respect to the catalyst regeneration, we conducted the reaction of the intermediate 1a-OAc with HBpin in toluene-d8 at room temperature (eqn (2)). Through monitoring the 1H NMR and 31P NMR spectra of the reaction mixture, it is found that as the intermediate 1a-OAc gradually disappeared in about one hour (see ESI† for details), 1a P–H bond was formed synchronously. This confirmed the effective regeneration of 1a from HBpin. Moreover, when 20 mol% 1a-OAc was used as the catalyst, the reduction could also work quite well to furnish the desired product in 63% yield (eqn (3)). Therefore, 1a-OAc can be regarded as an intermediate in the catalytic cycle to regenerate 1a. In addition, to exclude the possibility of a radical chain process, that is, a direct oxygen abstraction from benzoin by the phosphinyl radical, DFT calculations were conducted (see ESI† for details). The results showed that 1a-[P]˙ and 1b-[P]˙ have a comparable ability in abstracting the oxygen atom (with an energy difference of 0.78 kcal mol−1, eqn (4)). This failed to explain the disparate yields for 1a and 1b systems (90% vs. <10%). Besides, the difference in the oxidation potentials of 1a-[P]˙ (Eox = −2.39 V) and of 1b-[P]˙ (Eox = −1.94 V)17a is consistent well with the observed diverse reduction results. All these preferentially support an electron-transfer initiated reduction.
 | (1) |
 | (2) |
 | (3) |
 | (4) |
Based on the above control experiments and the computation, we outlined the catalytic cycle for reductive cleavage of C–O bonds in Scheme 6. The reaction is turned on by the isobutyronitrile radical, which abstracts a hydrogen-atom from diazaphosphinane 1a to produce the actual reductant phosphinyl radical. This potent electron donor (Eox = −2.39 V) then transfers an electron to 2a (Ered = ∼−2.3 V),3c,4 furnishing the ketyl radical anion 6 and the corresponding phosphonium cation. The σ-C–O bond of the intermediate 6 is readily cleaved to afford the ketyl 7 and acetate. The ketyl 7 would not be further reduced into the corresponding enolate, but instead abstracts a hydrogen-atom from 1a and simultaneously triggers the next catalytic cycle. Meanwhile, a combination of the stable phosphonium cation with acetate produces 1a-OAc, which could regenerate the catalyst 1a from the terminal reductant HBpin. Accordingly, the success of the present deoxygenation primarily attributes to two crucial factors: the mechanism switch from the originally hydridic reduction to a kinetically more accessible electron reduction by the initiator azodiisobutyronitrile, and the “upconversion” of weakly reducing HBpin into strongly reducing diazaphosphinane by catalytic phosphorus species.20 Moreover, in our systems, HBpin serves as both the electron and hydrogen-atom sources, namely the apparent hydride donor. This is different from what was known for the previous SED systems, in which the reductants only provide the electron, and hence, extraneous hydrogen sources are necessarily employed.
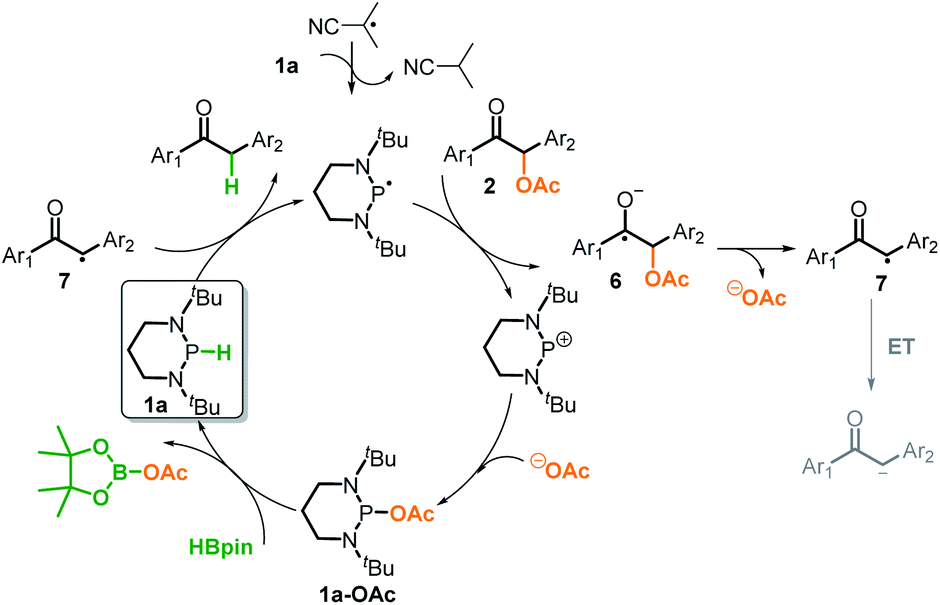 | ||
| Scheme 6 Proposed mechanism of C–O bond cleavage. | ||
Conclusions
In conclusion, we conducted the catalytic deoxygenation of benzoin substrates under transition-metal-free conditions with 1,3-di-tert-butyl-1,3,2-diazaphosphinane 1a as the effective catalyst and HBpin as the terminal reductant. In contrast to the previously established hydridic reduction of carbonyl bonds by diazaphosphinane, this selective reduction of benzyl σ-C–O bonds was achieved through mechanism regulation. The present reaction features high efficiency, low cost and good chemo-selectivity, and is the first diazaphosphinane-catalyzed electron reduction, which not only enriched the reaction patterns of NHP chemistry, and also demonstrated promising application potential in the deprotection of amino acids and degradation of sugars and biopolymeric lignins. Further attempts to realize catalytic electron reductions by using analogous systems are currently under investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial grants from National Natural Science Foundation of China (No. 21973052, 21933008, 21602116, 91745101), and National Science & Technology Fundamental Resource Investigation Program of China (No. 2018FY201200).Notes and references
- (a) D. H. R. Barton and S. W. McCombie, J. Chem. Soc., Perkin Trans. 1, 1975, 1574–1585 RSC; (b) A. Studer and S. Amrein, Synthesis, 2002, 835–849 CrossRef CAS; (c) R. M. Lopez, D. S. Hays and G. C. Fu, J. Am. Chem. Soc., 1997, 119, 6949–6950 CrossRef CAS; (d) J. M. Herrmann and B. König, Eur. J. Org. Chem., 2013, 7017–7027 CrossRef CAS; (e) M. M. Heravi, A. Bakhtiari and Z. Faghihi, Curr. Org. Synth., 2014, 11, 787–823 CrossRef CAS.
- (a) J. D. Nguyen, B. S. Matsuura and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed; (b) H. Lange, S. Decina and C. Crestini, Eur. Polym. J., 2013, 49, 1151–1173 CrossRef CAS; (c) C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed; (d) K. K. Meier, S. M. Jones, T. Kaper, H. Hansson, M. J. Koetsier, S. Karkehabadi, E. I. Solomon, M. Sandgren and B. Kelemen, Chem. Rev., 2018, 118, 2593–2635 CrossRef CAS PubMed.
- (a) H. R. Diéguez, A. López, V. Domingo, J. F. Arteaga, J. A. Dobado, M. M. Herrador, J. F. Quílez del Moral and A. F. Barrero, J. Am. Chem. Soc., 2010, 132, 254–259 CrossRef PubMed; (b) E. Speckmeier, C. Padié and K. Zeitler, Org. Lett., 2015, 17, 4818–4821 CrossRef CAS PubMed; (c) E. Speckmeier and K. Zeitler, ACS Catal., 2017, 7, 6821–6826 CrossRef CAS; (d) Y. Mostinski, D. Lankri, Y. Konovalov, R. Nataf and D. Tsvelikhovsky, Chem. Sci., 2019, 10, 9345–9350 RSC; (e) G. L. Lackner, K. W. Quasdorf and L. E. Overman, J. Am. Chem. Soc., 2013, 135, 15342–15345 CrossRef CAS PubMed; (f) M. M. Pichon, D. Hazelard and P. Compain, Eur. J. Org. Chem., 2019, 6320–6332 CrossRef CAS; (g) I. Chatterjee, D. Porwal and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 3389–3391 CrossRef CAS PubMed; (h) Sandeep, P. Venugopalan and A. Kumar, Eur. J. Org. Chem., 2020, 2530–2536 CrossRef CAS.
- S. P. Y. Cutulic, N. J. Findlay, S.-Z. Zhou, E. J. T. Chrystal and J. A. Murphy, J. Org. Chem., 2009, 74, 8713–8718 CrossRef CAS PubMed.
- (a) C. A. Rose, S. Gundala, C.-L. Fagan, J. F. Franz, S. J. Connon and K. Zeitler, Chem. Sci., 2012, 3, 735–740 RSC; (b) I. Piel, M. D. Pawelczyk, K. Hirano, R. Fröhlich and F. Glorius, Eur. J. Org. Chem., 2011, 5475–5484 CrossRef CAS.
- (a) B. Xu, S.-F. Zhu, X.-D. Zuo, Z.-C. Zhang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2014, 53, 3913–3916 CrossRef CAS PubMed; (b) Y. Wei, B. Rao, X. Cong and X. Zeng, J. Am. Chem. Soc., 2015, 137, 9250–9253 CrossRef CAS PubMed; (c) Y. Zhao, A. Aguilar, D. Bernard and S. Wang, J. Med. Chem., 2015, 58, 1038–1052 CrossRef CAS PubMed; (d) J. Ke, Y. Tang, H. Yi, Y. Li, Y. Cheng, C. Liu and A. Lei, Angew. Chem., Int. Ed., 2015, 54, 6604–6607 CrossRef CAS PubMed; (e) C. C. C. Johansson and T. J. Colacot, Angew. Chem., Int. Ed., 2010, 49, 676–707 CrossRef CAS PubMed; (f) J. M. Fox, X. Huang, A. Chieffi and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 1360–1370 CrossRef CAS.
- (a) S. Chakraborty and H. Guan, Dalton Trans., 2010, 39, 7427–7436 RSC; (b) S. Chakraborty, P. Bhattacharya, H. Dai and H. Guan, Acc. Chem. Res., 2015, 48, 1995–2003 CrossRef CAS PubMed.
- (a) M. Heshmat and T. Privalov, Chem.–Eur. J., 2017, 23, 9098–9113 CrossRef CAS PubMed; (b) C. C. Chong, H. Hirao and R. Kinjo, Angew. Chem., Int. Ed., 2015, 54, 190–194 CrossRef CAS PubMed; (c) S. Burck, D. Gudat, M. Nieger and W.-W. Du Mont, J. Am. Chem. Soc., 2006, 128, 3946–3955 CrossRef CAS PubMed.
- J. Chen, Z. Zhang, D. Liu and W. Zhang, Angew. Chem., Int. Ed., 2016, 55, 8444–8447 CrossRef CAS PubMed.
- (a) B. Lu, J. Li, G. Lv, Y. Qi, Y. Wang, T. Deng, X. Hou and Y. Yang, RSC Adv., 2016, 6, 93956–93962 RSC; (b) X. Wang and R. Rinaldi, ChemSusChem, 2012, 5, 1455–1466 CrossRef CAS PubMed.
- A. Fuerstner, A. Hupperts, A. Ptock and E. Janssen, J. Org. Chem., 1994, 59, 5215–5229 CrossRef CAS.
- L. Zhang and L. Jiao, Chem. Sci., 2018, 9, 2711–2722 RSC.
- (a) J. A. Murphy, J. Org. Chem., 2014, 79, 3731–3746 CrossRef CAS PubMed; (b) E. Doni and J. A. Murphy, Chem. Commun., 2014, 50, 6073–6087 RSC; (c) S. Rohrbach, R. S. Shah, T. Tuttle and J. A. Murphy, Angew. Chem., Int. Ed., 2019, 58, 11454–11458 CrossRef CAS PubMed; (d) J. Broggi, T. Terme and P. Vanelle, Angew. Chem., Int. Ed., 2014, 53, 384–413 CrossRef CAS PubMed.
- D. Gudat, A. Haghverdi and M. Nieger, Angew. Chem., Int. Ed., 2000, 39, 3084–3086 CrossRef CAS PubMed.
- (a) C. C. Chong, B. Rao and R. Kinjo, ACS Catal., 2017, 7, 5814–5819 CrossRef CAS; (b) M. R. Adams, C. H. Tien, B. S. N. Huchenski, M. J. Ferguson and A. W. H. Speed, Angew. Chem., Int. Ed., 2017, 56, 6268–6271 CrossRef CAS PubMed; (c) C. C. Chong and R. Kinjo, Angew. Chem., Int. Ed., 2015, 54, 12116–12120 CrossRef CAS PubMed; (d) S. Miaskiewicz, J. H. Reed, P. A. Donets, C. C. Oliveira and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 4039–4042 CrossRef CAS PubMed; (e) M. R. Adams, C. H. Tien, R. McDonald and A. W. H. Speed, Angew. Chem., Int. Ed., 2017, 56, 16660–16663 CrossRef CAS PubMed; (f) J. H. Reed, P. A. Donets, S. Miaskiewicz and N. Cramer, Angew. Chem., Int. Ed., 2019, 58, 8893–8897 CrossRef CAS PubMed; (g) D. Gudat, Dalton Trans., 2016, 45, 5896–5907 RSC; (h) Y.-C. Lin, E. Hatzakis, S. M. McCarthy, K. D. Reichl, T.-Y. Lai, H. P. Yennawar and A. T. Radosevich, J. Am. Chem. Soc., 2017, 139, 6008–6016 CrossRef CAS PubMed; (i) B. Rao, C. C. Chong and R. Kinjo, J. Am. Chem. Soc., 2018, 140, 652–656 CrossRef CAS PubMed.
- J. Zhang, J.-D. Yang and J.-P. Cheng, Angew. Chem., Int. Ed., 2019, 58, 5983–5987 CrossRef CAS PubMed.
- (a) J. Zhang, J.-D. Yang and J.-P. Cheng, Chem. Sci., 2020, 11, 3672–3679 RSC; (b) J. Zhang, J.-D. Yang and J.-P. Cheng, Chem. Sci., 2020, 11, 4786–4790 RSC.
- S. T. Sivanandan, A. Shaji, I. Ibnusaud, C. C. C. J. Seechurn and T. J. Colacot, Eur. J. Org. Chem., 2015, 38–49 CrossRef CAS.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- M. A. Syroeshkin, F. Kuriakose, E. A. Saverina, V. A. Timofeeva, M. P. Egorov and I. V. Alabugin, Angew. Chem., Int. Ed., 2019, 58, 5532–5550 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03220d |
| This journal is © The Royal Society of Chemistry 2020 |