
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Programmable synthesis of multiply arylated cubanes through C–H metalation and arylation†
Ryo
Okude
a,
Genki
Mori
b,
Akiko
Yagi
 *a and
Kenichiro
Itami
*ac
*a and
Kenichiro
Itami
*ac
aGraduate School of Science, Nagoya University, Chikusa, Nagoya 464-8602, Japan. E-mail: yagi.akiko@d.mbox.nagoya-u.ac.jp
bCentral Pharmaceutical Research Institute, Japan Tobacco Inc., 1-1 Murasaki-cho, Takatsuki, Osaka 569-1125, Japan
cInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Chikusa, Nagoya 464-8602, Japan
First published on 24th April 2020
Abstract
Cubane (C8H8), a cubic alkane, has long attracted attention owing to its unique 3D structure. In order to utilize the cubane scaffold widely in science and technology, a powerful method for synthesizing diverse cubane derivatives is required. Herein, we report the synthesis of mono-, di-, tri-, and tetra-arylated cubanes. Directed ortho-metalation with lithium base/alkyl zinc and subsequent palladium-catalyzed arylation enabled C–H metalation and arylation of cubane. This reaction allows the late-stage and regioselective installation of a wide range of aryl groups, realizing the first programmable synthesis of diverse multiply arylated cubanes.
Introduction
Structurally unique molecules have always fascinated scientists, which has driven investigations into their synthesis, functionalization, and applications. Cubane, which is a cubic alkane represented by C8H8, has particularly attracted attention over the years.1 The eight sp3-hybridized carbon atoms form C–C bonds with a small bond angle (ca. 90°), constructing a rigid cubic structure with high strain energy (161.5 kcal mol−1). Since the landmark synthesis by Eaton in 1964,2 the synthetic route has been greatly improved,3 enabling kilogram-scale synthesis to date.4 A number of derivatives have been created, and some of them have been found to work as high-energy materials,5 bioactive molecules,6 and ligands for metal–organic frameworks.7 Despite the high demand, the synthesis of cubane derivatives has been a very difficult task. Most of the reported derivatives have substituents at their 1- and 4-positions because these molecules are easily accessible from commercially available 1,4-cubanedicarboxylic acid, which is a synthetic precursor of cubane.8Late-stage C–H transformation of cubane is a potentially powerful strategy for rapidly generating molecular diversity in cubane derivatives, but the installable groups have mainly been limited to halogeno,9 carboxylic,10 phenyl,11 and hydroxy12 groups. The high reactivity of the C–C bonds can also cause decomposition of the cubane scaffold under various reaction conditions.13 To open a new vista of cubane-based molecular science, we began our studies of cubane C–H transformation chemistry, for which we selected arylation of cubane as the first and most valuable target reaction. Arylcubanes are cubane derivatives that have recently attracted attention as bioisosteres of pharmacologically important biaryls.14 Multiply arylated cubanes, unprecedented cubane derivatives, are also interesting for their unique, 3D, and diverse chemical spaces constructed with rigidly directed aryl groups. Herein, we report a palladium-catalyzed arylation of amidocubanes through directed ortho-C–H metalation. This method permits the regioselective, late-stage installation of a wide variety of aryl groups onto the cubane framework, culminating in the first synthesis of multiply arylated cubanes (Fig. 1).
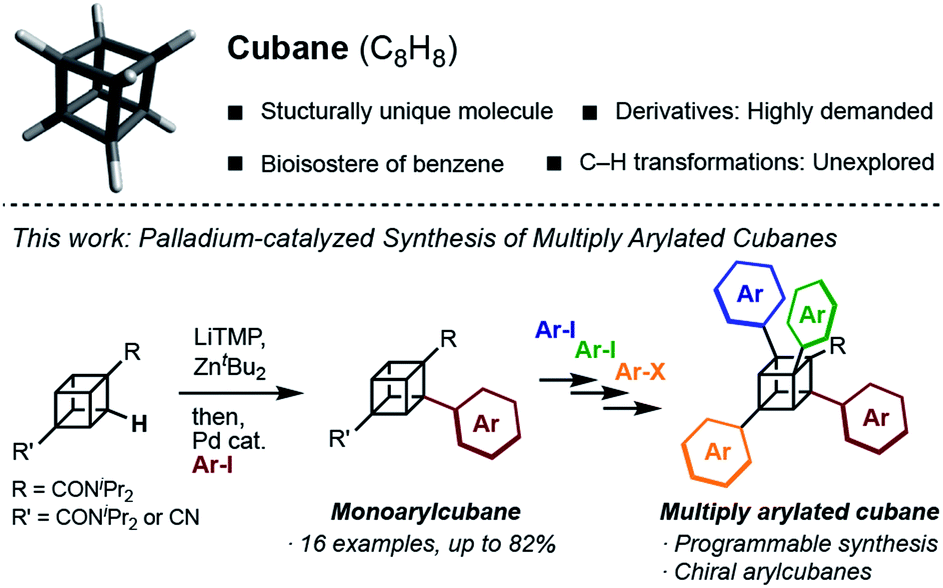 | ||
| Fig. 1 Schematic illustration of this work. | ||
In 1988, Bashir-Hashemi reported C–H phenylation of cubane, in which cubylmagnesium bromide was generated through directed ortho-lithiation of cubane-1,4-bis(N,N-diisopropyl-amide) (1a) and then treated with benzyne to give phenylcubane.11 In reference to this reaction, we focused on the development of arylation through directed ortho-metalation of 1a. Based on the pKa value of cubane (calculated pKa = 40.8)15 and that of benzene (pKa = 43), the metalation and arylation methods for arenes were investigated. In 1999, Kondo and Uchiyama developed a palladium-catalyzed arylation of ethyl benzoate by using lithium tetramethyl-piperidide (LiTMP) and ZntBu2.16 In this reaction, biaryls are most likely obtained through the generation of aryllithium zincate directed by an ester group.
Results and discussion
When we applied the arylation developed by Kondo and Uchiyama to 1a and 4-iodobenzonitrile with Pd2(dibenzylideneacetone(dba))3·CHCl3 and PPh3 as catalysts, monoarylcubane 2a was successfully obtained in 23% yield (see Table S1,† entry 1). The ligands for the palladium-catalyzed arylation were then investigated, and 2a was obtained in 76% isolated yield by using triphenylphosphite. The regioselectivity of arylation was confirmed by X-ray single-crystal structure analysis. Parameters such as catalyst loading, reaction time, concentration, and leaving group on the arene were also investigated, leading to the conclusion that the conditions described in Scheme 1 are optimal (see Table S1† for details). Because no multiply arylated cubanes were obtained under these conditions, we presumed that a monocubyllithium zincate species was generated and reacted with an aryl palladium complex to afford the monoarylcubane. With the optimal reaction conditions in hand, the installable aryl groups were examined. Aryl iodides with electron-withdrawing groups at the para-position (4-CN and 4-CF3) gave monoarylated cubanes in excellent yields (2a and 2b). Other groups, such as ester, amide, sulfonamide, Ph, tBu, and Me, at the para-position were also installed in moderate yields (2c–2h). Aryl iodides with meta-substituents reacted in acceptable yields, whereas the reactions of ortho-substituted arenes resulted in low yields (2i–2m). Heteroarenes such as pyridine, quinolone, and thiophene are also applicable in the reaction (2n–2p). X-ray crystal structures were successfully obtained for the monoarylcubanes 2a, 2e, 2f, 2k, and 2p (see Fig. S1–S5†).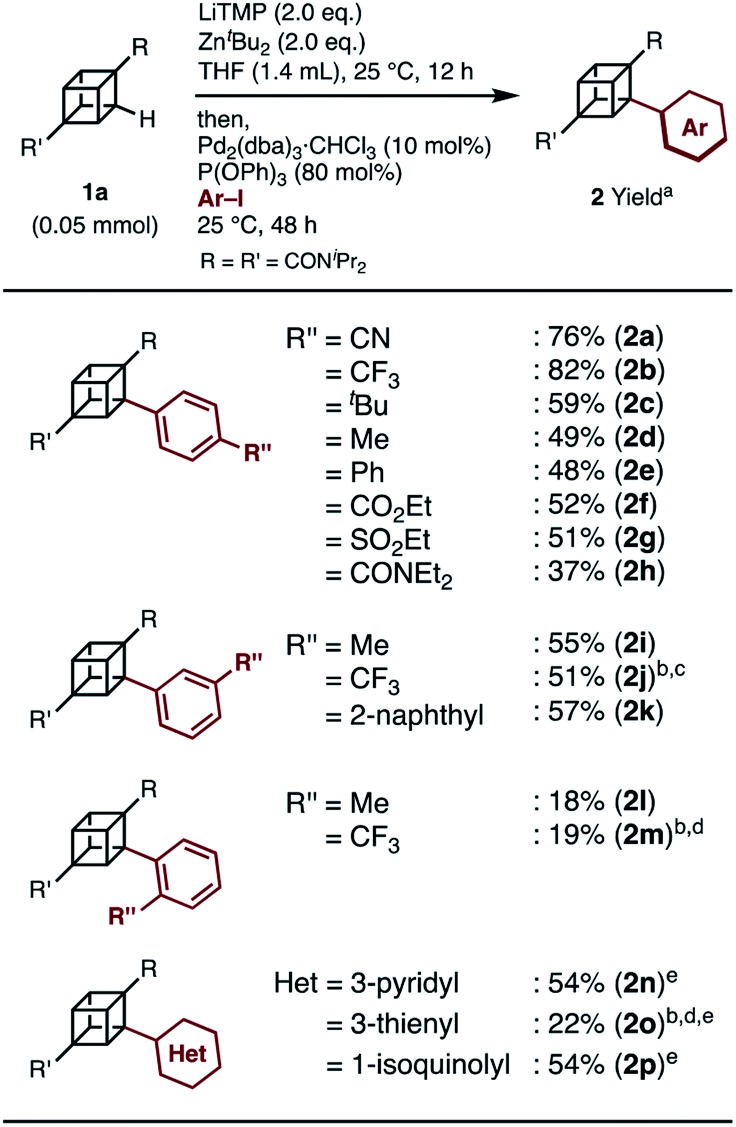 | ||
| Scheme 1 Substrate scope of iodoarenes in palladium-catalyzed arylation of cubane. a Isolated yield. b 0.05 M, Pd2(dba)3·CHCl3/P(OPh)3 = 1/4, THF solution (10 mol%), 25 °C, 24 h. c1a: 0.10 mmol. d1a: 0.10 mmol, ortho-metalation: 1 h. e Pd2(dba)3·CHCl3 (20 mol%), P(OPh)3 (160 mol%). | ||
Next, the synthesis of diarylcubanes was investigated. We expected the arylation of monoarylcubanes to give diarylcubanes. However, no further arylated product was obtained from 2b and a 4-iodoarene (Scheme 2a). The product arylated on the trifluoromethylaryl group of 2b was not identified, and about 40% of 2b was decomposed. Deuteration after directed ortho-metalation of 2b did not proceed on the cubane framework, indicating that the cubyllithium zincate was not generated from 2b (Scheme 2b). In a multiple iodination reaction of a cyanocubane derivative reported by Eaton, enhanced acidity of cubane was the key for multiple functionalization.17 Following this principle, cyanocubane-amide 1b′ was synthesized and utilized for multiple arylation (Scheme 2c). While the reaction of 1b′ at 25 °C (Condition A) resulted in a low yield of 2b′, the metalation of 1b′ at low temperature (−78 °C to 0 °C, Condition B) followed by arylation at 25 °C afforded 2b′ in 68% yield. Notably, 2b′ can be obtained even on a gram scale (1.1 g). The conditions used for the arylation of 1b′ also enabled the arylation of 2b′ to give diarylcubane 3a in excellent yield (73%). The structure of 3a was confirmed by X-ray crystal structure analysis, which proved the regioselectivity of the second aryl group (Fig. S6†). Iodoarenes with bromo and ester groups, as well as iodonaphthalenes, reacted well to give the corresponding diarylcubanes 3b–3f. The second arylation reaction took place regioselectively at the C–H bonds near the amido group, demonstrating that the aryl-installing positions are predictable and programmable. It is notable that the diarylcubanes are chiral cubanes, which are a few cubane derivatives.8a,18 The racemic products 3a and 3f were separated with a chiral column to afford the chiral molecules (Fig. S9 and S10†).
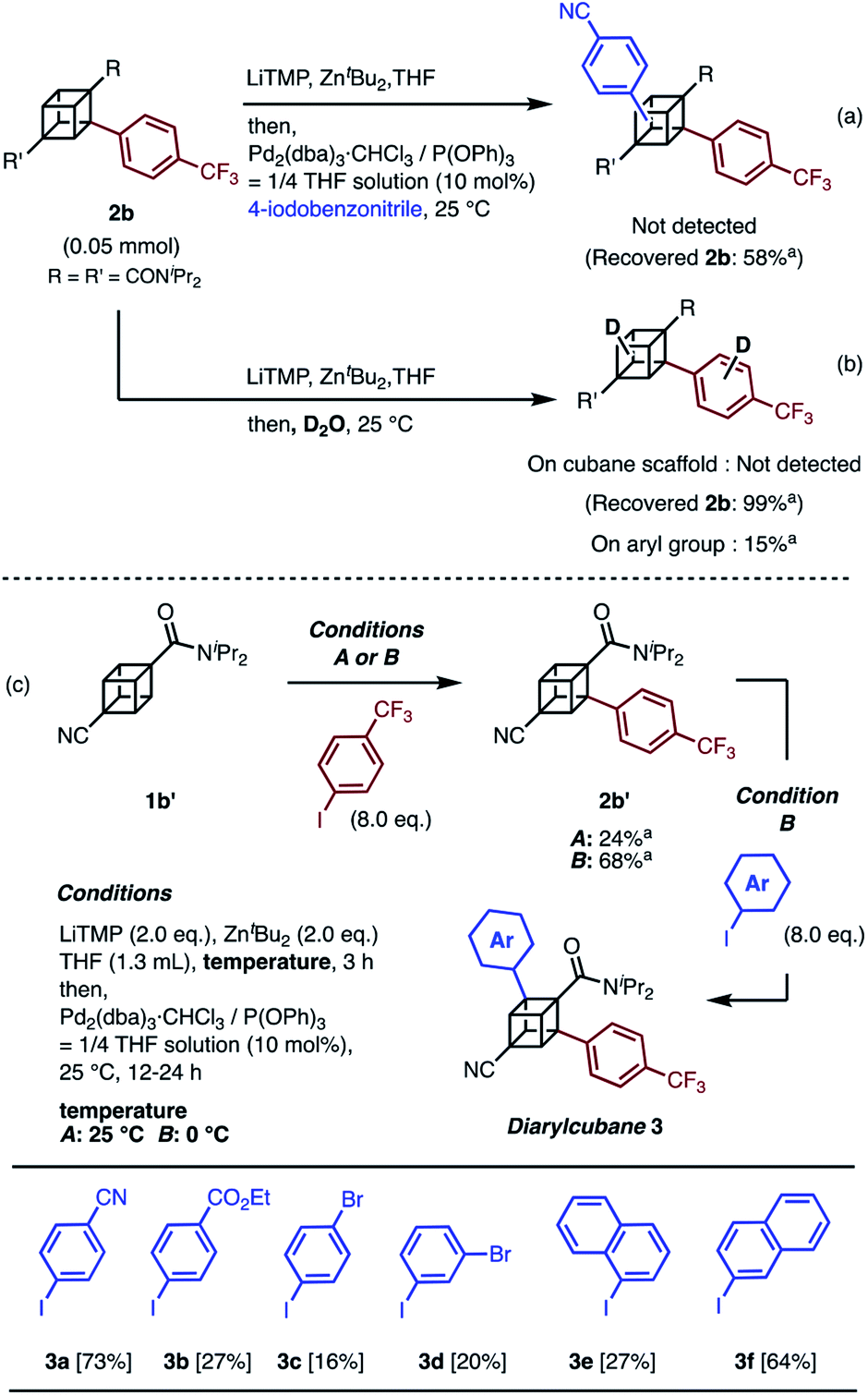 | ||
| Scheme 2 Attempts at (a) arylation of 2b and (b) deuteration of 2b through directed ortho-metalation. (c) Synthesis of diarylcubanes and scope for iodoarenes. a 1H NMR yield. | ||
The next challenge in our campaign was to achieve the first synthesis of triarylcubanes (Scheme 3a). The arylation of 3f under our optimal conditions (Condition B) proceeded to give triarylcubanes 4a, 4b, and 4c, respectively. Therefore, we envisioned that the programmable synthesis of triarylcubanes is accessible (Scheme 3b and c). Through a combination of our methods and those of Baran,19 the arylation and subsequent hydrolysis/esterification/decarboxylation of 3f afforded 4d as a racemic mixture. Triarylcubanes with three different aryl groups have several regioisomers depending on the substituent positions; one of the regioisomers of 4d can also be accessed by transformation of the cyano group of 3f. In accordance with a previous report,20 a sequence of hydrolysis, esterification, and decarboxylative arylation afforded the racemic product 4e. The four isomers of triarylcubanes have different chemical spaces constructed by differently directed aryl groups, realizing rapid access to diverse chemical spaces through C–H arylations of cubane.
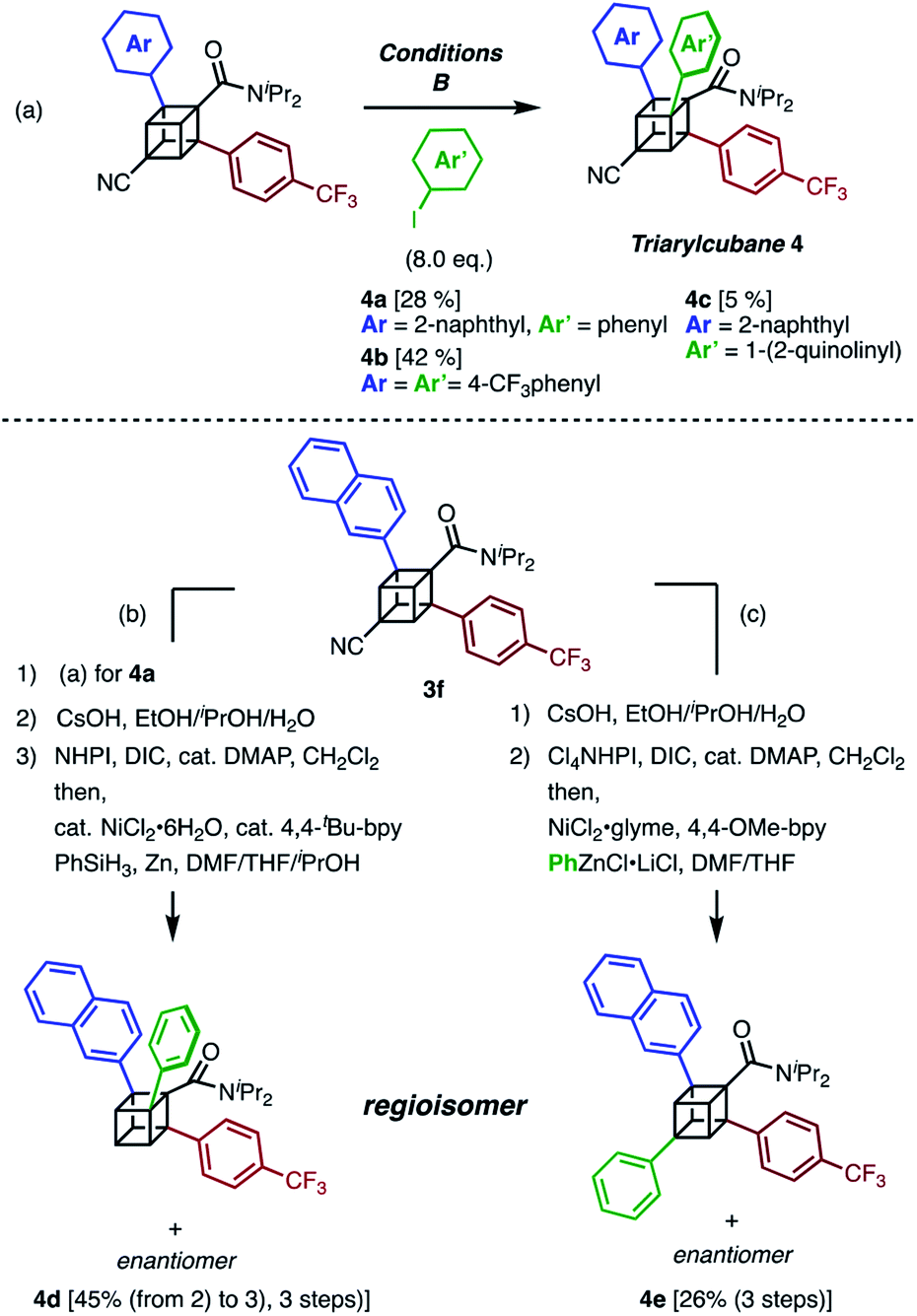 | ||
| Scheme 3 Synthesis of triarylcubanes and programmable synthesis of triarylcubane regioisomers. (a) Synthesis of triarylcubanes 4a, 4b, and 4c. (b and c) Regioselective synthesis of triarylcubane isomers. (b) Arylation, hydrolysis, and decarboxylation. | ||
We finally synthesized a tetraarylcubane by combining our arylation and Senge's decarboxylation. Hydrolysis and subsequent decarboxyphenylation of 4b successfully furnished tetraarylcubane 5a (Scheme 4), the structure of which was confirmed by X-ray crystal structure analysis (Fig. 2). The four aryl groups were introduced at the expected positions, directing four different vectors. This rigid and unique structure has potential as a novel tetrahedron unit widely used in the creation of 3D organic frameworks.
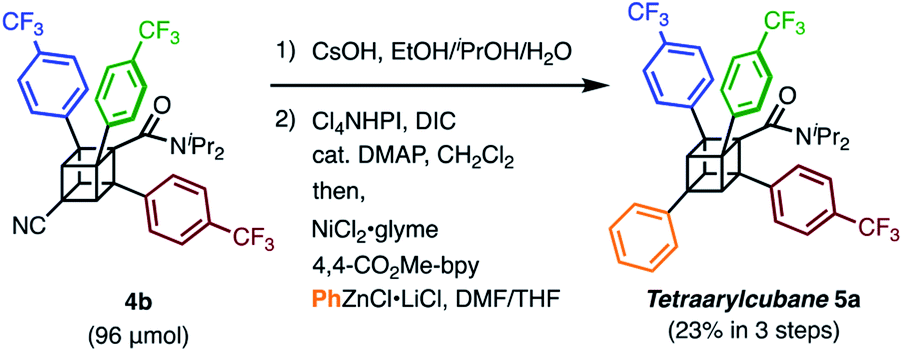 | ||
| Scheme 4 The synthesis of tetraarylcubane 5a. | ||
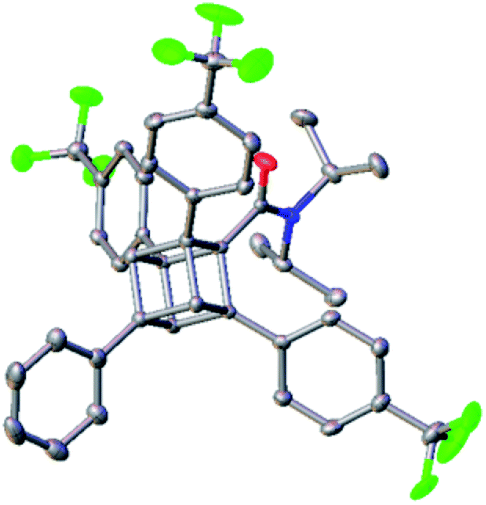 | ||
| Fig. 2 X-ray crystal structure of 5a at 50% thermal stability. | ||
Conclusions
In summary, we have achieved a programmable synthesis of multiply arylated cubanes through directed ortho-C–H metalation of amidocubanes and subsequent palladium-catalyzed cross-coupling. A wide range of aryl groups were installable regioselectively, and several kinds of mono-, di-, tri-, and tetra-arylated cubanes were synthesized. The regioselective synthesis of triarylcubane isomers is also possible, demonstrating that diverse multiply arylated cubanes are accessible by the current method. Arylcubanes are a new family of cubane derivatives, which have unique 3D chemical spaces and significant potential as functional materials and pharmaceuticals. The applications of arylcubanes in materials science and biology are now being explored in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ERATO program of the JST (JPMJER1302 to K. I.), CREST program of the JST (JPMJCR19R1 to A. Y.) and the JSPS KAKENHI (grant numbers 19H05463 to K. I. and 19K15537 to A. Y.). R. O. acknowledges the Graduate Program of Transformative Chem-Bio Research at Nagoya University, supported by the MEXT (WISE program). We thank Dr Yasutomo Segawa for the X-ray crystal structure analysis and Dr Ryosuke Takise for the fruitful discussion. ITbM is supported by the World Premier International Research Center Initiative (WPI), Japan.Notes and references
- (a) G. W. Griffin and A. P. Marchand, Chem. Rev., 1989, 89, 997 CrossRef CAS; (b) P. E. Eaton, Angew. Chem., Int. Ed. Engl., 1992, 31, 1421 CrossRef; (c) K. F. Biegasiewicz, J. R. Griffiths, G. P. Savage, J. Tsanaktsidis and R. Priefer, Chem. Rev., 2015, 115, 6719 CrossRef CAS PubMed.
- P. E. Eaton and T. W. Cole, J. Am. Chem. Soc., 1964, 86, 962 CrossRef CAS.
- (a) C. H. DePuy, B. W. Ponder and J. D. Fitzpatrick, J. Org. Chem., 1964, 29, 3508 CrossRef CAS; (b) P. E. Eaton and R. A. Hudson, J. Am. Chem. Soc., 1965, 87, 2769 CrossRef CAS; (c) N. B. Chapman, J. M. Key and K. J. Toyne, J. Org. Chem., 1970, 35, 3860 CrossRef CAS; (d) M. Bliese and J. Tsanaktsidis, Aust. J. Chem., 1997, 50, 189 CrossRef CAS.
- M. J. Falkiner, S. W. Littler, K. J. McRae, G. P. Savage and J. Tsanaktsidis, Org. Process Res. Dev., 2013, 17, 1503 CrossRef CAS.
- (a) G. J. Piermarini, S. Block, R. Damavarapu and S. Iyer, Propellants, Explos., Pyrotech., 1991, 16, 188 CrossRef CAS; (b) M. A. Dallaston, J. S. Brusnahan, C. Wall and C. M. Williams, Chem.–Eur. J., 2019, 25, 83442 Search PubMed.
- (a) T. A. Reekie, C. M. Williams, L. M. Rendina and M. Kassiou, J. Med. Chem., 2019, 62, 1078 CrossRef CAS PubMed; (b) S. D. Houston, T. Fahrenhorst-Jones, H. Xing, B. A. Chalmers, M. L. Sykes, J. E. Stok, S. C. Farfan, J. M. Burns, P. V. Bernhardt, J. J. De Voss, G. M. Boyle, M. T. Smith, J. Tsanaktsidis, G. P. Savage, V. M. Avery and C. M. Williams, Org. Biomol. Chem., 2019, 17, 6790 RSC.
- L. K. Macreadie, E. J. Mensforth, R. Babarao, K. Konstas, S. G. Telfer, C. M. Doherty, J. Tsanaktsidis, S. R. Batten and M. R. Hill, J. Am. Chem. Soc., 2019, 141, 3828 CrossRef CAS PubMed.
- (a) Y. Kato, C. M. Williams, M. Uchiyama and S. Matsubara, Org. Lett., 2019, 21, 473 CrossRef CAS PubMed; (b) M. Oi, R. Takita, J. Kanazawa, A. Muranaka, C. Wang and M. Uchiyama, Chem. Sci., 2019, 10, 6107 RSC.
- (a) P. E. Eaton and G. Castaldi, J. Am. Chem. Soc., 1985, 107, 724 CrossRef CAS; (b) A. Bashir-Hashemi, H. L. Ammon and C. S. Choi, J. Org. Chem., 1990, 55, 416 CrossRef CAS; (c) A. A. Fokin, O. Lauenstein, P. A. Gunchenko and P. R. Schreiner, J. Am. Chem. Soc., 2001, 123, 1842 CrossRef CAS PubMed.
- (a) P. E. Eaton, G. T. Cunkle, G. Marchioro and R. M. Martin, J. Am. Chem. Soc., 1987, 109, 949 CrossRef; (b) P. E. Eaton, H. Higuchi and R. Millikan, Tetrahedron Lett., 1987, 28, 1055 CrossRef CAS; (c) J. C. Bottaro, P. E. Penwell and R. J. Schmitt, J. Org. Chem., 1991, 56, 1305 CrossRef CAS.
- A. Bashir-Hashemi, J. Am. Chem. Soc., 1988, 110, 7234 CrossRef CAS.
- C. Annese, L. D'Accolti, C. Fusco, R. Gandolfi, P. E. Eaton and R. Curci, Org. Lett., 2009, 11, 3574 CrossRef CAS PubMed.
- (a) S. Plunkett, K. J. Flanagan, B. Twamley and M. O. Senge, Organometallics, 2015, 34, 1408 CrossRef CAS; (b) S. D. Houston, B. A. Chalmers, G. P. Savage and C. M. Williams, Org. Biomol. Chem., 2019, 17, 1067 RSC.
- (a) B. A. Chalmers, H. Xing, S. Houston, C. Clark, S. Ghassabian, A. Kuo, B. Cao, A. Reitsma, E. P. Murray, J. E. Stok, G. M. Boyle, C. J. Pierce, S. W. Littler, D. A. Winkler, P. V. Bernhardt, C. Pasay, J. J. D. Voss, J. McCarthy, P. G. Parsons, G. H. Walter, M. T. Smith, H. M. Cooper, S. K. Nilsson, J. Tsanaktsidis, G. P. Savage and C. M. Williams, Angew. Chem., Int. Ed., 2016, 6, 3580 CrossRef PubMed; (b) Y. P. Auberson, C. Brocklehurst, M. Furegati, T. C. Fessard, G. Koch, A. Decker, L. LaVecchia and E. Briard, ChemMedChem, 2017, 12, 590 CrossRef CAS PubMed.
- B. A. Chalmers, A. P.-J. Chen, G. P. Savage and C. M. Williams, Aust. J. Chem., 2010, 63, 1108 CrossRef CAS.
- (a) Y. Kondo, M. Shilai, M. Uchiyama and T. Sakamoto, J. Am. Chem. Soc., 1999, 121, 3539 CrossRef CAS; (b) M. Uchiyama, Y. Matsumoto, D. Nobuto, T. Furuyama, K. Yamaguchi and K. Morokuma, J. Am. Chem. Soc., 2006, 128, 8748 CrossRef CAS PubMed; (c) M. Yasui, R. Ota, C. Tsukano and Y. Takemoto, Org. Lett., 2018, 20, 7656 CrossRef CAS PubMed.
- P. E. Eaton, Y. Xiong and J. P. Zhou, J. Org. Chem., 1992, 57, 4277 CrossRef CAS.
- (a) P. E. Eaton and G. Castaldi, J. Am. Chem. Soc., 1985, 107, 724 CrossRef CAS; (b) P. E. Eaton, C. H. Lee and Y. Xiong, J. Am. Chem. Soc., 1989, 111, 8016 CrossRef CAS; (c) P. E. Eaton, Y. Xiong and C.-H. Lee, J. Chin. Chem. Soc., 1991, 38, 303 CrossRef CAS; (d) P. E. Eaton, Y. Xiong and R. Gilardi, J. Am. Chem. Soc., 1993, 115, 10195 CrossRef CAS; (e) B. L. May, P. Clements, J. Tsanaktsidis, C. J. Easton and S. F. Lincoln, J. Chem. Soc., Perkin Trans. 1, 2000, 463 RSC; (f) A. A. Fokin, P. R. Schreiner, R. Berger, G. H. Robinson, P. Wei and C. F. Campana, J. Am. Chem. Soc., 2006, 128, 5332 CrossRef CAS PubMed.
- T. Qin, L. R. Malins, J. T. Edwards, R. R. Merchant, A. J. E. Novak, J. Z. Zhong, R. B. Mills, M. Yan, C. Yuan, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 6, 260 CrossRef PubMed.
- (a) F. Toriyama, J. Cornella, L. Wimmer, T.-G. Chen, D. D. Dixon, G. Creech and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 11132 CrossRef CAS PubMed; (b) S. S. R. Bernhard, G. M. Locke, S. Plunkett, A. Meindl, K. J. Flanagan and M. O. Senge, Chem.–Eur. J., 2018, 24, 1026 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and spectral data for all compounds, including scanned images of the 1H and 13C NMR spectra. CCDC 1993519 (2a), 1993515 (2e), 1993518 (2f), 1993517 (2k), 1993513 (2p), 1993512 (3a), 1993516 (4b) and 1993514 (5a). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01909g |
| This journal is © The Royal Society of Chemistry 2020 |