
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrathin two-dimensional conjugated metal–organic framework single-crystalline nanosheets enabled by surfactant-assisted synthesis†
Zhiyong
Wang‡
 ab,
Gang
Wang‡
a,
Haoyuan
Qi
ad,
Mao
Wang
c,
Mingchao
Wang
a,
SangWook
Park
a,
Huaping
Wang
a,
Minghao
Yu
a,
Ute
Kaiser
d,
Andreas
Fery
b,
Shengqiang
Zhou
c,
Renhao
Dong
*a and
Xinliang
Feng
*a
ab,
Gang
Wang‡
a,
Haoyuan
Qi
ad,
Mao
Wang
c,
Mingchao
Wang
a,
SangWook
Park
a,
Huaping
Wang
a,
Minghao
Yu
a,
Ute
Kaiser
d,
Andreas
Fery
b,
Shengqiang
Zhou
c,
Renhao
Dong
*a and
Xinliang
Feng
*a
aCenter for Advancing Electronics Dresden (cfaed) and Faculty of Chemistry and Food Chemistry, Technische Universität Dresden, 01062 Dresden, Germany. E-mail: renhao.dong@tu-dresden.de; xinliang.feng@tu-dresden.de
bLeibniz-Institut für Polymerforschung Dresden e. V. (IPF), 01069 Dresden, Germany
cHelmholtz-Zentrum Dresden-Rossendorf, Institute of Ion Beam Physics and Materials Research, 01328 Dresden, Germany
dCentral Facility for Electron Microscopy, Electron Microscopy Group of Materials Science, Universität Ulm, 89081 Ulm, Germany
First published on 21st April 2020
Abstract
Two-dimensional conjugated metal–organic frameworks (2D c-MOFs) have recently emerged for potential applications in (opto-)electronics, chemiresistive sensing, and energy storage and conversion, due to their excellent electrical conductivity, abundant active sites, and intrinsic porous structures. However, developing ultrathin 2D c-MOF nanosheets (NSs) for facile solution processing and integration into devices remains a great challenge, mostly due to unscalable synthesis, low yield, limited lateral size and low crystallinity. Here, we report a surfactant-assisted solution synthesis toward ultrathin 2D c-MOF NSs, including HHB-Cu (HHB = hexahydroxybenzene), HHB-Ni and HHTP-Cu (HHTP = 2,3,6,7,10,11-hexahydroxytriphenylene). For the first time, we achieve single-crystalline HHB-Cu(Ni) NSs featured with a thickness of 4–5 nm (∼8–10 layers) and a lateral size of 0.25–0.65 μm2, as well as single-crystalline HHTP-Cu NSs with a thickness of ∼5.1 ± 2.6 nm (∼10 layers) and a lateral size of 0.002–0.02 μm2. Benefiting from the ultrathin feature, the synthetic NSs allow fast ion diffusion and high utilization of active sites. As a proof of concept, when serving as a cathode material for Li-ion storage, HHB-Cu NSs deliver a remarkable rate capability (charge within 3 min) and long-term cycling stability (90% capacity retention after 1000 cycles), superior to the corresponding bulk materials and other reported MOF cathodes.
Introduction
Metal–organic frameworks (MOFs) represent a class of crystalline coordination polymers consisting of metal ions/clusters connected by organic ligands.1 Profiting from their well-defined structures, high specific surface area and structural/chemical diversity, MOFs have attracted great attention as advanced materials for gas storage/separation,1a,2 catalysis,3 and sensors.4 However, the conventional three-dimensional (3D) MOFs showed extremely low electrical conductivity (<10−10 S cm−1), due to the large separation of metal centres by multi-atom and insulating organic ligands. This led to the low utilization of built-in redox centres and slow reaction kinetics, which significantly limited their applications in electrochemical systems.5 Recent advances disclosed that the designs of layer-stacked two-dimensional conjugated MOFs (2D c-MOFs) by employing planar organic ligands and square-planar metal–complex linkages inducing high delocalization of π-electrons in the 2D plane have led to improved electrical conductivity6 (as high as ∼60 S cm−1 in the powder form7). Besides the intrinsic conductivity, 2D c-MOFs also possess inherent porosity, abundant active sites, and tunable redox states, which lay the foundation for their applications in electrochemical energy storage/conversion,8 such as Li-ion batteries,9 Li-sulfur batteries,10 Na-ion batteries,11 supercapacitors,5 water splitting, and oxygen reduction reactions.12 For instance, a nickel–hexaiminobenzene (Ni–HIB) 2D c-MOF was reported to be utilized as a cathode material in Li-ion batteries and displayed a high specific capacity of 155 mA h g−1 and a stable cycling performance up to 300 cycles, which are comparable to those of other commercially used cathode materials in Li-ion batteries (e.g., transition-metal compound cathodes possess a capacity of <200 mA h g−1).9,13However, the currently developed 2D c-MOFs are generally synthesized in a bulk powder form by the solvothermal method. Accordingly, a large number of active sites are buried and inaccessible for charge carriers in these powder samples, leading to sluggish ion diffusion and low utilization of active sites. The apparent electrochemical performance of bulk 2D c-MOFs has been therefore limited compared with the theoretical values (for example, the theoretical specific capacity of Ni–HIB can reach ∼312 mA h g−1).14 In contrast, ultrathin nanosheets (NSs, <10 nm in thickness) possess a range of fascinating attributes including higher surface areas and sufficient accessible active sites,15 and their nanoscale thicknesses provide a short ion/electron migration length owing to the quantum confinement effect.16 As a result, the development of ultrathin 2D c-MOF NSs is an attractive avenue for enhancing the electrochemical energy storage/conversion performance. To date, a great effort has been dedicated to synthesizing ultrathin non-conjugated 2D MOF NSs, including top-down physical and chemical delamination of 2D layered bulk crystals,2,17 as well as bottom-up interface-assisted synthesis1b and template-assisted synthesis.15b,18 Particularly, surfactant-assisted solution synthesis has been regarded as an efficient bottom-up method to prepare ultrathin MOF NSs.15d,19 Compared with the role of the interface and other templates, the surfactant (e.g., polyvinylpyrrolidone (PVP)15d and hexadecyltrimethylammonium bromide (CTAB)20) can not only restrict the growth of the MOFs along the layer-stacking direction but also help to stabilize the as-synthesized MOF NSs in solution for facile processing. Nevertheless, despite the above progress of non-conjugated MOF materials, the synthesis of 2D c-MOF NSs by scalable solution processing or even the exfoliation of a layer-stacked bulk 2D c-MOF into NSs has been unexplored considering the strong interlayer π–π stacking interaction. Thus, the large-sized, ultrathin 2D c-MOF single-crystalline NSs remain an appealing synthetic target.
Herein, we report a surfactant-assisted solution synthesis of ultrathin 2D c-MOF NSs (>500 mg, 80–90% yield) (Table S1†). The resultant 2D c-MOF NSs are single-crystalline; the HHB-Cu(Ni) NSs have a thickness of 4–5 nm (8–10 layers) and a lateral size of 0.25–0.65 μm2, while the HHTP-Cu NSs possess a thickness of ∼5.1 ± 2.6 nm (∼10 layers) and a lateral size of 0.002–0.02 μm2. As one typical example, few-layered HHB-Cu NSs present a Brunauer–Emmett–Teller (BET) surface area (385 m2 g−1), much higher than that of the corresponding bulk particles (119 m2 g−1). To this end, HHB-Cu NSs are utilized as a cathode material for Li-ion batteries and present a specific capacity of 153 mA h g−1 in the potential window of 1.3–2.6 V (the specific capacity reached 400 mA h g−1 in the potential window of 1.3–4.0 V) and exhibit high rate capability (charge within 3 min, 81%, 1.0 A g−1vs. 0.1 A g−1) and long-term cycling stability (90% capacity retention after 1000 cycles), which are superior to those of its bulk powder sample (capacity, 40 mA h g−1, rate capability, 25%, 1.0 A g−1vs. 0.1 A g−1, and cycling stability, 65% capacity retention after 50 cycles) and the thus-far reported MOF cathodes (Table S2†). Our work paves the way to develop conductive 2D c-MOF NSs by scalable synthesis that enhances the electrochemical performance and allows future facile solution-processing and integration into electronic devices.
Results and discussion
The synthesis of ultrathin HHB-Cu NSs by a surfactant-assisted solution synthesis method is illustrated in Fig. 1A. For the first time, the anionic surfactant sodium dodecyl sulfate (SDS) was employed to produce uniform ultrathin 2D c-MOF nanosheets. In a typical synthesis, HHB-Cu NSs were obtained through the preparation of Cu(OAc)2 salts and SDS aqueous solution (1.7 mM) (Fig. 1B), followed by the addition of tetrahydroxy-1,4-quinone (THQ) ligands and then sonication of the mixture at 50 °C for 30 minutes (see the ESI for experimental details†). The negatively charged hydrophilic tail of the SDS molecules will preferentially anchor onto the surface of MOFs through electrostatic interaction.15d Thus, the adsorbed SDS molecules can serve as structure directing agents to significantly alleviate the layer stacking, thus leading to the anisotropic growth of the MOFs in the 2D direction and consequently the formation of ultrathin and large-sized nanosheets. In addition, the hydrophobic chains of SDS could not only weaken the interactions of interlayers and disassemble to form ultrathin nanosheets during the sonication treatment but also help to stabilize the as-synthesized HHB-Cu MOF NSs in solution to prevent further agglomeration.20,21 The solid product was collected and washed with water and ethanol in an ultrasonic ice bath for 30 min. Then, the upper colloidal suspension was collected and dried under vacuum for 12 h at 80 °C to afford dark samples. The diluent colloidal suspension of the obtained HHB-Cu NSs exhibited a Tyndall effect (inset in Fig. 1A); and due to their ultrathin nature, the colloid possessed excellent stability in the aqueous solution for at least 6 months.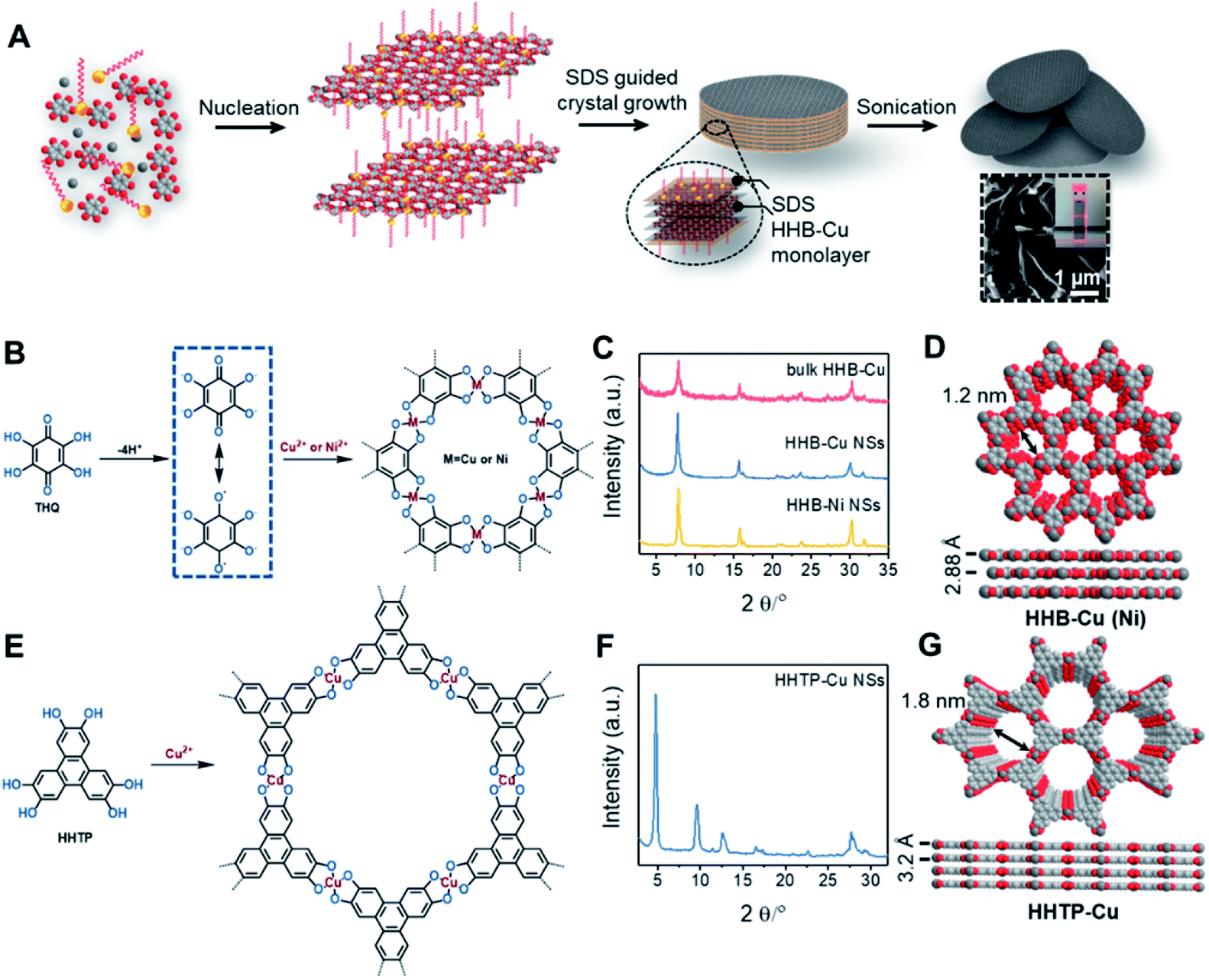 | ||
| Fig. 1 (A) Synthetic scheme of ultrathin HHB-Cu NSs using a surfactant-assisted solution synthesis method. Inset: SEM image of HHB-Cu NSs and Tyndall effect of a diluent colloidal suspension. (B) Reaction scheme for the synthesis of HHB-Cu and HHB-Ni NSs. (C) PXRD patterns of HHB-Cu NSs, HHB-Ni NSs, and bulk HHB-Cu particles. (D) The unit-cell structures of HHB-Cu derived using AB slipped-parallel stacking models. Grey, red, and white spheres represent Cu, O and C atoms, respectively. Na+ counter-ions are omitted for clarity. (E) Reaction scheme for the synthesis of HHTP-Cu NSs. (F) PXRD patterns of HHTP-Cu NSs. (G) The unit-cell structures of HHTP-Cu derived using AA stacking models. Grey, red, and white spheres represent Cu, O and C atoms, respectively. | ||
Powder X-ray diffraction (PXRD) patterns in Fig. 1C exhibit strong reflections at 2θ = 7.8°, 15.7°, and 27.5°, which can be indexed to the (100), (200) and (300) crystallographic planes of the HHB-Cu structure, respectively. This result reveals that the obtained product is highly crystalline with hexagonal packing in ab planes (a = b = 1.2 nm).6c The peak at 2θ = 30.2°, corresponding to the (001) reflection, suggests an ordered stacking with an interlayer spacing of ∼2.88 Å. Thus, an AB slipped-parallel model is built according to the PXRD results,6c as shown in Fig. 1D. In Fig. 2A, the atomic force microscopy (AFM) image demonstrates a sheet structure and smooth surface (the arithmetic average roughness (Ra) is about 0.23 nm) for HHB-Cu NSs. The thickness of the NSs was determined to be 4.2 ± 1.1 nm and the lateral size was 0.30–0.65 μm2 (Fig. 2B and S1†). SEM images also show an inherent sheet structure of HHB-Cu NSs, which is consistent with the AFM observation (Fig. S2†).
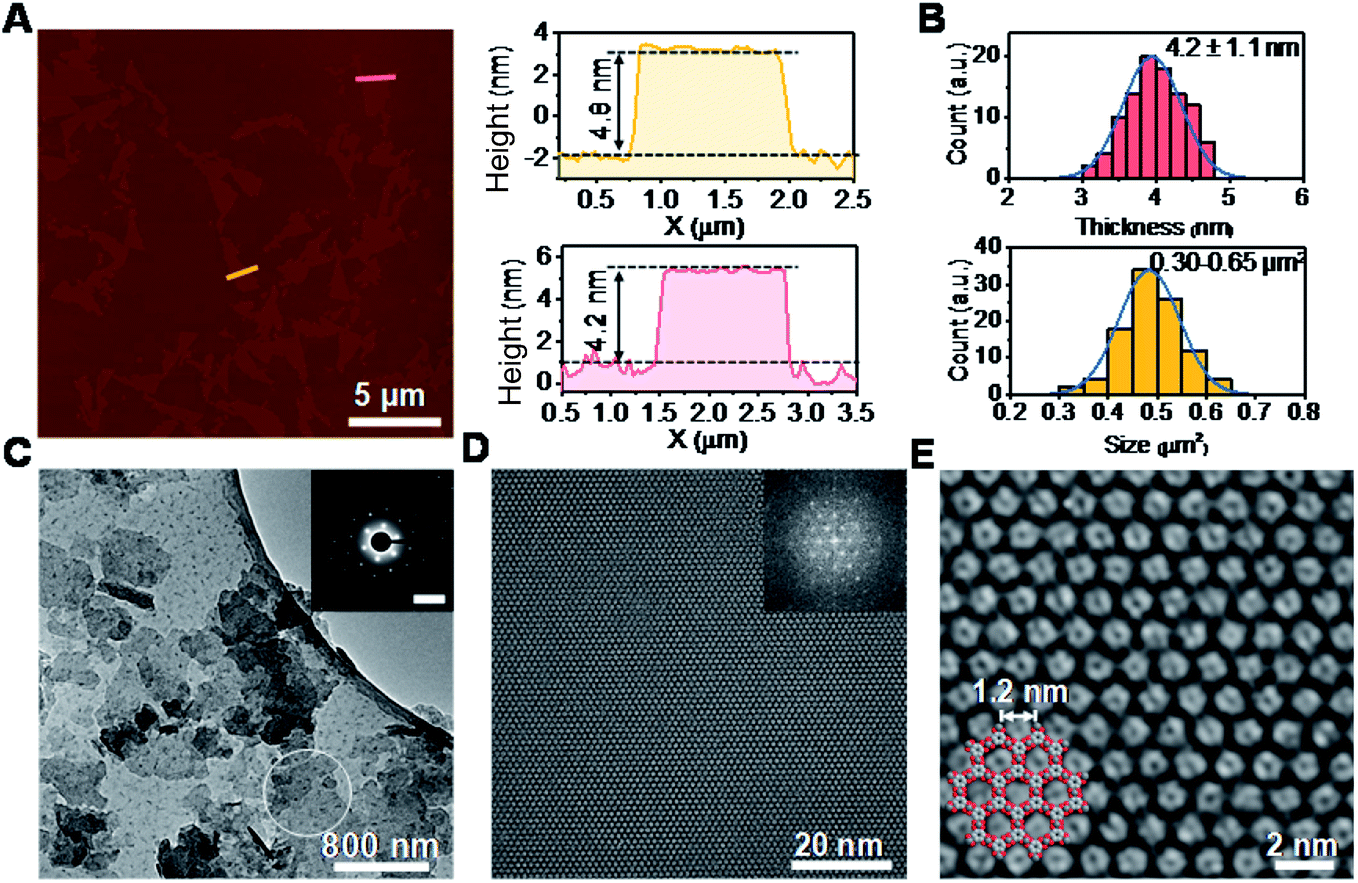 | ||
| Fig. 2 Morphology and crystal structural characterization of HHB-Cu NSs. (A) AFM image of HHB-Cu NSs and the height profile along the marked lines in the image. (B) HHB-Cu NS thickness and size distributions as determined by AFM. (C) TEM image of HHB-Cu NSs. Inset: SAED pattern of HHB-Cu NSs. Scale bar: 1.85 nm−1. (D) AC-HRTEM image of HHB-Cu NSs. Inset: corresponding FFT image with reflections up to 4.6 nm−1 (i.e., image resolution 0.22 nm). (E) Enlarged image of (D) with the structure model overlaid. | ||
The EDX mapping and spectrum (Fig. S3 and S4†) demonstrate a uniform distribution of C, Cu, O, and Na in the achieved HHB-Cu NSs. A Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O ratio of 1.2:4 was determined, which is consistent with X-ray photoelectron spectroscopy (XPS) characterization (1.1:4) and very close to the calculated value for CuO4 units (1:4) (Fig. S5†). The XPS Cu 2p3/2 peak at 933 eV and the satellite peaks at 932 and 943 eV indicate that Cu exhibits two oxidation states of +2 and +1 with a Cu2+/Cu+ ratio of about 9.44. As shown in Fig. S6A,† the Cu K-edge of X-ray absorption near-edge structure (XANES) in HHB-Cu NSs is completely different from those of CuO and Cu foil. The pre-edge feature in HHB-Cu NSs originates from the transition of 1s core electrons to hybridized orbitals of 3d. In addition, it can be seen that the white-line intensity of HHB-Cu NSs is slightly lower than that of CuO, indicating that Cu species are in two oxidation forms in HHB-Cu NSs, consistent with the XPS results. Accordingly, the framework of HHB-Cu NSs possesses several redox states with up to two electron transfers per CuO4 unit (Scheme S2†), which act as active sites for a promising energy storage function. The recorded extended X-ray absorption fine structure (EXAFS) spectra show that there is one prominent peak at ∼1.52 Å from the Cu–O contribution in HHB-Cu NSs (Fig. S6B†), while another sample CuO clearly exhibits two different predominant peaks at ∼1.56 Å and ∼2.49 Å, which arise from Cu–O and Cu–Cu bonds, respectively. These results provide strong proof for the formation of square planar complexes via the coordination of THQ and Cu ions and confirm the absence of metal oxides such as CuO and Cu foil in the HHB-Cu NSs.
:O ratio of 1.2:4 was determined, which is consistent with X-ray photoelectron spectroscopy (XPS) characterization (1.1:4) and very close to the calculated value for CuO4 units (1:4) (Fig. S5†). The XPS Cu 2p3/2 peak at 933 eV and the satellite peaks at 932 and 943 eV indicate that Cu exhibits two oxidation states of +2 and +1 with a Cu2+/Cu+ ratio of about 9.44. As shown in Fig. S6A,† the Cu K-edge of X-ray absorption near-edge structure (XANES) in HHB-Cu NSs is completely different from those of CuO and Cu foil. The pre-edge feature in HHB-Cu NSs originates from the transition of 1s core electrons to hybridized orbitals of 3d. In addition, it can be seen that the white-line intensity of HHB-Cu NSs is slightly lower than that of CuO, indicating that Cu species are in two oxidation forms in HHB-Cu NSs, consistent with the XPS results. Accordingly, the framework of HHB-Cu NSs possesses several redox states with up to two electron transfers per CuO4 unit (Scheme S2†), which act as active sites for a promising energy storage function. The recorded extended X-ray absorption fine structure (EXAFS) spectra show that there is one prominent peak at ∼1.52 Å from the Cu–O contribution in HHB-Cu NSs (Fig. S6B†), while another sample CuO clearly exhibits two different predominant peaks at ∼1.56 Å and ∼2.49 Å, which arise from Cu–O and Cu–Cu bonds, respectively. These results provide strong proof for the formation of square planar complexes via the coordination of THQ and Cu ions and confirm the absence of metal oxides such as CuO and Cu foil in the HHB-Cu NSs.
The Raman spectra of HHB-Cu NSs manifest the elimination of OH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peaks at 3370 cm−1 and 1649 cm−1, respectively, which confirm the efficient coordination between Cu and O (Fig. S7†). The UV/vis absorption spectra of the dispersed HHB-Cu NSs on a quartz substrate show a broad absorption band in the near-IR region, suggesting an absorption edge of ∼1.7 eV (Fig. S8;† also see Tauc plots of (ahν)2vs. hν). Based on low-pressure N2 adsorption measurements at 77 K, the BET surface area of HHB-Cu NSs was estimated to be as high as 385 m2 g−1 (Fig. S9†), due to the existence of more exposed surface between layers as a result of the high crystallinity, large domain size, and nanoscale thicknesses. Without using SDS (Fig. S10†), the contrast synthesis under the same conditions resulted in layer-stacked HHB-Cu powders with lower crystallinity and smaller domain size (Fig. S11†), in which the surface was deeply buried, leading to a lower accessible surface area (BET surface area, 119 m2 g−1) (Fig. S9†).
O peaks at 3370 cm−1 and 1649 cm−1, respectively, which confirm the efficient coordination between Cu and O (Fig. S7†). The UV/vis absorption spectra of the dispersed HHB-Cu NSs on a quartz substrate show a broad absorption band in the near-IR region, suggesting an absorption edge of ∼1.7 eV (Fig. S8;† also see Tauc plots of (ahν)2vs. hν). Based on low-pressure N2 adsorption measurements at 77 K, the BET surface area of HHB-Cu NSs was estimated to be as high as 385 m2 g−1 (Fig. S9†), due to the existence of more exposed surface between layers as a result of the high crystallinity, large domain size, and nanoscale thicknesses. Without using SDS (Fig. S10†), the contrast synthesis under the same conditions resulted in layer-stacked HHB-Cu powders with lower crystallinity and smaller domain size (Fig. S11†), in which the surface was deeply buried, leading to a lower accessible surface area (BET surface area, 119 m2 g−1) (Fig. S9†).
To gain insight into the crystallinity of the resultant HHB-Cu NSs, we performed selected-area electron diffraction (SAED) and aberration-corrected high-resolution transmission electron microscopy (AC-HRTEM) imaging. The SAED pattern reveals a hexagonal symmetry with sharp diffraction spots (Fig. 2C). The first-order reflections were found at 0.93 nm−1, corresponding to a hexagonal unit cell with a side length of 1.24 nm (inset in Fig. 2C), agreeing with the lattice parameters of HHB-Cu.6c The SAED patterns acquired on isolated HHB-Cu NSs manifest single crystallographic orientation (Fig. S12†), revealing the single-crystalline nature of the HHB-Cu NSs. Furthermore, AC-HRTEM imaging unambiguously illustrates the highly ordered hexagonal MOF network with a near-atomic resolution of 0.22 nm. Strikingly, no point or line defects have been observed within the field-of-view, further evidencing the high crystallinity of the HHB-Cu NSs (Fig. 2D). The measured pore size on the magnified AC-HRTEM image was 1.2 nm, which is in perfect agreement with the structural model (Fig. 2E).
The versatility of the surfactant-assisted solution synthesis method using SDS was investigated through the preparation of a variety of 2D c-MOF NSs including HHB-Ni NSs and HHTP-Cu NSs (HHTP = 2,3,6,7,10,11-hexahydroxytriphenylene). Structural and morphological characterization by PXRD, TEM, SAED, and AFM measurements revealed that the synthetic HHB-Ni presented single-crystalline ultrathin NSs with a lateral size of 0.25–0.56 μm2 and a thickness of 4.5 ± 1.4 nm (∼9 layers) (Fig. 1C and S13†). In addition, the obtained HHTP-Cu NSs synthesized with the surfactant show a single-crystalline feature with a lateral size of 0.002–0.02 μm2 and a thickness of 5.1 ± 2.6 nm (∼10 layers) (Fig. 1E and F and S14†), whereas bulk powder with a rod like morphology was achieved in the absence of the surfactant.
The electrical conductivity of HHB-Cu NSs was derived to be 1.5 × 10−7 S cm−1 through the van der Pauw method at 300 K (Fig. S15 and S16†). The variable-temperature conductivity measurements displayed a non-linear increase of electrical conductivity from 243 to 310 K, indicating a typical semiconducting feature. As shown in Fig. S17,† a hole concentration of ∼1.4 × 1014 cm−3 and charge carrier mobility of 2.4 ± 0.3 cm2 V−1 s−1 at 300 K were further evaluated from the Hall resistance (RHall) under a magnetic field (H), suggesting a p-type semiconducting behavior of HHB-Cu NSs. In contrast, the bulk HHB-Cu shows a lower conductivity of 8.95 × 10−8 S cm−1 in the pellet form at 300 K (Fig. S18†). Benefiting from the fact that HHB-Cu NSs possess single crystallinity, a high aspect ratio, and electrically conductive features, this material was used as a model system for determining the role of few-layered 2D c-MOF NSs in improving electrochemical Li-ion storage.
The electrochemical performance of HHB-Cu NSs was then evaluated in coin cells with Li metal as the counter electrode. The electrolyte is 1 M LiPF6 in ethylene carbonate (EC)/ethyl methyl carbonate (EMC) (v/v = 1:1). The cyclic voltammetry (CV) profiles of HHB-Cu NSs exhibit a couple of redox peaks within the potential window of 1.3 to 2.6 V (vs. Li/Li+) (Fig. 3A and S19†), which suggest reversible transformation between the neutral and negatively charged (reduced) states of HHB-Cu NSs with the insertion/desertion of Li ions. In the galvanostatic charge–discharge test at 1.3–2.6 V, the HHB-Cu NS electrode delivers a specific capacity of 153 mA h g−1 at 0.1 A g−1 (Fig. S20†). The charge–discharge curves present a single charge/discharge plateau, matching very well with the CV result. After 50 cycles at 0.1 A g−1, the reversible capacity could be stabilized at 122 mA h g−1 (Fig. S21†). Under extended potential windows of 1.3–3.3 V and 1.3–4.0 V, the specific capacity of the HHB-Cu NS electrode can be pushed up to 300 and 400 mA h g−1, respectively, which is superior to those of other reported MOFs (66–155 mA h g−1) (Table S2 and S22†). These capacities correspond to the full utilization of CuO4 units (CuO40/CuO42−; Scheme S3†) and CuO4 (CuO40/CuO42−)/Cu center (Cu2+/Cu+).22 However, after tens of cycles, the capacity decayed to below 200 mA h g−1, corresponding to a low capacity retention of 36.0–66.7%. The above results suggest that the HHB-Cu NS electrode exhibits better electrochemical reversibility under a one-electron redox process at 1.3–2.6 V (CuO4−/CuO42−; Scheme S2†) than under a multi-electron process at 1.3–3.3 V and at 1.3–4.0 V. This can be explained by the fact that less strain will be generated in the framework of HHB-Cu NSs under the one-electron process, thus leading to higher structural/cycling stability.
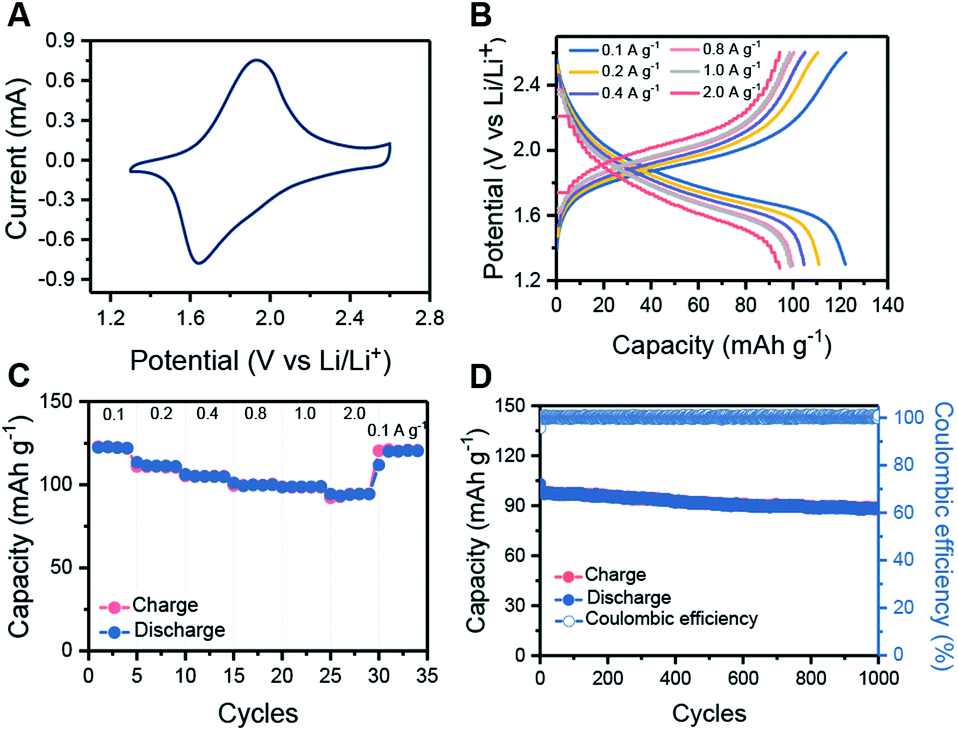 | ||
| Fig. 3 Electrochemical performance of HHB-Cu NS electrodes. (A) CV profiles collected at 1 mV s−1 for HHB-Cu NSs. (B) Charge–discharge curves of HHB-Cu NSs at specific current densities. (C) The rate capability of HHB-Cu NSs at different current densities. (D) Long-term cycling performance of HHB-Cu NSs at 1 A g−1. | ||
The rate capabilities of the HHB-Cu NS electrode were further evaluated under various current densities. Clearly, increasing the current rate from 0.1 to 2.0 A g−1 slightly increases the charge–discharge polarization (Fig. 3B). Specific capacities of 122, 111, 105, 100, 99 and 94 mA h g−1 could still be retained at 0.1, 0.2, 0.4, 0.8, 1.0 and 2.0 A g−1, respectively (Fig. 3C). We attribute the high rate performance of HHB-Cu NSs to their ultrathin morphology, which provides short pathways for fast ion/electron diffusion in the electrode. The long-term cycling stability of the HHB-Cu NS electrode was also surveyed at 1.0 A g−1 (charge in 6 min). After 1000 cycles, a high capacity retention of 90% was achieved with 100% coulombic efficiency (Fig. 3D), suggesting robust structural stability of HHB-Cu NSs under the electrochemical environment and high redox reversibility of active sites. The cycling stability of our HHB-Cu NSs is the record performance among the reported MOFs (Table S2, ESI†).
For comparison, the electrochemical behavior of bulk HHB-Cu particles was also evaluated under the same conditions. Compared to HHB-Cu NSs, the bulk HHB-Cu represented a much smaller CV area and larger peak voltage gaps, indicating lower capacity/active site utilization and higher polarization (Fig. 4A). At 1.3–2.6 V (Scheme S2†), the bulk HHB-Cu exhibited a limited capacity of 40–50 mA h g−1 (Fig. S20†), which could be ascribed to the sluggish ion diffusion and low utilization (13.4%) of active sites resulting from a long-range parallel-stacking structure (Fig. 4B). The rate capability of bulk HHB-Cu exhibits huge polarization upon increasing the current density (Fig. S23A†), thereby sacrificing most of the capacity. A low capacity of 4.6 mA h g−1 for bulk HHB-Cu was retained at 2.0 A g−1, which was only 11.5% of the value obtained at 0.1 A g−1 (Fig. S23B†).
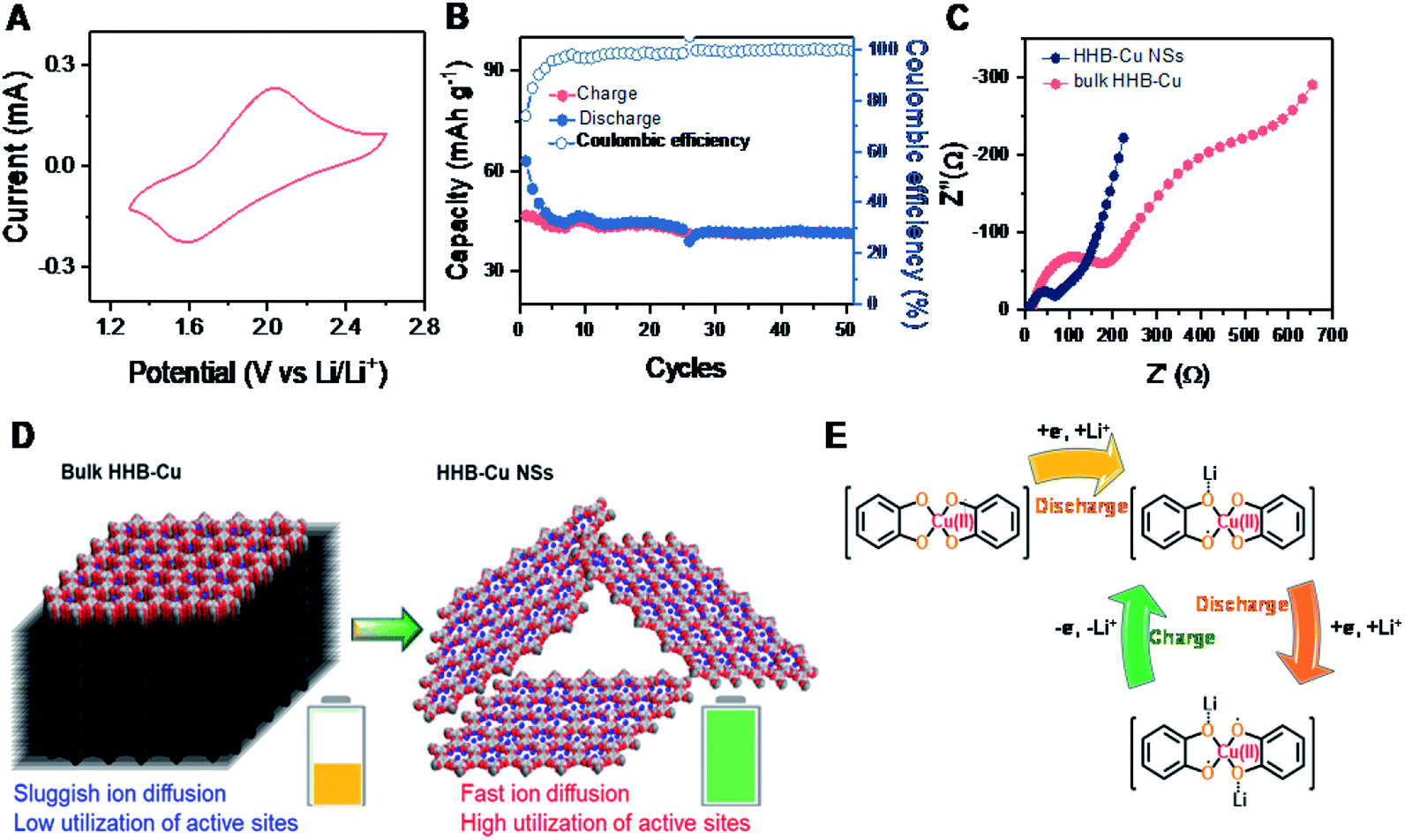 | ||
| Fig. 4 (A) CV profiles collected at 1 mV s−1 for bulk HHB-Cu. (B) Cycling performance of bulk HHB-Cu at 0.1 A g−1. (C) The EIS data represented in a Nyquist plot. (D) Schematic comparison of bulk HHB-Cu and HHB-Cu NSs as cathodes for Li-ion storage. (E) The evolution of the electronic states of the CuO4 unit during the charge/discharge process. | ||
To further understand the electrochemical performance and to evaluate the merit of HHB-Cu NSs in ion/electron diffusion, we performed electrochemical impedance spectroscopy (EIS) measurements on HHB-Cu NSs and bulk HHB-Cu. The obtained Nyquist plots are shown in Fig. 4C. Obviously, HHB-Cu NSs display a much lower charge transfer resistance (Rct) of ∼85 Ω when compared to bulk HHB-Cu (Rct = ∼216 Ω), and an almost vertical low-frequency region represents a lower ionic resistance. These results confirm the substantial contribution of the ultrathin nature of HHB-Cu NSs in the fast charge/discharge process and the high utilization of active sites (Fig. 4D). Due to the open porosity and thin sheet morphology, the Li+ ion diffusion along the channels most probably dominates as the main path for Li+ insertion into HHB-Cu NSs rather than diffusion between layers. In support of the redox mechanism, we further compared the electrochemical properties of HHB-Cu NSs and HHB-Ni NSs. Clearly, the CV profile of HHB-Cu NSs is characterized by two distinct peaks at 1.64 and 1.93 V, whereas the HHB-Ni NSs have a substantially broadened peak region in the potential range from 1.9 to 2.5 V (Fig. S24†). As a result, we highlight that the active site should be mainly situated on the CuO4 unit, which dominates the electrochemical behavior of HHB-Cu MOFs.5 On the other hand, detailed studies were further performed by ex situ Fourier transform infrared (FT-IR) spectroscopy. As shown in Fig. S25,† the peak at ∼1515 cm−1, assigned to the CC bond from the benzene ring, disappeared in the discharged state, suggesting the disappearance of the conjugation of the C6 ring.23 In addition, a reversible change of a peak at ∼1068 cm−1 (attributed to the C–O bond) during the charge and discharge processes was observed, confirming the Li-storage activity of CuO4 units. The proposed evolution of the electronic states of the CuO4 unit during the charge/discharge process is shown in Fig. 4E.
Conclusions
In summary, we demonstrated a universal strategy through a surfactant-assisted solution synthesis method to prepare ultrathin 2D c-MOF nanosheets with few-layered structures, large lateral sizes (aspect ratio exceeding 200), and single-crystalline features on a large scale and in high yield. The synthetic HHB-Cu NSs as a showcase were utilized as a cathode material in Li-ion batteries, which facilitated the utilization efficiency of redox sites and allowed faster ion/electron kinetics due to the improved surface area and significantly shortened ion/electron diffusion pathways compared to bulk HHB-Cu particles. Thus, HHB-Cu NSs presented greatly improved Li-ion storage performance with a specific capacity of 153 mA h g−1 and exhibited high rate capability (charge within 3 min, 81%, 1.0 A g−1vs. 0.1 A g−1) and long-term cycling stability (90% capacity retention after 1000 cycles), which are superior to those of the state-of-the-art MOFs. Our work highlights an approach towards solution-processible ultrathin crystalline nanosheets of 2D c-MOFs and paves the way for the development of other devices, such as electronics and sensors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the EU Graphene Flagship (No. 785219), ERC starting grant (FC2DMOF, No. 852909), ERC Consolidator Grant (T2DCP), DFG project (2D polyanilines, No. 426572620), Coordination Networks: Building Blocks for Functional Systems (SPP 1928, COORNET), and H2020-MSCA-ITN (ULTIMATE, No. 813036), as well as the German Science Council and Center of Advancing Electronics Dresden (cfaed). Zhiyong Wang gratefully acknowledges funding from the China Scholarship Council. The authors thank Dr Markus Löffler, Dr Petr Formanek, Baokun Liang, Dr Jichao Zhang, Dr Xubing Li, and Dr Tao Zhang for SEM, TEM, XANES, XPS and Raman measurements. The authors acknowledge the cfaed and Dresden Center for Nanoanalysis (DCN) at TUD.Notes and references
- (a) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed; (b) R. Makiura, S. Motoyama, Y. Umemura, H. Yamanaka, O. Sakata and H. Kitagawa, Nat. Mater., 2010, 9, 565–571 CrossRef CAS PubMed.
- Y. Peng, Y. Li, Y. Ban, H. Jin, W. Jiao, X. Liu and W. Yang, Science, 2014, 346, 1356–1359 CrossRef CAS PubMed.
- Q. Lin, X. Bu, A. Kong, C. Mao, X. Zhao, F. Bu and P. Feng, J. Am. Chem. Soc., 2015, 137, 2235–2238 CrossRef CAS PubMed.
- D. Maspoch, D. RuizMolina, K. Wurst, N. Domingo, M. Cavallini, F. Biscarini, J. Tejada, C. Rovira and J. Veciana, Nat. Mater., 2003, 2, 190–195 CrossRef CAS PubMed.
- D. Feng, T. Lei, M. R. Lukatskaya, J. Park, Z. Huang, M. Lee, L. Shaw, S. Chen, A. A. Yakovenko, A. Kulkarni, J. Xiao, K. Fredrickson, J. B. Tok, X. Zou, Y. Cui and Z. Bao, Nat. Energy, 2018, 3, 30–36 CrossRef CAS.
- (a) R. W. Day, D. K. Bediako, M. Rezaee, L. R. Parent, G. Skorupskii, M. Q. Arguilla, C. H. Hendon, I. Stassen, N. C. Gianneschi, P. Kim and M. Dincă, ACS Cent. Sci., 2019, 5, 1959–1964 CrossRef CAS PubMed; (b) X. Huang, P. Sheng, Z. Tu, F. Zhang, J. Wang, H. Geng, Y. Zou, C.-a. Di, Y. Yi, Y. Sun, W. Xu and D. Zhu, Nat. Commun., 2015, 6, 7408 CrossRef CAS PubMed; (c) J. Park, A. C. Hinckley, Z. Huang, D. Feng, A. A. Yakovenko, M. Lee, S. Chen, X. Zou and Z. Bao, J. Am. Chem. Soc., 2018, 140, 14533–14537 CrossRef CAS PubMed; (d) T. Kambe, R. Sakamoto, K. Hoshiko, K. Takada, M. Miyachi, J.-H. Ryu, S. Sasaki, J. Kim, K. Nakazato, M. Takata and H. Nishihara, J. Am. Chem. Soc., 2013, 135, 2462–2465 CrossRef CAS PubMed; (e) M. Hmadeh, Z. Lu, Z. Liu, F. Gándara, H. Furukawa, S. Wan, V. Augustyn, R. Chang, L. Liao, F. Zhou, E. Perre, V. Ozolins, K. Suenaga, X. Duan, B. Dunn, Y. Yamamto, O. Terasaki and O. M. Yaghi, Chem. Mater., 2012, 24, 3511–3513 CrossRef CAS; (f) R. Dong, P. Han, H. Arora, M. Ballabio, M. Karakus, Z. Zhang, C. Shekhar, P. Adler, P. S. Petkov, A. Erbe, S. C. B. Mannsfeld, C. Felser, T. Heine, M. Bonn, X. Feng and E. Cánovas, Nat. Mater., 2018, 17, 1027–1032 CrossRef CAS PubMed.
- L. Sun, B. Liao, D. Sheberla, D. Kraemer, J. Zhou, E. A. Stach, D. Zakharov, V. Stavila, A. A. Talin, Y. Ge, M. D. Allendorf, G. Chen, F. Léonard and M. Dincă, Joule, 2017, 1, 168–177 CrossRef CAS.
- (a) D. Sheberla, L. Sun, M. A. BloodForsythe, S. l. Er, C. R. Wade, C. K. Brozek, A. n. AspuruGuzik and M. Dincă, J. Am. Chem. Soc., 2014, 136, 8859–8862 CrossRef CAS PubMed; (b) T. Kambe, R. Sakamoto, K. Hoshiko, K. Takada, M. Miyachi, J. Ryu, S. Sasaki, J. Kim, K. Nakazato and M. Takata, J. Am. Chem. Soc., 2013, 135, 2462–2465 CrossRef CAS PubMed.
- K. Wada, K. Sakaushi, S. Sasaki and H. Nishihara, Angew. Chem., Int. Ed., 2018, 57, 8886–8890 CrossRef CAS PubMed.
- F. Li, X. Zhang, X. Liu and M. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 15012–15020 CrossRef CAS PubMed.
- F. Wang, Z. Liu, C. Yang, H. Zhong, G. Nam, P. Zhang, R. Dong, Y. Wu, J. Cho, J. Zhang and X. Feng, Adv. Mater., 2020, 32, 1905361 CrossRef CAS PubMed.
- (a) H. Zhong, K. H. Ly, M. Wang, Y. Krupskaya, X. Han, J. Zhang, J. Zhang, V. Kataev, B. Buchner, I. M. Weidinger, S. Kaskel, P. Liu, M. Chen, R. Dong and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 10677–10682 CrossRef CAS PubMed; (b) X. Li, X. Yang, H. Xue, H. Pang and Q. Xu, EnergyChem, 2020, 2, 100027 CrossRef.
- E. Pomerantseva, F. Bonaccorso, X. Feng, Y. Cui and Y. Gogotsi, Science, 2019, 366, eaan8285 CrossRef CAS PubMed.
- J. Park, M. Lee, D. Feng, Z. Huang, A. C. Hinckley, A. Yakovenko, X. Zou, Y. Cui and Z. Bao, J. Am. Chem. Soc., 2018, 140, 10315–10323 CrossRef CAS PubMed.
- (a) Y. Wang, M. Zhao, J. Ping, B. Chen, X. Cao, Y. Huang, C. Tan, Q. Ma, S. Wu, Y. Yu, Q. Lu, J. Chen, W. Zhao, Y. Ying and H. Zhang, Adv. Mater., 2016, 28, 4149–4155 CrossRef CAS PubMed; (b) M. Zhao, Y. Huang, Y. Peng, Z. Huang, Q. Ma and H. Zhang, Chem. Soc. Rev., 2018, 47, 6267–6295 RSC; (c) Y. Huang, M. Zhao, S. Han, Z. Lai, J. Yang, C. Tan, Q. Ma, Q. Lu, J. Chen, X. Zhang, Z. Zhang, B. Li, B. Chen, Y. Zong and H. Zhang, Adv. Mater., 2017, 29, 1700102 CrossRef PubMed; (d) M. Zhao, Y. Wang, Q. Ma, Y. Huang, X. Zhang, J. Ping, Z. Zhang, Q. Lu, Y. Yu, H. Xu, Y. Zhao and H. Zhang, Adv. Mater., 2015, 27, 7372–7378 CrossRef CAS PubMed.
- (a) W. Liao, J. Zhang, S. Yin, H. Lin, X. Zhang, J. Wang, H. Wang, K. Wu, Z. Wang, Y. Fan, M. Pan and C. Su, Nat. Commun., 2018, 9, 2401 CrossRef PubMed; (b) S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- (a) P.-Z. Li, Y. Maeda and Q. Xu, Chem. Commun., 2011, 47, 8436–8438 RSC; (b) J. Huang, Y. Li, R. Huang, C. He, L. Gong, Q. Hu, L. Wang, Y. Xu, X. Tian and S. Liu, Angew. Chem., Int. Ed., 2018, 130, 4722–4726 CrossRef; (c) V. K.-M. Au, K. Nakayashiki, H. Huang, S. Suginome, H. Sato and T. Aida, J. Am. Chem. Soc., 2019, 141, 53–57 CrossRef CAS PubMed.
- J. Duan, S. Chen and C. Zhao, Nat. Commun., 2017, 8, 15341 CrossRef CAS PubMed.
- Q. Zuo, T. Liu, C. Chen, Y. Ji, X. Gong, Y. Mai and Y. Zhou, Angew. Chem., Int. Ed., 2019, 131, 10304–10309 CrossRef.
- A. Pustovarenko, M. G. Goesten, S. Sachdeva, M. Shan, Z. Amghouz, Y. Belmabkhout, A. Dikhtiarenko, T. Rodenas, D. Keskin, I. K. Voets, B. M. Weckhuysen, M. Eddaoudi, L. C. P. M. de Smet, E. J. R. Sudhölter, F. Kapteijn, B. Seoane and J. Gascon, Adv. Mater., 2018, 30, 1707234 CrossRef PubMed.
- (a) J. Landman, S. Ouhajji, S. Prévost, T. Narayanan, J. Groenewold, A. P. Philipse, W. K. Kegel and A. V. Petukhov, Sci. Adv., 2018, 4, eaat1817 CrossRef PubMed; (b) Y. Wang, M. Zhao, J. Ping, B. Chen, X. Cao, Y. Huang, C. Tan, Q. Ma, S. Wu, Y. Yu, Q. Lu, J. Chen, W. Zhao, Y. Ying and H. Zhang, Adv. Mater., 2016, 28, 4149–4155 CrossRef CAS PubMed.
- Z. Zhang, H. Yoshikawa and K. Awaga, J. Am. Chem. Soc., 2014, 136, 16112–16115 CrossRef CAS PubMed.
- Q. Jiang, P. Xiong, J. Liu, Z. Xie, Q. Wang, X.-Q. Yang, E. Hu, Y. Cao, J. Sun, Y. Xu and L. Chen, Angew. Chem., Int. Ed., 2020, 132, 5311–5315 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Sample characterization, supplementary XRD, XPS and HRTEM data, and battery experimental results. See DOI: 10.1039/d0sc01408g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |