
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA germaaluminocene†
Lena
Albers
 *,
Patrik
Tholen
,
Marc
Schmidtmann
and
Thomas
Müller
*
*,
Patrik
Tholen
,
Marc
Schmidtmann
and
Thomas
Müller
*
Institute of Chemistry, Carl von Ossietzky University Oldenburg, Carl von Ossietzky-Straße 9-11, D-26129 Oldenburg, Federal Republic of Germany. E-mail: lena.albers@uni-oldenburg.de; thomas.mueller@uni-oldenburg.de
First published on 10th February 2020
Abstract
The reactions of dipotassium germacyclopentadienediide with two Group 13 dichlorides, Cp*BCl2 and Cp*AlCl2, yield two structurally different products. In the case of boron a borole complex of germanium(II) is obtained. The aluminium halide gives an unprecedented neutral germaaluminocene. Both compounds were fully characterised by multinuclear NMR spectroscopy supported by DFT computations. The molecular structure of the germaaluminocene was determined by XRD.
Introduction
The aim of utilizing readily available and environmentally benign main group element compounds for activation of unreactive materials and strong bonds instead of transition metal-based complexes became increasingly popular during the last decade.1 We attempted to follow this lead by establishing polarised heteroalkenes I as they mimic the electronic situation in transition metal complexes (Fig. 1). The polarised frontier orbitals of these alkenes I provide the needed amphiphilic reactivity. Examples for small molecule activation by polarised heteroalkenes have already been reported.2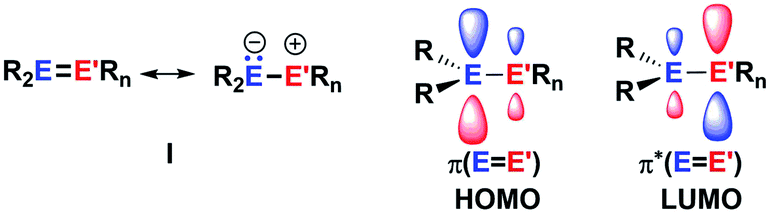 | ||
| Fig. 1 Polarised heteroalkenes I and a sketch of their frontier orbitals. | ||
Our initial idea was to obtain access to polarised heterofulvenes III in which the Group 14 element is partnered with Group 13–15 elements (Scheme 1). For this reason, we studied the reaction of dipotassium germa- or silacyclopentadienediides II3 with a series of main group element dihalides. In several cases, we isolated rearranged products with rather unusual structures in high yields.4 For example, the reaction of aminoboron dichlorides with germacyclopentadienediide K2[1] yielded unprecedented borole complexes of germanium(II) 2.4a The recent publication by Ruth and Sindlinger who reported the synthesis of a borole-based aluminocene 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 (Fig. 2) prompted us to communicate our own results on the reactivity of pentamethylcyclopentadiene-substituted boron and aluminium dichlorides versus dipotassium germacyclopentadienediide K2[1] (Scheme 2).
5 (Fig. 2) prompted us to communicate our own results on the reactivity of pentamethylcyclopentadiene-substituted boron and aluminium dichlorides versus dipotassium germacyclopentadienediide K2[1] (Scheme 2).
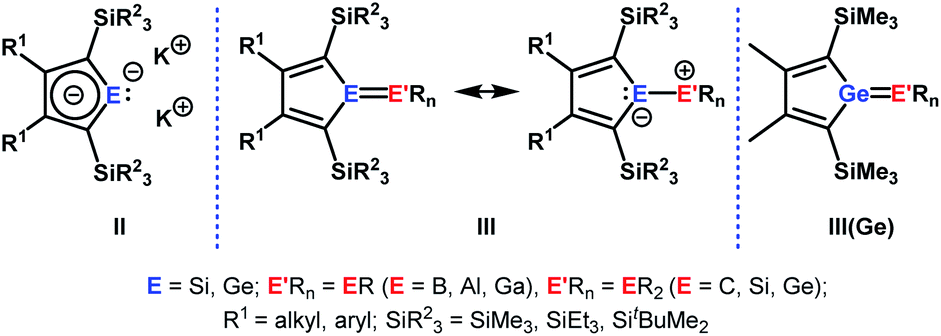 | ||
| Scheme 1 Dipotassium sila- or germacyclopentadienediides II, polarised heterofulvenes III and III(Ge). | ||
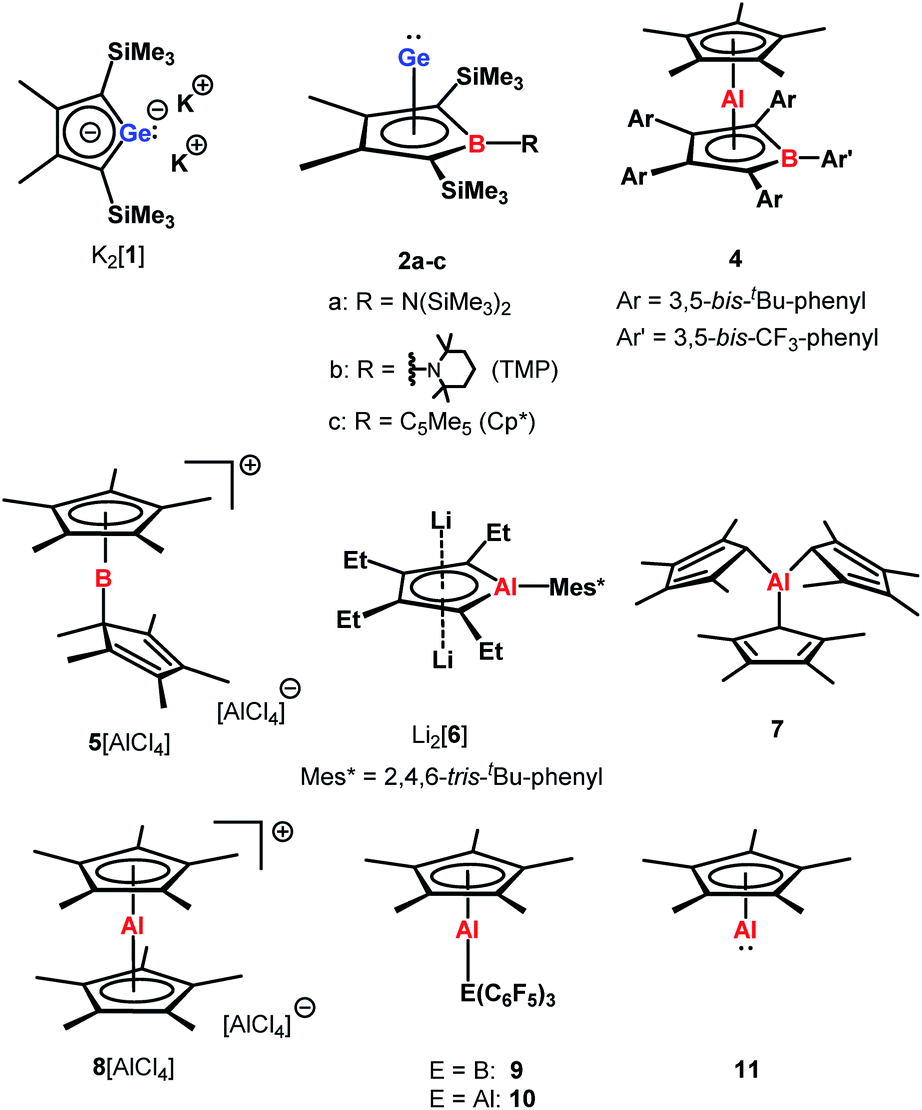 | ||
| Fig. 2 Main group compounds relevant for the discussion. | ||
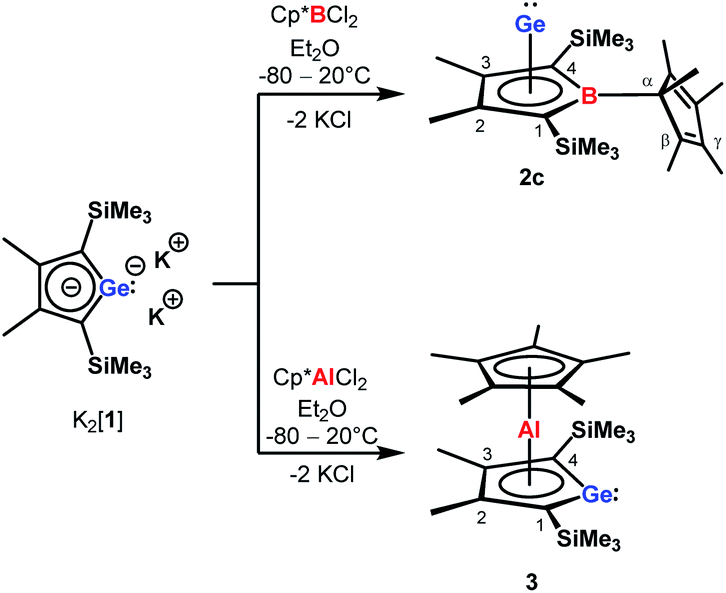 | ||
| Scheme 2 Synthesis of borole germanium complex 2c and germaaluminocene 3 from dipotassium germacyclopentadienediide K2[1] by salt metathesis. | ||
Results and discussion
The reaction of K2[1]3 with Cp*BCl26 gave the expected borole complex 2c. The NMR spectra suggested quantitative conversion and after work-up complex 2c was isolated as a brown oil in 35% yield (Scheme 2). NMR spectroscopy evidenced the presence of the expected borole ring with a η1-bound cyclopentadienyl substituent. Interestingly, the NMR data indicated frozen rotation around the B-Cα single bond, giving rise to ten 13C NMR signals for the cyclopentadienyl substituent (Table S3, ESI†). In addition, all four carbon atoms of the borole ring are magnetically non-equivalent. Even at T = 70 °C the NMR signals show no detectable line broadening. Diagnostic for the structure of the product 2c are the high-field shifted 13C NMR resonances of carbon atoms C1/C4 compared to germole dianion 12− (δ13C(C1/C4) = 156.23 (1); 99.8, 99.3 (2c)). In addition, the 13C NMR signals of C1/C4 in borole 2c are markedly broadened (full width at half height, ω½ = 60 Hz), due to the quadrupole moment of the neighbouring boron atom. The position of the 11B NMR resonance (δ11B = 37, ω½ = 283 Hz) indicates tri-coordination for the boron atom. The boron resonance is significantly deshielded compared to that of borocenium tetrachloroaluminate 5[AlCl4] (δ11B = −42)7 which discards the possibility of η5-coordination. These NMR chemical shifts are all close to those reported previously for amino-substituted borole complexes 2a and 2b4a (Table S3†). In addition, NMR chemical shift calculations using a DFT-optimised molecular structure of complex 2c provide 13C and 11B chemical shifts that are close to the experiment (i.e. δ11Bcalc = 30) and strongly support our structural proposal for 2c (Table S3†).
Applying the same conditions to the reaction of K2[1] with Cp*AlCl2,8 we noticed complete conversion of both reactants to a single product according to NMR spectroscopy. Due to its high solubility, we isolated compound 3 by crystallization in only moderate yields of 28% as yellow solid. The NMR data indicated a molecular structure very different from that of the germanium borole complex 2c. Compound 3 is characterised by a relative sharp, high-field shifted 27Al NMR signal at δ27Al = −77 (ω½ = 703 Hz). The number of signals in the 1H and 13C NMR spectra suggests a highly symmetric structure for compound 3 and the two 13C NMR resonances of the Cp* ligand are in the typical range of η5-coordination (δ13C = 114.9, 10.8) (Table S3†). Even at temperatures as low as T = −90 °C, all Cp*-carbon atoms are magnetically equivalent, which supports the presence of a η5-bound Cp*-substituent (see ESI†). The 13C NMR chemical shifts of the heterole ring are decisively different from that of the borole ring in 2c. In particular, the carbon atoms C1/C4 are significantly deshielded compared to the borole complex 2c or compared to the germole dianion 12− (δ13C(C1/4) = 167.0 (3); 99.8, 99.3 (2c) and 156.2 (12−)). These 13C and in particular 27Al NMR data are also different from those reported for the dilithium aluminacyclopentadiendiide Li2[6], (δ27Al = 198 (ω½ = 7000 Hz), Table S3†)9 or for the tris-η1-cyclopentadienyl-substituted alane 7 (δ27Al = 64).10 They are however close to that of η5-coordinated cyclopentadienyl aluminium compounds 8+–11 (δ27Al = −59 to −150)11 which suggests a η5-coordination of the cyclopentadienyl substituent to the aluminium atom for complex 3. The NMR data is consistent with a sandwich structure of compound 3 as depicted in Scheme 2. Further support comes from NMR chemical shift calculations for a DFT-optimised sandwich structure 3 which predict aluminium and carbon NMR chemical shifts that are very close to the experimental values (δ27Alcalc = −71, Table S3†).
Yellow crystals of 3, suitable for X-ray diffraction (XRD) analysis were obtained from hexanes. In the solid state compound 3 adopts a sandwich structure with a η5-coordinated Cp* ligand and with close contacts to all five atoms of the germole ring (Fig. 3). All germole-C–Al distances (213–228 pm) and the Ge/Al separation (248.8 pm) are significantly shorter than the respective sum of the van der Waals radii ΣvdW (ΣvdW(C/Al) = 354 pm; ΣvdW(Ge/Al) = 395 pm).12 The Ge–Al distance matches even that of Ge–Al single bonds in germyl-alanes13 and -alanates14 (244.9–254.5 pm, theoretically predicted: 247 pm15). The distance between the centre of the Cp* ring and the aluminium atom (186.6 pm) is only slightly larger than that found for the Cp*Al(I) complex 911c but significantly smaller than found for monomeric Cp*Al(I) 1116 (see Table S2†).
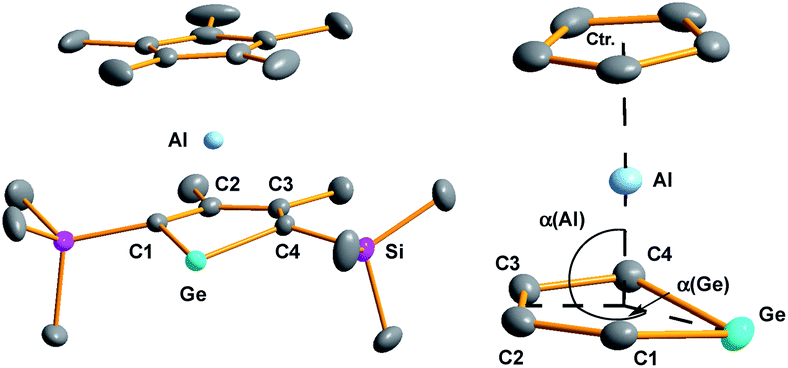 | ||
| Fig. 3 Left: Molecular structure of 3 in the crystal (Hydrogen atoms are omitted; thermal ellipsoids at 50% probability). Right: Coordination environment of the aluminium atom. Selected atom distances [pm] and angles [°]: Ge–C1 199.91(10), Ge1–C4 200.02(10), Al–C1 212.87(10), Al–C2 227.18(10), Al–C3 227.58(10), Al–C4 214.16(11), Ge–Al 248.78(4), C1–C2 144.70(13), C2–C3 142.12(14), C3–C4 144.56(13), Al–Ctr. 186.64(3), Al–C(Cp*) 217–228, α(Ge) 165.638(9); α(Al) 91.628(10). | ||
The planes defined by the Cp* ring and by the four carbon atoms of the germole ligand are aligned almost parallel (inter planar angle 3.3°). The C–C bond lengths in the germacycle indicate delocalization (142.1–144.7 pm). Compared to the germole dianion 12− in the triple ion pair K2[1] (139.5–142.0 pm),3 or in the germole dianion hafnium complexes 12 (134.7–142.8 pm)17 or 13 (140.7–144.5 pm) (Fig. 4),4b all bonds in the germole ring of 3 are slightly elongated, indicating electron transfer from the germole ring to the aluminium atom (Fig. 3).
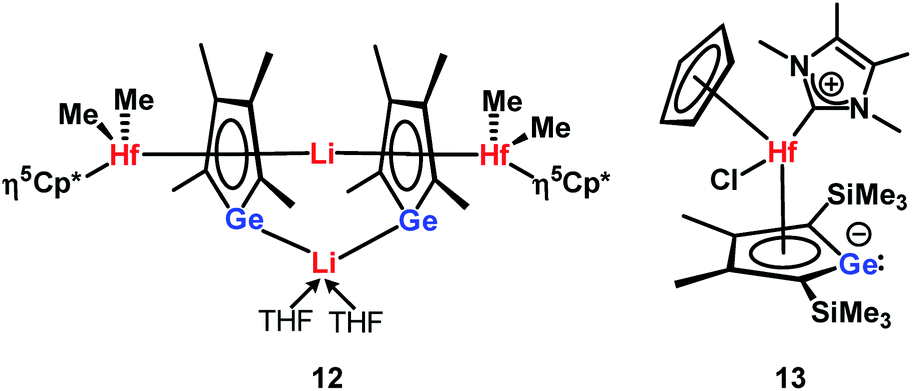 | ||
| Fig. 4 Selected germole dianion metal complexes. | ||
Quantum mechanical calculations at the M06-2X/6-311+G(d,p) level of theory were performed to investigate the bonding situation in neutral germaaluminocenes 3 and 3M (3M is a close model to 3, in which all substituents at the germole ring and at the cyclopentadiene ring have been replaced by hydrogen atoms).18 The analysis of the frontier molecular orbitals suggests a substantial germylene character of the sandwich complex 3M. This is shown by surface diagrams of the frontier orbitals which indicate a significant contribution of an in-plane lone pair at germanium to HOMO−1 (Fig. 5). In addition, the 4p(Ge) atomic orbital dominates the LUMO and the LUMO+1. The HOMO is essentially a combination of an π-orbital of the germole cycle that is polarised towards the germanium atom and atomic orbitals of the aluminium. The results of a natural bond orbital (NBO) analysis for complex 3 indicate significant charge transfer from the germole ring to the aluminium atom as shown by the calculated NBO group charges (Fig. 6). The covalent character of the aluminium/germole interaction is supported by the calculated Wiberg bond indices (WBI).19 These exceed those calculated for the ionic interaction between the potassium ions and the germole ring for K2[1] (Fig. 6). In particular, the WBIs for the C1/C4–Al linkages are larger than those of C(Cp*)–Al bonds in 8+ and close to those predicted for the Al–C bond in Me–Al(I) (Fig. 6).
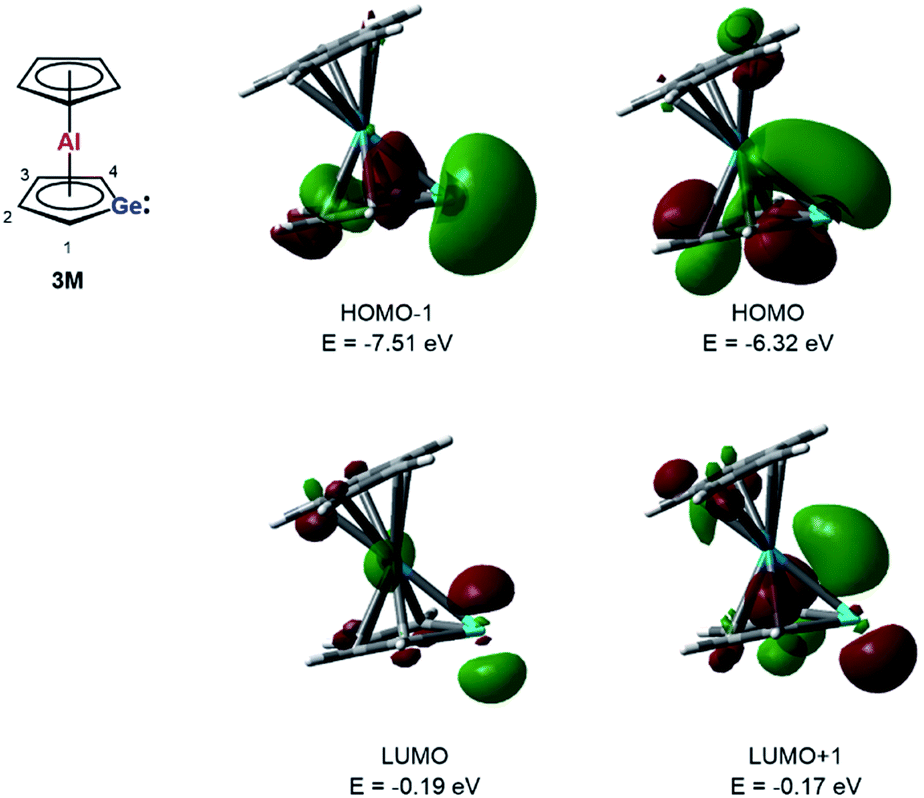 | ||
| Fig. 5 Calculated surface diagrams of frontier molecular orbitals of germaaluminocene 3M (M06-2X/6-311+G(d,p), isodensity value 0.04). | ||
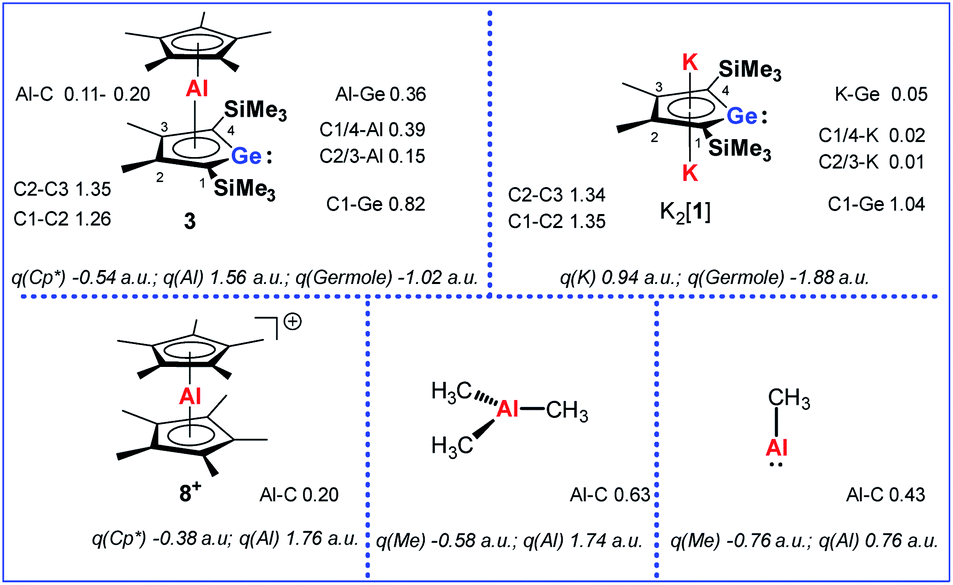 | ||
| Fig. 6 Calculated WBIs of pertinent bonds and NBO group charges q of complexes 3, K2[1], 8+ and aluminium methyl compounds (M06-2X/6-311+G(d,p)). | ||
This covalent bonding is visualised by a natural localised molecular orbital (NLMO, Fig. 7a), which indicates the electron delocalisation between atomic orbitals of the aluminium and π-type orbitals of the germole ligand. The complementary analysis of the calculated electron density of germaaluminocene 3M in the framework of the quantum theory of atoms in molecules (QTAIM) finally reveals a consistent picture with bond paths between the aluminium atom and the carbon atoms C1/4 and a valence shell charge concentration (VSCC) between the aluminium atom and the germanium atom (Fig. 7b).
 | ||
| Fig. 7 (a) Surface diagram of the NLMO of 3M showing the interaction between the germole ring and the CpAl fragment (surface value 0.04, hydrogen atoms omitted). (b) Molecular graph of 3M projected on a contour plot of the Laplacian of the electron density in the molecular mirror plane (M06-2X/6-311+G(d,p)). | ||
On the basis of our theoretical results we suggest a delocalised bonding scheme for model germaaluminocene compound 3M and likewise for the experimentally investigated compound 3. Scheme 3 shows the three extreme Lewis forms of the delocalisation of the bonding situation in the germaaluminocene.
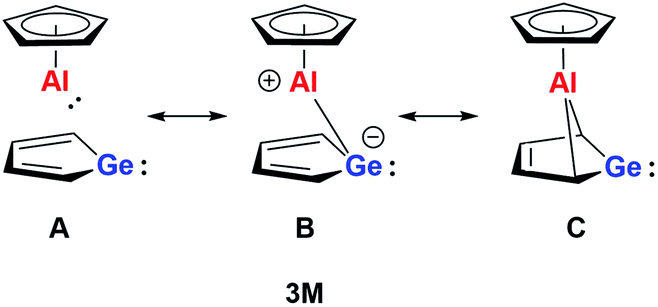 | ||
| Scheme 3 Resonance representations of neutral germaaluminocene 3M. | ||
In the course of our computational investigation of possible reaction channels for the formation of germanium borole or alumole complexes (2c and 14) or germaborocene or -aluminocene sandwich compounds (15 and 3), we found that structures with an Y-shaped arrangement20 around the two heteroatoms, as it is shown by boraalkene 1621 or borasilene 17,22 are only high lying transition states (see ESI† for a more extensive description of the corresponding potential energy surfaces). This is surprising, in view of the recent isolation of an almost linear boragermene 182c by Rao and Kinjo, who used an amino substituent at the boron atom (Fig. 9).
As reported previously we synthesised borole complexes of germanium(II) 2a and 2b from the reaction of a germacyclopentadienediide 12− with amino boron dichlorides.4a Now, we found the same structural motif when using the Cp* substituent at the boron atom. This suggests that the germacyclopentadienyl fragment, used in the chemistry presented here, has a significant effect on the reaction outcome.
In agreement with the exclusive formation of the borole germanium(II) complex 2c, we found that 2c is significant more stable than the isomeric germaborocene 15 (ΔE = 63 kJ mol−1, Fig. 8). The situation is different for the isomeric aluminium compounds 3 and 14. In qualitative agreement with the isolation of the germaaluminocene 3 (Fig. 8), it is more stable than the alumole germanium(II) complex 14 by ΔE = 24 kJ mol−1. We notice however that their energy difference is significantly smaller. Moreover, the barrier for the interconversion 3 → 14 (78 kJ mol−1) is relatively small (Fig. 8). These computational results for the aluminium compounds indicate the possibility to influence the product formation significantly by substituent effects. Decisive for the higher stability of the germametallocene 3vs. the metallole Ge(II) complex 14 is the larger size of the aluminium atom compared to the boron atom in the isomer pair 2c/15. Its inclusion into the delocalised metallole cycle is disfavoured due to the misfit of 3p(Al) and 2p(C) orbitals but its larger size facilitates the η5-coordination to the Cp* ligand.7,9,23
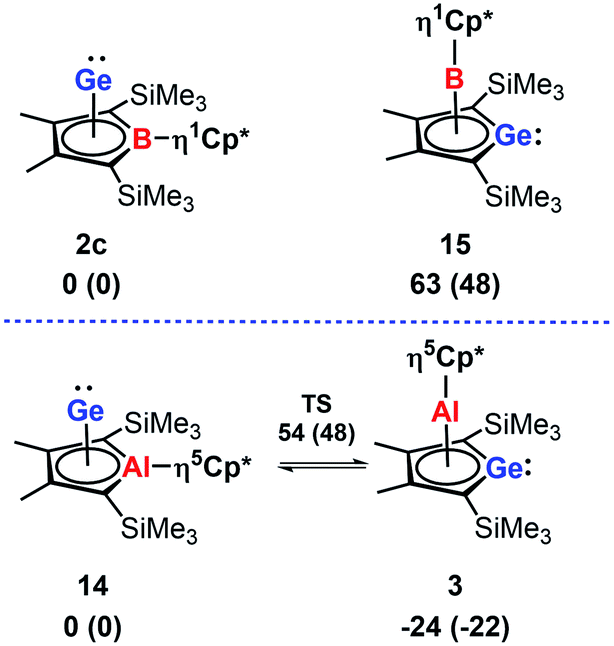 | ||
| Fig. 8 Relative energies ΔE and relative free Gibbs enthalpies at T = 298 K ΔG298 (in brackets) of isomeric borole 2c and alumole 14 complexes and germole complexes 15 and 3. | ||
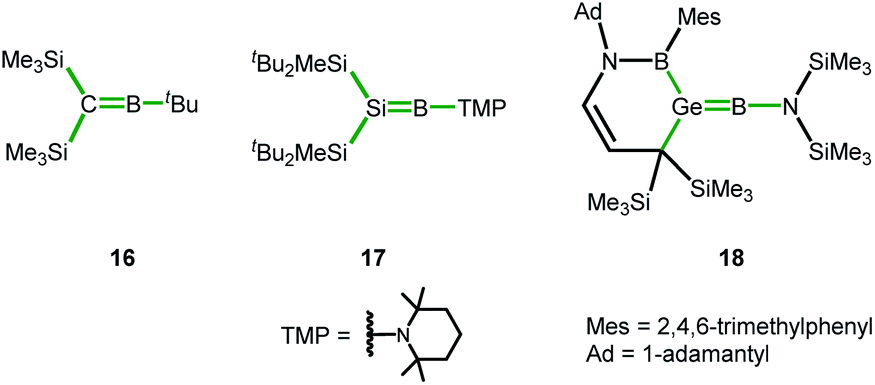 | ||
| Fig. 9 Y-shaped Group 14 boraalkenes relevant for the discussion (Y-shape arrangement indicated by green bonds). | ||
Conclusions
In agreement with previous results, the reaction of K2[1] Cp*BCl2 gives a borole complex of Ge(II) 2c. The respective reaction of K2[1] with Cp*AlCl2 provides access to the neutral germaaluminocene 3, which represents a new class of aluminium π-complexes. Similar to aluminocenium cation 8+11a,24 and boraaluminocene 4,5 germaaluminocene 3 shows a η5,η5-coordination of both five-membered rings. In contrast to these previous examples, germaaluminocene 3 exhibits a free electron pair and a low-lying acceptor orbital at the germanium atom, which suggests additional germylene-like reactivity.
The small energy difference between the germole 3 and isomeric alumole 14 structures and the small energetic barriers for their interconversion imply that also the synthesis of alumole complexes of Ge(II) is in reach, when substituent effects are employed advantageously.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG-Mu1440/13-1, INST 184/108-1 FUGG).Notes and references
- (a) P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed; (b) G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439 CrossRef CAS PubMed.
- (a) T. Kosai and T. Iwamoto, Chem.–Eur. J., 2018, 24, 7774–7780 CrossRef CAS PubMed; (b) F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 13159–13163 CrossRef CAS PubMed; (c) B. Rao and R. Kinjo, Angew. Chem., Int. Ed., 2019, 59, 3147–3150 CrossRef PubMed.
- Z. Dong, C. R. W. Reinhold, M. Schmidtmann and T. Müller, Organometallics, 2018, 37, 4736–4743 CrossRef CAS.
- (a) P. Tholen, Z. Dong, M. Schmidtmann, L. Albers and T. Müller, Angew. Chem., Int. Ed., 2018, 57, 13319–13324 CrossRef CAS PubMed; (b) Z. Dong, K. Bedbur, M. Schmidtmann and T. Müller, J. Am. Chem. Soc., 2018, 140, 3052–3060 CrossRef CAS PubMed; (c) Z. Dong, C. R. W. Reinhold, M. Schmidtmann and T. Müller, J. Am. Chem. Soc., 2017, 139, 7117–7123 CrossRef CAS PubMed; (d) Z. Dong, C. R. W. Reinhold, M. Schmidtmann and T. Müller, Angew. Chem., Int. Ed., 2016, 55, 15899–15904 CrossRef CAS PubMed.
- C. P. Sindlinger and P. N. Ruth, Angew. Chem., Int. Ed., 2019, 58, 15051–15056 CrossRef CAS PubMed.
- P. Jutzi, B. Krato, M. Hursthouse and A. Howes, Chem. Ber., 1987, 120, 565 CrossRef CAS.
- A. Voigt, S. Filipponi, C. L. B. Macdonald, J. D. Gorden and A. H. Cowley, Chem. Commun., 2000, 911–912 RSC.
- H.-J. Koch, S. Schulz, H. W. Roesky, M. Noltemeyer, H.-G. Schmidt, A. Heine, R. Herbst-Irmer, D. Stalke and G. M. Sheldrick, Chem. Ber., 1992, 125, 1107–1109 CrossRef CAS.
- T. Agou, T. Wasano, P. Jin, S. Nagase and N. Tokitoh, Angew. Chem., Int. Ed., 2013, 52, 10031–10034 CrossRef CAS PubMed.
- J. D. Fisher, P. H. M. Budzelaar, P. J. Shapiro, R. J. Staples, G. P. A. Yap and A. L. Rheingold, Organometallics, 1997, 16, 871–879 CrossRef CAS.
- (a) R. W. Schurko, I. Hung, C. L. B. Macdonald and A. H. Cowley, J. Am. Chem. Soc., 2002, 124, 13204–13214 CrossRef CAS PubMed; (b) J. D. Gorden, C. L. B. Macdonald and A. H. Cowley, Chem. Commun., 2001, 75–76 RSC; (c) J. D. Gorden, A. Voigt, C. L. B. Macdonald, J. S. Silverman and A. H. Cowley, J. Am. Chem. Soc., 2000, 122, 950–951 CrossRef CAS; (d) C. Dohmeier, H. Schnöckel, U. Schneider, R. Ahlrichs and C. Robl, Angew. Chem., Int. Ed., 1993, 32, 1655–1657 CrossRef; (e) J. Gauss, U. Schneider, R. Ahlrichs, C. Dohmeier and H. Schnöckel, J. Am. Chem. Soc., 1993, 115, 2402–2408 CrossRef CAS.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- J. D. Erickson, J. C. Fettinger and P. P. Power, Inorg. Chem., 2015, 54, 1940–1948 CrossRef CAS PubMed.
- (a) T. Habereder, H. Nöth and M. Suter, Z. Naturforsch., B: Chem. Sci., 2009, 64, 1387 CAS; (b) T. Habereder, K. Knabel and H. Nöth, Eur. J. Inorg. Chem., 2001, 2001, 1127–1129 CrossRef.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- A. Haaland, K.-G. Martinsen, S. A. Shlykov, H. V. Volden, C. Dohmeier and H. Schnöckel, Organometallics, 1995, 14, 3116–3119 CrossRef CAS.
- J. M. Dysard and T. D. Tilley, J. Am. Chem. Soc., 2000, 122, 3097–3105 CrossRef CAS.
- (a) The Programme Gaussian 16 Revision A.03 Gaussian Inc., Wallingford CT Search PubMed, 2016 was used; (b) For details see the ESI.†.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- The term “Y-shaped arrangement” indicates the spatial arrangement of the three substituents around the double bond, for example, of vinyl cations or boragermenes..
- R. Boese, P. Paetzold, A. Tapper and R. Ziembinski, Chem. Ber., 1989, 122, 1057–1060 CrossRef CAS.
- N. Nakata and A. Sekiguchi, J. Am. Chem. Soc., 2006, 128, 422–423 CrossRef CAS PubMed.
- (a) O. Kwon and M. L. McKee, J. Phys. Chem. A, 2001, 105, 10133–10138 CrossRef CAS; (b) J. O. C. Jimenez-Halla, E. Matito, M. Solà, H. Braunschweig, C. Hörl, I. Krummenacher and J. Wahler, Dalton Trans., 2015, 44, 6740–6747 RSC.
- C. L. B. Macdonald, J. D. Gorden, A. Voigt, S. Filipponi and A. H. Cowley, Dalton Trans., 2008, 1161–1176 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1969078. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc00401d |
| This journal is © The Royal Society of Chemistry 2020 |