
A polymorphic fluorescent material with strong solid state emission and multi-stimuli-responsive properties†
Ji-Yu
Zhu
a,
Chun-Xiang
Li
b,
Peng-Zhong
Chen
a,
Zhiwei
Ma
c,
Bo
Zou
 c,
Li-Ya
Niu
a,
Ganglong
Cui
*b and
Qing-Zheng
Yang
*a
c,
Li-Ya
Niu
a,
Ganglong
Cui
*b and
Qing-Zheng
Yang
*a
aKey Laboratory of Radiopharmaceuticals, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: qzyang@bnu.edu.cn
bKey Laboratory of Theoretical and Computational Photochemistry, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: ganglong.cui@bnu.edu.cn
cState Key Laboratory of Superhard Materials, College of Physics, Jilin University, Changchun 130012, China
First published on 30th September 2019
Abstract
A bright difluoroboron β-diketonate derivative 1 showing four emission colors (green, yellow, orange and red) with high quantum yields (41–74%) in four polymorphs and one amorphous state is reported. Green-emissive crystals (1-G and 1-G′) exhibit dimeric aggregation structures due to the strong molecular π–π interaction but exhibit hypsochromic emission compared to yellow-emissive crystals (1-Y) with monomeric aggregation because of lacking such π–π interactions. These novel emission phenomena are rationalized by theoretical calculations. High fluorescence sensitivity of compound 1 to its molecular packing modes results in excellent responsive behavior to multiple external stimuli thereby showing reversible change of emission colors under mechanical grinding, heating, solvent fuming and hydrostatic pressure.
Introduction
Organic fluorescent materials have attracted increasing attention due to their potential applications in optoelectronics, bioimaging and fluorescent sensors.1–10 It is of great importance to tune the emission color of the fluorescent materials to meet the requirements in various fields such as OLEDs, imaging and chemosensing.1–10 Chemists usually utilize covalent modification of chromophores to prepare fluorescent materials with various emission colors.11–15 Sophisticated synthesis and separation are mostly involved for modifying structures of chromophores to tune the photophysical properties of the formed materials.Self-assembly of organic chromophores through non-covalent interactions offers an alternative and promising strategy for preparing organic fluorescent materials with multiple emission colors.16–44 Impressive fluorescent assemblies such as organic–inorganic hybrids, supramolecular polymers, organic gels, and organic cocrystals prepared by a self-assembly strategy have been reported.16–37 Organic fluorophores in the aggregation state may exhibit excited properties different from that in solution as a result of interchromophore electronic interaction.38–40 The inter-chromophore energy and electron-transfers can be regulated in these materials through modulating interchromophore interaction to achieve different emission colors.41–44 On the contrary, it is a challenge to achieve different emission colors from assemblies only containing a single chromophore. Several chromophores have been reported to exhibit intense emission upon aggregation, but most of them merely show two or three emission colors.45–56 In addition, one of their aggregation states shows a drastic reduction of fluorescence quantum yield in many reported works.55–60 For example, the fluorescence quantum yields (QYs) of thiazolothiazole derivatives decrease from 47% to 7% in different states.55 A difluoroboron fluorophore reported by Sket et al. also showed more than five-fold decrement in the fluorescence efficiency upon grinding its pristine crystals.56 To our best knowledge, no example of organic fluorescent materials assembled from a single chromophore manifesting more than four aggregation states with high fluorescence quantum yields in all phases has been reported to date.
Herein we report a bright tetraphenylene (TPE)-dioxaborine derivative 1 with four polymorphs and one amorphous solid state (1-G, 1-G′, 1-Y, 1-O and 1-R) exhibiting four emission colors from green to red (Fig. 1). All five emissive aggregation states exhibit high fluorescence quantum yields over 40%. The origin of this emission behavior has been experimentally and theoretically elucidated. Compound 1 shows reversible changes in emission colors in response to multiple external stimuli of grinding, heating, solvent fuming, and hydrostatic pressure.
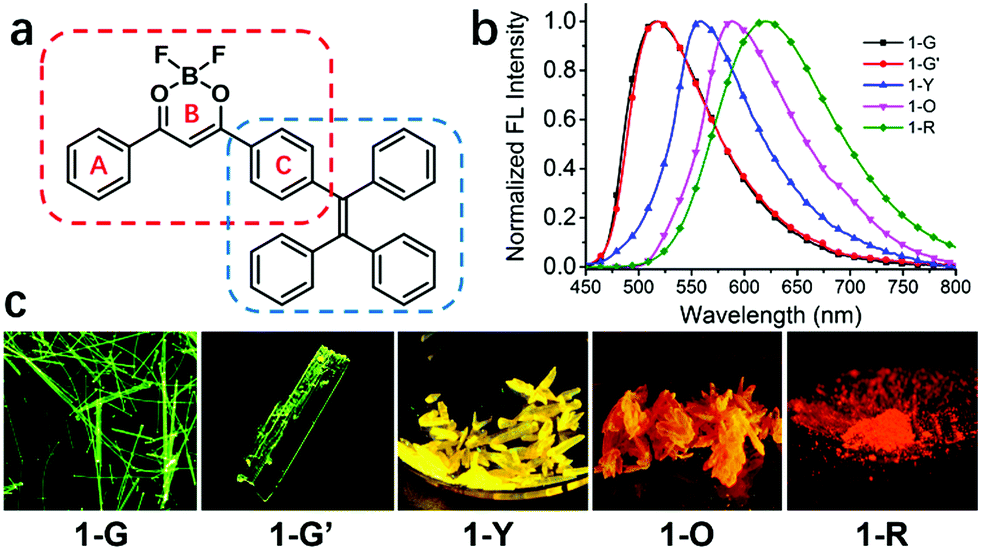 | ||
| Fig. 1 (a) Chemical structure of compound 1; (b) normalized emission spectra of 1-G, 1-G′, 1-Y, 1-O and 1-R. λex = 420 nm. (c) The fluorescent images of compound 1 in its four polymorphs and one amorphous solid state (microscopy images of 1-G and 1-G′; the photographs of 1-Y, 1-O and 1-R taken under 365 nm UV lamp). | ||
Result and discussion
Compound 1 was synthesized in 55% yield by Claisen condensation of TPE ketone with benzoate, followed by the complexation with BF3 (see ESI†).61–64 Its photophysical properties in solution and property of aggregation induced emission are shown in the ESI† (Fig. S2 and S3).Single crystal structures and analysis of 1
Four types of single crystals of 1 were obtained by slow solvent evaporation of its solution in dichloromethane/hexane (see Fig. 1 and Fig. S1, ESI†). Green emissive crystals (1-G and 1-G′) exhibit strong fluorescence centered at 520 nm with fluorescence quantum yields of 49% and 46%. The fluorescence peaks of yellow and orange emissive crystals are at 557 and 586 nm (QY: 46% and 74%). Its amorphous solid state (1-R) obtained by fast precipitation from THF/H2O shows red emission centered at 618 nm (QY: 41%). Detailed data for photophysical properties are summarized in Table 1 and Table S1 (ESI†).| 1 | 1-G | 1-G′ | 1-Y | 1-O | 1-R |
|---|---|---|---|---|---|
| λ em (exp.) | 520 | 520 | 557 | 586 | 618 |
| ϕ (%) | 49 | 46 | 74 | 47 | 41 |
| λ em (cal.) | 533 | 536 | 555 | 581 | — |
Single-crystal X-ray diffraction analysis reveals that compound 1 adopts quite different molecular packing modes in four crystals (Fig. 2, Fig. S4–S7 and Tables S3–S6, ESI†). The twisting angles among aromatic moieties A, B, and C play important roles in tuning assembly modes (Fig. 1 and Table S2, ESI†). In 1-G, the central dioxaborine ring B is almost coplanar with both phenyl rings A and C with the torsion angles of 4.21° and 7.21° while the phenyl rings of the TPE unit are highly twisted. These features of the structure benefit the formation of dimer structures through head-to-tail antiparallel molecular packing with multiple intermolecular interactions such as π–π stacking, dipole–dipole interactions, and C–H⋯π and C–H⋯F hydrogen bonds between adjacent molecules61,63 (Fig. S8, ESI†). 1-G molecules are layered along the b-axis direction. In each layer molecules are assembled in a zig-zag way along the a-axis direction (Fig. S9, ESI†). 1-G′ forms a dimeric structure with similar conformation and assembly modes as 1-G. The torsion angle between the central dioxaborine ring B and the phenyl unit C (6.18°) is slightly bigger than that in 1-G (4.21°). On the other hand, the phenyl ring C and the other three phenyl rings of the TPE unit in 1-G′ adopt twisted conformations with the large dihedral angles of 86.45°, 69.05° and 58.59°, respectively, while the corresponding angles in 1-G are 85.37°, 80.71° and 55.64°, respectively (Table S2, ESI†). With respect to 1-Y, the central dioxaborine ring B is twisted with both phenyl rings A and C in torsion angles of 30.31° and 29.24°, respectively. It presents lamellar arrangements with an interlayer distance of 4.05 Å but without dimeric structures because of such highly twisted molecular conformation, which eventually leads to an unobvious intermolecular π–π interaction. In 1-O the central dioxaborine ring B is also coplanar with the phenyl ring C in a torsion angle of 7.98° but twisted with the phenyl ring A in a torsion angle of 26.47°, which to a certain extent favors the formation of dimeric structures. However, unlike the dimers in 1-G that exhibit centrosymmetric properties, those in 1-O display rotationally symmetric features. Strong intermolecular interactions such as CH⋯F and CH⋯π are observed among dimers in 1-O. It is worth stressing again that 1-G and 1-G′ with significant π–π stacking interaction show hypsochromic emission compared with 1-Y having no such interaction, which is not in accordance with the common rule.65–70
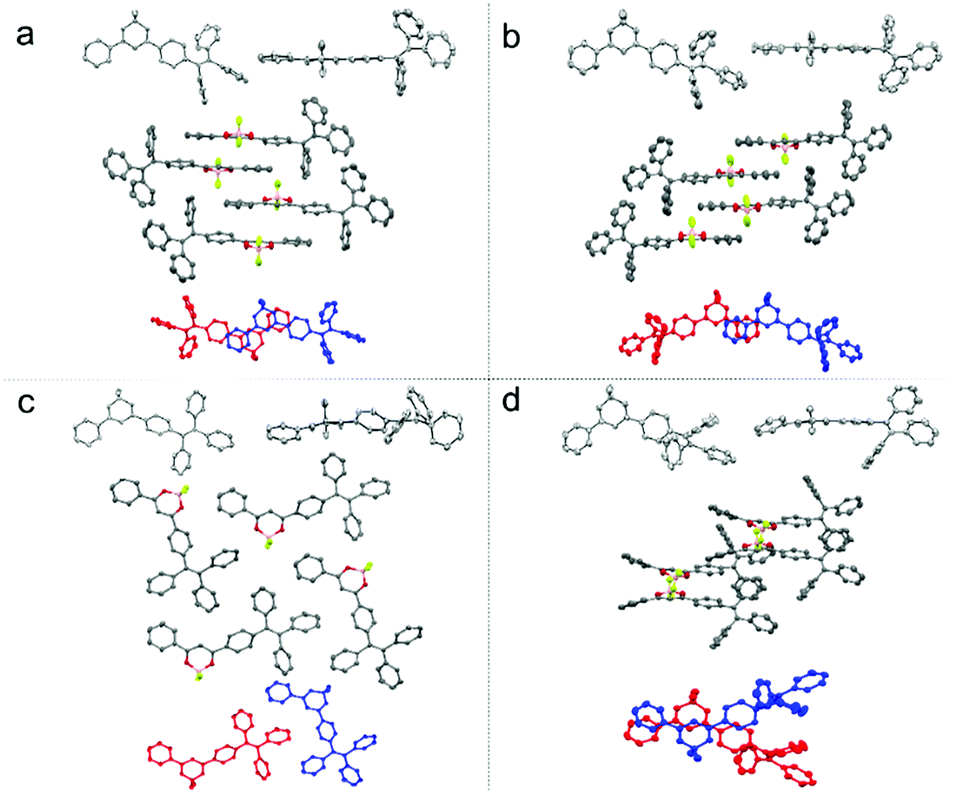 | ||
| Fig. 2 The molecular structures and stackings in the 1-G (a), 1-G′ (b), 1-Y (c), 1-O (d) crystals: the molecular conformation of compound 1 in front view and side view (top, in every group); molecular stacking structures along the long molecule axis and the intermolecular interactions between adjacent molecules (middle, in every group) and the molecular overlap between adjacent molecules (below, in every group). | ||
Time-dependent density functional theory calculation
To rationalize experimentally observed emission phenomena, we employed the time-dependent density functional theory (TD-DFT) method in combination with the quantum mechanics/molecular mechanics (QM/MM) approach to explore excited-state properties of 1-G, 1-G′, 1-Y and 1-O. As mentioned above 1-G, 1-G′, and 1-O contain side-by-side dimeric structures, which are not found in 1-Y. The QM region includes two monomers for the former three crystals but only one monomer for the latter; the MM region includes all the remaining monomers in clusters. Table 1 and Table S7 (ESI†) collect TD-DFT calculated vertical emission energies for 1-G, 1-G′, 1-Y and 1-O. The calculated emission wavelengths of 1-G and 1-G′ are close to each other, 533 nm versus 536 nm, and are hypsochromic in relation to 555 nm of 1-Y. The calculated emission wavelength of 1-O, 581 nm, is bathochromic. Obviously, all calculated results are in excellent agreement with experiments.Luminescence in all four studied crystals comes from the singlet excited state (S1), which corresponds to moving an electron from the LUMO to HOMO (Fig. 3 and Table S7, ESI†). In 1-G and 1-O, the HOMO and LUMO are primarily located on both the TPE and phenyl dioxaborine fragments belonging to different monomers; while, in 1-Y, both are located within the same monomer due to the nonexistence of dimer structures (Fig. 3). Thus, the S1 state is an intermolecular excited-state charge transfer electronic state in the former two but an intramolecular state in the latter one. These different excited-state electronic properties are caused by distinct molecular packing modes.
 | ||
| Fig. 3 Frontier molecular orbitals (HOMO, HOMO−1, LUMO, and LUMO+1) of the dimer structures of (left) 1-G and (right) 1-O in their S1 minima. Also shown is the HOMO and LUMO of the monomer of (middle) 1-Y in its S1 minimum. See text for discussion. Those for 1-G′ are very similar to those of 1-G (Fig. S12, ESI†). | ||
One interesting finding is that the emission colors of 1-G and 1-G′ are hypsochromic but that of 1-O is bathochromic in relation to that of 1-Y. This result is unexpected because the strong intermolecular interaction can induce the bathochromic shift in emission compared with that of the monomeric state. To figure out why 1-G, 1-G′, and 1-O have distinct hypsochromic and bathochromic emissions we have analyzed the Frontier molecular orbitals of the dimers in 1-G, 1-G′, and 1-O (Fig. 3). The fluorescence mainly originates from the LUMO-to-HOMO electronic de-excitation transition and one thus focuses on the variation of energy levels of the HOMO and LUMO. In 1-G, both the HOMO and LUMO are stabilized compared with 1-Y due to the existence of π–π interaction between two neighboring monomers. Nonetheless, the HOMO is stabilized more significantly than the LUMO (0.046 vs. 0.146 eV). So, the HOMO–LUMO energy gap is increased by 0.1 eV leading to a hypsochromic emission in 1-G. In 1-O both the HOMO and LUMO are also stabilized; however, the LUMO is stabilized more remarkably than the HOMO, 0.092 vs. 0.062 eV. Thereby a bathochromic emission color is observed in 1-O.
Solid state emission of 1 upon external stimuli
Compound 1 in the solid state exhibits excellent multi-stimuli-responsive behavior on account of its emission being highly sensitive to the molecular packing. After grinding, four types of crystalline solids produce 1-R with red emission centered at 618 nm (Fig. 4c, Fig. S13–S15, ESI†). Taking 1-G as an example its emission peak is shifted from 520 to 618 nm after grinding (Fig. 4a). Powder X-ray diffraction (PXRD) of 1-G crystalline powder manifests intense and sharp peaks while broad and featureless reflections are observed for ground samples. This result indicates that grinding causes the change of the molecular packing mode of 1-G from the crystalline to amorphous state (Fig. S16, ESI†).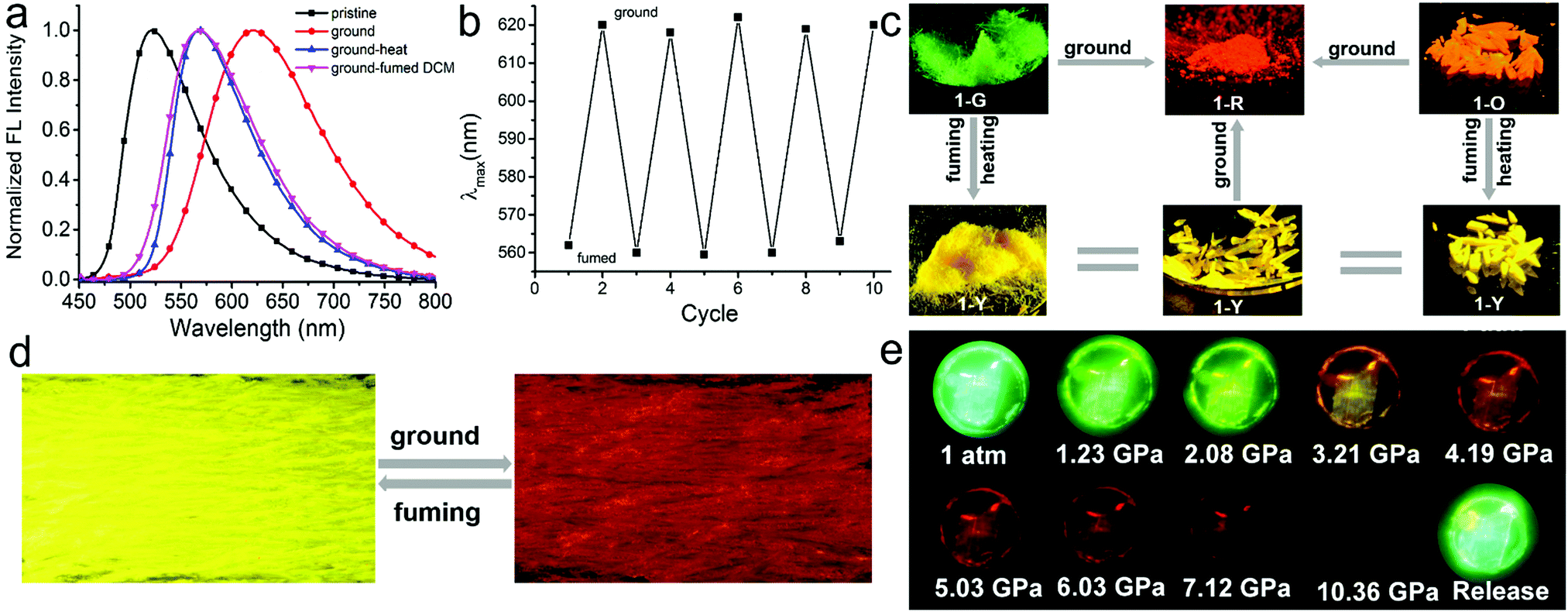 | ||
| Fig. 4 (a) Normalized emission spectra of 1-G as a pristine crystalline powder (black line), ground (red line), heated after being ground (blue line), and fumed with DCM vapor after being ground (pink line). λex = 420 nm. (b) Reversible switching emission of a pristine powder of 1-Y by repeating grinding–fuming cycles, and there is the obvious color change in every cycle as shown in photographs (d) taken under UV irradiation at 365 nm. (c) Transition among five different aggregates with diverse luminescent colors upon stimuli: 1-G and 1-O crystalline powders can transform into 1-Y crystalline powders by heating or fuming with DCM vapor; all crystalline powders can transform into 1-R aggregates by fully grinding; (d) 1-R aggregates also can transform into 1-Y crystalline powders by fuming with DCM vapor. The images of the photographs were taken under UV irradiation at 365 nm. The detailed photographs of specific transformations for every single crystal form were summarized in Fig. S19–S21 (ESI†). (e) Fluorescence images of a 1-G crystal under different isotropic pressures. | ||
After heating at 220 °C for 30 min, red emissive amorphous 1-R changes to yellow emissive crystalline 1-Y (Fig. S18, ESI†). This change is supported by the fact that PXRD of annealed samples manifests sharp peaks. This result suggests that the heating induces the structural rearrangement from 1-R to 1-Y. The formation of 1-Y rather than other types of crystalline states after heating of 1-R suggests that 1-Y might be thermodynamically more stable than the other crystalline forms. In addition, after heating at 220 °C for 30 min, the emission colors of crystalline powders of 1-G, 1-G′ and 1-O also change from their initial green and orange to yellow with the emission band centered at 560 nm (Fig. 4, Fig. S17 and S22, ESI†). Differential scanning calorimetry (DSC) is conducted to gain insight into the thermochromic behavior (Fig. S18, ESI†). All DSC curves of crystalline powders of 1-G, 1-G′, 1-Y and 1-O exhibit the same strong endothermic peak at about 270 °C corresponding to their melting points. In addition, both 1-G and 1-O also show an endothermic peak at 204 °C and 218 °C, respectively, which could belong to the temperature for phase-to-phase transition but is not found in the curve of 1-Y. These results provide further evidence to support that 1-Y is the most stable thermodynamic state.
In addition to heating, treating 1-R with dichloromethane (DCM) vapor also induces phase transformation from 1-R to 1-Y accompanied by a change of fluorescence color from red to yellow (Fig. 4c and Fig. S20, ESI†). This responsive behavior to solvent fuming is fast and the process completes within 1 min (supporting video, ESI†). Further grinding solvent-fumed samples (λem = 560 nm) produces red emissive amorphous state (λem = 618 nm). It should be noted that repeated grinding–fuming cycles of these materials induces reversible changes of the emission colors (Fig. 4b).
In addition to mechanical grinding, the emission of the four crystals is also extremely sensitive to hydrostatic pressure stimulus. Red-shifted emission colors are observed with increasing pressure for all samples. Taking 1-G as an example its emission color gradually shifts from green (520 nm) to yellow (574 nm), and eventually to red (651 nm) with a monotonic decrease in intensity, when the pressure increases from atmospheric pressure to approximately 5.0 GPa. The red shift is ascribed to the pressure-induced decrease of inter-molecular distances and revolving of the molecular planes. It is confirmed by the gradually broad and red-shifted absorption spectra with increment of pressure. The emission signal is fully quenched at the pressure of 10.36 GPa. Subsequently, releasing the pressure to atmospheric pressure recovers the emission color to the initial one. In addition, such pressure-induced color changes are also apparent under daylight. The 1-G crystal changed its color from pale green to dark red on compression and recovered after decompression (Fig. S23–S25, ESI†).
Conclusions
In summary we have reported a tetraphenylene derivative 1 containing a difluoroboron β-diketonate moiety which exhibits strong and multiple emission colors from green to red in its four polymorphic states and one amorphous solid state (1-G, 1-G′, 1-Y, 1-O and 1-R). Among them 1-G and 1-G′ emit counterintuitive hypsochromic fluorescence emission compared to 1-Y without intermolecular π–π interaction. On the basis of single crystal structural analysis and theoretical calculations, these novel emitting phenomena were rationalized. Our present results demonstrate the importance of both molecular conformation and packing modes in regulating the emission colors of organic fluorophores in aggregation states. The emission of 1 being highly sensitive to the molecular packing enables it to exhibit highly efficient and stable reversible emission changes in response to multiple external stimuli such as mechanical grinding, heating, solvent fuming and hydrostatic pressure. These excellent photophysical properties of compound 1 make it potentially useful in optoelectronics, optical sensors and smart materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21525206, 21725304, 21561130149).Notes and references
- A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS.
- T.-H. Han, Y. Lee, M.-R. Choi, S.-H. Woo, S.-H. Bae, B. H. Hong, J.-H. Ahn and T.-W. Lee, Nat. Photonics, 2012, 6, 105 CrossRef CAS.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC.
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- L.-Y. Niu, Y.-S. Guan, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, J. Am. Chem. Soc., 2012, 134, 18928–18931 CrossRef CAS.
- X. Yang, G. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2015, 44, 8484–8575 RSC.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- D.-H. Kim, A. D’Aléo, X.-K. Chen, A. D. S. Sandanayaka, D. Yao, L. Zhao, T. Komino, E. Zaborova, G. Canard, Y. Tsuchiya, E. Choi, J. W. Wu, F. Fages, J.-L. Brédas, J.-C. Ribierre and C. Adachi, Nat. Photonics, 2018, 12, 98–104 CrossRef CAS.
- T. Senthilkumar, L. Zhou, Q. Gu, L. Liu, F. Lv and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 13114–13119 CrossRef CAS.
- Y. Matsuo, Y. Wang, H. Ueno, T. Nakagawa and H. Okada, Angew. Chem., Int. Ed., 2019, 58, 8762–8767 CrossRef CAS.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS.
- Z. Zhang, B. Xu, J. Su, L. Shen, Y. Xie and H. Tian, Angew. Chem., Int. Ed., 2011, 50, 11654–11657 CrossRef CAS.
- W. Sun, S. Guo, C. Hu, J. Fan and X. Peng, Chem. Rev., 2016, 116, 7768–7817 CrossRef CAS.
- Z. Lei, X. Li, X. Luo, H. He, J. Zheng, X. Qian and Y. Yang, Angew. Chem., Int. Ed., 2017, 56, 2979–2983 CrossRef CAS.
- J. Wang, Q. Ma, Y. Wang, Z. Li, Z. Li and Q. Yuan, Chem. Soc. Rev., 2018, 47, 8766–8803 RSC.
- B. Song, S. Kandapal, J. Gu, K. Zhang, A. Reese, Y. Ying, L. Wang, H. Wang, Y. Li, M. Wang, S. Lu, X.-Q. Hao, X. Li, B. Xu and X. Li, Nat. Commun., 2018, 9, 4575 CrossRef.
- T. Buchecker, P. Schmid, I. Grillo, S. Prévost, M. Drechsler, O. Diat, A. Pfitzner and P. Bauduin, J. Am. Chem. Soc., 2019, 141, 6890–6899 CrossRef CAS.
- R. Abbel, C. Grenier, M. J. Pouderoijen, J. W. Stouwdam, P. E. L. G. Leclère, R. P. Sijbesma, E. W. Meijer and A. P. H. J. Schenning, J. Am. Chem. Soc., 2009, 131, 833–843 CrossRef CAS.
- H.-Q. Peng, L.-Y. Niu, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Chem. Rev., 2015, 115, 7502–7542 CrossRef CAS.
- H.-Q. Peng, C.-L. Sun, L.-Y. Niu, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Adv. Funct. Mater., 2016, 26, 5483–5489 CrossRef CAS.
- E. E. Greciano, B. Matarranz and L. Sánchez, Angew. Chem., Int. Ed., 2018, 57, 4697–4701 CrossRef CAS.
- Y. Chang, Y. Jiao, H. E. Symons, J.-F. Xu, C. F. J. Faul and X. Zhang, Chem. Soc. Rev., 2019, 48, 989–1003 RSC.
- X. Zhu, J.-X. Wang, L.-Y. Niu and Q.-Z. Yang, Chem. Mater., 2019, 31, 3573–3581 CrossRef CAS.
- W. Zheng, G. Yang, N. Shao, L.-J. Chen, B. Ou, S.-T. Jiang, G. Chen and H.-B. Yang, J. Am. Chem. Soc., 2017, 139, 13811–13820 CrossRef CAS.
- T. Gorai and U. Maitra, Angew. Chem., Int. Ed., 2017, 56, 10730–10734 CrossRef CAS.
- D. Spitzer, V. Marichez, G. J. M. Formon, P. Besenius and T. M. Hermans, Angew. Chem., Int. Ed., 2018, 57, 11349–11353 CrossRef CAS.
- H. Wang, C. N. Zhu, H. Zeng, X. Ji, T. Xie, X. Yan, Z. L. Wu and F. Huang, Adv. Mater., 2019, 31, 1807328 CrossRef.
- J. D. Tang, C. Mura and K. J. Lampe, J. Am. Chem. Soc., 2019, 141, 4886–4899 CrossRef CAS PubMed.
- W. Tanaka, H. Shigemitsu, T. Fujisaku, R. Kubota, S. Minami, K. Urayama and I. Hamachi, J. Am. Chem. Soc., 2019, 141, 4997–5004 CrossRef CAS PubMed.
- H.-G. Fu, Y. Chen and Y. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 16117–16122 CrossRef.
- C. Zhang, Y. Yan, Y. S. Zhao and J. Yao, Acc. Chem. Res., 2014, 47, 3448–3458 CrossRef CAS.
- Y.-L. Lei, Y. Jin, D.-Y. Zhou, W. Gu, X.-B. Shi, L.-S. Liao and S.-T. Lee, Adv. Mater., 2012, 24, 5345–5351 CrossRef CAS.
- P.-Z. Chen, Y.-X. Weng, L.-Y. Niu, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Angew. Chem., Int. Ed., 2016, 55, 2759–2763 CrossRef CAS.
- P.-Z. Chen, H. Zhang, L.-Y. Niu, Y. Zhang, Y.-Z. Chen, H.-B. Fu and Q.-Z. Yang, Adv. Funct. Mater., 2017, 27, 1700332 CrossRef.
- X. Yan, T. R. Cook, P. Wang, F. Huang and P. J. Stang, Nat. Chem., 2015, 7, 342 CrossRef CAS.
- M. Zuo, W. Qian, T. Li, X.-Y. Hu, J. Jiang and L. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 39214–39221 CrossRef CAS.
- W.-C. Geng, S. Jia, Z. Zheng, Z. Li, D. Ding and D.-S. Guo, Angew. Chem., Int. Ed., 2019, 58, 2377–2381 CrossRef CAS.
- Q. Gao, L.-H. Xiong, T. Han, Z. Qiu, X. He, H. H. Y. Sung, R. T. K. Kwok, I. D. Williams, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2019, 141, 14712–14719 CrossRef CAS.
- G. Niu, X. Zheng, Z. Zhao, H. Zhang, J. Wang, X. He, Y. Chen, X. Shi, C. Ma, R. T. K. Kwok, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, K. S. Wong, P. Wang and B. Z. Tang, J. Am. Chem. Soc., 2019, 141, 15111–15120 CrossRef CAS.
- F. Song, Y. Cheng, Q. Liu, Z. Qiu, J. W. Y. Lam, L. Lin, F. Yang and B. Z. Tang, Mater. Chem. Front., 2019, 3, 1768–1778 RSC.
- P. Harvey, A. Nonat, C. Platas-Iglesias, L. S. Natrajan and L. J. Charbonnière, Angew. Chem., Int. Ed., 2018, 57, 9921–9924 CrossRef CAS.
- M. Sarma and K.-T. Wong, ACS Appl. Mater. Interfaces, 2018, 10, 19279–19304 CrossRef CAS.
- L. Chen, D. Chen, Y. Jiang, J. Zhang, J. Yu, C. C. DuFort, S. R. Hingorani, X. Zhang, C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2019, 58, 7008–7012 CrossRef CAS.
- J. Xue, Q. Liang, R. Wang, J. Hou, W. Li, Q. Peng, Z. Shuai and J. Qiao, Adv. Mater., 2019, 1808242 CrossRef.
- T. Mutai, H. Satou and K. Araki, Nat. Mater., 2005, 4, 685–687 CrossRef CAS.
- S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS.
- G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS.
- Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2011, 50, 9128–9132 CrossRef CAS.
- Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782–10785 CrossRef CAS.
- H. Ito, M. Muromoto, S. Kurenuma, S. Ishizaka, N. Kitamura, H. Sato and T. Seki, Nat. Commun., 2013, 4, 2009 CrossRef.
- P. Zhang, W. Dou, Z. Ju, X. Tang, W. Liu, C. Chen, B. Wang and W. Liu, Adv. Mater., 2013, 25, 6112–6116 CrossRef CAS PubMed.
- L. Liu, X. Wang, N. Wang, T. Peng and S. Wang, Angew. Chem., Int. Ed., 2017, 56, 9160–9164 CrossRef CAS.
- C. Wang and Z. Li, Mater. Chem. Front., 2017, 1, 2174–2194 RSC.
- G. Huang, Y. Jiang, S. Yang, B. S. Li and B. Z. Tang, Adv. Funct. Mater., 2019, 29, 1900516 CrossRef.
- K. Wang, H. Zhang, S. Chen, G. Yang, J. Zhang, W. Tian, Z. Su and Y. Wang, Adv. Mater., 2014, 26, 6168–6173 CrossRef CAS.
- P. Galer, R. C. Korošec, M. Vidmar and B. Šket, J. Am. Chem. Soc., 2014, 136, 7383–7394 CrossRef CAS.
- K. Devi and R. J. Sarma, CrystEngComm, 2019, 21, 4811–4819 RSC.
- Y. Zhou, L. Qian, M. Liu, X. Huang, Y. Wang, Y. Cheng, W. Gao, G. Wu and H. Wu, J. Mater. Chem. C, 2017, 5, 9264–9272 RSC.
- M. Jin, T. Seki and H. Ito, J. Am. Chem. Soc., 2017, 139, 7452–7455 CrossRef CAS.
- S. Mukherjee and P. Thilagar, J. Mater. Chem. C, 2016, 4, 2647–2662 RSC.
- P.-Z. Chen, L.-Y. Niu, Y.-Z. Chen and Q.-Z. Yang, Coord. Chem. Rev., 2017, 350, 196–216 CrossRef CAS.
- Y. Qi, Y. Wang, Y. Yu, Z. Liu, Y. Zhang, G. Du and Y. Qi, RSC Adv., 2016, 6, 33755–33762 RSC.
- P.-Z. Chen, J.-X. Wang, L.-Y. Niu, Y.-Z. Chen and Q.-Z. Yang, J. Mater. Chem. C, 2017, 5, 12538–12546 RSC.
- L. Zhai, F. Zhang, J. Sun, M. Liu, M. Sun and R. Lu, Dyes Pigm., 2017, 145, 54–62 CrossRef CAS.
- E. E. Jelley, Nature, 1936, 138, 1009 CrossRef CAS.
- A. S. Davydov, Phys.-Usp., 1964, 7, 145 CrossRef.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- A. Eisfeld and J. S. Briggs, Chem. Phys., 2006, 324, 376–384 CrossRef CAS.
- S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863–873 CrossRef CAS.
- C. Kaufmann, D. Bialas, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2018, 140, 9986–9995 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1921291 and 1921299–1921301. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qm00518h |
| This journal is © the Partner Organisations 2020 |