
A polymorphic fluorescent material with strong solid state emission and multi-stimuli-responsive properties†

Abstract
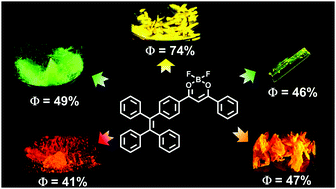
A bright difluoroboron β-diketonate derivative 1 showing four emission colors (green, yellow, orange and red) with high quantum yields (41–74%) in four polymorphs and one amorphous state is reported. Green-emissive crystals (1-G and 1-G′) exhibit dimeric aggregation structures due to the strong molecular π–π interaction but exhibit hypsochromic emission compared to yellow-emissive crystals (1-Y) with monomeric aggregation because of lacking such π–π interactions. These novel emission phenomena are rationalized by theoretical calculations. High fluorescence sensitivity of compound 1 to its molecular packing modes results in excellent responsive behavior to multiple external stimuli thereby showing reversible change of emission colors under mechanical grinding, heating, solvent fuming and hydrostatic pressure.
 Please wait while we load your content...
Please wait while we load your content...

