
Identifying competitive tin- or metal-free catalyst combinations to tailor polyurethane prepolymer and network properties†
Priscilla
Arnould
a,
Lionel
Bosco
a,
Federico
Sanz
b,
Frédéric N.
Simon
b,
Stéphane
Fouquay
b,
Guillaume
Michaud
b,
Jean
Raynaud
 *a and
Vincent
Monteil
*a
*a and
Vincent
Monteil
*a
aLaboratory of Chemistry, Catalysis, Polymers and Processes C2P2, équipe CPP, Université de Lyon 1- Claude Bernard, CNRS-CPE Lyon, UMR 5265, Villeurbanne, France. E-mail: jean.raynaud@univ-lyon1.fr; vincent.monteil@univ-lyon1.fr
bBostik Smart Technology Center, ZAC du bois de Plaisance, Venette, France
First published on 25th August 2020
Abstract
The influences of selected catalysts on the structures and properties of polyurethane prepolymers and networks are investigated to adjust the catalyst/structure/property relationship to a targeted application. This study highlights the necessity of catalysis for polyurethane synthesis, both at the prepolymer and at the crosslinking stages, and emphasizes on the catalyst-dependency of each stage. We also suggest some tin-free and overall metal-free alternatives to ubiquitous tin-based catalysts with metals such as Bi, Ti, Zn and organic catalysts such as DABCO, DMDEE. In polyurethane formulations without fillers, the strong interwoven urethane- and urea-devired H-bonding network is mainly responsible for the mechanical properties of the material and tends to overshadow the catalyst effects. Nonetheless, in the presence of fillers such as those used in industrial polyurethane formulations, tensile tests evidenced that mechanical properties are affected and can be tailored by the choice of catalyst.
1. Introduction
Polyurethanes represent an important class of polymers owing to their versatility arising from their large range of properties which can be harnessed in many applications such as coatings, foams, building infrastructures, adhesives, sealants, and elastomers.1–4 Polyurethane synthesis, established by Otto Bayer in 1937, occurs spontaneously between aromatic or aliphatic multifunctional isocyanates, often diisocyanates, such as 4,4-methylenebis(phenylisocyanate), tolylene diisocyanate and isophorone diisocyanate and polyether- or polyester-based polyols.1,5 Nevertheless, even when aromatic isocyanates are employed, kinetic rates can be very low and reaching full conversion can prove challenging in an industrially viable reaction time. It is well-known that the catalyst choice influences kinetics6–17 but only a few studies deal with the systematic impact of said catalyst on polyurethane structures, molar masses, remaining free isocyanate and properties (viscosity, thermal, mechanical etc.).10,13–15,18–25Mercury and tin catalysts have been widely used as catalysts in the fields of polyurethane synthesis and crosslinking due to their efficiency/selectivity towards the reaction between isocyanate and alcohol moieties. Even though tin-based systems remain the strategy of choice efficiency-wise, because of health and environmental concerns, they have slowly been replaced by other catalysts to provide more sustainable alternatives. Researchers have made efforts in order to find alternative catalysts, either based on metals such as iron,6,9,13,16 copper,9,13 zinc,6,10,16 bismuth,6,10 titanium6,10,16 and cobalt,6,10 or strong organic bases such as 1,4-diazabicyclo[2.2.2]octane (DABCO),7,11,12,15,17 2,2′-dimorpholinodiethylether (DMDEE),7,11,14 various guanidines15,17 and also organic acids.17
Despite thorough investigations of catalysts’ kinetics for polyurethane syntheses, few reports deal with the impact of catalysts on polyurethane properties such as molar masses, residual volatile isocyanate/corresponding amine or aniline, viscosities, mechanical and thermal properties of final materials, which are crucial parameters regarding regulations and processability of polymers.10,13–15,18–25 In foams12,22 and hybrid materials,18–25 the impact of catalysts on structure, morphologies and consequently on material properties have already been partially investigated in order to design polyurethanes by either using a single catalyst or an combination of several catalysts.
In the present work NCO-terminated prepolymers were synthesized in bulk with 4,4-methylenebis(phenylisocyanate) (4,4-MDI) and two poly(propylene glycol)s (PPG) (global functionality of polyol blend was equal to 2.45, characteristics are given in the Experimental section) in the presence of different metallic and organic catalysts (Fig. 1 and 2). Crosslinking of these prepolymers then occurred with air moisture exposure. Kinetics and prepolymers’ features as well as crosslinked materials’ properties were studied in order to identify the impact of catalysts on polyurethanes and suggest efficient tin-free systems. However, in polyurethane-urea materials, H-bonding which is mainly responsible for the mechanical properties tend to shield the impact of catalyst on crosslinked materials. This article provides a comprehensive methodology to pinpoint the catalyst/structure/property relationship and appreciate the impact of catalysis on the properties of crosslinked polyurethanes.
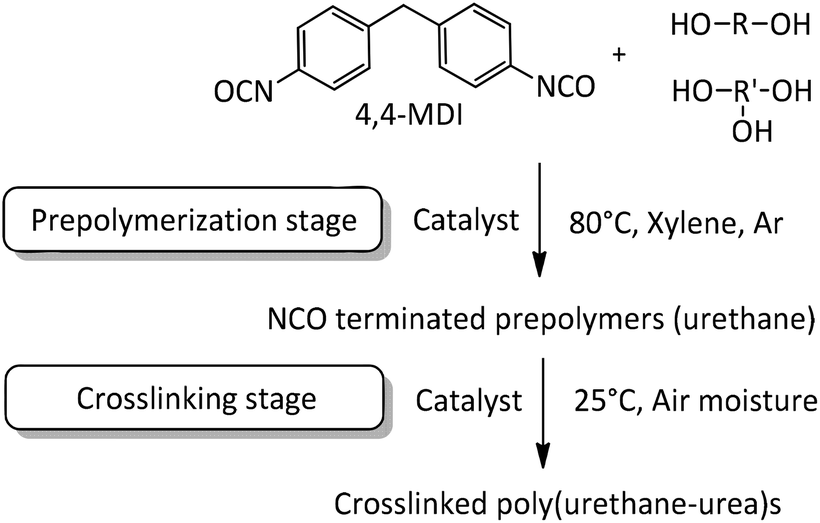 | ||
| Fig. 1 Process for NCO terminated prepolymers and crosslinked polyurethanes. | ||
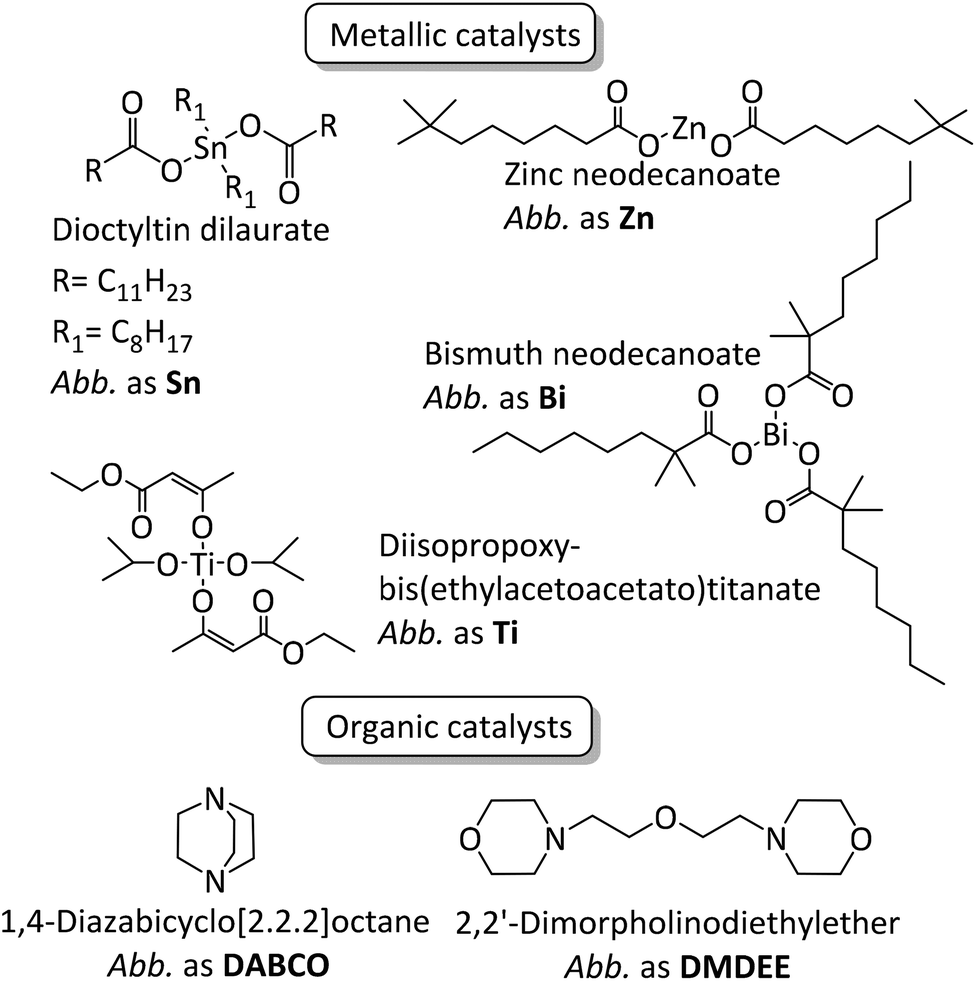 | ||
| Fig. 2 Structures of the studied catalysts and their designation in the present article. | ||
2. Results and discussion
2.1. Prepolymerization stage
 | ||
| Fig. 3 Influence of metallic and organic catalysts on the kinetics of prepolymerization reaction at 80 °C determined by FTIR-ATR. | ||
| Catalyst | Time of reaction a (min) | Time to reach 50% conversion of NCO functions by FTIR b (min) | Molar amount of catalysts compared to tin-catalysts (mol eq. Sn 6 ppm) |
|---|---|---|---|
| a Time to reach a constant value of the absorption of the stretching band of NCO determined by FTIR. NB: [NCO]/[OH] = 1.83. b Time to reach 50% of NCO overall decrease (from max. value to min. value of the stretching band at 2270 cm−1) corresponding to the complete conversion of [OH]. (Fig. S1, ESI). | |||
| Uncatalysed | 1080 | 220 | 0 |
| Sn 6 ppm | 120 | 9 | 1 |
| Ti 7.2 ppm | 120 | 13 | 2 |
| Bi 5.8 ppm | 120 | 11 | 1 |
| Zn 24 ppm | 150 | 24 | 8 |
| DABCO 45 ppm | 360 | 44 | 50 |
In the presence of a catalyst we elected to work with molar quantities so that reactivities/kinetics could be comparable. We did a pre-screen of the various catalysts and adjusted the molar quantity so that both ppm (in weight) and activities would be on the same order of magnitude. In order to find a potential alternative to tin catalysts, the amount of each catalyst in the reaction are compared to the amount of Sn catalyst which is required to significantly increase reaction rate (Table 1). The amount of competing catalysts is selected in order to feature similar kinetics as tin catalysts (reference here: Sn catalyst) in order to assess the different catalyst activities (Fig. S4, ESI†). Thus, compared to tin catalysts, the amount of Ti catalyst is multiplied by two, Zn catalysts by eight and DABCO by fifty in order to achieve similar reaction times and kinetics. Our two criteria are expressed in Table 1: total time of reaction and time to reach 50% conversion of NCO functions. Most metallic catalysts have a lower catalytic activity than the Sn catalyst. Only Bi catalyst had similar catalytic activity compared to Sn catalysts on this system with the same molar amount. Organic catalysts are significantly less effective than metallic catalysts (DABCO as a nonetheless potent organic was chosen as reference in our study). The kinetic study demonstrated the influence of catalysts on prepolymerization rate: the reaction time is divided by three with the organic catalyst DABCO 45 ppm, by nine with Sn 6 ppm, Zn 24 ppm, Bi 5,8 ppm, and by seventeen with Ti 7,2 ppm (Table 1). In terms of kinetics, Bi, Ti and Zn catalysts seem to be interesting alternative to tin-based catalysts.
In order to characterize prepolymers, various analytical techniques were used such as rheometry for viscosity, SEC-THF for molar masses and for the titration of residual isocyanate at the end of reaction (calibration of the SEC technique detailed in ESI, Fig. S6 and 7†). In this work, an excess of isocyanate was used at the prepolymerization stage and the remaining MDI is a major drawback regarding the regulation since it is volatile and potentially harmful. In fact, strict regulation has been established in order to limit the amount of remaining isocyanate monomers due to their toxicity (and that of their derivatives). We consequently monitored the impact of catalysts on this crucial parameter by SEC-THF. In order to avoid side reaction during SEC analysis, which can alter molar masses, a pretreatment of prepolymers is necessary. Prepolymers are quenched with an excess of anhydrous methanol to obtained MeOH-capped urethanes at the chain ends instead of free isocyanate moieties.15 The reaction occurs at room temperature during 24 h to warrant complete quench (Fig. S8 in ESI†). Fig. 4 represents a typical chromatogram obtained by SEC-THF of NCO-terminated prepolymer quenched with methanol. At high molar masses (low elution volume), the chromatogram is representative of the distribution of molar masses. In addition, at high elution volume, we identified a characteristic signal representing the MeOH-quenched 4,4-MDI (noted di(MeOH)-MDI), which remains at the end of the reaction due to the ratio NCO/OH > 1 and of polyaddition typical reaction statistics.
 | ||
| Fig. 4 Typical SEC-THF chromatograms of NCO-terminated polyurethane prepolymers quenched with MeOH. | ||
Although the influence of catalysts on kinetics was investigated, only few articles deals with the properties of prepolymers such as molar masses, viscosity, residual monomer which is de facto catalyst-dependent.10,13–15 In this work, significant differences in prepolymer features were evidenced as function of the catalyst, noticeably in terms of molar masses and distributions (Table 2). Two groups of catalysts were observed and led to distinct features of prepolymers: groups A and B in Table 2. Catalysts of group A yielded prepolymers with lower molar masses than catalysts of group B.
| Catalyst | Molar amount of catalysts compared to tin catalyst (mol eq. DOTL 6 ppm) | Remaining isocyanate a (%) | Viscosity b (Pa s) |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (g mol−1) c (g mol−1) |
M
wc (g mol−1) |
Đc |
|
|---|---|---|---|---|---|---|---|
|
a Titration determined by SEC-THF.
b Zero-shear viscosity determinated by rheometry at 25 °C.
c Molar masses and dispersities determined by SEC-THF by integrating the entire distribution (between 3.0 and 6.0 for logM values) without any exclusion of oligomer signals.
|
|||||||
| A | Uncatalysed | 0 | 3.3 | 14 | 12000 |
26500 |
2.2 |
| DABCO 45 ppm | 50 | 3.2 | — | 12000 |
27600 |
2.3 | |
| Zn 24 ppm | 8 | 4.2 | 16 | 11900 |
27200 |
2.2 | |
| Ti 7.2 ppm | 2 | 3.5 | 15 | 12200 |
27700 |
2.2 | |
| B | Sn 6 ppm | 1 | 4.1 | 22 | 13700 |
36800 |
2.7 |
| Bi 5.8 ppm | 1 | 3.8 | — | 13400 |
31600 |
2.3 | |
Indeed, Fig. 5 demonstrated that chains with higher molar masses were promoted by group-B catalysts and consequently higher-viscosity and higher residual-isocyanate containing prepolymers were obtained. Therefore, polyurethane prepolymerization stage is catalyst-dependent and catalysts can tailor NCO-terminated prepolymers (Fig. 5). These differences in molar-mass distribution tendencies are systematically observed for different NCO-terminated sets of prepolymer syntheses in presence of A- or B-type catalysts (Fig. S9 for reproducibility, ESI†).
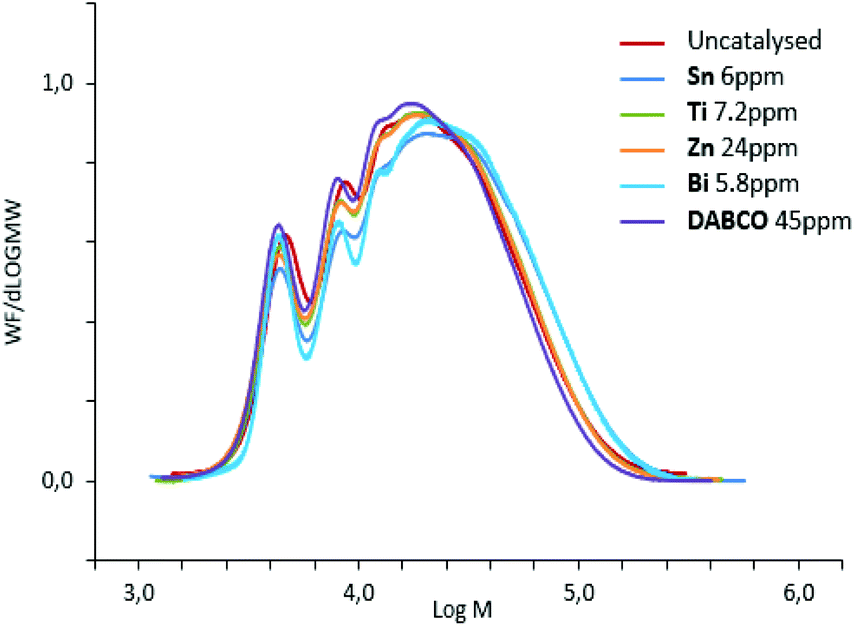 | ||
| Fig. 5 SEC-THF chromatograms of polyurethane NCO-terminated prepolymers, quenched with MeOH, obtained with different catalysts. | ||
We also studied the influence of catalysts at different NCO/OH ratio as well as the effect of catalyst concentration. At equal NCO/OH ratio, Table 3 demonstrated that the concentration of catalyst had almost no effect on prepolymer features (Table 3, entries 1–3 vs. 4–6 for Sn catalysts and entries 7–9 vs. 10–12 for DABCO).
| Entry | Catalyst | NCO/OH ratio | Concentration of catalysts (ppm) |
M
na (g mol−1) |
M
wa (g mol−1) |
Đa |
Remaining isocyanateb (%) |
|---|---|---|---|---|---|---|---|
| a Molar masses and dispersity determined by SEC-THF. b Titration evaluated by SEC-THF. | |||||||
| 1 | Sn | 1.7 | 6 | 16900 |
59000 |
3.5 | 2.7 |
| 2 | 1.83 | 6 | 13700 |
36800 |
2.7 | 4.1 | |
| 3 | 2.05 | 6 | 11900 |
26500 |
2.2 | 5.2 | |
| 4 | 1.7 | 600 | 16400 |
60900 |
3.7 | 3.0 | |
| 5 | 1.83 | 600 | 13700 |
37200 |
2.7 | 3.9 | |
| 6 | 2.05 | 600 | 11500 |
24900 |
2.2 | 5.5 | |
| 7 | DABCO | 1.7 | 45 | 14000 |
41500 |
2.9 | 2.3 |
| 8 | 1.83 | 45 | 12000 |
27600 |
2.3 | 3.2 | |
| 9 | 2.05 | 45 | 10500 |
20800 |
2.0 | 5.2 | |
| 10 | 1.7 | 4500 | 13300 |
37400 |
2.8 | 2.5 | |
| 11 | 1.83 | 4500 | 12200 |
27200 |
2.2 | 3.4 | |
| 12 | 2.05 | 4500 | 10300 |
20900 |
2.0 | 4.6 | |
When the ratio NCO/OH decreased, the effect of catalyst on prepolymer properties is easier to evidence. Indeed, at NCO/OH equal to 2.05, no significant difference was observed between the two group-representative catalysts Sn and DABCO whereas at a lower ratio (1.7), noticeable differences in molar masses were observed (Fig. 6). The results confirmed that metallic catalysts, Sn and Bi promote wider distributions of molar masses compared to the other metallic and organic catalysts and a low ratio NCO/OH is necessary to pinpoint this catalysis impact on the prepolymerization.
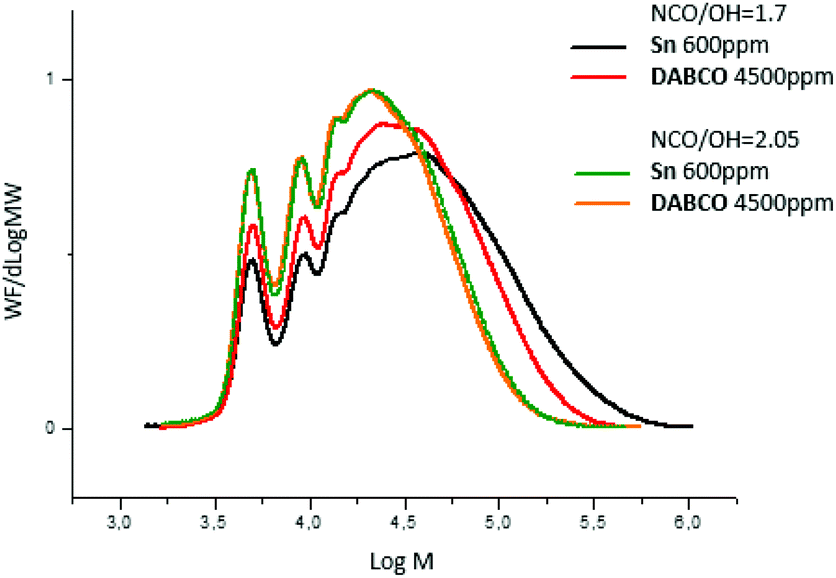 | ||
| Fig. 6 SEC-THF chromatograms of polyurethane prepolymers quenched with MeOH in function of catalyst (nature, amounts) and ratio NCO/OH. | ||
At low ratio NCO/OH, these significant differences in high molar-mass fractions have a marked impact on viscosity and residual isocyanate (Fig. 7). These experiments clearly evidenced that Sn and Bi catalysts behaviors were different than other catalysts and uncatalysed system. Both catalysts led to NCO-terminated prepolymers with higher molar masses, higher viscosity and higher residual isocyanate monomer. These experiments also demonstrated the obvious but major role of the ratio NCO/OH on the prepolymer features. Industrially, selecting the viscosity and residual MDI through appropriate choice of catalyst and NCO/OH is thus advantageous to adjust the prepolymer for its future processing/conditioning/storage.
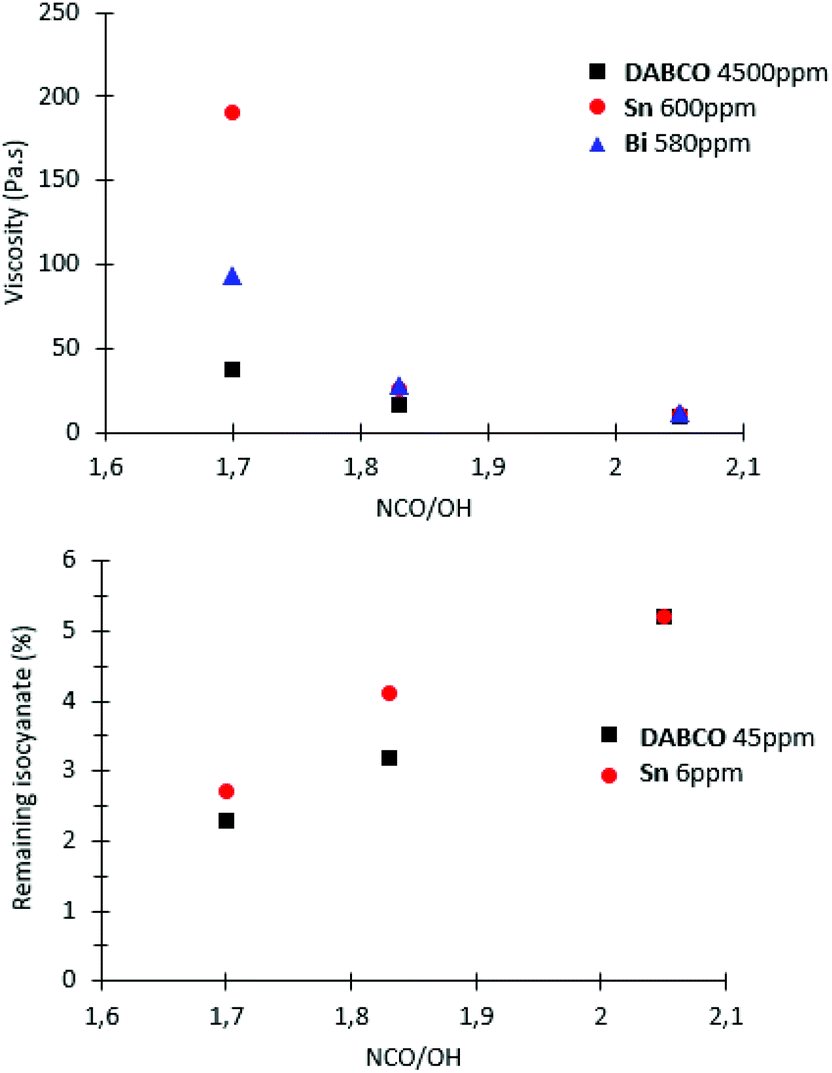 | ||
| Fig. 7 Evolution of remaining isocyanate and NCO-terminated prepolymers viscosity versus catalyst nature and NCO/OH ratio. NCO-terminated prepolymer viscosities were measured after one hour of reaction by rheometry. | ||
The catalysts’ varying behaviours could be directly correlated to their specific mode of activation. Indeed, metallic and organic catalysts have different modes of activation of isocyanate and alcohol moieties. Mechanism of organic catalysis depends on the acidity/basicity and nucleophilicity/electrophilicity of the catalyst.15,26 For DABCO,26 mechanism is based on an inherent and mostly unassisted nucleophilic addition of alcohol on isocyanate and then, prototropy between intermediates is sped up by the organic catalyst (Fig. S10, ESI†). This mechanism is favoured for DABCO and all organic catalysts which have lower pKa than alcohol moieties in the reaction medium (good approximation is given by the pKa26) and consequently, cannot formally deprotonate the alcohol moiety. Due to this mode of activation, organic catalysts play a key role in the increase of proton exchange rate but do not formally react with chain ends.
For metallic catalysts, mechanisms are less well-defined and researchers proposed two different pathways, depending on the metallic center: Lewis acid and/or insertion mechanism (Fig. S11, ESI†). On one hand, some metallic catalysts, such as Sn,6,9,27,28 Zr,10 Cu,9,29 Fe,9 Bi28 could exchange one of their ligands with the alcohol or interacted with the hydrogen of the alcohol and then isocyanate reacts with this active complex to synthesize urethane. On the other hand, some metallic catalysts, such as Sn,6,9,27,28 and Ti30 catalysts, interact with either the nitrogen or oxygen of the isocyanate via typical Lewis acidity favoring nucleophilic attack of alcohol. However, compared to organic catalysts, metallic catalysts create bonding with the chain ends, which could explain their particular influence on prepolymer properties. The different behaviors between metallic catalysts (such as Sn-, Ti- and Zn-based catalysts for instance) could then be the result of relative reactivity towards either alcohol or isocyanate (N and O) moieties but could also arise from the dual-activation capacity of some catalysts (Fig. S10, ESI†).31 Indeed, researchers evidenced by RMN study that metallic catalysts, such as Zr catalysts,10 could only react with the alcohol to initiate the polymerization whereas Sn, Ti catalysts6,9,27,28,30 demonstrated the capacity to be involved in reaction with both, isocyanate and alcohol.
2.2. Crosslinking of NCO-terminated prepolymers
The crosslinking kinetics between NCO-terminated prepolymers and air moisture was monitored by using the ATR mode of infrared spectroscopy. A thin film of 300 μm was directly realized on the ATR diamond with a hand-coater and an automatic sequence was created to record spectra during several hours. During the crosslinking, the decrease of NCO stretching band at 2270 cm−1 was monitored in order to represent the consumption of isocyanate functional groups versus reaction time (Fig. 8).
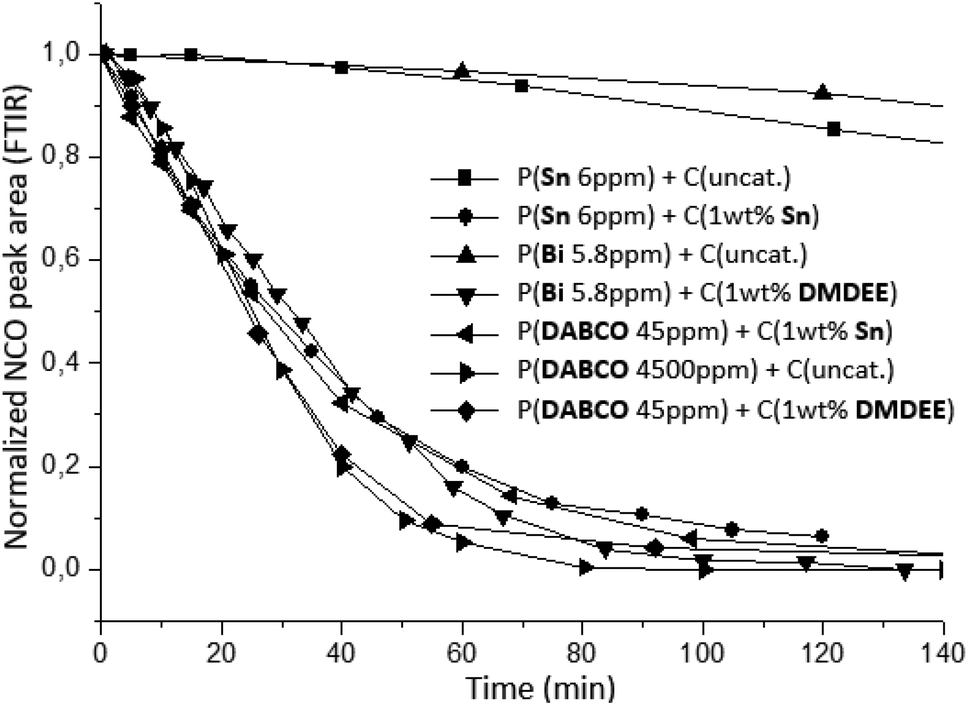 | ||
| Fig. 8 Kinetic of crosslinking between NCO-terminated prepolymers and air moisture using different associations of catalyst (monitored by FTIR at 25 °C). | ||
Fig. 8 indicates that systems with no further addition of catalysts at crosslinking stage featured low crosslinking rate. Amount of prepolymerization catalysts are not sufficient to accelerate crosslinking significantly. Due to the typical low concentration used during prepolymerization, crosslinking kinetics between NCO-terminated prepolymers and air moisture is too slow, probably due to diffusion limitations thus lower probability of encounter between H2O, isocyanate and catalyst molecules. These results evidenced the necessity to increase the concentration of the prepolymerization catalyst such as P(DABCO 4500 ppm) + C(uncat.) or add further catalysts at the crosslinking stage to significantly improve kinetics. Different synergies of catalysts were thereafter investigated: metallic/organic catalysts used at the prepolymerization stage either used in larger amounts alone or in conjunction with another metallic catalyst or an organic catalyst were used for the crosslinking stage. The selected organic catalyst was DMDEE here, which is reported to be effective for the H2O-isocyanate reaction and displayed the advantage to be liquid.7,11,14 In our case, using increased loadings of catalysts for the second stage, it was found that organic (DMDEE) and metallic systems (Sn) display similar efficiency and considerably increase the reaction rate (Fig. 8). The association of Bi or DABCO catalysts at the prepolymerization stage and DMDEE as crosslinking catalyst displayed the same kinetic profile as Sn catalyst used at both stages. These catalyst associations were therefore efficient tin-free and even metal-free alternative systems.
| NCO/OH prepolymers | Entry | P(catalyst) | C(catalyst) | Height tanδ |
T α (°C) | Width tanδ (°C) |
|---|---|---|---|---|---|---|
| 2.05 | 1 | DABCO 4500 ppm | — | 1.14 | −27.6 | 21.9 |
| 2 | Sn 600 ppm | — | 1.10 | −26.9 | 22.2 | |
| 3 | Bi 580 ppm | — | 1.07 | −28.1 | 22.0 | |
| 1.83 | 4 | DABCO 45 ppm | — | 1.26 | −30.2 | 19.1 |
| 5 | DABCO 4500 ppm | — | 1.27 | −30.1 | 18.8 | |
| 6 | DABCO 45 ppm | 1 wt% Sn | 1.24 | −30.3 | 19.7 | |
| 7 | Sn 6 ppm | — | 1.25 | −30.2 | 19.0 | |
| 8 | Sn 600 ppm | — | 1.27 | −30.2 | 19.1 | |
| 9 | Sn 6 ppm | 1 wt% Sn | 1.24 | −30.8 | 20.3 | |
| 10 | Bi 580 ppm | — | 1.25 | −30.0 | 18.9 | |
| 1.7 | 11 | DABCO 4500 ppm | — | 1.36 | −32.6 | 17.2 |
| 12 | Sn 600 ppm | — | 1.39 | −32.2 | 17.5 | |
| 13 | Bi 580 ppm | — | 1.39 | −32.4 | 17.0 |
Then, the impact of catalyst concentrations at the prepolymerization stage on the properties of the final material was evaluated. On a par with the results obtained at the prepolymerization stage, it was found that the catalyst concentrations of the prepolymerization stage have no impact on the properties of the final material (Table 4, entries 4 vs. 5 and 7 vs. 8). Nevertheless, at a given NCO/OH ratio, it was not expected that thermomechanical properties such as Tα of each material would be independent of the crosslinking catalyst (Table 4, entries 4 vs. 6 and 7 vs. 9). DSC analyses were also performed to determine the glass-transition temperature (Tg). As for DMA study, temperatures of glass transition of PU-networks were catalyst-independent and were consistently 15 °C lower than Tα (Table S1, ESI†). In poly(urethane–urea)s, hydrogen bonding has the major role on the mechanical properties. In fact, H-bonding enforces a specific conformation of chains, critical for the mechanical properties and could consequently preclude observation of the catalyst effects on the mechanical properties.
This phenomenon can be straightforwardly observed by 1H-NMR spectroscopy by using deuterated solvents of different polarities: benzened6, acetoned6 and DMSOd6. 1H-NMR spectra evidenced the impact of the polarity of the environment by the significant shift of the NH signal of the urethane in presence of a polar solvent such as DMSO (Fig. 9).
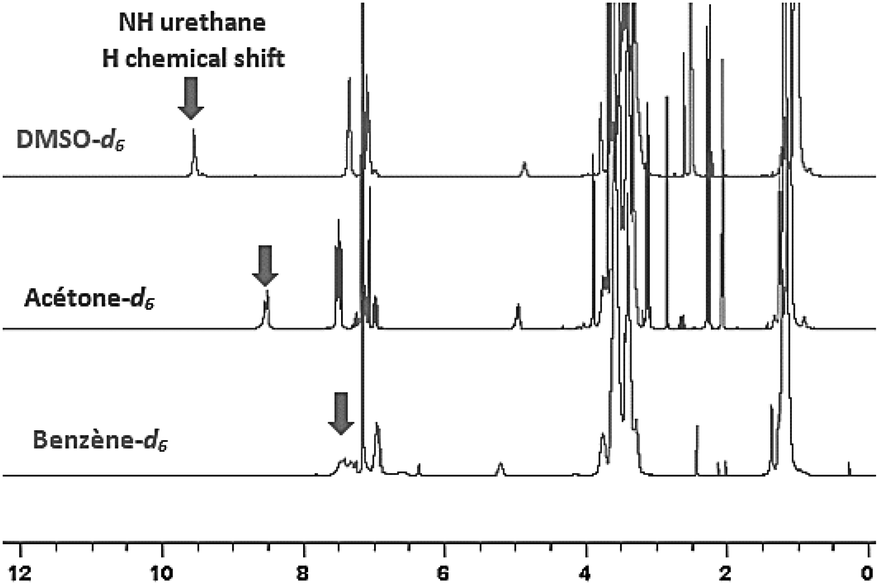 | ||
| Fig. 9 Evolution of chemical shift of urethane function on 1H-NMR spectrum in function of solvent polarity. | ||
| NCO/OH prepolymers | P(catalyst) + C(uncat.) | Solvent | T crosslinking (°C) | Height tanδ |
T α (°C) | Width tanδ (°C) |
Integral tanδa (°C) |
|
|---|---|---|---|---|---|---|---|---|
|
a Integration of tanδ curve in the temperature range of −85 to 35 °C.
b Done on two different experiments for reproducibility.
|
||||||||
| 1 | 2.05 | Sn 600 ppm | — | RT | 1.09 | −26.9 | 22.2 | — |
| 2 | 20 wt% DMSO | 36 h RT | 1.20 | −34.3 | 17.5 | — | ||
| 3 | 20 wt% DMSO | 36 h RT, 2 h 90 °C | 1.12 | −31.3 | 19.7 | — | ||
| 4 | 1.83 | Sn 600 ppm | — | RT | 1.27 | −30.2 | 19.1 | 29.87 |
| 5 | 20 wt% DMSO | 36 h RT, 12 h 90 °C | 1.21 | −32.7 | 17.4 | 27.15 | ||
| 6 | 1.83 | DABCO 4500 ppm | — | RT | 1.27 | −30.1 | 18.8 | 30.06 |
| 7 | 20 wt% DMSO | 36 h RT, 12 h 90 °C | 1.32 | −30.9 | 18.8 | 31.06/31.67b | ||
Indeed, the physical network was influenced by the presence of DMSO, which acts both as a plasticizer and as a H-bonding disrupter in our case (Table 5, entries 1 and 2). Moreover, it was found that this method is reversible, by removing DMSO under temperature/vacuum (at 90 °C), the material almost recovered its initial properties at NCO/OH equal to 2.05 (Table 5, entries 1–3). This observation was less pronounced at lower NCO/OH ratio due to the decrease of H-bonding within the materials (Table 5, entries 6–8).
Using this DMSO strategy, we could observe slight differences in the final properties of the materials from the different experiments (Table 5). Catalysts indeed have an impact on the thermomechanical properties of the final 3D-network, evidenced in presence of DMSO (Table 5, entries 5 and 7). In particular, the integral values of tanδ in presence of DMSO are significantly higher in the DABCO case than in the Sn case (31.37 °C vs. 27.15 °C, see integral-tanδ column, Table 5). This reflects a better damping ratio and therefore a more homogeneous network for the DABCO system. DMSO is a small organic molecule used in this formulation to shield H-bonding and plasticize at the same time. These formulations thus behave differently from actual PU-based mastics that also comprise inorganic fillers and additives with a higher filler/polyurethane ratio. Although DMSO infers a lowering of Tα, all fillers/additives/plasticizers used in a formulated mastic on the other hand aim at improving the mechanical properties via increasing the toughness, thus the elongation at break, and adjust the moduli of the materials. Industrial formulations have thus been investigated (see below, part 2.2.4).
δ without any conclusion on moduli due to the strong dependency of sample size for DMA measurement. In this part, tensile tests have been realized to discriminate the influence of each catalyst on the mechanical properties (moduli and elongation at break) of 3D-networks. Similarly to DMA analysis, tensile tests on pure PU mastics (without any additive/filler/plasticizer) confirmed that mechanical properties were almost catalyst-independent, with similar values of moduli (at 100% elongation), tensile strengths and elongations at break (respectively ∼1.2 MPa, ∼2 MPa and 255–260%; Table S2, ESI†). In order to evidence the impact of catalyst nature on the mechanical properties, actual industrial formulations comprising organic and inorganic fillers, such as PVC and calcium carbonate, as well as additives and plasticizers were used at the crosslinking stage (Table S3, ESI†).33 Although different catalyst concentrations were used in this part for practicality, results in Table 2 – which demonstrated that NCO-terminated prepolymers were independent of catalyst concentration (for a given catalyst) – allow us to compare the different experiments. Solid-state 1H and 13C NMR spectra of PU networks without fillers were recorded and no signal associated to side-products (isocyanurates, allophanates etc.) could be evidenced (Fig. S14 and 15, ESI†). Due to the absence of any side-product and the addition of same amount of fillers, all differences in mechanical properties result from the catalyst impact on PU networks. Potential interactions between fillers and catalysts were not studied. However, due to typically very high loadings of catalyst at the crosslinking stage used industrially in the presence of “treated” or rendered-mostly-inert organic and inorganic fillers, it is unlikely that most catalyst would become inefficient.
These formulations evidenced the catalyst-dependency of formulated materials’ properties, especially in the case of elongation-at-break values, and suggested that catalysts played a crucial role in building the 3D network thus modifying the final materials properties (Table 6 and Fig. S16, ESI†). The moduli and elongations at break were similar when the same catalyst was used at the crosslinking stage (regardless of the prepolymerization catalyst), which evidenced a marginal influence of the prepolymerisation catalyst on the materials properties (Table 6, entries 2 and 4).
| Entry | P(catalyst)a | C(catalyst) (0.2 wt%) | Elongation at break (%) | Modulus at 100% of elongation (MPa) | Ultimate tensile strength (at break) (MPa) |
|---|---|---|---|---|---|
| a NCO-terminated prepolymers obtained at NCO/OH ratio equal to 1.83. | |||||
| 1 | Sn 7.7 ppm | Sn | 544 | 1.02 | 1.32 |
| 2 | Bi 6.3 ppm | DMDEE | 391 | 1.32 | 1.60 |
| 3 | Bi 6.3 ppm | DABCO | 819 | 0.70 | 1.48 |
| 4 | DABCO 95 ppm | DMDEE | 376 | 1.29 | 1.54 |
The small concentration of the prepolymerization catalyst remaining at the crosslinking stage compared to the catalyst amount specifically added for crosslinking, could explain the P(catalyst)'s limited impact on the network construction. On the contrary, varying the catalyst nature at the crosslinking stage led to very different mechanical properties, adjusting them while keeping the component of the formulation strictly identical, thus highlighting the versatility of PU properties in that case through appropriate choice of catalyst. (Table 6, entries 1–3) These results are in agreement with industrial observations on PU mastics in the presence of fillers: catalysts change the 3D building of these formulated networks.
Indeed, materials crosslinked with DMDEE exhibited 376–391% of elongation at break, Sn went up to 544% whereas DABCO enhanced the elongation at break up to 819% and led to the most ductile material (Table 6, entries 1–3), confirmed by the lowest modulus at 100% of elongation. The elongation at break is directly related to the structure and the organization of the 3D-network, which allowed us to evidence the major influence of catalyst on these parameters. Values of integrals of tanδ over the range of temperature representative of tensile testing suggest that the damping ratio of formulations can be affected by the choice of catalyst (see Table S4 and corresponding discussion, ESI†).
These important differences could be the results of the relative homogeneity of the materials regarding the repartition of crosslinking knots and the average molar masses of in-between segments. Indeed, heterogeneous materials, with high and low crosslinking-density zones, induce an important local strain near crosslinking-dense zones, which consequently leads to a smaller elongation at break. In these formulations, DABCO led to the most homogenous chemical (see tanδ integrals, Table 5) and physical structure of the crosslinked materials compared to Sn and DMDEE respectively. Catalysts can effectively influence the 3D-network structure and lead to significant variations in mechanical properties. These catalyst associations (at both stages) provide competitive tin-free (Bi then either DABCO or DMDEE, entries 2 & 3 of Table 6) and overall metal-free (DABCO then DMDEE, entry 4 of Table 6) systems.
3. Conclusion
The reaction between 4,4′-MDI and polyether-based polyols was monitored and optimized in the presence of metallic and organic catalysts in terms on kinetics but also to highlight the catalyst influence on the polyurethane properties. At the prepolymerization stage metallic catalysts are more effective than organic catalysts (at the same molarity) but all improve significantly the reaction rate and can decrease the reaction time by an order of magnitude, when compared to the uncatalysed system. SEC and rheometry clearly indicated the catalyst dependence of NCO-terminated prepolymers. Two different catalyst behaviours were identified where tin and bismuth catalysts led to NCO-terminated prepolymers with higher molar masses, viscosity and residual isocyanate monomer than other catalysts (Ti-, Zn- and DABCO) and uncatalysed system. This study suggests efficient catalysts, which tin-free or even metal-free, to adjust kinetics and prepolymer features.At the crosslinking stage, associations of metallic and/or organic catalysts also demonstrated an important impact on crosslinking via hydrolysis/urea-bond formation reaction rate. In poly(urethane–urea)s, DMA analysis evidenced that H-bonding was mainly responsible for mechanical properties. A methodology is provided to shield H-bonding in these materials by addition/diffusion of polar molecules into the 3D-network. Although catalysts demonstrated a huge influence on NCO-terminated prepolymer structures, it was found that thermomechanical properties of crosslinked materials seemed to be almost independent of the choice of catalyst, in the absence of inorganic fillers. However, for formulations with inorganic fillers, we evidenced major differences in terms of elongations at break and moduli at 100% elongation, which demonstrates the crucial impact of catalyst nature on chemical and physical 3D-network structures. Harnessing catalysis enhanced the versatility of PU-based materials by adjusting and expanding thermal and mechanical PU properties only by appropriate choice of catalyst system. Finally, the catalyst associations suggested in this study can effectively be either tin-free or even metal-free systems.
4. Experimental
4.1. Chemical products
4,4′-Methylenebis(phenyl isocyanate) (4,4′-MDI, 98%), 1,4-Diazabicyclo[2.2.2]octane (DABCO), reagentPlus, ≥99%, 2,2′-dimorpholinodiethylether (DMDEE, 97%) and bismuth neodecanoate were purchased from Sigma-Aldrich and used without further purification. Dioctyltin dilaurate was purchased from TIB Chemicals AG and zinc neodecanoate from King Industries. Diisopropoxy-bis(ethylacetoacetato)titanate was purchased from ABCR chemicals. Xylene (99%) was purchased from Bisolve Chemicals. Poly(propylene glycol)s diol (Mn = 2000 g mol−1, IOH = 56 mg KOH per g) and triol (Mn = 3780 ± 100 g mol−1, IOH = 44.5 ± 1.5 mg KOH per g) were purchased from DOW and Bostik.4.2. Reagents and solvents
Poly(propylene glycol)s were dried by heating at 80 °C under vacuum during 16 hours. Xylene, methanol and deuterated acetone were dried over 4 Å molecular sieves. 4,4′-methylenebis(phenyl isocyanate) (4,4′-MDI) was conserved under argon at −20 °C.The preparation of polyurethane mastics was divided in two main steps: the prepolymerization and the crosslinking stage.
In the first one, an NCO-terminated prepolymer was synthetized and the second stage consisted in the crosslinking of these prepolymers with air moisture in order to obtain films as final material.
4.3. Preparation of polyurethane prepolymers
Polyurethanes prepolymers were synthetized at three NCO/OH ratio equal to 1.7; 1.83 and 2.05. Before the synthesis, jacketed reactor was dried at 80 °C under vacuum during 30 minutes. Then, reaction was carried out under argon atmosphere, by mechanical stirring at 80 °C. For NCO/OH equal to 1.83, 41.95 g of triol PPG, 27.54 g of diol PPG, 16.5 g of xylene and catalysts were slowly added to 13.98 g of 4,4′-MDI under stirring (120–130 rpm). At the end of reaction, polyurethanes prepolymers were kept under argon.4.4. Preparation of polyurethane thin films
Crosslinking occurred between NCO-terminated prepolymers and air moisture. A thin film of polymers (300 μm thick) was realized on glass plate using a film applicator. Films were crosslinked during one night and FTIR analysis were used to verified the disappearance of the NCO band.4.5. Preparation of polyurethane dumbbells
Crosslinking occurred between NCO-terminated prepolymers and air moisture in presence of additives and fillers such as calcium carbonate. In each formulation, 32.6 wt% of NCO-terminated prepolymers and 0.2 wt% of selected crosslinking catalysts were added to inorganic fillers33 (formulations were given in Table S3, ESI†). The dispersion of fillers was done by mechanical stirring at 500–1000 rpm. Regular dumbbells samples are prepared (reduced section size L × l × e: 20 × 4×0.5 mm) and respected the international standard ISO 37 (2011).334.6. Infrared spectroscopy
Reactions were monitoring by attenuated total reflectance module of infrared spectroscopy on a TGA-IR Nicolet iS50 from ThermoFischer, with 32 scans and a resolution of 4. For kinetics analysis, NCO peak at 2270 cm−1 was integrated in function of time.4.7. Size exclusion chromatography
Analysis are carried out using a Viscotek GPCMax from Malvern Instruments. Experiments occurred at 35 °C, in THF as eluent at a flow rate of 1 ml min−1. The sample injection is set at 100 μL. Samples were prepared at a concentration of 5 mg ml−1 with THF and filtered with a PTFE filter at 0.45 μm. Molar masses and remaining isocyanates were determined by using RI detection and polystyrene calibration.4.8. NMR spectroscopy
Liquid-state 1H and 13C spectra were recorded using a Bruker Advance III 300 spectrometer operating at 300 MHz for 1H (corresponding to ∼75 MHz for 13C) with a 5 mm tube in acetone-d6 or benzene-d6 at room temperature. The following conditions were used: for 1H NMR, pulse 90°; relaxation delay 2 s; 32 scans; for 13C NMR, relaxation delay 3 s; 8192 scans. Solid-state 1H and 13C spectra were recorded using a Bruker Advance III 700 spectrometer operating at 700 MHz for 1H (corresponding to ∼176 MHz for 13C) in a 3.2 mm rotor (rotational speed: 22 kHz). The following conditions were used: for 1H NMR, relaxation delay 5 s; 8 scans, sequence: zgse pulse 90° with solid echo; for 13C NMR, relaxation delay 2 s; 4096 scans, sequence HPDEC with solid echo.4.9. Differential scanning calorimetry (DSC)
DSC analyses were performed on a DSC 3 + StarSyst from Mettler Toledo in 40 μL aluminum pans. For these experiments, 2 heating ramps separated by a cooling ramp were realized. A first heating from −80 °C to 200 °C was realized at 10 K min−1; then the samples were cooled to −80 °C at 5 K min−1 and a second heating to 200 °C at 10 K min−1 was used to determine the glass transition temperature. All experiments occurred under 30 mL min−1 of nitrogen atmosphere.4.10. DMA
Experiments were realized on DMA 1 Mettler Toledo in the film tension geometry. Rectangular samples (size L × l × e: 5 × 5 × 0.13 mm) were heated from −90 °C to 40 °C at a rate of 2 K min−1 and tested at 1 Hz and 10 μm of amplitude with a preload force equal to 2N.4.11. Tensile test
Experiments were performed on Switch Swicki at an extention rate of 500 mm min−1 using a cell of 2.5 kN. Elongation at break, maximal modulus at break and modulus at 100% of elongation were measured according ISO 37 (2011) standard.334.12. Rheology
Rheological measurements were conducted on a Mars 60 apparatus from ThermoScientific using a plane–plane geometry of 20 mm of diameter. Viscosities were measured under nitrogen atmosphere at 25 °C using controlled shear rate from 100 s−1 to 0.1 s−1 and back from 0.1 s−1 to 100 s−1. All samples were found Newtonian over the whole range of shear rate.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Franck Collas for his expertise on thermomechanical analyses, Damien Montarnal for valuable discussions on mechanical properties of materials, Oliver Boyron and Manel Taam for their expertise on SEC characterization and David Gajan for solid-state NMR spectroscopy. The authors also acknowledge Bostik for financial support (PA's PhD fellowship). This article is a tribute to the 50-year anniversary of the French Polymer Group (Groupe Français des Polymères – GFP). We dedicate this work to the memory of Prof. Jean-Pierre Pascault.Notes and references
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- H. M. C. C. Somarathna, S. N. Raman, D. Mohotti, A. A. Mutalib and K. H. Badri, Constr. Build. Mater., 2018, 190, 995–1014 CrossRef CAS.
- H.-W. Engels, H.-G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., Int. Ed., 2013, 52, 9422–9441 CrossRef CAS.
- M. Szycher, Szycher's Handbook of Polyurethanes, CRC Press, New York, 2nd edn, 2013 Search PubMed.
- G. Wegener, M. Brandt, L. Duda, J. Hofmann, B. Klesczewski, D. Koch, R.-J. Kumpf, H. Orzesek, H.-G. Pirkl, C. Six, C. Steinlein and M. Weisbeck, Appl. Catal., A, 2001, 221, 303–335 CrossRef CAS.
- J. W. Britain and P. G. Gemeinhardt, J. Appl. Polym. Sci., 1960, 5(11), 207–211 CrossRef.
- J. Burkus, J. Org. Chem., 1961, 26(3), 779–782 CrossRef CAS.
- A. Farkas and G. A. Mills, Adv. Catal., 1962, 13, 393–446 CAS.
- R. A. Ligabue, A. L. Monteiro, R. F. de Souza and M. O. de Souza, J. Mol. Catal. A: Chem., 1998, 130, 101–105 CrossRef CAS.
- W. J. Blank, Z. A. He and E. T. Hessell, Prog. Org. Coat., 1999, 35, 19–29 CrossRef CAS.
- A. L. Silva and J. C. Bordado, Catal. Rev., 2004, 46, 31–51 CrossRef.
- R. V. Maris, Y. Tamano, H. Yoshimura and M. Gay, J. Cell. Plast., 2005, 41, 305–322 CrossRef.
- V. De Lima, N. Da Silva Pelissoli, J. Dullius, R. A. Ligabue and S. Einloft, J. Appl. Polym. Sci., 2010, 115, 1797–1802 CrossRef CAS.
- R. Gogoi, U. K. Niyogi, M. S. Alam and D. S. Mehra, J. Appl. Polym. Sci., 2012, 29, 451–462 CAS.
- J. Alsarraf, Y. A. Ammar, F. Robert, E. Cloutet, H. Cramail and Y. Landais, Macromolecules, 2012, 45, 2249–2256 CrossRef CAS.
- Y. Schellekers, B. Trimpont, P.-J. Goelen, K. Binnemans, M. Smet, M.-A. Persoons and D. De Vos, Green Chem., 2014, 16, 4401–4407 RSC.
- F.-Z. Belmokaddem, J. Dagonneau, J. Lhomme, R. Blanc, A. Garduno-Alva, C. Maliverney, A. Baceiredo, E. Maerten, E. Fleury and F. Méchin, Des. Monomers Polym., 2016, 19, 347–360 CrossRef CAS.
- M. A. Semsarzadeh and A. H. Navarchian, J. Appl. Polym. Sci., 2003, 90, 963–972 CrossRef CAS.
- S. K. Rath, A. M. Ishack, U. G. Suryavansi, L. Chandrasekhar and M. Patri, Prog. Org. Coat., 2008, 62, 393–399 CrossRef CAS.
- Z. Gao, D. Wu, W. Su and X. Ding, J. Appl. Polym. Sci., 2009, 111, 1293–1299 CrossRef CAS.
- X. Wei, P. N. Shah, K. Bagdi, K. Seethamraju and R. Faust, J. Macromol. Sci., Part A: Pure Appl.Chem., 2014, 51, 6–15 CrossRef CAS.
- S. Dworakowska, D. Bogdal, F. Zaccheria and N. Ravasio, Catal. Today, 2014, 223, 148–156 CrossRef CAS.
- Q. Zhang, X.-M. Hu, M.-Y. Wu, Y.-Y. Zhao and C. Yu, J. Appl. Polym. Sci., 2018, 46460, 1–11 Search PubMed.
- Y. V. Yakovlev, Z. O. Gagolkina, Eu. V. Lobko, I. Khalakhan and V. V. Klepko, Compos. Sci. Technol., 2017, 144, 208–214 CrossRef CAS.
- C. Tan, V. Luona, T. Tirri and C.-E. Wilen, Polymers, 2018, 10, 1–16 Search PubMed.
- K. Schewetlick, R. Noack and F. Stebner, J. Chem. Soc., Perkin Trans. 2, 1994, 3, 599–608 RSC.
- R. P. Houghton and A. W. Mulvaney, J. Organomet. Chem., 1996, 518, 21–27 CrossRef CAS.
- S.-G. Luo, H.-M. Tan, J.-G. Zhang, Y.-J. Wu, F.-K. Pei and X.-H. Meng, J. Appl. Polym. Sci., 1997, 65, 1217–1225 CrossRef CAS.
- S. D. Evans and R. P. Houghton, J. Mol. Catal. A: Chem., 2000, 164, 157–164 CrossRef CAS.
- C. Spino, M.-A. Joly, C. Godbout and M. Arbour, J. Org. Chem., 2005, 70, 6118–6121 CrossRef CAS.
- H. Sardon, A. C. Engler, J. M. W. Chan, J. M. Garcia, D. J. Coady, A. Pascual, D. Mercerreyes, G. O. Jones, J. E. Rice, H. W. Horn and J. L. Hedrick, J. Am. Chem. Soc., 2013, 135, 16235–16241 CrossRef CAS.
- E. Yilgör, E. Burgaz, E. Yurtserver and I. Yilgör, Polymers, 2000, 41, 849–857 CrossRef.
- F. Sanz, G. Michaud, F. Simon, J. Raynaud, V. Monteil and L. Bosco, WO2019/122706, 2019.
Footnote |
| † Electronic supplementary information (ESI) available: Determination of NCO-terminated prepolymers kinetics by FTIR/NMR; influence of catalyst concentration on prepolymerization rate monitored by FTIR; 1H NMR spectra of prepolymers terminated NCO structures with different catalysts; calibration of SEC-THF for residual isocyanate titration; mechanisms of organic or metallic catalysts at prepolymerization stage and of urea formation at the crosslinking stage; influence of catalysts on mechanical properties by DMA/DSC; 1H NMR study to determine the optimal DMSO concentration to shield urea H-bonding within PU 3D-network; formulations of crosslinked polyurethanes in presence of fillers; elucidation of crosslinked PU chemical structure by solid-state NMR; example of stress–strain curve for polyurethane materials. See DOI: 10.1039/d0py00864h |
| This journal is © The Royal Society of Chemistry 2020 |