
Impacts of performing electrolysis during organocatalyzed atom transfer radical polymerization†
Daniel A.
Corbin
 ,
Blaine G.
McCarthy
and
Garret M.
Miyake
*
,
Blaine G.
McCarthy
and
Garret M.
Miyake
*
Department of Chemistry, Colorado State University, Fort Collins, Colorado 80523-1872, USA. E-mail: garret.miyake@colostate.edu
First published on 30th June 2020
Abstract
An electrochemical variant of organocatalyzed atom transfer radical polymerization (O-ATRP) is developed and investigated. Inspired by electrochemically mediated atom transfer radical polymerization (eATRP), potentiostatic electrolysis is used to manipulate the catalyst's redox states in O-ATRP to understand whether deactivation in O-ATRP can be enhanced to improve polymerization control. During the course of this work, several possible side reactions are investigated, and the electrochemical apparatus is optimized to reduce side reactions at the counter electrode. This electrochemically modified O-ATRP method (eO-ATRP) is then studied at different applied potentials, under different irradiation conditions, and with two photoredox catalysts to understand the impact of electrolysis on polymerization control. Ultimately, although electrolysis was successfully used to improve polymerization control in O-ATRP, some additional challenges have been identified. Several key questions are postulated to guide future work in this area.
Introduction
First reported in 2014, organocatalyzed atom transfer radical polymerization (O-ATRP) is a controlled radical polymerization method employing organic photoredox catalysts (PCs) for the production of polymers with targeted molecular weights and architectures.1,2 The proposed mechanism of O-ATRP proceeds through absorption of light by a PC to access an excited state (PC*). This excited state then reduces the alkyl-halide(bromide) bond of an initiator or polymer chain-end to generate the PC radical cation (PC˙+), Br−, and ‘active’ radicals capable of polymerization propagation with vinyl monomers (Fig. 1). Importantly, the PC˙+ that forms mediates deactivation in O-ATRP, during which bromide is reinstalled on the chain-end of a polymer to generate a ‘dormant’ species and the ground state PC.1–4 It has been proposed that deactivation in O-ATRP could proceed through a termolecular reaction, in which PC˙+, Br−, and the radical chain-end react simultaneously to form the dormant polymer and ground state PC.5 While computational results support that this termolecular reaction is thermodynamically feasible, our working hypothesis is that deactivation proceeds through a bimolecular reaction, in which PC˙+ and Br− preassociate to form the PC˙+Br− ion pair that then reacts with the propagating radical. Regardless of the exact mechanism of this process, the effect is the same: deactivation reduces the concentration of radicals in solution and thereby suppresses radical-based termination reactions, which would otherwise hinder control over polymer structure.6–10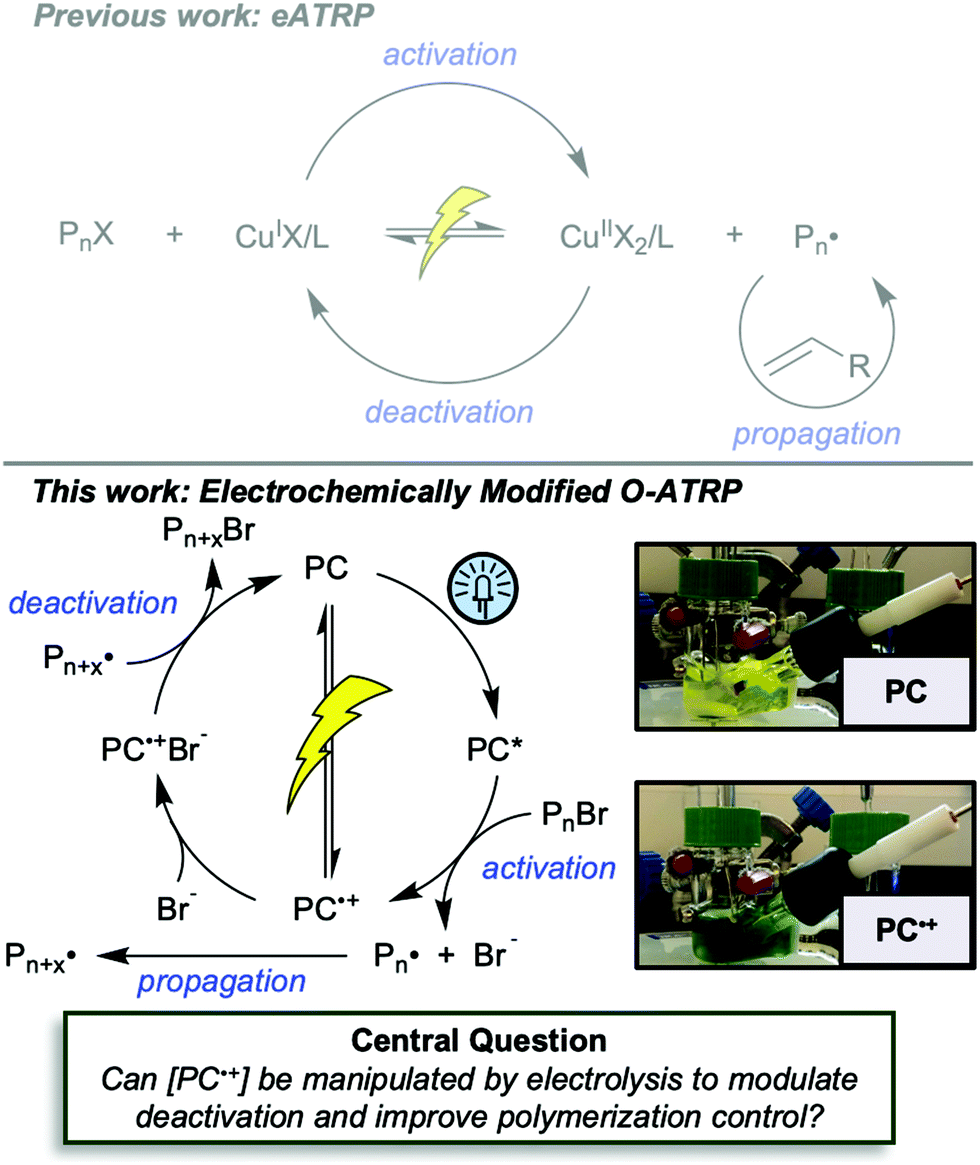 | ||
| Fig. 1 Previous work demonstrated the ability to mediate ATRP using electrochemistry (top). In this work (bottom), we ask whether this principle can be applied to O-ATRP to control the concentration of PC˙+ and thereby control deactivation in this polymerization method. Figure inset (bottom right) demonstrates the conversion of PC to PC˙+ using potentiostatic bulk electrolysis. | ||
Since the inception of this method, much work has focused on expanding the utility of O-ATRP through various approaches. Some strategies have focused on the development of new photoredox catalysts3,4,11–17 as a means to access the polymerization of new monomers, such as acrylonitrile11 and acrylates.17 Alternatively, other advancements have come through the application of O-ATRP for the synthesis of materials with advanced architectures18,19 and applications,20,21 while some investigations have focused on understanding the mechanism of O-ATRP5,22 and the structure–property relationships of the PCs3,4,15,23–25 employed therein. Despite these advancements, the monomer scope of O-ATRP and its ability to produce polymers of high molecular weight26–31 remains limited, especially in comparison to metal catalyzed ATRP.32
To overcome these limitations and further advance the O-ATRP method, more detailed investigation of the deactivation mechanism and methods to control this process are desirable. In some sense, modulation of deactivation has been attempted through the development of new PCs with more oxidizing radical cations,17 which might mediate a faster deactivation process than less oxidizing radical cations. However, we envisioned a more direct approach to study the deactivation process would be to manipulate the concentration of deactivator present rather than the oxidation potential of the species. To achieve this effect, we drew inspiration from electrochemically mediated ATRP (eATRP, Fig. 1), wherein electrochemistry has been used to control activator and deactivator concentrations in metal catalyzed ATRP and mediate controlled polymerizations under a range of different conditions.33–37 Analogously, one can envision manipulating the concentration of PC˙+ in solution by potentiostatic electrolysis of the PC (Fig. 1, inset) according to the Nernst equation (eqn (1)). By performing this process to generate a higher [PC˙+] in situ, it might be possible to increase the rate of deactivation to afford enhanced polymerization control in challenging systems. As such, this work probes whether electrolysis of the PC can be used during O-ATRP to increase the [PC˙+] to improve deactivation, as well as the impact of performing electrolysis on the polymerization solution.
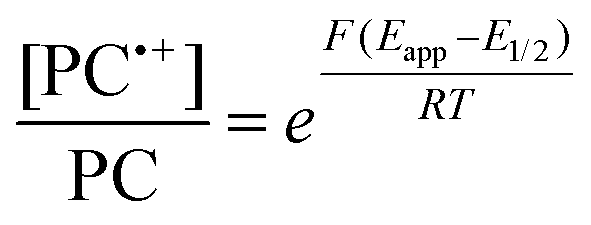 | (1) |
Results and discussion
Initial conditions and polymerization results
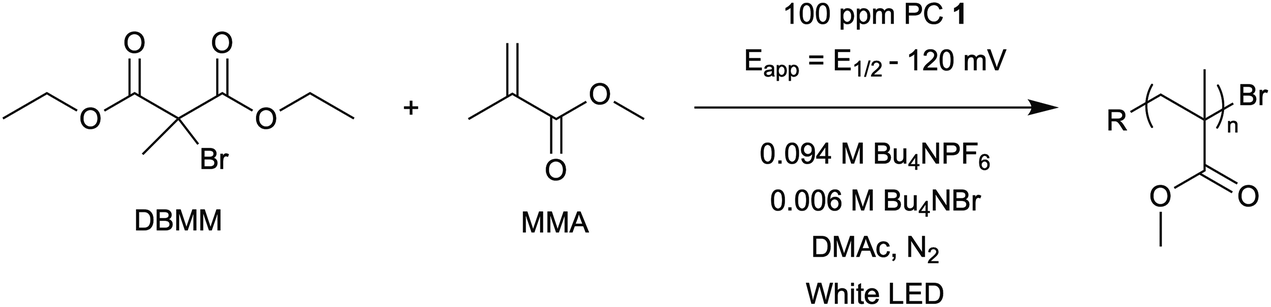
The central hypothesis of this work is that by applying an appropriate electrochemical potential (Eapp), the concentration of PC˙+ (the deactivator) in O-ATRP can be manipulated to improve polymerization control. Polymerization control in this work was determined by four criteria: (1) linear pseudo-first order kinetics of monomer conversion, (2) linear and increasing molecular weight (Mn) as a function of monomer conversion, (3) decreasing and low dispersity (Đ < 1.5) during the course of polymerization, and (4) achieving initiator efficiency near 100% (I* = Mn, theo/Mn, exp).To investigate the effects of increasing the concentration of PC˙+ in O-ATRP using electrolysis, a degree of polymerization (DP) of 200 was targeted since previous reports at this DP exhibited only moderate control relative to lower targeted DPs.3,4,16 Moreover, to minimize the possibility of introducing redox side reactions, dihydrophenazine PCs were employed, since this family of PCs would require application of the least oxidizing potential to achieve a higher concentration of PC˙+ relative to other PC families. However, within the dihydrophenazine family, radical addition to the phenazine core has been proposed as a possible side reaction leading to poor initiator efficiency in O-ATRP. As such, PC 1 was chosen because the core-positions of this PC are blocked by naphthyl substituents (Fig. 2), reducing the risk of this PC reacting undesirably with radicals in solution.16 With this PC chosen, all other polymerization conditions (Fig. 2) were selected based on published conditions for O-ATRP using PC 1.16
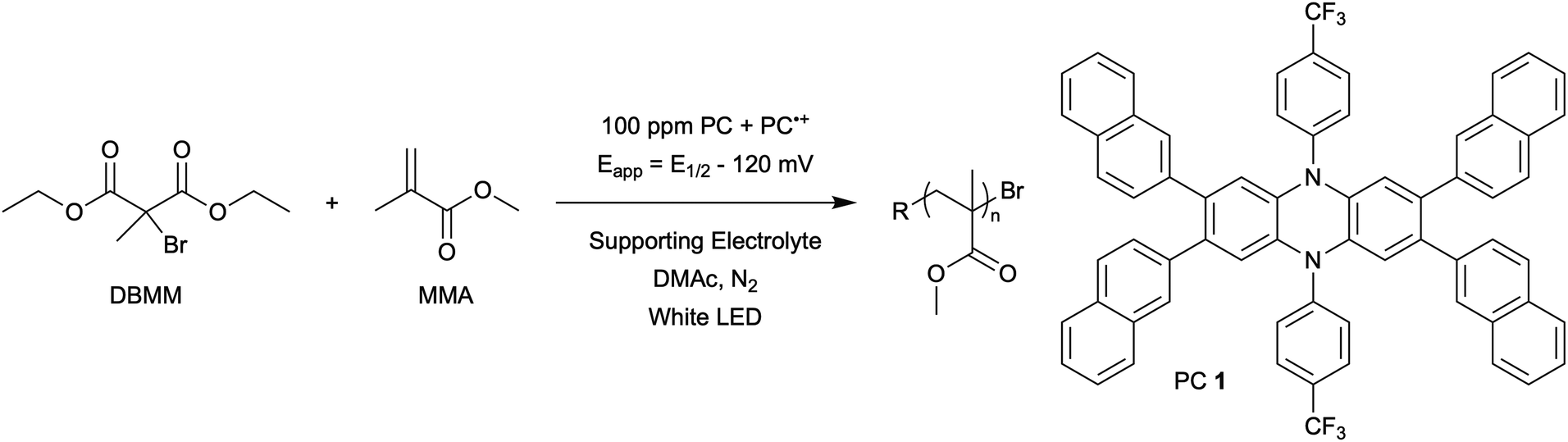 | ||
| Fig. 2 General scheme for the eO-ATRP of MMA using DBMM as the initiator and 100 ppm of PC 1. | ||
For the supporting electrolyte (SE), a 0.1 M mixture of tetra-n-butylammonium hexafluorophosphate (Bu4NPF6, 94%) and tetra-n-butylammonium bromide (Bu4NBr, 6%) was initially chosen based on conditions reported for eATRP33 and altered later. A lower Eapp was used relative to eATRP [Eapp = E1/2 − 120 mV vs. Eapp ∼ E1/2(CuII/CuI)]. In eATRP, both the concentrations of CuI and CuII can be manipulated by electrolysis, directly impacting both activation and deactivation. However, in O-ATRP activation is mediated by PC* (Fig. 1), the concentration of which is likely dependent on the intensity of the light source. In fact, based on published data38 for common PCs in O-ATRP, estimates indicate that only up to about 1% of the PC exists as PC* under steady state conditions (see Estimation of excited state PC concentration in ESI†). As such, electrolysis conditions were chosen to produce roughly 1% PC˙+ based on eqn (1). Finally, to prevent side reactions from occurring at the counter electrode, a U-cell was chosen with a very-fine glass frit separating the counter electrode from the polymerization solution (see Experimental equipment in ESI†).
To evaluate the impact of electrolysis on polymerization control, eO-ATRP was conducted in the presence of an oxidizing applied potential (Eapp = E1/2 − 120 mV) (Fig. 3). It was hypothesized that these eO-ATRP conditions would lead to a slower overall rate of polymerization and the synthesis of PMMA with lower Đ, yet neither effect was observed. The observed rate constants of the polymerizations (O-ATRP = 0.17 h−1, eO-ATRP = 0.14 h−1) and Đ of the PMMA synthesized (Đ = 1.23 for O-ATRP, Đ = 1.19 for eO-ATRP) were similar. Unexpectedly, eO-ATRP exhibited some loss of control, with I* values deviating significantly from 100%, especially at higher monomer conversions (I* = 72% at 66% conversion and I* = 61% at 94% conversion).
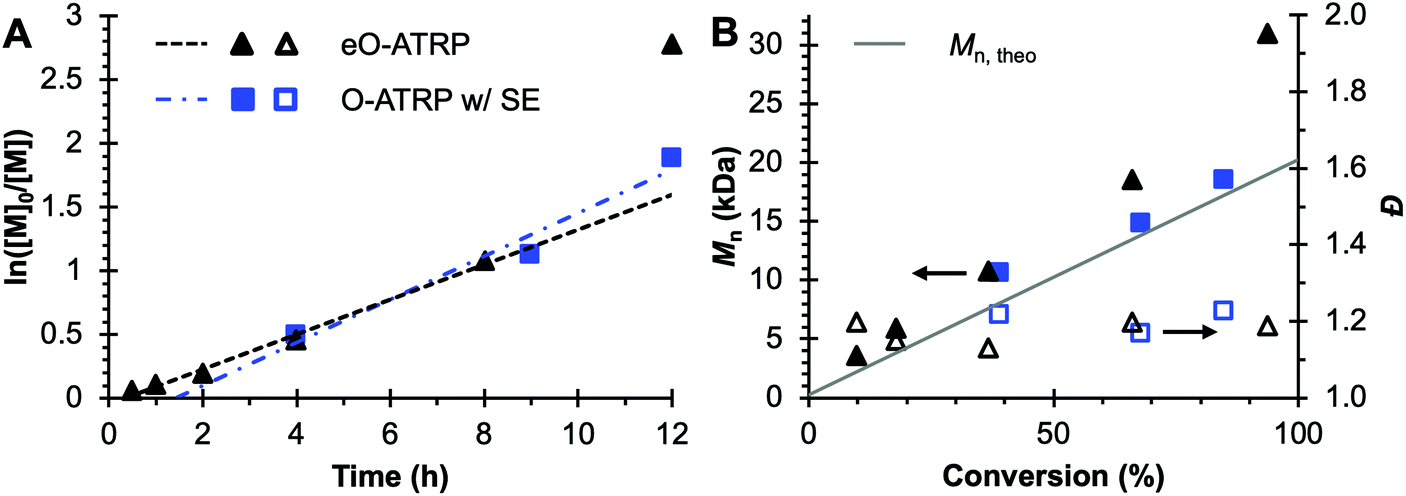 | ||
Fig. 3 Plot of the natural logarithm of monomer (M) consumption over time (A). Molecular weight (filled markers) and Đ (hollow markers) evolution (B) for eO-ATRP (black triangles) and O-ATRP with supporting electrolyte (blue squares). Conditions: [MMA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[DBMM]:[1] = [1000]:[5]:[0.1], 2 mL MMA, 2 mL DMAc, SE = 0.094 M Bu4NPF6 and 0.006 M Bu4NBr. Reactions performed in a U-cell and irradiated with a high-power white LED (see Experimental equipment in ESI†). For eO-ATRP, working electrode = glassy carbon, counter electrode = Pt wire, reference electrode = Ag wire quasi-reference electrode, and Eapp = E1/2 − 120 mV. :[DBMM]:[1] = [1000]:[5]:[0.1], 2 mL MMA, 2 mL DMAc, SE = 0.094 M Bu4NPF6 and 0.006 M Bu4NBr. Reactions performed in a U-cell and irradiated with a high-power white LED (see Experimental equipment in ESI†). For eO-ATRP, working electrode = glassy carbon, counter electrode = Pt wire, reference electrode = Ag wire quasi-reference electrode, and Eapp = E1/2 − 120 mV. | ||
Hypotheses for poor control
In total, nine hypotheses to explain the observed data were formulated. While all nine hypotheses are stated below, hypotheses 1–8 are also depicted schematically in Table 1.
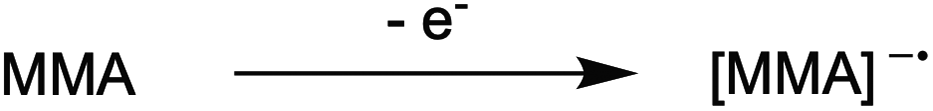
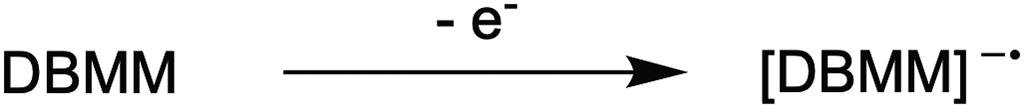

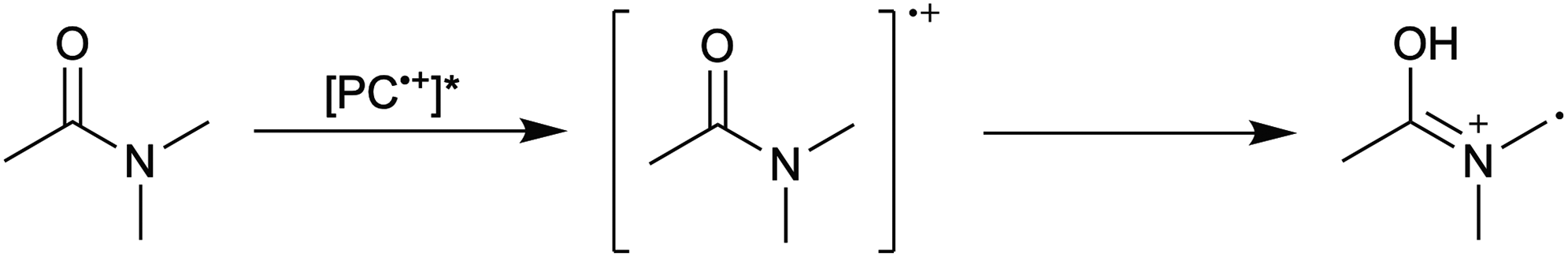

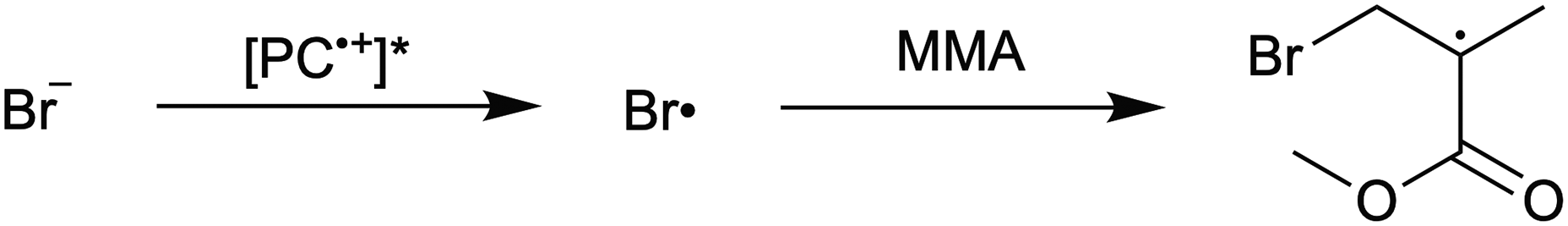

1. Due to the highly reducing nature of PC* [E°(1˙+/1*) ∼ −1.8 V vs. SCE],16 tetra-n-butylammonium cation (Bu4N+) is reduced to form a reactive species that hinders polymerization control. This process likely occurs through formation of Bu4N˙, which rapidly decomposes to tributylamine and butyl radical.39 The amine may act as an electron donor to quench PC˙+, leading to poor deactivation. Further, butyl radical formation could lead to unwanted initiation and termination events in the polymerization.
2. MMA is oxidized at the surface of the working electrode to generate a reactive species, which either reacts with the PC or interrupts the polymerization.
3. DBMM is oxidized at the surface of the working electrode to generate a reactive species, which either reacts with the PC or interrupts the polymerization. Consumption of the initiator through this side reaction would also lower I*.
4. Bromide ion, either from the supporting electrolyte or the activation of an alkyl-bromide bond, is oxidized at the working electrode, generating a bromine radical capable of initiating polymer chains.
5. Photoexcitation of PC˙+ generates a strongly oxidizing excited-state species, which oxidizes DMAc to generate a reactive radical capable of performing initiation and termination reactions.
6. Photoexcited PC˙+ oxidizes the radical chain-end of a propagating polymer, generating a reactive carbocation that rapidly and irreversibly terminates under O-ATRP conditions.
7. Photoexcited PC˙+ oxidizes Br−, either from the supporting electrolyte or from the activation of an alkyl-bromide bond, generating a bromine radical capable of initiating new polymer chains.
8. Hexafluorophosphate from the supporting electrolyte competitively ion-pairs with PC˙+ to form PC˙+PF6−, leading to poor polymerization control. If this process occurs to a significant extent, it would limit the formation of PC˙+Br− and thereby reduce the rate of deactivation.
9. Under current conditions, the counter electrode is insufficiently separated from the polymerization. As such, control is lost either as the PC and PC˙+ diffuse to the counter electrode and undergo degradation, or as reactive species produced at the counter electrode diffuse into the polymerization and cause side reactions.
With these hypotheses in hand, experiments were then devised to test and support or disprove each one. For example, in hypothesis 1, it is proposed that the reduction of Bu4N+ (hypothesis 1) could be responsible for side reactivity in eO-ATRP leading to poor control. If this hypothesis is true, changing the supporting electrolyte to LiPF6 (Table 2, entry 1; also see ESI†) should eliminate reduction of the cation and thereby improve the polymerization outcome, as 1* should not be able to reduce Li+. Of course, this experiment is based on the assumption that no other significant side reactivity would occur with LiPF6, but this assumption is supported by later experiments with this supporting electrolyte (vide infra). However, no improvement in polymerization control was observed in this experiment (Đ = 2.08, I* = 82%), disproving hypothesis 1.
To test hypotheses 2–4, cyclic voltammetry (CV) was performed to examine the redox behavior of each component of the polymerization solution. Since the CV of 1 in DMAc with 0.1 M Bu4NPF6 has previously been reported elsewhere,16 the redox stability of the solvent and this supporting electrolyte were not examined. To test the redox stability of MMA (hypothesis 2), CV was used to examine a mixture of MMA and DMAc in a ratio corresponding to that used in eO-ATRP (Fig. S35†). No current response was observed in the relevant potential range (−0.1–0.1 V vs. Ag/AgNO3), disproving hypothesis 2. Next, DBMM was added to the solution and its redox stability (hypothesis 3) was examined by CV, which revealed only a reduction peak around −1.2 V vs. Ag/AgNO3 (Fig. S36†). Since no response was observed in the range relevant to eO-ATRP, these data disprove hypothesis 3.
A similar experiment was performed to test for Br− oxidation at the working electrode (hypothesis 4), where CV was used to examine a solution of 0.1 M Bu4NPF6 (94%) and Bu4NBr (6%) in MMA and DMAc. This time, an irreversible oxidation followed by a quasi-reversible redox couple was observed (Fig. S37†), presumably corresponding to Br− oxidation to form Br3−, followed by oxidation of Br3− to form Br2.39–43 However, no current response was seen in the appropriate potential range for eO-ATRP, indicating this redox reaction is unlikely to interfere in these polymerizations. Further evidence to disprove this hypothesis was found in a control reaction excluding 1 and light (see Control experiments section in ESI†). If Br− oxidation at the working electrode could lead to unwanted polymerization of MMA, it should be observable under these conditions. However, proton NMR analysis of the reaction solution after 24 h of electrolysis showed no polymer formation (Table 2, entry 2), disproving hypothesis 4.
Since no evidence could be found for deleterious side reactivity at the working electrode, hypotheses 5–7 for possible side reactions involving photoexcited PC˙+ were considered next. Each hypothesis is based on the concept that PC˙+ might be able to access a strongly oxidizing excited state by absorption of visible light. In turn, photoexcitation of this species might lead to the oxidation of DMAc (hypothesis 5), the radical chain-end of a propagating polymer (hypothesis 6), or Br− (hypothesis 7). Currently, no evidence exists to disprove the oxidation of DMAc by this species (hypothesis 5), so this hypothesis will be revisited later in the text (vide infra).
With regard to the oxidation of the chain-end radical (hypotheses 6), the ground state of PC˙+ is not sufficiently oxidizing to directly cause this side reaction, necessitating photoexcitation to make the oxidation thermodynamically feasible. However, it seems unlikely that this reaction would occur to a significant extent considering that the components of this reaction should both be in low concentrations. Due to deactivation in O-ATRP, the formation of chain-end radicals should be suppressed to prevent radical-coupling reactions. In addition, it seems unlikely that the concentration of photoexcited PC˙+ would be sufficient to react with this species to a significant degree, since the lifetimes of photoexcited species are generally quite short (10−9–10−6 s) and most of the PC˙+ should exist in the ground state. Of course, this argument does not necessarily mean that this side reaction does not take place in eO-ATRP. However, based on these kinetics considerations as well as experiments related to hypothesis 9 (vide infra), this side reaction does not appear sufficient to explain the current issues observed in eO-ATRP.
With regard to the oxidation of Br− by photoexcited PC˙+ (hypothesis 7), while a bimolecular reaction between Br− and photoexcited PC˙+ could be considered unlikely based on the same kinetic argument that is presented above (at least in the absence of a bromide-containing supporting electrolyte), it is also possible that PC˙+ and Br− could associate prior to photoexcitation. If photoexcitation of the PC˙+Br− ion pair occurred, the oxidation of Br− would be more feasible given the close proximity of these species, which would reduce the necessity for a long-lived PC˙+ excited state. Currently, no evidence exists to rule out the photoexcitation of PC˙+Br−. However, to our knowledge, no evidence for this side reaction in O-ATRP has yet been found, as the prevalence of this reaction would hinder the production of well-defined polymers by O-ATRP. Further, since an improvement in polymerization control was observed in experiments related to hypothesis 9 (vide infra), where this side reaction would have still been present, this reaction does not appear to be a significant contributor to poor control in eO-ATRP.
Another possibility that was considered is competitive ion pairing between PC˙+ and either Br− or PF6− (hypothesis 8). Depending on the relative strengths of ion pairing in PC˙+Br− and PC˙+PF6−, it is possible that the formation of PC˙+PF6− might prevent the formation of PC˙+Br− and thereby lower the rate of deactivation. To test this hypothesis, O-ATRP was carried out in the presence of 0.1 M LiPF6 (Table 2, entry 3; also see Fig. S39†), yielding PMMA with low Đ (Đ = 1.17) and good molecular weight control (I* = 114%). While this experiment does not indicate whether competitive ion pairing is present in eO-ATRP, it does suggest this interaction does not limit polymerization control, disproving hypothesis 8.
Therefore, the remaining hypotheses that were considered are the oxidation of DMAc by photoexcited PC˙+ (hypothesis 5) and insufficient separation of the counter electrode from the reaction solution (hypothesis 9). To test hypothesis 9, a new apparatus (Fig. 4) was employed featuring a vycor-glass frit (pore size ∼ 4 nm (ref. 44)) to separate the counter electrode instead of the previously used U-cells with very-fine glass frits (pore size ∼ 2 μm (ref. 45)). Excitingly, eO-ATRP with 0.1 M LiPF6 as the supporting electrolyte exhibited excellent control (Table 3, entry 4: Đ = 1.17, I* = 110%), with I* near 100% and Đ below 1.2 for nearly the entire polymerization (Fig. 5). Further, while this experiment does not directly test hypothesis 5 for DMAc oxidation by photoexcited PC˙+, it does suggest this side reaction is less significant, as its effects should have been observable even under these new experimental conditions. Based on this result and complying with Ockham's razor,46 hypothesis 9 appears to be the simplest explanation for why eO-ATRP initially showed limited improvement over O-ATRP under similar conditions. As such, all future experiments were performed with this new apparatus using vycor-glass separators for the counter and reference electrodes.
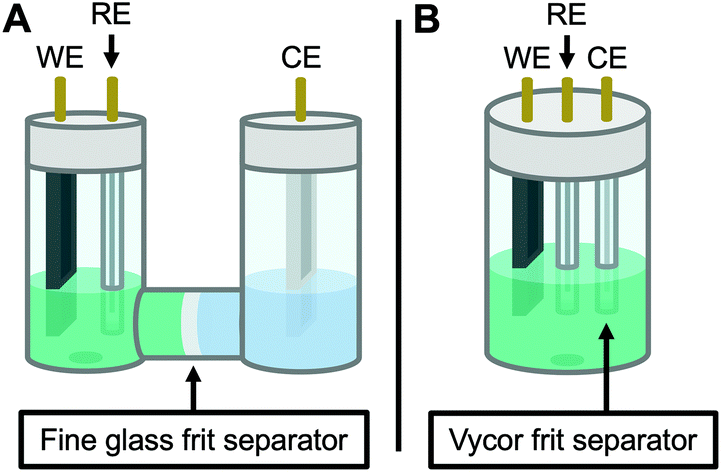 | ||
| Fig. 4 Diagram of the apparatus used in this work. Originally, a modified U-cell was employed to separate the working and counter electrode compartments (A). When this separator was found to be ineffective on the time scale of eO-ATRP, a new apparatus was developed using a 5-neck electrochemical flask (see Experimental equipment in ESI†) and vycor frit separators to isolate the counter electrode (B). WE = working electrode, RE = reference electrode, and CE = counter electrode. | ||
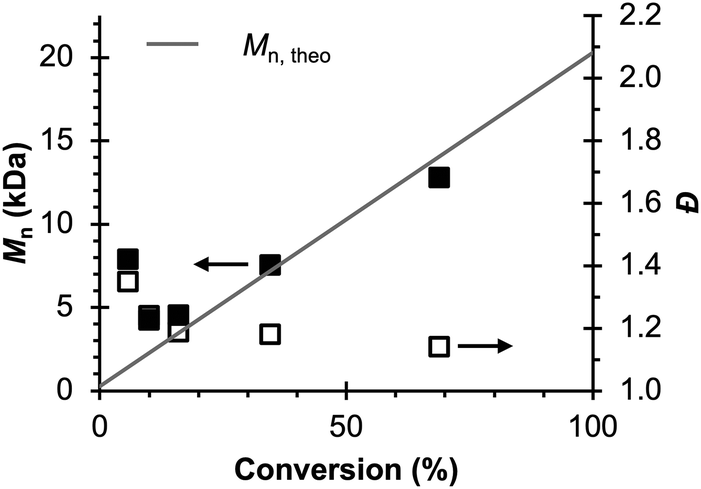 | ||
| Fig. 5 Evolution of molecular weight (filled squares) and Đ (hollow squares) for eO-ATRP using a vycor-glass frit to separate the counter electrode from the polymerization solution. Conditions: [MMA]:[DBMM]:[1] = [1000]:[5]:[0.1], 2 mL MMA, 2 mL DMAc, SE = 0.1 M LiPF6. Reaction performed in a 5-neck pear flask and irradiated with an 80 mm × 40 mm white LED well (9 LED segments, see Experimental Equipment in ESI†). Working electrode = glassy carbon, counter electrode = Pt wire, reference electrode = Ag/AgNO3, and Eapp = E1/2 − 120 mV. | ||
| Entry | Controla | Conv. (%) | M n, theo (kDa) | M n, GPC (kDa) | Đ |
I*c (%) |
|---|---|---|---|---|---|---|
|
a General conditions unless otherwise stated: [MMA]:[DBMM]:[1] = [1000]:[5]:[0.1], 2 mL MMA, 2 mL DMAc, SE = 0.1 M LiPF6. Reactions performed in a 5-neck pear flask and irradiated with an 80 mm × 40 mm white LED well (9 LED segments, see Experimental equipment in ESI†). Where applicable, working electrode = glassy carbon, counter electrode = Pt wire, reference electrode = Ag/AgNO3, and Eapp = E1/2 − 120 mV.
b Calculated by Mw/Mn.
c Calculated by Mn, theo/Mn, GPC.
|
||||||
| 4 | None | 69 | 14.0 | 12.8 | 1.17 | 110 |
| 5 | No electrolysis | 52 | 10.7 | 8.47 | 1.33 | 127 |
| 6 | No SE | 68 | 13.8 | 11.0 | 1.27 | 126 |
| 7 | No PC | 7 | 1.56 | 52.7 | 2.23 | 3.0 |
| 8 | No initiator | 63 | — | 239 | 1.94 | — |
| 9 | No light | 0 | — | — | — | — |
| 10 | No PC or light | 0 | — | — | — | — |
Impact of reaction parameters on control
To evaluate how each reaction component contributes to eO-ATRP, control polymerizations were performed (Table 3). In the absence of electrolysis (entry 5) or supporting electrolyte (entry 6), a controlled polymerization was still observed, but Đ and I* both rose (Đ = 1.33 and 1.27, I* = 127% and 126%, respectively) relative to eO-ATRP (Đ = 1.17, I* = 110%). These data demonstrate that improvement in polymerization control can be obtained by the application of an electrochemical potential. Reactions performed in the absence of PC (entry 7) or initiator (entry 8) exhibited characteristics of a free radical polymerization (Đ = 2.23 and 1.94, respectively), whereas reactions in the dark – with or without PC, entries 9 and 10, respectively – showed no conversion by 1H NMR after 24 hours.Further influences on polymerization control were studied by variation of the light source, application of a more oxidizing electrochemical potential, and use of a different PC (see Supplemental polymerization data in ESI†). Similar to previous O-ATRP systems,47 it was found that intensity of the light source had a significant impact on polymerization control. Lowering the intensity of the light caused a decrease in polymerization control, as observed by a gradual increase in Đ and deviation of I* from 100% (Fig. S43 and S44†). Interestingly, while a small increase in light intensity afforded similar control (Fig. S45†), large increases in light intensity from use of high-power LEDs resulted in a decrease in control (Fig. S48 and S49†). When a more oxidizing electrochemical potential was applied to this system (Eapp = E1/2 − 60 mV) to compensate for a possible increase in the rate of activation, further loss of control was observed (Fig. S50†). While this result is consistent with the possibility of a side reaction stemming from photoexcitation of PC˙+, further investigation of this possible reactivity is necessary. Finally, eO-ATRP was attempted with 3,7-di(4-biphenyl)-1-naphthyl-10-phenoxazine (2) as the PC. However, no improvement in polymerization control was observed, as electrolysis led to a significant increase in Đ and complete loss of molecular weight control (Fig. S52 and S53†).
Conclusion
In summary, through a number of control experiments, we have investigated the impact of performing electrolysis during O-ATRP to manipulate the concentration of deactivator in solution. Using cyclic voltammetry, several background reactions at the working electrode were evaluated and ruled out. The formation of bromine radical at the working electrode to initiate undesired polymerizations was further probed through a control polymerization, although this reaction did not appear operative under the conditions used in this work. Further, the impact of competitive ion pairing between the PC radical cation and PF6− from the supporting electrolyte was studied but found to be insignificant under these conditions. While the possibility of side reactivity originating from photoexcitation of the PC radical cation was also proposed, ultimately it was discovered that optimization of the electrochemical apparatus to prevent side reactions at the counter electrode was most important for establishing a controlled polymerization.Although some improvement in polymerization control was observed in eO-ATRP relative to O-ATRP, this work has revealed the complexity of performing electrolysis during O-ATRP. Based on these results, several questions arise that are the focus of our ongoing work and that we believe will further improve the results of eO-ATRP. These questions include:
1. What is the effect of the supporting electrolyte on PC redox and photophysical properties?
2. Is the PF6− anion truly inert, or does competitive ion-pairing occur to any degree that might impact polymerization control?
3. If competitive ion-pairing occurs, is this effect more prominent for certain PCs or PC families than others?
4. Are there any side reactions through which PC˙+ is consumed during O-ATRP, such that increasing the concentration of PC˙+ in eO-ATRP increases the occurrence of these degradation pathways?
5. Is PC˙+Br− truly the deactivator in O-ATRP, or is another species responsible for this process?
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors are grateful for support provided by Colorado State University. Research reported in this publication was also supported by the National Institute of General Medical Sciences (Award R35GM119702) of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. B. G. M. acknowledges support from the NSF GRFP. D. A. C. would also like to thank Dr Scott Folkman for helpful discussions on electrochemistry, as well as Max Kudisch and Tara Mensch for assistance with the design and assembly of 3D printed photoreactors used in this work.References
- G. M. Miyake and J. C. Theriot, Macromolecules, 2014, 47, 8255–8261 CrossRef CAS.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. R. de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed.
- J. C. Theriot, C. H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed.
- R. M. Pearson, C. H. Lim, B. G. McCarthy, C. B. Musgrave and G. M. Miyake, J. Am. Chem. Soc., 2016, 138, 11399–11407 CrossRef CAS PubMed.
- X. C. Pan, C. Fang, M. Fantin, N. Malhotra, W. Y. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewskit, J. Am. Chem. Soc., 2016, 138, 2411–2425 CrossRef CAS PubMed.
- J. C. Theriot, B. G. McCarthy, C. H. Lim and G. M. Miyake, Macromol. Rapid Commun., 2017, 38, 1700040 CrossRef PubMed.
- E. H. Discekici, A. Anastasaki, J. R. de Alaniz and C. J. Hawker, Macromolecules, 2018, 51, 7421–7434 CrossRef CAS.
- G. Yilmaz and Y. Yagci, Polym. Chem., 2018, 9, 1757–1762 RSC.
- N. Corrigan, S. Shanmugam, J. T. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC.
- M. Chen, M. J. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- X. C. Pan, M. Lamson, J. J. Yan and K. Matyjaszewski, ACS Macro Lett., 2015, 4, 192–196 CrossRef CAS.
- S. Dadashi-Silab, X. C. Pan and K. Matyjaszewski, Chem. – Eur. J., 2017, 23, 5972–5977 CrossRef CAS PubMed.
- M. D. Ryan, J. C. Theriot, C. H. Lim, H. S. Yang, A. G. Lockwood, N. G. Garrison, S. R. Lincoln, C. B. Musgrave and G. M. Miyake, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3017–3027 CrossRef CAS PubMed.
- V. K. Singh, C. Yu, S. Badgujar, Y. Kim, Y. Kwon, D. Kim, J. Lee, T. Akhter, G. Thangavel, L. S. Park, P. C. Nandajan, R. Wannemacher, B. Milian-Medina, L. Luer, K. S. Kim, J. Gierschner and M. S. Kwon, Nat. Catal., 2018, 1, 794–804 CrossRef CAS.
- B. G. McCarthy, R. M. Pearson, C. H. Lim, S. M. Sartor, N. H. Damrauer and G. M. Miyake, J. Am. Chem. Soc., 2018, 140, 5088–5101 CrossRef CAS PubMed.
- J. P. Cole, C. R. Federico, C. H. Lim and G. M. Miyake, Macromolecules, 2019, 52, 747–754 CrossRef CAS PubMed.
- B. L. Buss, C. H. Lim and G. M. Miyake, Angew. Chem., 2020, 59, 3209 CrossRef CAS PubMed.
- B. L. Buss, L. R. Beck and G. M. Miyake, Polym. Chem., 2018, 9, 1658–1665 RSC.
- Y. C. Zhao, H. H. Gong, K. M. Jiang, S. J. Yan, J. Lin and M. Chen, Macromolecules, 2018, 51, 938–946 CrossRef CAS.
- E. H. Discekici, C. W. Pester, N. J. Treat, I. Lawrence, K. M. Mattson, B. Narupai, E. P. Toumayan, Y. D. Luo, A. J. McGrath, P. G. Clark, J. R. de Alaniz and C. J. Hawker, ACS Macro Lett., 2016, 5, 258–262 CrossRef CAS.
- S. B. Tan, J. Xiong, Y. F. Zhao, J. J. Liu and Z. C. Zhang, J. Mater. Chem. C, 2018, 6, 4131–4139 RSC.
- C. H. Lim, M. D. Ryan, B. G. McCarthy, J. C. Theriot, S. M. Sartor, N. H. Damrauer, C. B. Musgrave and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 348–355 CrossRef CAS PubMed.
- S. M. Sartor, Y. M. Lattke, B. G. McCarthy, G. M. Miyake and N. H. Damrauer, J. Phys. Chem. A, 2019, 123, 4727–4736 CrossRef CAS PubMed.
- D. Koyama, H. J. A. Dale and A. J. Orr-Ewing, J. Am. Chem. Soc., 2018, 140, 1285–1293 CrossRef CAS PubMed.
- S. Jockusch and Y. Yagci, Polym. Chem., 2016, 7, 6039–6043 RSC.
- R. N. Carmean, T. E. Becker, M. B. Sims and B. S. Sumerlin, Chem, 2017, 2, 93–101 CAS.
- J. T. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- H. H. Gong, Y. Gu, Y. C. Zhao, Q. Z. Quan, S. T. Han and M. Chen, Angew. Chem., 2020, 59, 919–927 CrossRef CAS PubMed.
- L. Mueller, W. Jakubowski, K. Matyjaszewski, J. Pietrasik, P. Kwiatkowski, W. Chaladaj and J. Jurczak, Eur. Polym. J., 2011, 47, 730–734 CrossRef CAS.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed.
- J. Rzayev and J. Penelle, Angew. Chem., Int. Ed., 2004, 43, 1691–1694 CrossRef CAS PubMed.
- K. Matyjaszewski, Adv. Mater., 2018, 30, 22 CrossRef PubMed.
- A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81–84 CrossRef CAS PubMed.
- S. Park, P. Chmielarz, A. Gennaro and K. Matyjaszewski, Angew. Chem., Int. Ed., 2015, 54, 2388–2392 CrossRef CAS PubMed.
- B. Li, B. Yu, W. T. S. Huck, W. M. Liu and F. Zhou, J. Am. Chem. Soc., 2013, 135, 1708–1710 CrossRef CAS PubMed.
- M. Fantin, A. A. Isse, A. Venzo, A. Gennaro and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 7216–7219 CrossRef CAS PubMed.
- J. R. Wang, M. Y. Tian, S. Q. Li, R. Wang, F. P. Du and Z. G. Xue, Polym. Chem., 2018, 9, 10 Search PubMed.
- Y. Du, R. M. Pearson, C. H. Lim, S. M. Sartor, M. D. Ryan, H. S. Yang, N. H. Damrauer and G. M. Miyake, Chem. – Eur. J., 2017, 23, 10962 CrossRef CAS PubMed.
- C. E. Dahm and D. G. Peters, J. Electroanal. Chem., 1996, 402, 91–96 CrossRef.
- A. I. Popov and D. H. Geske, J. Am. Chem. Soc., 1958, 80, 1340–1352 CrossRef CAS.
- H. E. Zittel and F. J. Miller, Anal. Chim. Acta, 1967, 37, 141–150 CrossRef CAS.
- T. Iwasita and M. C. Giordano, Electrochim. Acta, 1969, 14, 1045–1059 CrossRef CAS.
- G. D. Allen, M. C. Buzzeo, C. Villagran, C. Hardacre and R. G. Compton, J. Electroanal. Chem., 2005, 575, 311–320 CrossRef CAS.
- M. P. S. Mousavi, S. A. Saba, E. L. Anderson, M. A. Hillmyer and P. Buhlmann, Anal. Chem., 2016, 88, 8706–8713 CrossRef CAS PubMed.
- Frit porosity and sizes, https://adamschittenden.com/technical/frits/frit-size#EuroPorosities, (accessed April 2020).
- R. Hoffman, V. I. Minkin and B. K. Carpenter, Int. J. Philos. Chem., 1997, 3, 3–28 Search PubMed.
- M. D. Ryan, R. M. Pearson, T. A. French and G. M. Miyake, Macromolecules, 2017, 50, 4616–4622 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00643b |
| This journal is © The Royal Society of Chemistry 2020 |