
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of precursor structure on porous carbons produced by iron-catalyzed graphitization of biomass†
Robert D.
Hunter
a,
Jemma L.
Rowlandson
 b,
Glen J.
Smales
c,
Brian R.
Pauw
c,
Valeska P.
Ting
b,
Alexander
Kulak
d and
Zoe
Schnepp
*a
b,
Glen J.
Smales
c,
Brian R.
Pauw
c,
Valeska P.
Ting
b,
Alexander
Kulak
d and
Zoe
Schnepp
*a
aSchool of Chemistry, University of Birmingham, Birmingham, B15 2TT, UK. E-mail: z.schnepp@bham.ac.uk
bDepartment of Mechanical Engineering, University of Bristol, Bristol, BS8 1TR, UK
cBundesanstalt für Materialforschung und -prüfung (BAM), Unter den Eichen 87, Berlin 12205, Germany
dSchool of Chemistry, University of Leeds, Leeds, LS2 9JT, UK
First published on 13th October 2020
Abstract
This paper reports a systematic study into the effect of different biomass-derived precursors on the structure and porosity of carbons prepared via catalytic graphitization. Glucose, starch and cellulose are combined with iron nitrate and heated under a nitrogen atmosphere to produce Fe3C nanoparticles, which catalyze the conversion of amorphous carbon to graphitic nanostructures. The choice of organic precursor provides a means of controlling the catalyst particle size, which has a direct effect on the porosity of the material. Cellulose and glucose produce mesoporous carbons, while starch produces a mixture of micro- and mesopores under the same conditions and proceeds via a much slower graphitization step, generating a mixture of graphitic nanostructures and turbostratic carbon. Porous carbons are critical to energy applications such as batteries and electrocatalytic processes. For these applications, a simple and sustainable route to those carbons is essential. Therefore, the ability to control the precise structure of a biomass-derived carbon simply through the choice of precursor will enable the production of a new generation of energy materials.
Introduction
Porous carbon materials have a wide range of applications, finding use as anode materials in sodium and lithium-ion batteries,1 electrocatalyst supports for fuel cells,2 electrode materials for supercapacitors3 and adsorbents for water treatment.4 High performance in these applications is achieved through features such as high thermal and electrical conductivity and high surface area. Therefore, considerable effort has gone into producing carbon materials that have precise pore size distribution and connectivity, as well as a tailored bulk and surface structure. To achieve this, a wide range of synthetic methods have been developed, including hydrothermal carbonization,5 pyrolysis of saccharide aerogels (Starbon®)6 and pyrolysis of organic precursors followed by physical7 or chemical8 activation.Porous carbons with a high graphitic content are particularly attractive due to their high chemical stability and electronic conductivity. A good route to make these materials is through catalytic graphitization. Here, organic precursors are combined with a metal compound before heating in an inert atmosphere (typically N2 or Ar). Many examples use iron as the catalyst for graphitization, in the form of aqueous or ethanolic iron salts, such as iron nitrate and iron chloride,9 or organometallic species such as ferrocene.10 Other transition metals such as cobalt and nickel have also been used as catalysts for graphitization, however these elements introduce issues with toxicity and sustainability.11
The organic precursors also vary widely, and include small molecules such as sucrose,12 polymeric species such as phenolic resins,13 pure cellulose,14 raw biomass,15,16 or biomass-derived hydrochars.17 Depending on the precursors and conditions, catalytic graphitization has produced various graphitic nanostructures such as hollow ‘shells’,18 nanotubes19 and nanoribbons.11 The mechanism of formation of these nanostructures is generally agreed to proceed via in situ formation of Fe3C nanoparticles, which catalyze the conversion of amorphous carbon to graphitic carbon. In some systems, this process induces movement of the catalyst particles through the amorphous carbon intermediate,20 producing graphitic nanotubes. In other systems, the particles appear to remain stationary, forming graphitic shells. The underlying reason for this inconsistent behaviour is not known.
One of the challenges in exploiting catalytic graphitization as a route to porous carbons is the lack of understanding of the process. This fundamental understanding could not be obtained from the preceding studies, which have covered a wide range of precursors, but also employed diverse synthesis conditions. The variety of pyrolysis temperatures, times and heating rates provides no consistent picture of the different effects of each adjustable parameter to the synthesis. These factors mean that it is very difficult to compare reported properties of different carbons in the literature (e.g., porosity or electrocatalytic activity) or to make predictions of what systems might produce even better properties in the future. This is further complicated when biomass or biological molecules are used as precursors, due to the complexity and variability in these systems. For example, woody biomass contains cellulose polymers that are aligned and twisted into fibres and fibrils. Alongside the cellulose, the other main component of woody biomass is lignin, a complex polyphenolic molecule, as well as other smaller molecules. The composition of the lignin and the lignin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cellulose ratio can vary widely between different plant sources and also within certain parts of a plant.21 The ability to use biomass as a precursor, particularly agricultural or industrial biomass waste, means that catalytic graphitization has the potential to be an economical and sustainable route to valuable nanostructured carbons. However, before this can happen, there needs to be more understanding of how different precursors affect the structure and properties of the resulting carbon.
:cellulose ratio can vary widely between different plant sources and also within certain parts of a plant.21 The ability to use biomass as a precursor, particularly agricultural or industrial biomass waste, means that catalytic graphitization has the potential to be an economical and sustainable route to valuable nanostructured carbons. However, before this can happen, there needs to be more understanding of how different precursors affect the structure and properties of the resulting carbon.
In this paper we explore the effect of different biomass-derived materials on the structure of carbons produced by iron-catalyzed graphitization. Specifically, we compare glucose, starch and cellulose, which are very similar in terms of chemical composition but have different physical properties. Glucose is a water-soluble monosaccharide with the molecular formula C6H12O6 (Fig. 1a). In comparison, starch is a mixture of amylose (Fig. 1b) and amylopectin (Fig. 1c), the ratio of which depends on the botanical origin of the starch (typically 20–35% amylose but can range from 3–50%).22 Amylose is a linear polymer with a molecular weight of 1 × 105–1 × 106, consisting of α(1 → 4) linked glucose units. Amylopectin is also comprised of α(1 → 4) linked glucose units but approximately 5% of the glucose units have an α(1 → 6) linked chain, leading to a highly branched structure. Starch is commonly used as a food thickener and gelling agent, due to its ability to produce viscous solutions or gels by absorbing water and swelling when heated.23 The swelling is believed to proceed via initial breakdown of hydrogen bonding within the semicrystalline outer branches of the amylopectin molecules, followed by absorption of water within the amorphous regions and, eventually, complete separation of amylose and amylopectin.24 Finally, cellulose (Fig. 1d) is also a polysaccharide that consists of glucose units, but these are β(1 → 4) linked in linear chains, which typically align into fibrils and fibres, often with a crystalline microstructure.25
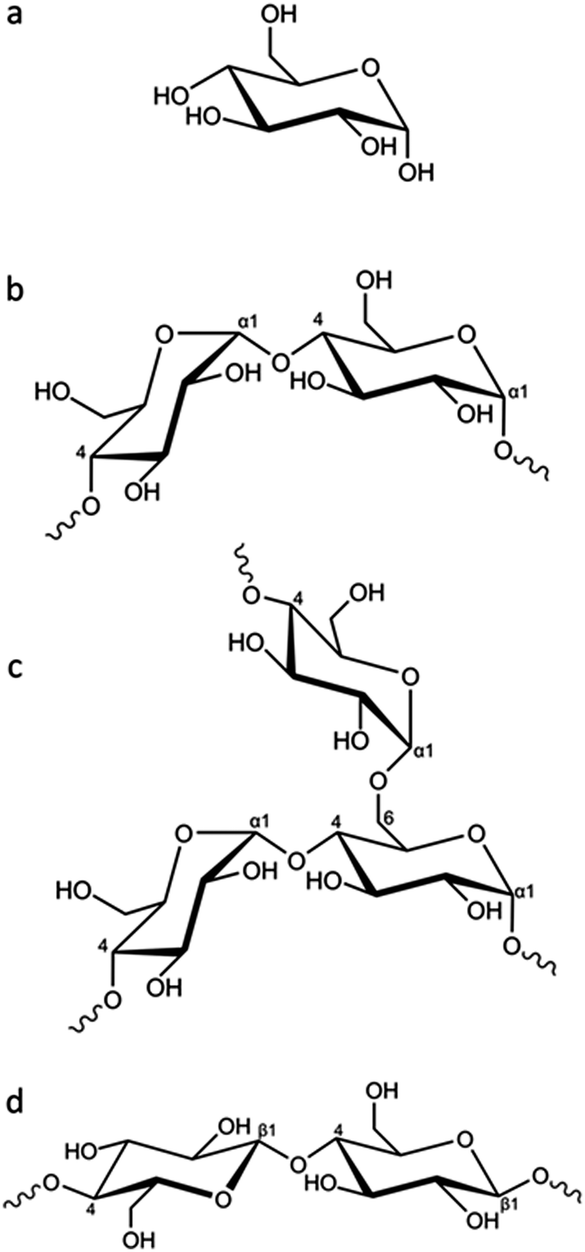 | ||
| Fig. 1 (a) Structure of glucose and partial representative structures of (b) amylose, (c) amylopectin and (d) cellulose. | ||
Results and discussion
Graphitization of glucose, starch and cellulose
Porous carbon samples were prepared by catalytic graphitization using D-glucose, potato starch (typically 20–30% amylose) and cellulose fibres as the sources of carbon and iron nitrate as the catalyst. Fig. 2 shows powder X-ray diffraction (PXRD) data of the resulting black powders with characteristic peaks for Fe3C (ICDD 00-035-0772) and graphite (ICDD 01-071-4630) in all three samples. The presence of a more intense peak at 45° 2θ in the cellulose sample suggests a mixture of Fe (ICDD 00-001-1262) and Fe3C exists in this system. The graphite peak intensity relative to the iron phase appears to be lower in the starch sample, suggesting that the degree of graphitization is lower in this sample. Furthermore, the main graphite peak in starch-derived carbon appears to overlay a much broader peak at a smaller 2θ. Broad peaks at a smaller diffraction angle than graphite (26° 2θ) are characteristic of turbostratic carbons and indicate the presence of more disordered graphene layers with a larger interlayer spacing than graphite. This suggests that the starch-derived carbon may consist of a mixture of graphitic and turbostratic carbon. | ||
| Fig. 2 PXRD patterns for carbon/Fe3C samples produced from glucose, starch and cellulose (5 g) and iron nitrate (0.68 mmol), held at 800 °C for 1 h. (*) indicates presence of minor α-Fe phase. | ||
Further characterization of the porous carbons was carried out using Raman spectroscopy, which showed the presence of two prominent peaks at approximately 1325 and 1600 cm−1 corresponding to the D and G bands respectively. The G band signifies the presence of graphitic carbon and is present in all graphitic materials, while the D band is forbidden in perfect graphite so indicates that all three carbons consist of a more disordered structure. Deconvolution of the spectra assuming a 4 peak Voigt function in which the peaks were attributed to the G1, D1, D2 and D3 bands (Fig. S1 and Table S1, ESI†) was used to extract peak positions and FWHM values.7 In all three carbons, the G band is shifted from 1581 cm−1 in perfect graphite to a higher value of approximately 1600 cm−1, more consistent with nanocrystalline graphitic domains.26 Deconvolution of the spectra highlights the broader peaks in the starch-derived carbon, suggesting a greater degree of disorder and a higher proportion of turbostratic carbon than the glucose- and cellulose-derived carbons, consistent with the results from PXRD.
Scanning electron microscopy (SEM) images of the porous carbons (Fig. 3a–c), recorded with a backscattered electron detector, show that all are comprised of complex nanostructures, with the electron-dense iron carbide catalyst particles visible as bright spots throughout the samples. All samples show some evidence of what may be tubular systems, as observed previously in the catalytic graphitization of raw biomass,19 but these are most clearly apparent in the sample prepared from cellulose fibres (Fig. 3c). Transmission electron microscopy (TEM) images are also similar to those seen previously in samples of graphitized biomass19 and cellulose,14 with graphitic ‘tubes’ and ‘shells’ present in all three samples (Fig. 3d–f) alongside the catalyst particles (visible as dark spots). Importantly, there is evidence for catalyst particle movement in all three samples, with many graphitic pores not containing a catalyst nanoparticle (example indicated by an arrow in Fig. 3e). Such catalyst particle movement has been previously observed using environmental TEM and is believed to involve travelling of the catalyst nanoparticle through an amorphous carbon matrix.20
 | ||
| Fig. 3 SEM images (backscattered electron detector) of porous carbons derived from (a) glucose, (b) starch and (c) cellulose (5 g) and iron nitrate (0.68 mmol), held at 800 °C for 1 h (scale bar = 200 nm) and TEM images of the carbons derived from (d) glucose, (e) starch and (f) cellulose (scale bar = 100 nm). | ||
Small angle X-ray scattering (SAXS) gives an indication of the Fe3C particle size distribution in the samples, which, in turn, can offer information about porosity. Fig. 4a shows the SAXS data for the glucose, starch and cellulose samples. The data was fitted and analysed using McSAS, a Monte Carlo method to extract form-free size distributions (fit was removed for clarity and can been seen in Fig. S2, ESI†).27 The particle size histograms for the glucose and cellulose samples (Fig. 4b and d) show a trimodal (volume-weighted) distribution of scattering features, similar to results in a previous study of porous carbon from gelatin and iron nitrate.18 As SAXS relies on scattering features with a contrasting electron density compared to their surroundings, there are various possible scattering contrasts to consider within the samples. The peak between 10 < r (nm) < 100 in the histograms of all three samples, can be ascribed to the Fe3C particles in the carbon matrix, and is consistent with the Fe3C nanoparticle sizes observed in TEM. The peak for cellulose has a greater relative volume fraction and is positioned towards larger radii, suggesting that the Fe3C nanoparticles are larger in the cellulose-derived carbon. This could be because cellulose does not dissolve or swell in the cold water, meaning the iron nitrate precursor is deposited only on the hydroxyl-rich surface of the cellulose particles. During pyrolysis, the iron nitrate first decomposes to iron oxide particles, which then react with the underlying carbon to produce Fe3C nanoparticles. As the nanoparticles are on the surface of the carbon particles (produced by decomposition of the cellulose itself), they can grow freely. In the glucose and starch samples, the iron salt is combined in solution with the organic precursor, producing a more homogeneous mixture. The resulting iron carbide particles would be embedded in a matrix of amorphous carbon, meaning nanoparticle growth is more restricted.
 | ||
| Fig. 4 (a) SAXS data fits for carbons produced from glucose, starch and cellulose (5 g) and iron nitrate (0.68 mmol), held at 800 °C for 1 h. Particle size histograms coupled with visibility limits (black dots, left y-axis) and cumulative distribution functions (right y-axis) for carbons from (b) glucose, (c) starch and (d) cellulose. | ||
The peaks in the particle distribution histograms between 3–10 nm radius are likely due to the carbon/air interface of the numerous mesopores within the sample, while the smallest distributions (<1 nm) may be attributed to surface roughness due to micropores.18 Besides the TEM results, additional evidence for these distribution mode assignments comes from XRD and SAXS data of samples where the majority of the Fe3C component has been etched away through acid washing (Fig. S3a–f, ESI†). The size distribution histograms for cellulose and glucose show that the particles of largest radius (10–100 nm) are largely removed from the acid-washed samples (Fig. S3g and i, ESI†). Starch presents a slightly more complex picture. In the starch-derived carbon sample, the particle size histogram (Fig. 4c) shows a more significant contribution to the scattering from very small features in the sample, suggesting the presence of more micropores, possibly linked to the significant amounts of turbostratic carbon in this sample (as indicated by XRD data).28 Furthermore, upon acid washing, the size distribution histogram changes shape significantly (Fig. S3h, ESI†), with a drop in the relative volume fraction across many length-scales. This suggests a wider distribution of Fe3C nanoparticle sizes in the starch-derived carbon. XRD data shows that a considerable amount of the Fe3C is still present after acid washing the starch-derived carbon (Fig. S3b, ESI†), which is consistent with the SAXS observations and suggests that the Fe3C is only partially removed.
Nitrogen sorption isotherms for glucose, starch and cellulose samples (Fig. 5a) display a type IV shape with hysteresis due to capillary condensation, which is characteristic of mesoporous materials. The type of hysteresis loop appears to be most consistent with type H4. Sharp changes in adsorption and desorption branches associated with type H1 and H2 due to pore blocking are not observed.29,30 Isotherms exhibit a distinct region for micropore filling at low p/p0 and a sharp decrease at approximately 0.4p/p0 consistent with the type-H4 loop. The three isotherms do not saturate at high p/p0, suggesting that the pore size distribution may extend past the mesopore range and into the macropore range, consistent with the particle sizes and pores observed in electron microscopy. Interestingly, the shape of the isotherm for the starch-derived carbon is very different to those of the carbons derived from cellulose and glucose. This is similar to the observations from SAXS and suggests that the graphitization process is different in starch-derived carbon. Adsorptive characteristics were derived from the N2 isotherms (Fig. 5b), including the maximum quantity of gas adsorbed (max Qads), the total (Vtot) micro- (Vmicro) and meso- (Vmeso) pore volumes, the BET (SBET), micropore (Smicro) and mesopore (Smeso) surface area, and the volume-weighted average pore size (wavg:2D-NLDFT). These data mirror the adsorption isotherms and show comparative performance between the glucose- and cellulose-derived carbons but significantly different properties for the carbon from starch. Cellulose- and glucose-derived carbons exhibit BET surface areas of 358 m2 g−1 and 343 m2 g−1 respectively, compared to starch-derived carbon with a BET surface area of 450 m2 g−1. The increase in surface area for the starch sample is explained by the presence of a greater number of micropores in this system, with micropores accounting for 60% of the total pore volume and 76% of the total surface area. This is compared to ∼20% of the total volume and ∼50% of the total surface area for the cellulose- and glucose-derived carbons. Full values can be found in Table 1. These results are consistent with previous observations of the broad ‘graphite-like’ peak in the XRD and suggest that the carbon in the starch sample is comprised of a mixture of turbostratic carbon (micropores) and graphitic nanostructures (mesopores).
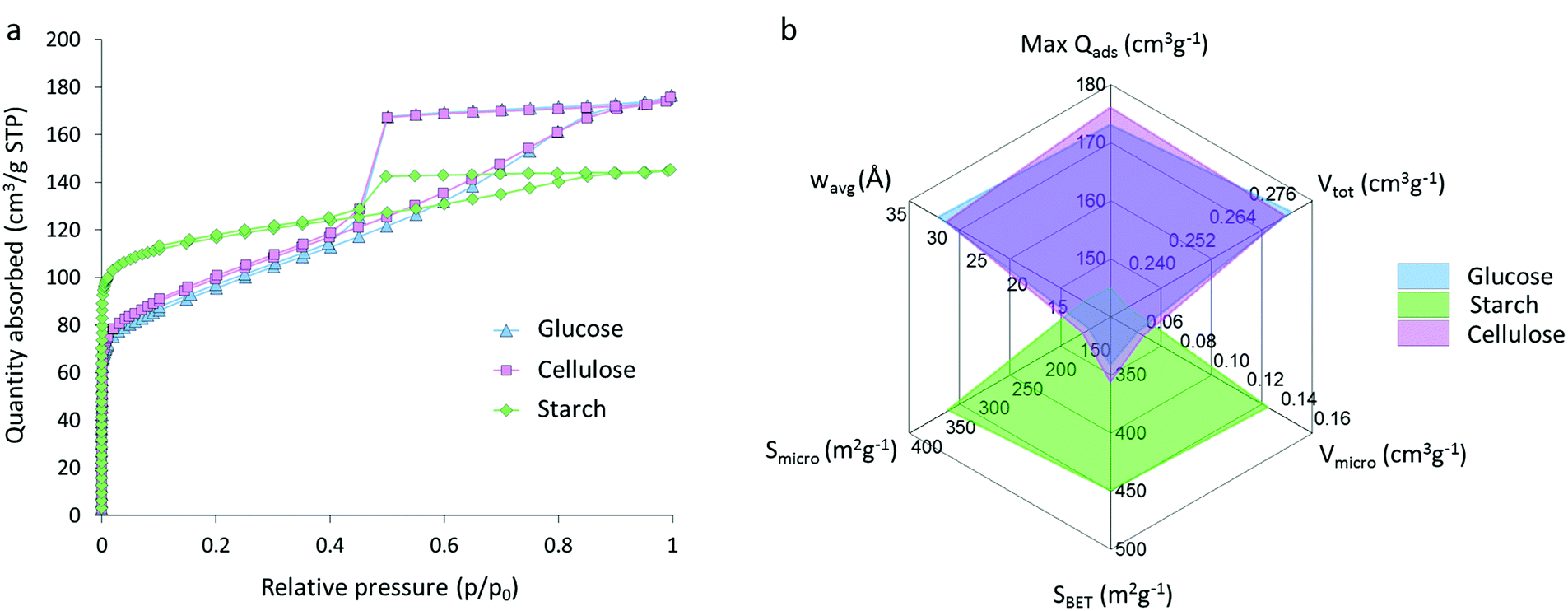 | ||
| Fig. 5 (a) Nitrogen sorption isotherms for carbons derived from glucose, starch and cellulose (5 g) and iron nitrate (0.68 mmol), held at 800 °C for 1 h and (b) adsorptive characteristics for the carbons, calculated from the isotherms. | ||
| Precursor | Max Qads (cm3 g−1) | V tot (cm3 g−1) | V micro (cm3 g−1) | S micro (m2 g−1) | V meso (cm3 g−1) | S meso (cm3 g−1) | w avg:2D-NLDFT (Å) | S BET (m2 g−1) |
|---|---|---|---|---|---|---|---|---|
| Glucose | 177 ± 1 | 0.27 ± <0.01 | 0.06 ± <0.01 | 133 ± 1 | 0.19 ± <0.01 | 174 ± 1 | 30.9 ± 0.6 | 343 ± 1 |
| Starch | 145 | 0.22 | 0.13 | 342 | 0.07 | 73 | 13.2 | 450 |
| Cellulose | 176 ± 1 | 0.27 ± <0.01 | 0.06 ± <0.01 | 143 ± 1 | 0.19 ± 0.01 | 175 ± 1 | 29.7 ± 0.7 | 358 ± 1 |
Effect of iron concentration
Varying the amount of Fe(NO3)3 added to the organic precursors affects the amount and crystallinity of Fe3C present in the samples and the degree of graphitization. Fig. 6 shows the XRD patterns for three samples of carbon prepared from starch (5 g) with 0.34, 0.68 and 3.4 mmol of Fe(NO3)3. An increase in iron leads to sharper and well-resolved Fe3C peaks, suggesting larger Fe3C particles. The characteristic peak for graphite also becomes sharper, but is still high at the lowest iron concentration. This suggests that only a small amount of iron is necessary to drive graphitization, consistent with previous reports on the graphitization of biomass.19 The peak for turbostratic carbon in this sample is high, but this is possibly just due to the relative Fe3C and graphite peaks being smaller. Particle size histograms (Fig. 6b and c) derived from SAXS data (Fig. S4, ESI†) clearly show a large increase in the relative volume fraction of particles in the 10–100 nm radius range with increasing iron nitrate concentration, consistent with the conclusions from the XRD data. The peak centres in the particle size histograms also shift to a higher radius, as might be expected from a higher Fe3C particle size. This is reasonable, given that there is less biomass and less carbon and therefore more growth and sintering of the Fe3C particles. Corresponding samples prepared from glucose (Fig. S5, ESI†) and cellulose (Fig. S6, ESI†) show a similar trend of a high graphite peak at low iron concentration, despite the very small peaks for Fe3C. Interestingly, the graphite peak for the carbon from starch with the lowest iron concentration is much broader than the equivalent cellulose and glucose graphite peaks. This suggests a more disordered and less graphitized system in starch-derived carbons, reflecting the results of porosimetry above. The particle size histograms for glucose and cellulose carbons also show a large increase in the peak for particles in the 10–100 nm radius range with increasing iron concentration. | ||
| Fig. 6 (a) PXRD data for carbon samples prepared from starch (5 g) with various amounts (mmol) of iron nitrate, held at 800 °C for 1 h. Particle size histograms coupled with visibility limits (black dots, left y-axis) and cumulative distribution functions (right y-axis) for carbons from starch (5 g) with (a) 3.4, (b) 0.68 and (c) 0.34 mmol of iron nitrate. | ||
Effect of synthesis conditions
The starch system appears to display some unique properties and so this was examined further with a study of synthesis conditions. In previous reports of the synthesis of Fe3C from a biomass precursor (gelatin), the mechanism was found to proceed via initial formation of very small iron oxide nanoparticles (Fe3O4) followed by carbothermal reduction to FeO and Fe3N and finally transformation to Fe3C.31 To investigate if a similar mechanism operates in the glucose, starch and cellulose systems, samples were heated to various temperatures between 500 °C and 800 °C and held for 1 hour. Fig. S7 (ESI†) shows XRD patterns of the resulting carbon samples, indicating that Fe3C is produced at around 700 °C. The starch and cellulose samples show some evidence of an oxide phase but even the glucose sample has some evidence of very broad peaks for FeO (ICDD 01-074-1886) if the data is magnified (Fig. S8, ESI†). This suggests that the same mechanism of iron oxide nanoparticle reduction is operating in glucose, cellulose and starch-derived samples. Interestingly, the sample of starch-derived carbon at 700 °C has a much broader graphite peak, suggesting that graphitization is occurring more slowly in this sample. In order to investigate this further, a series of samples were prepared where glucose, starch and cellulose were combined with iron nitrate, heated to 800 °C and then held at the maximum temperature for different amounts of time. Fig. S9 (ESI†) shows XRD data for glucose and cellulose samples, where the graphite and Fe3C/Fe peaks do not change significantly from 0–2 hours, indicating that the onset and progression of graphitization in these systems is very fast and likely to start below 800 °C. In the glucose system, there is still a considerable amount of iron oxide in the system at 0 h, suggesting that graphitization occurs immediately after the first Fe3C nanoparticles are formed. In contrast, the effect of hold time for starch is substantial. At 0 h hold time, the XRD pattern (Fig. 7a) shows that Fe3C has fully formed, but the graphite peak is extremely broad and shifted to a low 2θ, suggesting that only turbostratic carbon is present in the system.28 The very broad, merged Fe3C peaks suggest that the Fe3C particle size is very small at 0 h hold time and grows over the course of the 2 hours hold, indicated by the Fe3C peaks becoming sharper. This is confirmed by SAXS data (Fig. S10, ESI†) and particle size histograms (Fig. S11, ESI†), which show a gradual emergence of a peak for particles in the 10–100 nm radius range. As the Fe3C nanoparticles grow, the graphite peak in the XRD becomes more pronounced and sharp. This data suggests that there is a critical size of Fe3C that is needed for graphitization to proceed rapidly. Similar effects of catalyst particle size on carbon nanotube growth rate have been observed in chemical vapour deposition.32 Significantly, this result shows that Fe3C nanoparticle growth rate is much more controlled in the starch system, potentially offering more control over the properties of the resulting porous graphitic carbon. | ||
| Fig. 7 (a) XRD diffraction patterns for carbons synthesized from starch (5 g) and iron nitrate (3.4 mmol) and held at 800 °C for different times. (b) N2 adsorption isotherms and (c) adsorptive characteristics of these starch-derived carbons. | ||
To further investigate this effect of hold time on the properties of the starch-derived carbons, we carried out nitrogen porosimetry. Nitrogen sorption isotherms (Fig. 7b) of the samples held for 0 and 0.5 h display little hysteresis and appear to give a type I-like isotherm, suggesting that the pores are predominantly in the micropore range. This is consistent with turbostratic carbon being the dominant contribution to sample porosity. The samples held for 1 and 2 h, however, display more of a type IV isotherm shape indicating the presence of mesopores. This difference is observed as a decrease in BET surface area (Fig. 7c) from 566 m2 g−1 (0 h) to 422 m2 g−1 (2 h) as the hold time is increased and micropore widening leads to the formation of mesopores. This trend is further highlighted by a decrease in micropore volume and surface area with increasing hold time along with an increase in mesopore volume and surface area and volume-weighted pore size (full data shown in Table S2, ESI†). The jump to mesopores observed in nitrogen sorption correlates with the increase in particle size observed in SAXS and further supports the proposal that Fe3C nanoparticles must reach a critical size before they become mobile and produce graphitic nanotubes.
Mechanism
The data indicates that starch offers a lot more control over Fe3C nanoparticle size, thereby influencing the graphitization process. In previous in situ synchrotron PXRD studies of Fe3C formation (from a gelatin–iron nitrate system), the reaction was shown to proceed via nucleation of very small Fe3O4 nanoparticles, followed by reduction to similarly small FeOx nanoparticles (∼2 nm diameter).31 This was followed by carbothermal reduction to Fe3N and then Fe3C, accompanied by a sharp increase in particle size (∼20 nm diameter). In a mechanistic study of graphitization of ashless cellulose filter paper, the reaction was shown to proceed in a similar way, and very small (∼2 nm diameter) iron oxide nanoparticles could be seen to coat the surface of the carbonized filter paper fibres.20 These also underwent carbothermal reduction to produce much larger Fe3C nanoparticles that drove graphitization. Given that iron oxide nucleation and growth does not appear to be significant (i.e., no sharp peaks in the XRD patterns) for glucose, starch or cellulose, it seems that the carbothermal reduction step is the point at which the starch system is unique in being able to restrict growth of the Fe3C phase. This may be due to packing of the carbonized matrix around the Fe3C nanoparticles before the onset of graphitization, which is linked to how iron interacts with the organic precursor and also how the different organic precursors decompose. In cellulose fibres, molecules of cellulose are tightly packed in crystalline arrays that in turn twist into fibrils and fibres. The surfaces of these are rich in polar functional groups and so it is highly likely that iron is adsorbed to the surface of the fibres rather than penetrating far into the fibres (Fig. 8a). On heating, the cellulose will decompose into carbon, with thermogravimetric analysis (TGA) showing that this occurs in a single step between 300 °C and 350 °C.33 The iron nitrate precursor will also decompose to produce iron oxide nanoparticles, coating the surface of the cellulose-derived carbon fibres. These intermediate iron oxide nanoparticles subsequently react with the carbon to produce Fe3C nanoparticles. As these Fe3C nanoparticles are able to freely move on the surface of the carbonized cellulose fibre, they can grow quickly to the critical size required for the onset of graphitization, allowing fast graphitization to occur. | ||
| Fig. 8 Schematics of the mechanism of graphitization for (a) cellulose fibres, (b) glucose and (c) starch. | ||
In glucose, the precursor is a homogeneous mixture of glucose molecules and Fe3+ ions (Fig. 8b), which would decompose on heating to produce a carbon-rich matrix with embedded iron oxide nanoparticles (Fig. S8, ESI†). TGA of glucose shows that the onset of mass loss occurs much earlier (∼200 °C) than for cellulose, reflecting the reduced thermal stability of the glucose monomer compared to the aligned cellulose polymers.33 The result of the early decomposition of glucose could be more significant growth of intermediate iron oxide nanoparticles as there is less carbon-rich matter present around the iron oxide to prevent growth and sintering. What is probably more significant, though, is that the carbon matrix during the FeOx to Fe3C transition appears to be mainly amorphous. An amorphous carbon matrix may allow for easier mass transport of iron during the iron carbide growth phase.
The structure of starch is considerably more complicated. Starch is comprised of ‘granules’, which are made up of a lamellar structure of alternating amorphous and crystalline regions.34 The amorphous regions contain linear amylose and the branching points of the amylopectin, while the crystalline regions contain double helices of amylopectin side-chains packed laterally into a crystalline lattice (Fig. 8c). On heating in water, the starch granules swell, lose crystallinity and begin to leach amylose. It is at this point that Fe3+ ions could diffuse into the granules, generating a ‘gel’ where the Fe3+ ions are held within the amylose/amylopectin matrix. TGA of starch shows that the major thermal decomposition step occurs at around 300 °C, much later than for glucose.33 Given that iron nitrate decomposes to iron oxide between 130 °C and 160 °C,35 this means that the covalent polymer network of starch persists for a lot more of the early growth stage of the iron oxide than the loosely bound glucose monomers. This is likely to restrict the growth of the iron oxide. In addition, the XRD, SAXS and nitrogen sorption data for the starch system suggest that a considerable amount of turbostratic carbon is present before the onset of graphitization. The presence of regions of disordered graphene sheets could ‘block’ the mass transport of iron and thus restrict the growth of the Fe3C nanoparticles. This means they take a lot longer to reach the critical size needed for graphitization. Whether each individual graphitization process (i.e., movement of a single catalyst particle) is then fast or whether the carbon matrix from starch also slows down the movement of the catalyst particles remains unknown.
The origin of the starch appeared to have a limited effect on the structure of the carbon product. Starch derived from potato and corn with amylose:amylopectin ratios of approximately 20:80 and 25:75 respectively were compared. A carbon produced by graphitization of corn starch showed a similar mixture of turbostratic and graphitic carbon to potato starch (Fig. S12, ESI†). This is to be expected due to the similar amylose: amylopectin ratios. The use of waxy corn starch, however, which consists of amylopectin with only a trace amount of amylose, resulted in a single sharp peak for graphitic carbon. PXRD data of an amylose-derived carbon did not show any evidence of graphitization (Fig. S12, ESI†). This result lends credence to the argument that the combination of both amylose and amylopectin is the key to restricting the growth of the Fe3C nanoparticles. It may be that the different decomposition profiles of the two polysaccharides combine to produce the right amount of control over Fe3C particle growth. Or it is possible that chemical reactions within the starch granule, such as formation of cross-links between the different components, only occur under certain combinations of amylose and amylopectin. Such crosslinking is believed to be critical to the formation of ‘hard’ (non-graphitizable) carbons. The lack of graphitization of amylose could be due to the low solubility of this component of starch when in its pure form. While these final points are speculative, the data clearly shows that there is huge scope for controlling starch graphitization through use of different precursors.
Conclusions
In conclusion, porous carbon materials containing tubular graphitic structures can be produced from glucose, starch and cellulose by iron-catalyzed graphitization. While all three organic precursors are comprised of the same sugar monomer, the structural properties of the resulting porous carbons are quite different. Glucose and cellulose produced primarily mesoporous carbons, while starch generated carbons with a range of micro- and mesoporous features. This is understood to be due to the much slower graphitization step that occurs in starch-derived samples. Cellulose and glucose both produce intermediate systems where the fast formation and growth of Fe3C catalyst particles is facilitated. This means that the onset and progression of graphitization is rapid. In contrast, starch produces an intermediate that is rich in turbostratic carbon. This is believed to block the growth of the Fe3C nanoparticles, meaning it takes longer for them to reach the critical size needed for graphitization. The results of this paper show that the starch-derived carbon offers considerably more scope for controlling the porous and microstructural properties of graphitic carbons through iron-catalyzed graphitization, demonstrating the importance of the physical properties of the organic precursor. Further work is needed to fully understand the complex starch system but it opens up the possibility of investigating more cross-linked polymeric precursors for iron-catalysed graphitization as these may ultimately provide more control over microstructure. This is similar to observations from the field of hard carbons and should offer a way for iron-catalyzed graphitization to be investigated more widely and systematically for the production of carbons for various applications.Experimental
Materials
D-(+)-Glucose (CAS 50-99-7), iron(III) nitrate nonahydrate (CAS 7782-61-8), cellulose fibres (C6288), potato starch (S2004), corn starch (S4180), waxy corn starch (S9679) and amylose from potato (A0512) were all sourced from Sigma-Aldrich.Catalytic graphitization
For glucose samples, 5 g of glucose was dissolved in 20 ml deionised (DI) water in a beaker with gentle heating to 40 °C and stirring for 10 min, 0.27 g (0.68 mmol) of Fe(NO3)3·9H2O was dissolved in 4 ml of DI water and this was added to the glucose solution, followed by stirring for 10 min. The mixture was dried in an air oven at 70 °C for 24 h. The resulting brown, sponge-like sample was placed in an alumina boat crucible then heated in a tube furnace at a rate of 5 °C min−1 under a nitrogen atmosphere with a flow rate of 1 l min−1 to 800 °C. The samples were held at 800 °C for 1 h before cooling completely to room temperature. For experiments with starch the procedure was the same except 5 g of potato/corn or waxy corn starch was dissolved in 45 ml of DI water at 70 °C and stirred for 10 min. The iron nitrate solution was added and the mixture stirred with continued heating for 10 minutes. The mixture was dried (as above) to produce an orange solid. For cellulose samples, 0.27 g (0.68 mmol) of Fe(NO3)3·9H2O was dissolved in 15 ml of DI water at room temperature and the resulting solution added to a beaker containing 5 g of cellulose fibres (powder). The mixture was manually stirred until the solution had been absorbed. The sample was dried to give an orange/yellow solid and pyrolyzed as above. The amount of water was kept to a minimum in each of these samples to reduce the energy requirements in the drying step (further detail in ESI†).Acid-washing
0.2 g of carbon/Fe3C sample was sonicated in 20 ml of 0.1 M HCl for 1 h. The mixture was then magnetically stirred for 24 h. The solid sample was collected by centrifugation and washed three times with deionised water and once with ethanol, then left to dry at room temperature in air for 24 h.Powder X-ray diffraction
Samples were ground into a fine powder and placed on low-background silicon wafer sample holders. PXRD experiments were performed using a PANalytical Empyrean diffractometer with a copper anode (wavelengths: Kα1 = 1.5406 Å, Kα2 = 1.5443 Å) and a Pixel 2D detector. The diffractometer did not have a monochromator but the Kβ radiation was removed with a nickel filter.Raman spectroscopy
Samples were ground into a fine powder and placed on a glass slide. Raman spectroscopy measurements were collected using a Renishaw inVia Raman microscope using a red laser at 10% power with a wavelength of 633 nm. Peak fitting was performed assuming a 4 peak Voigt function.Scanning electron microscopy
The morphologies of the carbon derivatives were determined using SEM, samples were mounted on an SEM stub using an adhesive copper tape. Samples were viewed with a FEG-SEM FEI Nova 450 using CBS detector (detector of backscattered electrons), operating at 5 kV with deceleration mode.Transmission electron microscopy
Small portions of sample (≈50 mg) were dispersed in ethanol (∼1 ml) by sonication for 10 min. One drop of the dispersion was pipetted on to a carbon-coated copper TEM grid. The images were obtained using a JEOL 2100 TEM containing a tungsten filament and a charge coupled device (CCD) detector.Small angle X-ray scattering
Samples were ground into a fine powder and distributed across a hole in a paper sample holder between two pieces of Scotch® Magic™ tape. The wide-range SAXS experiments were performed using the Multi-scale Analyser for Ultrafine Structures (MAUS) at the Federal Institute for Materials Research and Testing (BAM), Berlin. Copper and molybdenum anodes (8 eV and 17 eV photons, respectively) were used to measure over a wide q-range. Full details on the data collection, correction and analysis procedures can be found in the ESI.†Nitrogen sorption
Nitrogen sorption measurements were carried out at 77 K using a 3Flex volumetric gas sorption analyser from Micromeritics. 100–300 mg of sample was degassed at 300 °C for 4 h under 10−7 mbar vacuum. Nitrogen (AirProducts, 99.999%) isotherms were collected with filler rods over the range p/p0 10−8–1.0 and helium was used to calculate the free space following isotherm collection. BET surface areas were calculated by applying the Rouquerol correction to select the range p/p0 0.01–0.05 (for glucose/starch/cellulose) using the method recommended by the International Organization for Standardization (ISO) 9277.36 Adsorptive parameters and pore size distributions were calculated from N2 isotherms. Repeat measurements of the upper N2 isotherm (p/p0 ∼ 0.1–1.0) were performed for all samples and the full N2 isotherm for glucose and cellulose samples. The total pore volume measurable by gas sorption was obtained from the isotherm plateau at p/p0 0.99 and the micropore volume and micropore surface area using the t-plot method, according to ISO 15901-3.37 The mesopore volume and mesopore surface area was calculated using the BJH method according to ISO 15901-2.29Pore size distributions were calculated by the fitting of two-dimensional nonlinear density functional theory (2D-NLDFT) kernels to the N2 isotherms using the 3Flex (Micromeritics) software. A graphitic slit pore shape was assumed with a pore aspect ratio of 6 or 12 was chosen to minimize the standard deviation of fit between the model and experimental data. The volume-weighted average pore size was calculated from pore size distributions according to Laudisio et al.38
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the University of Birmingham for funding (RH PhD studentship).References
- X. Hu, X. Sun, S. J. Yoo, B. Evanko, F. Fan, S. Cai, C. Zheng, W. Hu and G. D. Stucky, Nano Energy, 2019, 56, 828–839 CrossRef CAS.
- Y. Hu, J. O. Jensen, W. Zhang, L. N. Cleemann, W. Xing, N. J. Bjerrum and Q. Li, Angew. Chem., Int. Ed., 2014, 53, 3675–3679 CrossRef CAS.
- P. Schlee, O. Hosseinaei, D. Baker, A. Landmer, P. Tomani, M. J. Mostazo-Lopez, D. Cazorla-Amoros, S. Herou and M. M. Titirici, Carbon, 2019, 145, 470–480 CrossRef CAS.
- B. Li, L. Yang, C. Q. Wang, Q. P. Zhang, Q. C. Liu, Y. D. Li and R. Xhao, Chemosphere, 2017, 175, 332–340 CrossRef CAS.
- M. M. Titirici, R. J. White, C. Falco and M. Sevilla, Energy Environ. Sci., 2012, 5, 6796–6822 RSC.
- V. Budarin, J. H. Clark, J. E. Hardy, R. Luque, K. Mikowski, S. J. Tavener and A. J. Wilson, Angew. Chem., Int. Ed., 2006, 45, 3782–3786 CrossRef CAS.
- J. L. Rowlandson, K. J. Edler, M. Tian and V. P. Ting, ACS Sustainable Chem. Eng., 2020, 8, 2186–2195 CrossRef CAS.
- S. A. Sajjadi, A. Meknati, E. C. Lima, G. L. Dotto, D. I. Mendoza-Castillo, I. Anastopoulos, F. Alakhras, E. I. Unuabonah, P. Singh and A. Hosseini-Bandegharaei, J. Environ. Manage., 2019, 236, 34–44 CrossRef CAS.
- A. Gomez-Martin, J. Martinez-Fernandez, M. Ruttert, A. Heckmann, M. Winter, T. Placke and J. Ramirez-Rico, ChemSusChem, 2018, 11, 2776–2787 CrossRef CAS.
- H. Duan, T. Yan, G. Chen, J. Zhang, L. Shi and D. Zhang, Chem. Commun., 2017, 53, 7465–7468 RSC.
- J. Hoekstra, A. M. Beale, F. Soulimani, M. Versluijs-Helder, J. W. Geus and L. W. Jenneskens, J. Phys. Chem. C, 2015, 119, 10653–10661 CrossRef CAS.
- J. Yang and S. Zuo, Diamond Relat. Mater., 2019, 95, 1–4 CrossRef CAS.
- H. Rastegar, M. Bavand-Vandchali, A. Nemati and F. Golestani-Fard, Phys. E, 2018, 101, 50–61 CrossRef CAS.
- J. Hoekstra, A. M. Beale, F. Soulimani, M. Versluijs-Helder, D. van de Kleut, J. M. Koelewijn, J. W. Geus and L. W. Jenneskens, Carbon, 2016, 107, 248–260 CrossRef CAS.
- M. Sevilla, C. Sanchís, T. Valdés-Solís, E. Morallón and A. B. Fuertes, J. Phys. Chem. C, 2007, 111, 9749–9756 CrossRef CAS.
- A. Gutiérrez-Pardo, J. Ramírez-Rico, R. Cabezas-Rodríguez and J. Martínez-Fernández, J. Power Sources, 2015, 278, 18–26 CrossRef.
- K. Lotz, A. Wütscher, H. Düdder, C. M. Berger, C. Russo, K. Mukherjee, G. Schwaab, M. Havenith and M. Muhler, ACS Omega, 2019, 4, 4448–4460 CrossRef CAS.
- Z. Schnepp, Y. Zhang, M. J. Hollamby, B. R. Pauw, M. Tanaka, Y. Matsushita and Y. Sakka, J. Mater. Chem. A, 2013, 1, 13576–13581 RSC.
- E. Thompson, A. E. Danks, L. Bourgeois and Z. Schnepp, Green Chem., 2015, 17, 551–556 RSC.
- S. Glatzel, Z. Schnepp and C. Giordano, Angew. Chem., Int. Ed., 2013, 52, 2355–2358 CrossRef CAS.
- R. C. Pettersen, in The Chemistry of Solid Wood, ed. R. Rowell, ACS, Washington, 1984, ch. 2, pp. 57–126 Search PubMed.
- N. Singh, J. Singh, L. Kaur, N. Singh Sodhi and B. Singh Gill, Food Chem., 2003, 81, 219–231 CrossRef CAS.
- P. J. Jenkins and A. M. Donald, Carbohydr. Res., 1998, 308, 133–147 CrossRef CAS.
- W. S. Ratnayake and D. S. Jackson, J. Agric. Food Chem., 2006, 54, 3712–3716 CrossRef CAS.
- K. Leppänen, S. Andersson, M. Torkkeli, M. Knaapila, N. Kotelnikova and R. Serimaa, Cellulose, 2009, 16, 999–1015 CrossRef.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- I. Bressler, B. R. Pauw and A. F. Thunemann, J. Appl. Crystallogr., 2015, 48, 962–969 CrossRef CAS.
- C. L. Burket, R. Rajagopalan and H. C. Foley, Carbon, 2017, 45, 2307–2320 CrossRef.
- ISO. Pore size distribution and porosity of solid materials by mercury porosimetry and gas adsorption – Part 2: Analysis of mesopores and macropores by gas adsorption. ISO 15901-2, 2006, pp. 1–30.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report), 2015, DOI: 10.1515/pac-2014-1117.
- Z. Schnepp, A. E. Danks, M. J. Hollamby, B. R. Pauw, C. A. Murray and C. C. Tang, Chem. Mater., 2015, 27, 5094–5099 CrossRef CAS.
- Z. Yu, D. Chen, B. Tøtdal and A. Holmen, Catal. Today, 2005, 100, 261–267 CrossRef CAS.
- S. Kudo, Z. Zhou, K. Yamasaki, K. Norinaga and J. Hayashi, Catalysts, 2013, 3, 757–773 CrossRef.
- J. Blazek and E. P. Gilbert, Carbohydr. Polym., 2011, 85, 281–293 CrossRef CAS.
- K. Deshpande, A. Mukasyan and A. Varma, Chem. Mater., 2004, 16, 4896–4904 CrossRef CAS.
- ISO. Determination of the Specific Surface Area of Solids by Gas Adsorption. BET Method. ISO 9277, 2010, pp. 1–24.
- ISO. Pore size distribution and porosity of solid materials by mercury porosimetry and gas adsorption – Part 3: Analysis of micropores by gas adsorption. ISO 15901-3, 2007, pp. 1–27.
- G. Laudisio, R. K. Dash, J. P. Singer, G. Yushin, Y. Gogotsi and J. E. Fischer, Langmuir, 2006, 22, 8945–8950 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00692k |
| This journal is © The Royal Society of Chemistry 2020 |