
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Porous carbons from sustainable sources and mild activation for targeted high-performance CO2 capture and storage†
Abdulsalam M.
Aljumialy
 ab and
Robert
Mokaya
*a
ab and
Robert
Mokaya
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: r.mokaya@nottingham.ac.uk
bDepartment of Chemistry, College of Science, University of Anbar, Anbar, Iraq
First published on 29th October 2020
Abstract
Carbon activation typically involves corrosive activating reagents such as KOH and NaOH. In this report, a less corrosive activating agent, i.e., potassium oxalate (PO), was explored for the preparation of activated carbons from biomass (hydrochar from sawdust, SD), polypyrrole (Ppy), or pre-mixed (SD + PPy) precursors. The resulting activated carbons have surface area of up to 2740 m2 g−1 and pore volume up to 1.7 cm3 g−1, and depending on activation conditions and/or type of precursor, are either highly microporous (up to 91% surface area from micropores) or highly mesoporous (up to 96% of surface area from mesopores). Unlike hydroxide activation, varying the PO/precursor ratio, within the range of 2–6, does not have a significant effect on porosity; carbons activated at PO/precursor ratio of 2, 4, 5 or 6 have comparable surface area. In contrast, activation temperature plays a critical role in determining the textural properties at any PO/precursor ratio. In this regard, a low PO/precursor ratio of 2 can achieve the full range of porosity, which not only offers a more eco-friendly and sustainable activation process but also allows easier control of the porosity by simple choice of activation temperature or type of precursor. The porosity of the activated carbons may be tailored towards 6 to 8 Å pore channels, which are excellent for CO2 storage at low pressure (i.e., post-combustion CO2 capture) where at 25 °C, the carbons capture up to 1.4 and 4.4 mmol g−1 of CO2 at 0.15 bar and 1 bar, respectively. The porosity can also be tailored towards mesoporosity, which is suited for moderate to high pressure (pre-combustion) CO2 storage, which reaches 20.3 mmol g−1 at 20 bar and 30.1 mmol g−1 at 40 bar at 25 °C. For high surface area samples, the porosity is favourable for pre-combustion CO2 capture, via pressure swing adsorption processes at an adsorption pressure of up to 40 bar and desorption at 1 bar, from gas mixtures (e.g., 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 H2/CO2) where the carbons achieve excellent working capacity.
:40 H2/CO2) where the carbons achieve excellent working capacity.
1. Introduction
Carbonaceous materials such as activated carbon have been widely used in a range of applications including as electrodes in supercapacitors and in gas (CO2 and H2) storage.1–10 Activated carbons are attractive due to their ease of preparation, high thermal and mechanical stability and variable packing density.11–21 The performance of activated carbons in most of their current and potential applications is dependent on their textural properties. The ability to control the textural properties of activated carbon is therefore very important in efforts aimed at fully exploiting their potential, especially in energy related gas storage applications where there is currently a great deal of attention on the storage and transportation of hydrogen and CO2. The use of hydrogen for energy production can mitigate CO2 emissions by reducing reliance on fossil fuels.22,23 The on-going use of fossil fuels generates CO2, which is considered to be a primary contributing factor to global warming.24,25 Therefore, it is necessary to mitigate CO2 emissions, especially from flue gases emanating from fossil fuel power stations prior to their release into the atmosphere.22–25 One of the ideas being explored for the removal of CO2 involves adsorption of CO2 onto solid adsorbents,26–34 as an alternative to amines based industrial processes. The amine based processes suffer from corrosion issues and have high regeneration costs as CO2 binds strongly to amines. In general, the amount of gas (hydrogen or CO2) adsorbed and stored on a solid depends on the porosity of the absorbent. For this reason, there have been increasing efforts to prepare tailorable porous materials with optimised porosity and gas uptake.26–37The synthesis of activated carbon often requires several steps based on either physical or chemical activation. The former involves physical treatment of a carbonaceous precursor at high temperature (typically above 900 °C) under oxidising gas conditions (flow of steam or CO2) while in the later, the carbonaceous precursor is acted on by a chemical agent at medium to high temperature (typically up to 900 °C). Chemical activation involves the use of a reagent (such as H3PO3, ZnCl2 or KOH) as an activating agent, and is widely used because it can generate activated carbon with higher surface area and/or narrow pore size distribution. The advantages of chemical activation, i.e., lower temperature and shorter operation time, and which can offer high carbon yields and better control of porosity, have triggered interest that has so far explored a wide range of carbon precursors including polymers,38–40 lignin,16 carbon nanotubes,34 olive stones,41,42 rice husks,43 and other sources.36,37
One of most extensively explored activating agents for the synthesis of activated carbons is potassium hydroxide via the so-called hydroxide activation. The use of KOH, however, suffers from drawbacks related to a toxic and corrosive nature. Thus milder activating agents but which offer similar or better textural properties are highly desired. There is growing research interest in the use of less corrosive and less toxic activating agents or porogens that go beyond hydroxide activation.44–53 For example, Sevilla and co-workers reported on the use of a mixture of melamine and potassium oxalate (PO) as activating agents for the preparation of biomass derived carbons suitable for use as electrode materials for supercapacitors.46 We have shown that the presence of melamine, in addition to PO, is necessary for achieving high surface area (>1600 m2 g−1) carbons that are potentially useful for pre-combustion CO2 capture.47 The added melamine was thought to have the function of an activator, which is not unexpected as PO is a milder activating agent compared to KOH.46,47 Indeed, most studies that have used PO as activating agent report carbons with low to medium surface area (typically <1500 m2 g−1) for activation at 800 °C or below.44–51 The low to moderate surface is observed even when additives such as CaCO3,49,50 or CaSO4,51 are included as porogens or activation enhancers. One previous study52 reported higher surface area (up to 2500 m2 g−1) but other studies show surface area below 1500 m2 g−1 under similar activation conditions.47 The surface area for PO activated carbons may be increased if higher activation temperature (>800 °C) is used,47,53 but such a high temperature is undesirable from a sustainability point of view.
It is known that the amount of activating agent and the activation temperature play a role in determining the porosity generated in KOH activated carbons. Recently, we have shown that the O/C ratio of any carbonaceous solid used as starting material (i.e., precursor) is a key consideration in determining porosity of KOH activated carbons, with high O/C ratio favouring greater surface area and pore volume.54,55 Given the attraction of PO as a mild activating agent, there is a need for a systematic study to clarify on how activation with PO works under conditions that go beyond just varying the amount of PO and activation temperature to include a variety of precursors with a range of O/C ratios. Such a systematic study is needed as, so far, no clear trends are evident from previous studies, with some reporting very low surface area, while others report either an increase or a decrease in surface area for higher amounts of PO.44–53 This report, explores the preparation of PO activated carbons from biomass and pre-mixed precursors that contain a mix of carbonaceous materials that are known to respond differently to similar levels of activation. The full range of possible porosity development using PO as activator is investigated, and the resulting materials are explored as CO2 storage materials both for low pressure (post-combustion) and medium to high pressure (pre-combustion) conditions.
2. Experimental section
2.1 Material synthesis
The sawdust was converted to hydrochar via hydrothermal carbonisation as follows; 6.4 g of sawdust was dispersed in 20 ml deionized water, placed in a stainless-steel autoclave and heated at 250 °C for 2 h, and then cooled to room temperature. The resulting carbonaceous solid was recovered by filtration, washed with deionized water and dried in an oven at 120 °C to yield the sawdust derived hydrochar (designated as SD). Polypyrrole (Ppy) was synthesised by mixing 3 g of pyrrole with 200 ml of 0.5 M FeCl3 solution and stirring the mixture for 2 h at room temperature. The polypyrrole, at a yield close to 100%, was then recovered, washed with distilled water and dried in an oven at 120 °C.For activation, the required amount of potassium oxalate (PO) was added to the carbon precursor (sawdust derived hydrochar, SD or Ppy or a mixture of SD + Ppy) and thoroughly mixed at PO/precursor mass ratio of 2, 4, 5 or 6. For activation of pre-mixed precursors containing SD and Ppy, three sets of samples were prepared from mixtures of SD:Ppy at weight ratio of 2:1, 1:1, or 1:2. The resulting mixture of PO and the chosen precursor was placed in an alumina boat and inserted into a tubular furnace and heated at a ramp rate of 5 °C min−1 to 600, 700, 800 or 900 °C under a flow of nitrogen. It was held for 1 h at the target temperature and then cooled to room temperature under a flow of nitrogen. The resulting samples were recovered and washed several times by stirring in 10% hydrochloric acid at room temperature followed by filtration and washing with deionized water until the filtrate was free of Cl− ions according to the AGNO3 test, and then dried in an oven at 120 °C. SD derived samples were designated as SDxT, where x is PO/SD ratio, and T is activation temperature. Ppy derived samples were designated as PpyxT, where x is PO/Ppy ratio and T is activation temperature. Samples derived from pre-mixed precursors of SD and Ppy were designated as Ppy-SD(a − b)xT, where a − b is the Ppy-SD ratio, x is PO/precursor ratio, and T is activation temperature such that a carbon prepared at 800 °C and PO/precursor ratio of 2 from a pre-mixed precursor containing a 1:2 Ppy:SD ratio is designated as Ppy-SD(1–2)2800.
2.2 Characterization methods
Thermogravimetric analysis (TGA) was performed using a TA Instruments SDT Q600 analyzer at heating rate of 10 °C/min under flowing air conditions. Powder XRD patterns were obtained using a PANalytical X’Pert PRO diffractometer with CuKα radiation (λ = 1.5406 Å) (40 kV, 40 mA), 0.02 step size and 2 s step time for values of 2θ between 2° and 80°. Elemental analysis (CHNO) was performed using an Exeter Analytical CE-440 Elemental Analyser. Nitrogen sorption isotherms and textural properties were determined at −196 °C using nitrogen sorption in a conventional volumetric technique on a Micrometrics 3Flex sorptometer. Before analysis, the samples were outgassed under vacuum at 230 °C for 16 h. Surface areas of the carbons were determined using the BET method based on adsorption data in the relative pressure range (P/P0) range of 0.01–0.2. The relative pressure range was selected to ensure a positive y-axis intercept from multipoint BET fitting (such that C > 0) and that Vads(1 − P/P0) would increase with P/P0.56 The total pore volume was calculated from the amount of nitrogen adsorbed at relative pressure (P/P0) of ca. 0.99. The micropore surface area and micropore pore volume were determined using t-plot analysis. The pore size distributions were obtained via Nonlocal density functional theory (NLDFT) methods using nitrogen adsorption data. Scanning electron microscopy (SEM) analysis was performed using a JEOL 7100F microscope. The carbon samples were loaded on a carbon conductive sticky tape prior to analysis.2.3 CO2 uptake measurements
CO2 uptake was determined using a Hiden Isochema XEMIS instrument at 25 °C and pressure of up to 40 bar. The carbons were outgassed for 3 h under vacuum at 240 °C prior to performing the CO2 uptake measurements. Our measurements directly determined the excess CO2 uptake. For low pressure measurements (up to 1 bar), we report the excess uptake as it is very close to the total uptake. At 20 bar and above, we report both the excess and total uptake, wherein the latter is obtained by factoring in the density of the CO2 under the measurement conditions and the total pore volume of the sample according to the following equation; θT = θExc+ dCO2 × VT, where; θT is total CO2 uptake, θExc is excess CO2 uptake, dCO2 is density (g cm−3) of CO2 gas at 25 °C and relevant pressure, and VT is pore volume (cm3 g−1) of the carbon. The CO2 density is available from the National Institute of Standards and Technology (NIST) website (http://www.nist.gov/).3. Results and discussion
3.1 Carbons derived from sawdust hydrochar
| Sample | Yield (wt%) | Elemental composition (wt%) | |||
|---|---|---|---|---|---|
| C | H | N | O | ||
| Sawdust | 48.5 | 5.7 | 0.3 | 45.5 | |
| Hydrochar | 57.5 | 5.2 | 0.4 | 36.9 | |
| SD2600 | 43 | 78.9 | 1.4 | 0.7 | 19.0 |
| SD2700 | 41 | 81.1 | 0.2 | 0.2 | 18.5 |
| SD2800 | 33 | 87.7 | 0.0 | 0.1 | 12.2 |
| SD2900 | 21 | 90.7 | 0.0 | 0.2 | 9.1 |
| SD4600 | 46 | 77.4 | 0.9 | 0.5 | 21.2 |
| SD4700 | 41 | 83.7 | 0.3 | 0.3 | 15.7 |
| SD4800 | 36 | 82.7 | 0.3 | 0.1 | 16.9 |
| SD4900 | 18 | 87.3 | 0.0 | 0.0 | 12.7 |
| SD5800 | 37 | 83.9 | 0.3 | 0.1 | 15.7 |
| SD6800 | 37 | 88.9 | 0.0 | 0.0 | 11.1 |
The thermal stability of the SD-derived activated carbons was investigated by thermogravimetric analysis (TGA) as shown in Fig. 1. The TGA curves show a first mass loss below 120 °C due to removal of water, followed by a significant mass loss at between 550 and 600 °C due to combustion of the carbon. The residual mass is lower than 4 wt%, and typically <1 wt%, which indicates that the activated carbons were generally free of inorganic residues, with the exception of sample SD4900 that had residual mass of ca. 7%. Carbons activated at higher temperature show higher resistance to combustion, with sample SD2900 and SD4900 having the highest thermal stability. The effect of activation temperature on thermal stability is exemplified by the fact that sample SD2600 had maximum burn-off at 500 °C compared to 600 °C for sample SD2900. Samples activated at PO/SD ratio of 4 (Fig. 1B) show slightly higher thermal stability than analogous samples activated at PO/SD ratio of 2 (Fig. 1A). In order to assess the effect of the PO/SD ratio on the thermal stability of the activated carbons, the stability of carbons activated at 800 °C and PO/SD ratio of 2, 4, 5 or 6 was investigated. The TGA curves (Fig. S1, ESI†) indicate that thermal stability slightly rises when the PO/SD ratio increases, such that the carbon activated at highest PO/SD ratio of 6 is the most resistant to combustion.
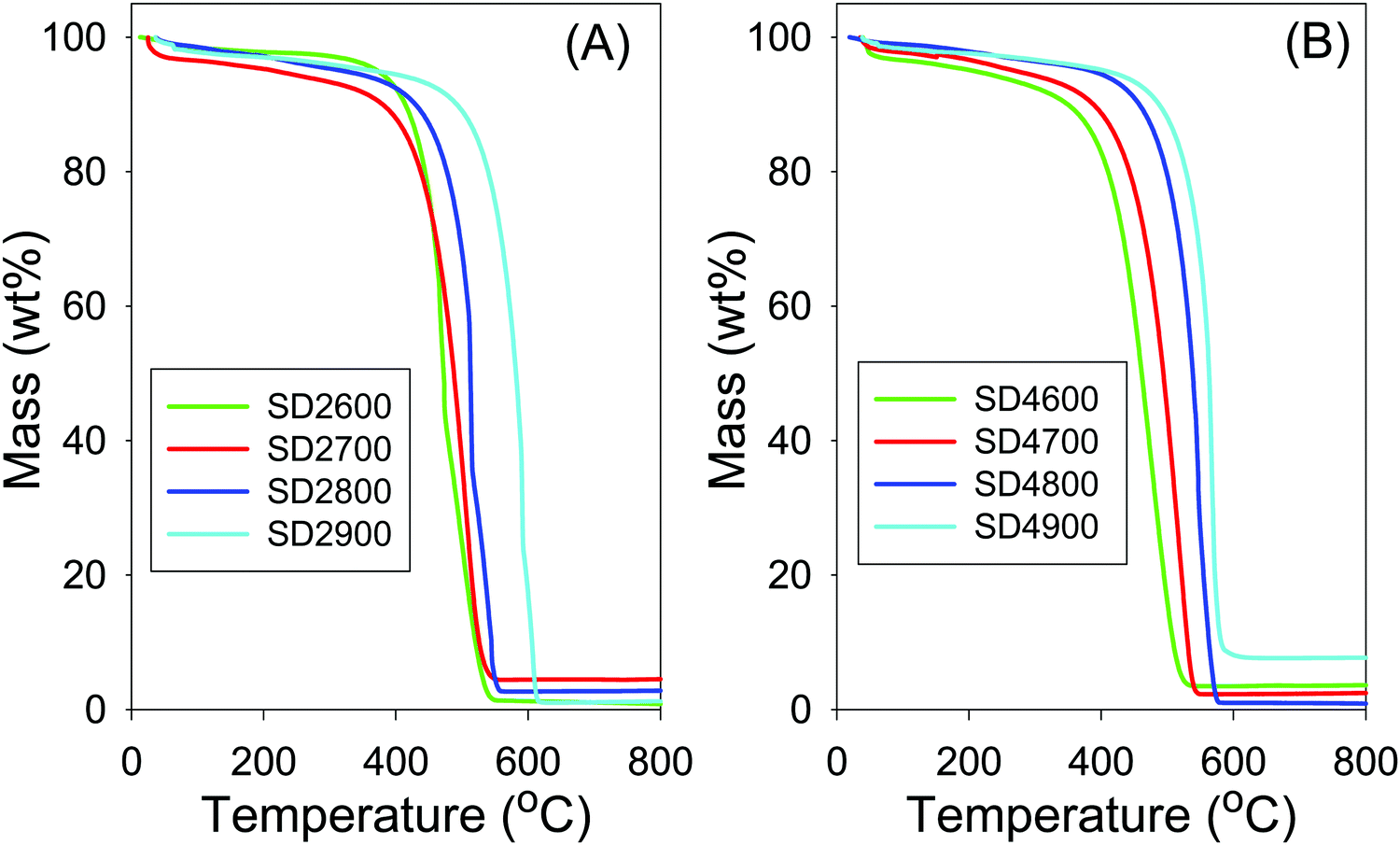 | ||
| Fig. 1 Thermogravimetric analysis curves of carbons activated at PO/SD ratio of (A) 2 or (B) 4. | ||
The powder XRD patterns of the activated carbons (Fig. S2, ESI†) show very broad peaks at 2θ = 26° and 43°, which relate to the graphitic/turbostratic nature of the carbons.56 The XRD patterns indicate that the PO/SD ratio does not significantly affect the nature of the carbons. The XRD patterns of some of the carbons show sharp peaks that may suggest the presence of inorganic residues from the activating agent, although the corresponding TGA curves (Fig. 1) show that such amounts are very low. Where present, the sharp peaks appear to arise from residual KCl from the washing process. The sharp but low intensity peak at 2θ = 26.5° may be ascribed to (200) diffraction of KCl but the absence of other expected diffractions suggest that the amount of the inorganic residue is very low, which is consistent with the TGA data in Fig. 1. In general, the XRD patterns of the carbon samples are consistent with their being amorphous but with some graphitic domains, which is typical of activated carbons.14–21 The XRD patterns and TGA curves are consistent with the expectation that higher activation temperature favours the presence of graphitic domains in the activated carbons and thus enhances thermal stability. In this respect, the trend in thermal stability of the PO activated carbons as a function of activation temperature is similar to what has previously been observed for KOH activated carbons.14–21 Scanning electron microscopy (SEM) was used to image and monitor changes in morphology of the sawdust biomass following hydrothermal carbonisation and activation. The SEM images of the sawdust, sawdust-derived hydrochar and representative activated carbons are shown in Fig. 2. The sawdust has a cellular-like structure that is typical of biomass, and following the hydrothermal carbonisation process, where the biomass is converted to hydrochar, some of the morphology of the sawdust appears to be retained.13,46 On the other hand, the activated carbons have morphology of sheet-like and irregularly shaped particles.14 It is interesting to note that the activated carbons have some particles with a morphology that is similar to that of the sawdust-derived hydrochar. This observation suggests that the potassium oxalate is a much milder activating agent compared to KOH; the latter significantly disrupts the morphology and structure of the precursor starting material even when mild activation is performed.12–20
 | ||
| Fig. 2 SEM images of sawdust, sawdust-derived hydrochar and representative activated carbons. | ||
Effect of activation temperature. The nitrogen sorption isotherms and the corresponding pore size distribution (PSD) curves of carbons activated at PO/SD ratio of 2 at 600 to 900 °C are shown in Fig. 3. All the activated carbons, except SD2900, show type I isotherms that are typical of microporous materials. High nitrogen sorption occurs at low relative pressure (P/P0 < 0.1) due to micropore filling,57 and no significant adsorption occurs at P/P0 greater than 0.1 except for sample SD2900. For activation at 800 °C, there is a slight broadening of the adsorption ‘knee’ in the isotherm, which suggests limited widening in the pore size distribution. The ‘knee’ broadening is particularly evident for sample SD2900, which shows an almost linear increase in adsorption up to P/P0 of 0.4 suggesting the presence of larger micropores and a significant proportion of mesopores.58,59
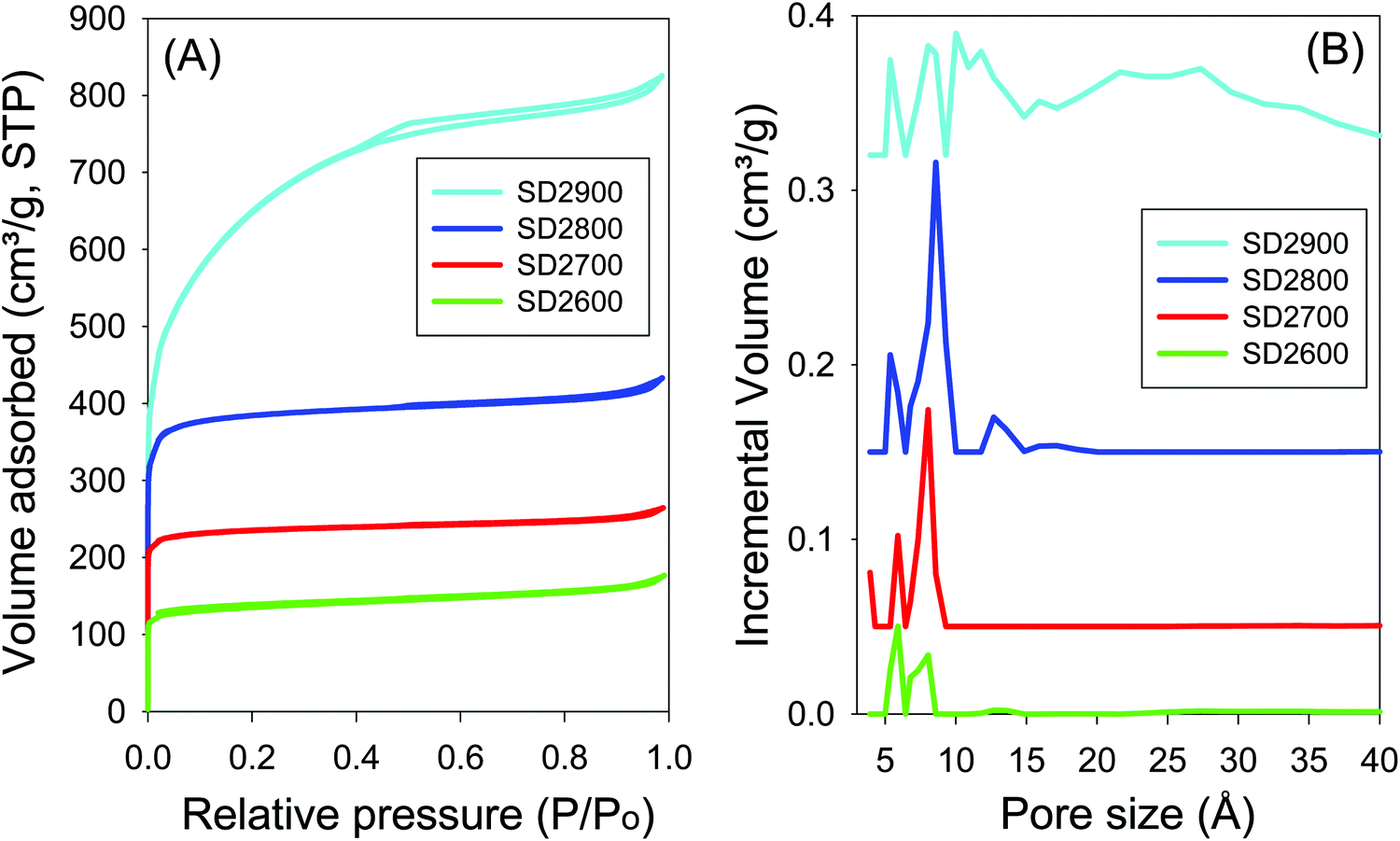 | ||
| Fig. 3 Nitrogen sorption isotherms (A) and pore size distributions curves (B) for carbon samples activated at temperatures between 600 and 900 °C and PO/SD ratio of 2. | ||
As shown in Table 2, the surface area and pore volume of carbons prepared at PO/SD ratio of 2 increases with activation temperature. The sample activated at 600 °C (SD2600) has surface area of 506 m2 g−1 and pore volume of 0.27 cm3 g−1, while the sample activated at 700 °C (SD2700) has surface area of 894 m2 g−1 and pore volume of 0.41 cm3 g−1. Sample SD2800 has surface area (1463 m2 g−1) and pore volume (0.67 cm3 g−1) that is 64% higher compared to that of sample SD2700. At the highest activation temperature of 900 °C (sample SD2900), the surface area and pore volume increase further, respectively, by 60% (up to 2350 m2 g−1) and 89% (up to 1.27 cm3 g−1) compared to sample SD2800. There seems to be a step change in the surface area and pore volume between 800 and 900 °C, which emphasises the role of activation temperature. In contrast, the micropore surface area decreases as the activation temperature rises from 800 to 900 °C (Table 2); the proportion of the micropore surface area is 34% for sample SD2900, compared to 90% for sample SD2800. According to the pore size distribution (PSD) curves shown in Fig. 3B, the pore size of carbons prepared at PO/SD ratio of 2, at 600 °C and 700 °C, is almost exclusively dominated by 6 and 8 Å pores (Table 2), while sample SD2800 shows a noticeable proportion of 13 Å pores. With rise in activation temperature to 900 °C, sample SD2900 shows a further widening of the pore size with a significant proportion of 27 Å pores (Fig. S3, ESI†). The presence of larger pores for samples activated at higher temperatures may be attributed to a higher level of activation caused by greater gasification.60
| Sample | Surface areaa (m2 g−1) | % μSA | Pore volumeb (cm3 g−1) | Pore size (Å) | CO2 uptake (mmol g−1) at 25 °C | ||
|---|---|---|---|---|---|---|---|
| 0.15 bar | 1 bar | 20 barc | |||||
| a Values in parenthesis are micropore surface area. b Values in parenthesis are micropore volume. c Values in parenthesis are total CO2 uptake. | |||||||
| SD2600 | 506 (411) | 81 | 0.27 (0.16) | 6/8 | 1.2 | 2.6 | 4.6 (4.8) |
| SD2700 | 893 (813) | 91 | 0.41 (0.33) | 6/8 | 1.4 | 3.8 | 7.6 (8.0) |
| SD2800 | 1463 (1311) | 90 | 0.67 (0.53) | 6/8/13 | 1.2 | 4.3 | 11.9 (12.5) |
| SD2900 | 2350 (800) | 34 | 1.27 (0.32) | 6/8/12/27 | 0.7 | 3.4 | 18.0 (19.1) |
| SD4600 | 575 (499) | 87 | 0.30 (0.20) | 6/8 | 1.2 | 2.9 | 5.1 (5.4) |
| SD4700 | 972 (875) | 90 | 0.46 (0.35) | 6/8 | 1.2 | 3.7 | 7.6 (8.0) |
| SD4800 | 1441 (1257) | 87 | 0.68 (0.51) | 6/9/13 | 1.0 | 3.8 | 11.2 (11.8) |
| SD4900 | 2205 (714) | 32 | 1.33 (0.29) | 6/8/12/27 | 0.7 | 3.2 | 17.2 (18.4) |
| SD5800 | 1498 (1321) | 88 | 0.70 (0.50) | 6/9/13 | 1.2 | 4.3 | 12.8 (13.4) |
| SD6800 | 1470 (1294) | 88 | 0.71 (0.52) | 6/8/13 | 1.2 | 4.4 | 12.6 (13.2) |
For samples activated at PO/SD ratio of 4, the nitrogen sorption isotherms and the corresponding pore size distribution (PSD) curves are shown in Fig. 4, and the textural properties are summarised in Table 2. The isotherms of carbons activated at 600 to 800 °C are type I, typical of microporous materials, where high nitrogen sorption occurs at low relative pressure (P/P0 < 0.1) due to micropore filling. The isotherms of samples SD4600 and SD4700 exhibit a sharp adsorption knee, and plateau at relative pressure higher than 0.1, i.e., no noticeable adsorption of nitrogen occurs at relative pressure higher than 0.1. The isotherm of sample SD4800 shows a gentle adsorption knee, and at the highest activation temperature of 900 °C (sample SD4900), the isotherm shows a broad adsorption knee with an almost linear increase in adsorption up to P/P0 of 0.4 that suggests the presence of significant mesoporosity.58,59 Regarding textural properties, sample SD4600 has surface area of 575 m2 g−1 and pore volume of 0.3 cm3 g−1, which increase, respectively, to 972 m2 g−1 and 0.46 cm3 g−1 for sample SD2700. Sample SD4800 has surface area and pore volume of 1441 m2 g−1 and 0.0.68 cm3 g−1, respectively, while at the highest activation temperature of 900 °C (sample SD4900), the surface area and pore volume are higher, respectively, by 53% (up to 2205 m2 g−1) and 95% (up to 1.33 cm3 g−1) compared to the sample activated at 800 °C (SD4800). On the other hand, the proportion of micropore surface area decreases from 87% for the sample SD2800 to 32% for SD2900. Fig. 4B shows the pore size distributions (PSDs) of SD4T carbons, and Table 2 summarises the pore size maxima values. The carbons activated at 600 and 700 °C are dominated by pores centred at ca. 6–8 Å, while sample SD4800 shows a small proportion of pores of size 14 Å. At the highest activation temperature of 900 °C, the PSD of sample SD4900 shows a significant proportion of pores of size 30 Å and above (Fig. S3, ESI†).
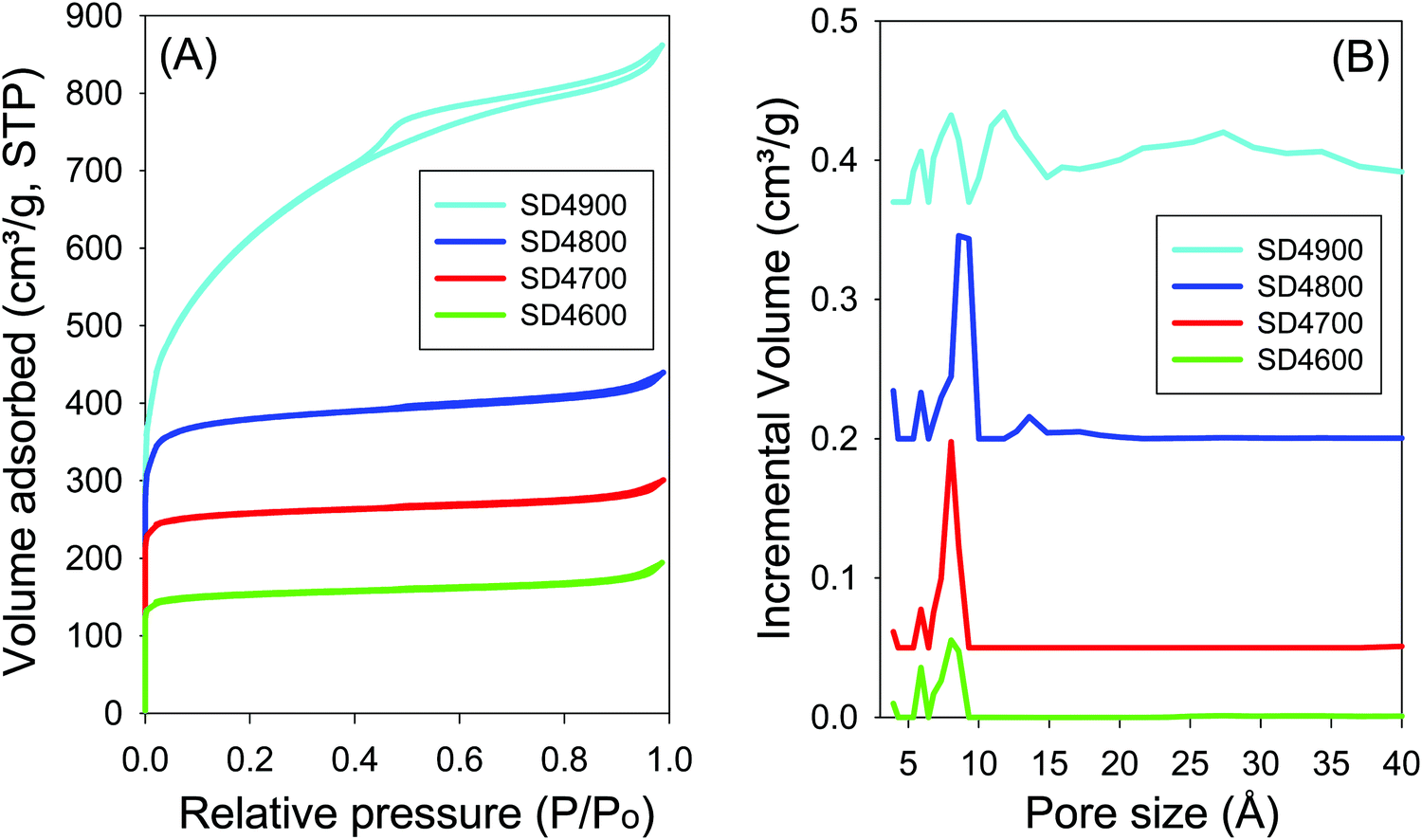 | ||
| Fig. 4 Nitrogen sorption isotherms (A) and pore size distribution curves (B) for carbon samples activated at temperatures between 600 and 900 °C and PO/SD ratio of 4. | ||
Effect of PO/SD ratio. In order to more fully explore the effect of increasing the amount of activating agent, additional samples were activated at PO/SD ratios of 5 or 6 at 800 °C. Fig. 5 shows the nitrogen sorption isotherms and pore size distribution curves for samples activated at 800 °C and PO/SD ratio of 2, 4, 5 or 6. The samples show similar isotherms (i.e., type I) that are typical of microporous materials, with almost all the nitrogen adsorbed in the low relative pressure domain (P/P0 < 0.1). Increasing the PO/SD ratio has little effect on the shape of the isotherm and amount of nitrogen uptake. Additionally, increasing the PO/SD ratio does not significantly affect the surface area, which remains ca. 1450 m2 g−1. It is remarkable that sample SD2800 (activated at a low PO/SD ratio of 2) has surface area and pore volume (1470 m2 g−1 and 0.7 cm3 g−1, respectively), which is similar to that of sample SD6800 that was activated at much higher PO/SD ratio of 6. The pore size distribution (PSD) curves of carbons activated at PO/SD ratios of 2, 4, 5 or 6 at 800 °C are shown in Fig. 5B, and the pore maxima are summarised in Table 2. Increasing the PO/SD ratio from 2 to 4 or 5 does not affect the pore size distribution; samples SD2800, SD4800 and SD5800 all have three pore maxima at 6, 9 and 13 Å. However, the PSD curve of the sample activated with PO/SD ratio of 6 shows four pore maxima at ca. 6, 8, 9 and 13 Å. In general, therefore, increasing the amount of activating agent does not significantly affect the PSD; there are no pores larger than 13 Å for any of the samples (i.e. SD2800, SD4800, SD5800 and SD6800) activated at increasingly higher PO/SD ratios. These findings show that using potassium oxalate as an activating agent offers (in addition to the advantage of being a less corrosive and less toxic activating agent compared to KOH) control of porosity by simply modifying the activation temperature. The ability to strictly tailor pore size of activated carbons is one of the barriers to the wide use of carbons in energy-related applications.61 From what has been discussed above, and as shown in Table 2, varying the PO/SD ratios does not significantly affect the porosity of the obtained carbons. In contrast, the activation temperature plays a critical role in determining the textural properties of the activated carbons.
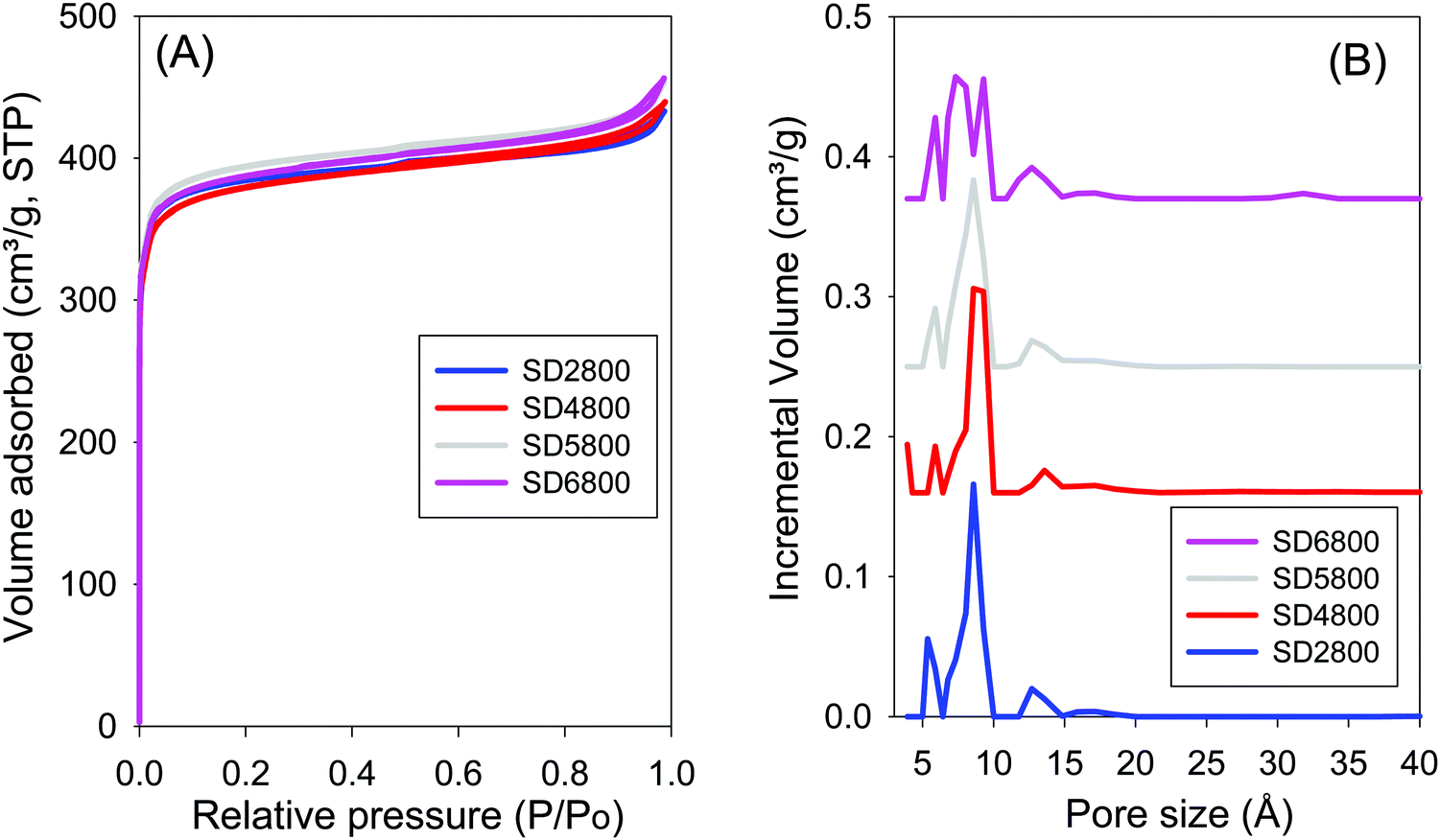 | ||
| Fig. 5 Nitrogen sorption isotherms (A) and pore size distribution curves (B) for carbon samples activated at 800 °C and PO/SD ratios of 2, 4, 5 and 6. | ||
Regarding the mechanism of activation,46,52 as the mixture of SD and PO is heated in the range of 500 to 600 °C, decomposition of PO occurs as follows; K2C2O4 → K2CO3 + CO. This is followed, at temperature of up to 700 °C, by reaction between C within the hydrochar and the generated potassium carbonate, which results in the etching of C atoms (i.e., pore formation) according to the following reaction; K2CO3 + 2C → 2K + 3CO. The potassium carbonate can also decompose according to the following reaction; K2CO3 → K2O + CO2. The gases from the decomposition of the carbonate cause pore formation via gasification of the hydrochar, along with the following reaction at temperature above 800 °C; C + CO2 → 2CO. Our findings on the effects of temperature and amount of PO are consistent with the above mechanism, which offer an explanation for why PO is a mild activating agent compared to KOH. It is likely that the milder nature of PO, compared to hydroxide activation, arises from the fact that PO activation occurs via oxalate decomposition to the carbonate followed by gasification reactions at high temperature, while the hydroxide reacts directly with C even at low temperature to generate the carbonate. Thus for KOH activation, the formation of the carbonate has an activating effect while for PO, the activating effect arises from decomposition of the carbonate and associated gasification processes that are accelerated at higher temperature. The series of reactions for PO activation are consistent with temperature being the more important variable as significant C etching requires high temperatures. In contrast, KOH reacts directly with the C in the carbonaceous precursor to generate the carbonate and K at lower temperature. The K is then intercalated into the carbonaceous matter causing further reactions and C etching, and gasification that lead to pore formation.
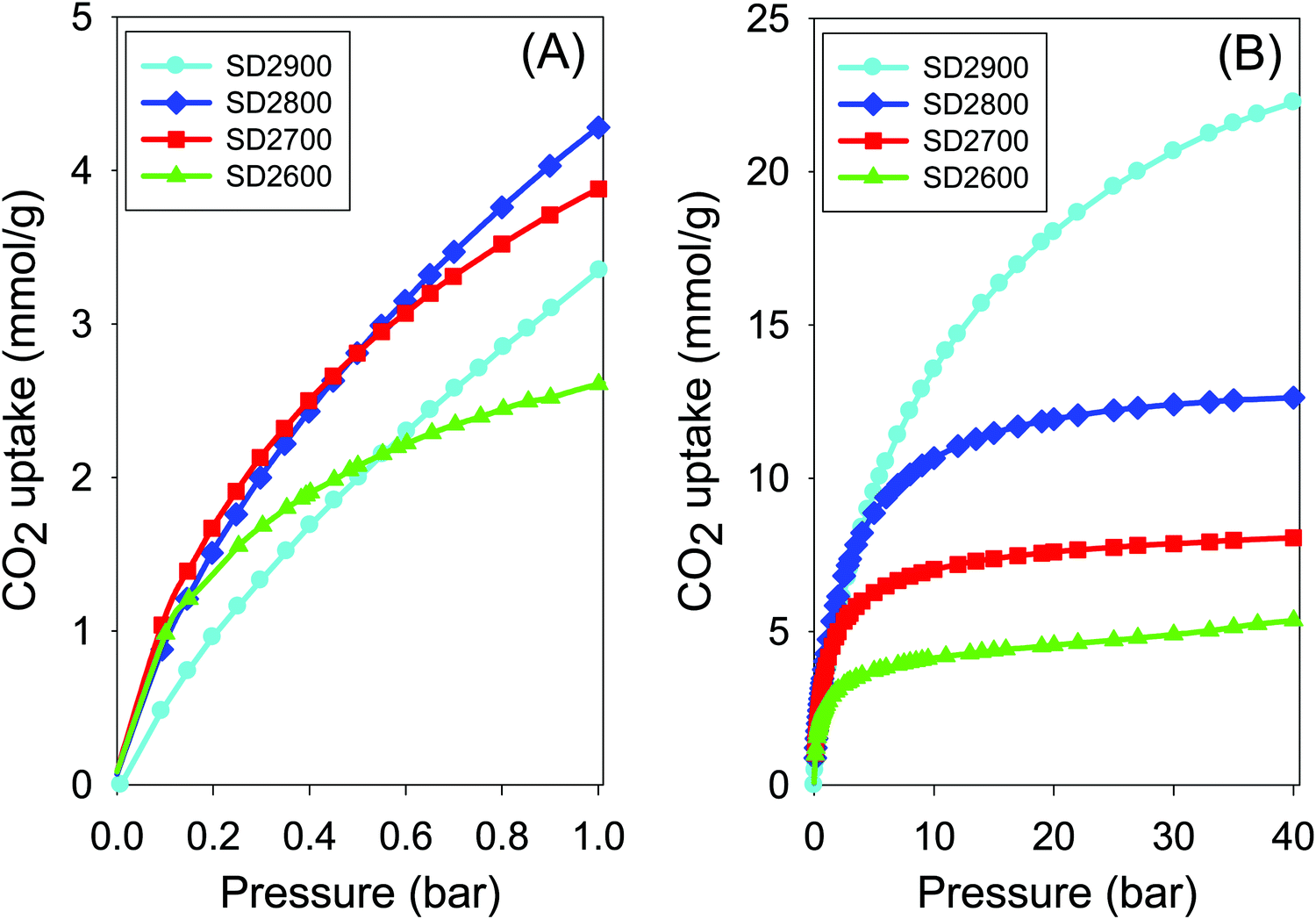 | ||
| Fig. 6 Carbon dioxide uptake isotherms at 25 °C and pressure of (A) 0–1 bar and (B) 0–40 bar of carbons activated at between 600 and 900 °C, and PO/SD ratio of 2. | ||
At lower pressure of 0.15 bar (Fig. 6A) the CO2 uptake is in the range 1.2–1.4 mmol g−1 for samples SD2600, SD2700 and SD2800, and is higher than that (0.7 mmol g−1) of sample SD2900 despite the later having the highest surface area. This trend emphasises the importance of microporosity for low pressure uptake. At higher pressure of 20 bar, the trend is reversed where the highest excess uptake of 18 mmol g−1 (total uptake of 19.1 mmol g−1) is for sample SD2900 compared to 4.6, 7.6, and 12 mmol g−1 for samples SD2600, SD2700 and SD2800, respectively. The data is consistent with the fact that the CO2 uptake at high pressure is dependent on the total surface area rather than pore size. As shown in Fig. 6B the carbons that capture high CO2 at low pressure (≤1 bar), i.e. SD2600, SD2700 and SD2800, due to their high microporosity, approach saturation at high pressure of 20 bar and there is no significant CO2 uptake at higher pressure of up to 40 bar. On the other hand, sample SD2900, which is much less microporous, does not approach saturation even at very high pressure of 40 bar, suggesting that greater CO2 uptake is possible at higher pressure. Interestingly, therefore, the activation temperatures can be altered to tailor the textural properties of the activated carbons towards CO2 captured at either low or high pressure.20 However, it would be more desirable to achieve good high pressure uptake for carbons prepared at lower temperature, e.g., 800 °C. Our data suggest that this is not possible for PO activation of sawdust hydrochar due to the mild nature of the activating agent that only achieves surface area of up to 1500 m2 g−1 for activation at 800 °C. The CO2 uptake of samples activated at PO/SD ratio of 4 was also investigated (Fig. S4 and Table 2). The uptake is in the range 2.9–3.8 mmol g−1 at 1 bar and 25 °C. At 0.15 bar the CO2 uptake is between 0.7 and 1.2 mmol g−1 with the highest CO2 uptake being for sample SD4700, while sample SD4900 has the lowest uptake despite having the highest surface area. At 20 bar, the excess uptake of sample SD4900 is 17.2 mmol g−1 (total uptake of 18.4 mmol g−1) compared to excess uptake of 5.1, 7.6, and 11.2 mmol g−1, respectively, for samples SD4600, SD4700 and SD4800. This once again illustrates the critical role of the pore size and surface area in determining the CO2 uptake depending on the measurement being at either low (≤1 bar) or moderate to high (>20 bar) pressure.
As discussed above, change in PO/SD ratio between 2 and 6 at 800 °C does not significantly affect the textural properties of the activated carbons. The CO2 uptake isotherms of carbons activated at PO/SD ratio between 2 and 6, at temperature of 800 °C are shown in Fig. 7, and the uptake values are summarised in Table 2. The CO2 uptake is in the range 3.8–4.4 mmol g−1 at 1 bar, where the lowest CO2 uptake is for the sample activated at ratio of 4 (SD4800) and which has slightly lower micropore surface area (Table 2) compared to samples activated at PO/SD ratio of 2, 5 or 6. Additionally, at very low pressure of 0.15 bar, the CO2 uptake is between 1.0 and 1.2 mmol g−1 (Table 2). At higher pressure of 20 bar, there is a noticeable increase in the CO2 uptake for samples activated at PO/SD ratio 5 and 6, where the excess CO2 uptake is 12.7 mmol g−1 compared to 11.2 and 11.9 mmol g−1 for samples activated at PO/SD ratios of 2 and 4, respectively. This is consistent with the fact that samples activated at PO/SD ratios of 5 or 6 have slightly higher surface area and pore volume than samples activated at ratio 2 or 4. Our findings show that there is very limited benefit, in terms of CO2 uptake, from significantly increasing the amount of PO, and that the temperature has a far greater effect on the uptake at high pressure (20 bar and above). However, it is undesirable to have to go to 900 °C in order to achieve significant storage capacity at high pressures that simulate pre-combustion CO2 capture.
 | ||
| Fig. 7 Carbon dioxide uptake isotherms at 25 °C and pressure of (A) 0–1 bar and (B) 0–40 bar of carbons activated at 800 °C and PO/SD ratio of 2, 4, 5 and 6. | ||
The CO2 uptake capacity of carbons activated at PO/SD ratio of 2 or 4 and temperature ranging between 600 and 900 °C, and that of the carbons activated at various PO/SD ratios between 2 and 6 (Table 2), suggest that the activation temperature is more effective than the PO/SD ratio in optimising or tailoring the textural properties of activated carbon for targeted CO2 uptake applications. For example, at pressure of 1 bar the CO2 uptake is 4.3 mmol g−1 for samples activated at 800 °C, and 2.6 mmol g−1 for samples activated at 600 °C. At low pressure of 0.15, the CO2 uptake of samples activated at 600 and 700 °C (at either ratio 2 or 4) is higher than that of samples activated at 900 °C. At all PO/SD ratios explored, surface area higher than 1500 m2 g−1 is only achieved for activation at 900 °C. This is due to the fact that PO is a mild activating agent compared to KOH, which does offer activated carbons with high surface at 700 and 800 °C. We therefore explored other avenues for improving the surface area of PO activated carbons prepared at temperatures of up to 800 °C, and investigated the consequences on CO2 uptake.
3.2 Carbons derived from polypyrrole and pre-mixed precursors
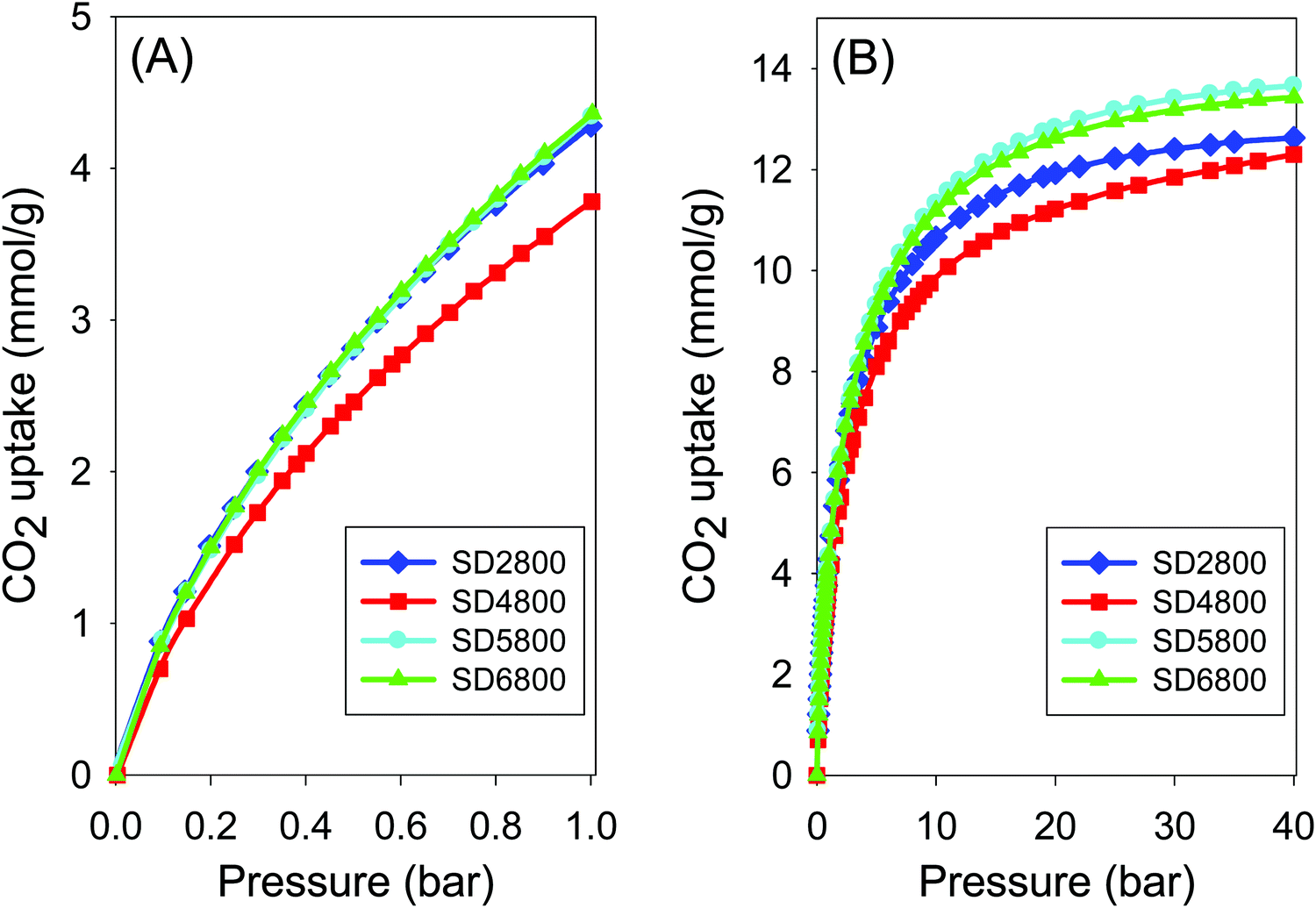
We, firstly, performed PO activation of Ppy to explore our hypothesis that it would generate activated carbons with higher surface area than sawdust hydrochar, and also to confirm that Ppy acts as a true carbonaceous precursor rather than as an activation enhancer as noted in the case of melamine.46,47 Activation of polypyrrole at PO/Ppy ratio of 2 and temperature of between 600 and 900 °C, offers activated carbon yields of 21–43 wt% (Table S1, ESI†), which are comparable to the yield from sawdust hydrochar (Table 1 and Table S1, ESI†). The C, H, N and O content, as a function of activation temperature, shows a trend similar to that of sawdust hydrochar, with C increasing while H, N and O decrease significantly (Table S1, ESI†). The porosity of the resulting carbons (Fig. S5, ESI†) in much higher than that of the SD derived carbons (Table 3). The surface area of Ppy derived samples activated at 800 °C is between 800 m2 g−1 and 2750 m2 g−1 compared to 500 m2 g−1 to 1500 m2 g−1 for SD derived carbons. The pore volume (0.4 to 1.5 cm3 g−1) is also higher than that of SD-derived samples (0.3–1.3 cm3 g−1). The Ppy derived carbons prepared at 800 or 900 °C have a much greater proportion of mesopores (Fig. S5 and Table 3, ESI†) compared to equivalent SD derived carbons. This is the expected trend based on the O/C ratio of sawdust hydrochar and polypyrrole, and starts to validate our hypothesis on the role played by the O/C ratio of the precursor in PO activation. We note that the surface area and pore volume observed for the PO activation of polypyrrole are, as expected, lower than those observed for KOH activation of polypyrrole under similar conditions.8,17,38,54
| Sample | Surface areaa (m2 g−1) | % μSA | Pore volumeb (cm3 g−1) | Pore size (Å) | CO2 uptake (mmol g−1) at 25 °C | ||
|---|---|---|---|---|---|---|---|
| 0.15 | 1 bar | 20 barc | |||||
| a Values in parenthesis are micropore surface area. b Values in parenthesis are micropore volume. c Values in parenthesis are total CO2 uptake. | |||||||
| SD2600 | 506 (411) | 81 | 0.27 (0.16) | 6/8 | 1.2 | 2.6 | 4.6 (4.8) |
| SD2700 | 893 (813) | 91 | 0.41 (0.33) | 6/8 | 1.4 | 3.8 | 7.6 (8.0) |
| SD2800 | 1463 (1311) | 90 | 0.67 (0.53) | 6/8/13 | 1.2 | 4.3 | 11.9 (12.5) |
| SD2900 | 2350 (800) | 34 | 1.27 (0.32) | 6/8/12/27 | 0.7 | 3.4 | 18.0 (19.1) |
| Ppy-SD(1–2)2600 | 678 (591) | 87 | 0.32 (0.24) | 6/8 | 1.4 | 3.3 | 5.9 (6.2) |
| Ppy-SD(1–2)2700 | 1174 (1007) | 86 | 0.54 (0.40) | 6/8/12 | 1.2 | 3.8 | 10.4 (10.9) |
| Ppy-SD(1–2)2800 | 2042 (1512) | 74 | 0.94 (0.59) | 6/9/13/20 | 0.9 | 3.9 | 16.6 (17.5) |
| Ppy-SD(1–2)2900 | 2436 (330) | 14 | 1.52 (0.14) | 6/8/12/27/34 | 0.5 | 2.4 | 18.2 (19.6) |
| Ppy-SD(1–1)2600 | 612 (527) | 86 | 0.29 (0.21) | 6/8 | 1.2 | 2.9 | 5.3 (5.6) |
| Ppy-SD(1–1)2700 | 1612 (1387) | 86 | 0.73 (0.56) | 6/8/9/13 | 1.2 | 4.1 | 12.5 (13.2) |
| Ppy-SD(1–1)2800 | 2291 (1688) | 74 | 1.06 (0.67) | 6/9/12/16/20 | 0.8 | 3.7 | 17.0 (18.0) |
| Ppy-SD(1–1)2900 | 2601 (273) | 11 | 1.67 (0.12) | 6/8/11/27/34 | 0.5 | 2.5 | 18.0 (19.5) |
| Ppy-SD(2–1)2600 | 655 (569) | 87 | 0.31 (0.23) | 6/7/9 | 1.3 | 3.1 | 5.6 (5.9) |
| Ppy-SD(2–1)2700 | 1665 (1435) | 86 | 0.75 (0.57) | 6/9/13 | 1.1 | 4.0 | 13.5 (14.2) |
| Ppy-SD(2–1)2800 | 2423 (1843) | 76 | 1.08 (0.72) | 6/8/9/12/20 | 0.8 | 3.6 | 17.8 (18.8) |
| Ppy-SD(2–1)2900 | 2612 (99) | 4 | 1.70 (0.05) | 6/8/13/27/34 | 0.5 | 2.4 | 18.8 (20.3) |
| Ppy2600 | 817 (711) | 87 | 0.39 (0.30) | 6/8 | 1.4 | 3.5 | 6.8 (7.2) |
| Ppy2700 | 1779 (1492) | 84 | 0.80 (0.59) | 6/9/13 | 1.0 | 3.8 | 13.7 (14.4) |
| Ppy2800 | 2747 (1841) | 67 | 1.23 (0.70) | 6/9/12/16/21 | 0.7 | 3.3 | 19.2 (20.3) |
| Ppy2900 | 2542 (263) | 10 | 1.45 (0.10) | 6/8/11/21/26 | 0.5 | 2.5 | 18.5 (19.8) |
PO activation was performed for pre-mixed precursors containing Ppy and SD at various ratios, i.e., 1:2, 1:1 or 2:1 Ppy:SD ratio. The selected Ppy:SD ratios offer a scenario where the amount of Ppy in the pre-mixed precursors is increased in a systematic manner such that it is possible to explore the effect of such an increase on the resulting activated carbons, and to relate it to observations for single PO activation of SD or Ppy. The yield of activated carbon from PO activation of all pre-mixed precursors, regardless of their composition, is similar to that from single activation of SD or Ppy (Table S1, ESI).† The trend in C, H, N and O content, as a function of activation temperature, is similar to that observed for single use of SD or Ppy, i.e., C increases while H, N and O decrease significantly (Table S1, ESI†). As expected, the amount of N is generally higher for activated carbons prepared from pre-mixed precursors with greater amounts of Ppy, especially for activation at 600 and 700 °C. It is interesting to note that the surface area and pore volume of the carbons prepared from pre-mixed precursors is generally higher than that of equivalent samples prepared from SD but lower that of samples from Ppy. Thus, in terms of the porosity generated, carbons prepared from SD and Ppy can be considered to be end members of a series with SD at the bottom and Ppy at the top. This is especially the case for samples activated at 700 and 800 °C. For example, the surface area of samples activated at 700 °C rises, with increasing amounts of Ppy in the precursor, in the order; SD (893 m2 g−1) < Ppy-SD(1–2) (1174 m2 g−1) < Ppy-SD(1–1) (1612 m2 g−1) < Ppy-SD(2–1) (1665 m2 g−1) < Ppy (1779 m2 g−1). For activation at 800 °C, a similar order is observed as follows; SD (1463 m2 g−1) < Ppy-SD(1–2) (2042 m2 g−1) < Ppy-SD(1–1) (2291 m2 g−1) < Ppy-SD(2–1) (2423 m2 g−1) < Ppy (2747 m2 g−1). Similar trends are observed for the pore volume. Samples activated at 600 °C also show similar trends but it should be noted that the level of activation at 600 °C is quite low.46 Sevilla and co-workers found that heating sawdust hydrochar (with no activating agent) at between 500 and 800 °C generates carbons with surface area of up to 420 m2 g−1.46 The surface area of carbons derived from SD containing precursors activated at 600 °C is 500 to 650 m2 g−1, which means that PO achieves only limited activation. This low level of activation may explain why a clear step-wise trend is not observed as is the case for activation at 700 and 800 °C. For activation at 900 °C, a step-wise rise in surface area is observed with increase in Ppy content in the precursor except for a decrease for sample Ppy2900, which we attribute to over activation. Such over activation is known to reduce surface area,21 and is consistent with polypyrrole having a relatively high O/C ratio, which means that it is more easily activated and thus likely to suffer from over activation at 900 °C.54
It is also clear from the porosity data (Table 3) and nitrogen sorption isotherms and pore size distribution curves of the samples prepared from pre-mixed precursors (Fig. S6–S8, ESI†) that there is a shift towards greater mesoporosity as the amount of Ppy in the precursor increases. This shift towards mesoporosity is also evident from the variations in proportion of surface area that arises from micropores, and the pore size maxima values summarised in Table 3. Our results show that by exploring PO activation of pre-mixed precursors, which are made up of carbonaceous matter (SD and Ppy) that is known to respond differently to similar levels of activation, we are able to modulate the porosity of the resulting carbons in a systematic manner and achieve higher values of surface area and pore volume. This approach can enable optimising of the porosity to generate microporous or mesoporous carbons to match specific applications, and we according explored the carbons for CO2 storage.
The much higher porosity of carbons derived from Ppy and pre-mixed precursor has a much greater effect on the CO2 uptake at higher pressure (>20 bar). As shown in Tables 2 and 3, the excess CO2 uptake at 20 bar of SD derived carbons reaches a maximum of ca. 12.9 mmol g−1 (total uptake of 13.5 mmol g−1), and for samples prepared at PO/SD ratio of 2, the maximum is 11.9 and 12.5 mmol g−1, respectively, for excess and total uptake. Carbons activated at 900 °C reach higher uptake, but as stated above it is desirable to avoid such a high activation temperature. The Ppy2800 sample achieves very high total CO2 uptake of 20.3 mmol g−1 at 20 bar, which is 60% higher than that (12.5 mmol g−1) of the equivalent SD derived carbon (SD2800). The equivalent pre-mixed precursor samples also achieve total uptake of between 17.5 and 18.8 mmol g−1. Our findings, therefore, show that by careful choice of precursor, it is possible to prepare carbons highly suited for medium to high pressure CO2 storage via mild PO activation.
Carbons that have excellent medium to high pressure CO2 uptake are potentially very well suited for CO2 separation from H2 before the latter is used as fuel for power stations that operate the Integrated Gasification Combined Cycle (IGCC). The separation of the gases in fuel streams with CO2 concentration of ca. 40 vol% (with H2 at 60 vol%) occurs at a pressure of up to 40 bar, which means that the CO2 partial pressure in the stream is up to ca. 16 bar. The CO2 is typically removed via pressure swing adsorption (PSA) processes, wherein capture takes place at the fuel stream pressure of up to 40 bar (meaning CO2 partial pressure of 16 bar) followed by desorption at 1 bar (i.e., CO2 partial pressure of 0.4 bar). The most important indicator of performance for CO2 capture in such pre-combustion scenarios is the PSA working capacity, which is the difference in storage capacity between the adsorption and desorption pressure. We therefore determined the PSA working capacity of the present carbons for adsorption pressure of 5 to 40 bar, which is typical for PSA systems used in pre-combustion CO2 for a 60% H2:40% CO2 gas stream. The excess PSA working capacity for SD derived carbons, shown in Fig. 8, is dependent on activation temperature, and while it is low for both sample SD2600 (1.2–2.5 mmol g−1) and SD2700 (2.5–4.9 mmol g−1), in the adsorption pressure of 5 to 40 bar, it increases for sample SD2800 (3.7–9.0 mmol g−1), and is much higher for SD2900 (3.7–14.9 mmol g−1). Furthermore, due to their higher pore volume, sample SD2800 and SD2900, show significant difference between excess and total PSA working capacity, especially for higher adsorption pressure (Fig. S13, ESI†). The PSA working capacity for the SD derived carbons is clearly related to porosity meaning that attractive working capacity is only observed for the sample activated at the highest temperature (900 °C). On the other hand, Ppy derived carbons prepared at lower activation temperature exhibit good to excellent PSA working capacity (Fig. 8B and Fig. S14, ESI†). The excess working capacity for Ppy2600 (1.2–2.6 mmol g−1) is similar to that of SD2600, while it is much higher (Fig. S15, ESI†) for Ppy2700 (3.7–12 mmol g−1), Ppy2800 (3.9–16.2 mmol g−1), and Ppy2900 (3.2–15.4 mmol g−1). A similar trend is observed for the total PSA working capacity (Fig. S16, ESI†).
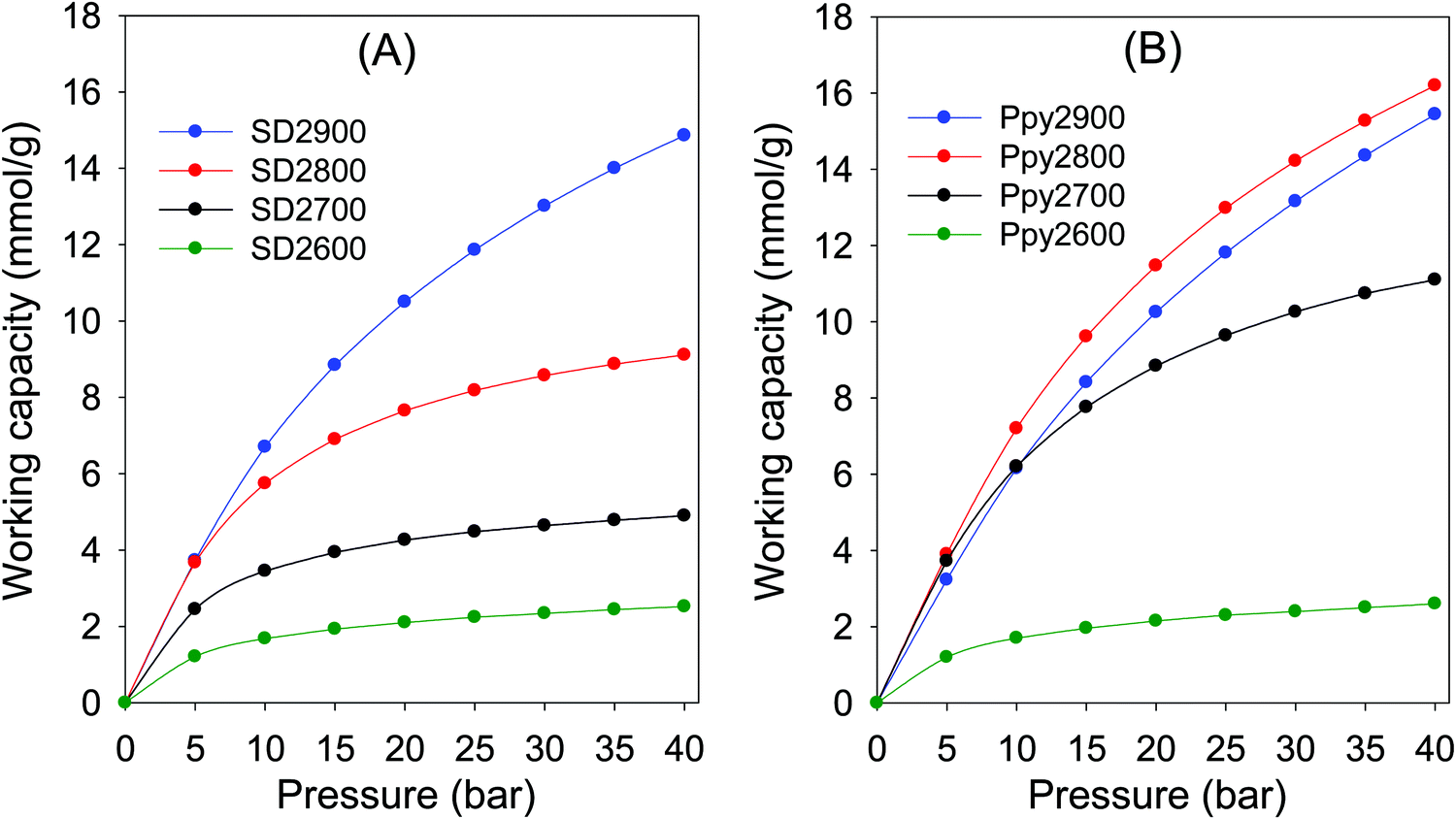 | ||
| Fig. 8 Excess pressure swing adsorption (PSA) CO2 working capacity for a 60:40 H2/CO2 mixture for (A) sawdust hydrochar (SD) or polypyrrole (Ppy) derived carbons activated at between 600 and 900 °C, and PO/precursor ratio of 2. | ||
As shown in Fig. 9, the carbons prepared from pre-mixed precursors at activation temperature of 800 °C exhibit excellent PSA working capacity, and in general outperform those prepared at 900 °C due to a better mix of microporosity and mesoporosity. The PSA excess working capacity of carbons derived from pre-mixed precursor at 800 °C (up to 15 mmol g−1 at adsorption pressure of 40 bar), is much higher than that of the equivalent SD derived sample, which only achieves 9 mmol g−1). Our findings, and particularly the clear role played by the temperature and type of precursor and consequences on applications such as CO2 storage, offer new insights beyond what is currently reported in the literature on the practicalities of using mild activating agents such as PO. To get a fuller picture of the performance of the present PO activated carbons, we compared their PSA working capacity to porous materials with much higher surface area, namely, a carbon (G-2.7-2) prepared via activation with PO and melamine,47 and metal organic frameworks (Nu-100 and MOF210).62,63 These porous materials are reported to have high CO2 uptake at high pressure due to their large surface area, and are claimed to be potentially useful for pre-combustion CO2 capture. The working capacity of the best performing PO activated carbons (Ppy2800 and Ppy-SD(2–1)2800) is comparable or better than these benchmark materials (Fig. S17, ESI†). The PO activated carbons offer better PSA working capacity than NU-100 and MOF210 despite the later having nearly twice the surface area. Furthermore, the PO activated carbons are expected to have higher volumetric PSA working capacity than the MOFs given that, unlike activated carbons, the packing density of high surface area MOFs tends to be low and they lose their uptake capacity when compacted.
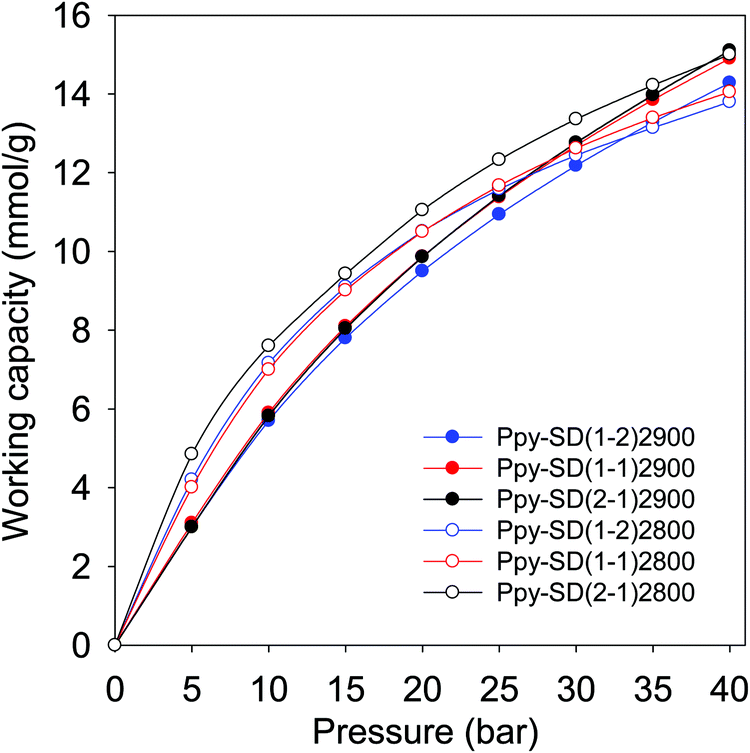 | ||
| Fig. 9 Excess pressure swing adsorption (PSA) CO2 working capacity for a 60:40 H2/CO2 mixture for carbons derived from pre-mixed precursors containing polypyrrole (Ppy) and sawdust hydrochar (SD) at various ratios. The carbons were prepared at activation temperature of 800 or 900 °C, and PO/precursor ratio of 2. | ||
4. Conclusion
We have shown that a less corrosive and toxic activating agent such as potassium oxalate (PO) can be successfully applied in a targeted manner to generate activated carbons from a range of precursors, including sawdust hydrochar, polypyrrole, or pre-mixed precursors that contain both biomass hydrochar and polypyrrole. By careful choice of activation conditions, it is possible to obtain activated carbons, with surface area of up to 2740 m2 g−1 and pore volume up to 1.7 cm3 g−1, which can be either highly microporous or highly mesoporous. The most important variables in controlling the porosity are the activation temperature and type of precursor based on their O/C ratio. Unlike what is observed for one of the most commonly used activating agents, KOH, changes in the PO/precursor ratio, within the range of 2–6, has no significant influence on the porosity of the PO activated carbons. This, therefore, unusually means that a relatively low PO/precursor ratio of 2, along with temperature variations, can be applied to generate a suite of carbons with the full range of porosity. This scenario offers a more eco-friendly and sustainable activation process in terms of amount of activating agent required, and also enables easier control of the porosity by simple choice of one variable, namely, activation temperature. We show that, due to their inherent O/C ratio, PO activation of sawdust hydrochar at temperatures up to 800 °C does not generate carbons with surface area higher than 1500 m2 g−1,46–51 while use of polypyrrole does achieve carbons with surface area up to 2700 m2 g−1 under similar activation conditions. Furthermore, the surface area and pore volume of the carbons prepared from pre-mixed precursors containing sawdust hydrochar and polypyrrole is higher than that of equivalent samples prepared from sawdust hydrochar but lower that of samples from polypyrrole. Thus, interestingly, in terms of the porosity generated, carbons prepared from sawdust hydrochar and polypyrrole may be considered to be end members of a series with the former at the bottom and latter at the top. It is, therefore, possible to direct the porosity of the activated carbons towards pores channels of size 6 to 8 Å that are efficient for CO2 storage at low pressure conditions that mimic post-combustion CO2 capture. At 25 °C, the carbons can store up to 1.4 and 4.4 mmol g−1 of CO2 at 0.15 bar and 1 bar, respectively. The micropore/mesopore mix of the carbons can also be controlled towards greater mesoporosity, which is favourable for moderate to high pressure CO2 storage conditions that mimic pre-combustion capture. At 25 °C, the carbons can store up to 20.3 mmol g−1 at 20 bar and 30.1 mmol g−1 at 40 bar. At a maximum activation temperature of 800 °C, the porosity of the carbons can be tailored so as to optimise pre-combustion CO2 capture via pressure swing adsorption (PSA) processes at adsorption pressure of up to 40 bar and desorption at 1 bar, from gas mixtures (e.g., 60:40 H2/CO2) where the carbons achieve excellent working capacity that is higher than that of benchmark porous materials that have much larger surface area.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the government of Iraq for funding a PhD studentship for Abdulsalam Aljumialy. RM thanks the Royal Society for a Research Grant, and for a Royal Society Wolfson Research Merit Award.References
- L. Wei and G. Yushin, Nano Energy, 2012, 1, 552 CrossRef CAS.
- M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250 RSC.
- Y. X. Xia, Z. X. Yang and Y. Zhu, J. Mater. Chem. A, 2013, 1, 9365 RSC.
- M. M. Titirici, R. J. White, C. Falco and M. Sevilla, Energy Environ. Sci., 2012, 5, 6796 RSC.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353 CrossRef CAS.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537 CrossRef CAS.
- J. C. Wang and S. Kaskel, J. Mater. Chem., 2012, 22, 23710 RSC.
- M. Sevilla, A. B. Fuertes and R. Mokaya, Energy Environ. Sci., 2011, 3, 1400 RSC.
- Z. Yang, Y. Xia and R. Mokaya, J. Am. Chem. Soc., 2007, 129, 1673 CrossRef CAS.
- T. S. Blankenship, N. Balahmar and R. Mokaya, Nat. Commun., 2017, 8, 1545 CrossRef.
- E. Masika and R. Mokaya, Energy Environ. Sci., 2014, 7, 427 RSC.
- L. Wei, M. Sevilla, A. B. Fuertes, R. Mokaya and G. Yushin, Adv. Energy Mater., 2011, 1, 356 CrossRef CAS.
- H. M. Coromina, D. A. Walsh and R. Mokaya, J. Mater. Chem. A, 2016, 4, 280 RSC.
- M. Sevilla and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 1765 RSC.
- M. Sevilla, W. Sangchoom, N. Balahmar, A. B. Fuertes and R. Mokaya, ACS Sustainable Chem. Eng., 2016, 4, 4710 CrossRef CAS.
- W. Sangchoom and R. Mokaya, ACS Sustainable Chem. Eng., 2015, 3, 1658 CrossRef CAS.
- N. Balahmar, A. C. Mitchell and R. Mokaya, Adv. Energy Mater., 2015, 5, 1500867 CrossRef.
- C. Robertson and R. Mokaya, Microporous Mesoporous Mater., 2013, 179, 151 CrossRef CAS.
- E. A. Hirst, A. Taylor and R. Mokaya, J. Mater. Chem. A, 2017, 6, 12393 RSC.
- N. Balahmar, A. Al-Jumialy and R. Mokaya, J. Mater. Chem. A, 2017, 5, 12330 RSC.
- T. S. Blankenship and R. Mokaya, Energy Environ. Sci., 2017, 10, 2552 RSC.
- J.-J. Hwang, Renewable Sustainable Energy Rev., 2013, 19, 220 CrossRef CAS.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796 CrossRef CAS.
- K. B. Tokarska and N. P. Gillett, Nat. Clim. Change, 2018, 8, 296 CrossRef CAS.
- R. J. Millar, J. S. Fuglestvedt, P. Friedlingstein, J. Rogelj, M. J. Grubb, H. D. Matthews, R. B. Skeie, P. M. Forster, D. J. Frame and A. R. Andmyles, Nat. Geosci., 2017, 10, 741 CrossRef CAS.
- B. Adeniran, E. Masika and R. Mokaya, J. Mater. Chem. A, 2014, 2, 14696 RSC.
- Z. J. Zhang, Z. Z. Yao, S. C. Xiang and B. L. Chen, Energy Environ. Sci., 2014, 7, 2868 RSC.
- B. Adeniran and R. Mokaya, Chem. Mater., 2016, 28, 994 CrossRef CAS.
- J. P. Marco-Lozar, M. Kunowsky, F. Suarez-Garcia, J. D. Carruthers and A. Linares-Solano, Energy Environ. Sci., 2012, 5, 9833 RSC.
- E. Haffner-Staton, N. Balahmar and R. Mokaya, J. Mater. Chem. A, 2016, 4, 13324 RSC.
- M. R. Hudson, Q. L. Queen, J. A. Mason, D. W. Fickel, R. F. Lobo and C. M. Brown, J. Am. Chem. Soc., 2012, 134, 1970 CrossRef CAS.
- Y. Xia, R. Mokaya, G. S. Walker and Y. Q. Zhu, Adv. Energy Mater., 2011, 1, 678 CrossRef CAS.
- Z. Zhang, J. Zhou, W. Xing, Q. Xue, Z. Yan, S. Zhuo and S. Z. Qiao, Phys. Chem. Chem. Phys., 2013, 15, 2523 RSC.
- B. Adeniran and R. Mokaya, J. Mater. Chem. A, 2015, 3, 5148 RSC.
- N. P. Wickramaratne and M. Jaroniec, J. Mater. Chem. A, 2013, 1, 112 RSC.
- A. Wahby, J. M. Ramos-Fernandez, M. Martnez-Escandell, A. Sepulveda-Escribano, J. Silvestre-Albero and F. Rodriguez-Reinoso, ChemSusChem, 2010, 3, 974 CrossRef CAS.
- N. P. Wickramaratne and M. Jaroniec, ACS Appl. Mater. Interfaces, 2013, 5, 1849 CrossRef CAS.
- B. Adeniran and R. Mokaya, Nano Energy, 2015, 16, 173 CrossRef CAS.
- M. Sevilla, A. B. Fuertes and R. Mokaya, Int. J. Hydrogen Energy, 2011, 36, 15658 CrossRef CAS.
- M. Sevilla, R. Mokaya and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 2930 RSC.
- M. Plaza, A. González, J. Pis, F. Rubiera and C. Pevida, Appl. Energy, 2014, 114, 551 CrossRef CAS.
- C. Moreno-Castilla, F. Carrasco-Marín, M. V. Lopez-Ramon and M. A. Alvarez-Merino, Carbon, 2001, 39, 1415 CrossRef CAS.
- A. R. Satayeva, C. A. Howell, A. V. Korobeinyk, J. Jandosov, V. J. Inglezakis, Z. A. Mansurov and S. V. Mikhalovsky, Sci. Total Environ., 2018, 630, 1237 CrossRef CAS.
- Y. Chen, W. Chen, B. Huang and M. Huang, Chem. Eng. Res. Des., 2013, 91, 1783 CrossRef CAS.
- J. Ludwinowicz and M. Jaroniec, Carbon, 2015, 82, 297 CrossRef CAS.
- M. Sevilla, G. A. Ferrero and A. B. Fuertes, Carbon, 2017, 114, 50 CrossRef CAS.
- M. Sevilla, A. M. Al-Jumialy, A. B. Fuertes and R. Mokaya, ACS Appl. Mater. Interfaces, 2018, 10, 1623 CrossRef CAS.
- J. Wei, S. Wan, P. Zhang, H. Ding, X. Chen, H. M. Xiong, S. Gao and X. Wei, New J. Chem., 2018, 42, 6763 RSC.
- M. Sevilla, G. A. Ferrero and A. B. Fuertes, Chem. Mater., 2017, 29, 6900 CrossRef CAS.
- J. Li, Q. Jiang, L. Wei, L. Zhong and X. Wang, J. Mater. Chem. A, 2020, 8, 1469 RSC.
- J. Wang, Q. Yang, W. Yang, H. Pei, L. Zhang, T. Zhang, N. Hu, Y. Suo and J. Wang, J. Mater. Chem. A, 2018, 6, 16690 RSC.
- Q. Jiang, Y. Wang, Y. Gao and Y. Zhang, Environ. Sci. Pollut. Res., 2019, 26, 30268 CrossRef CAS.
- H. Lin, C. Xu, Q. Wang, J. Wu, Y. Wang, Y. Zhang and G. Fan, Int. J. Hydrogen Energy, 2019, 44, 21527 CrossRef CAS.
- N. Balahmar and R. Mokaya, J. Mater. Chem. A, 2019, 7, 17466 RSC.
- A. Altwala and R. Mokaya, Energy Environ. Sci., 2020, 13, 2967 RSC.
- T. Ungár, J. Gubicza, G. Ribárik, C. Pantea and T. W. Zerda, Carbon, 2002, 40, 929 CrossRef.
- L. Zhang, J. Zhang, H. Fu, H. Zhang, H. Liu, Y. Wan, S. Zheng and Z. Xu, Microporous Mesoporous Mater., 2018, 260, 59 CrossRef CAS.
- J. Rodriguez-Mirasol, T. Cordero, L. Radovic and J. Rodriguez, Chem. Mater., 1998, 10, 550 CrossRef CAS.
- F. Su, X. Zhao, L. Lv and Z. Zhou, Carbon, 2004, 42, 2821 CrossRef CAS.
- J. Wang and S. Kaskel, J. Mater. Chem., 2012, 22, 23710 RSC.
- K. C. Bedin, A. C. Martins, A. L. Cazetta, O. Pezoti and V. C. Almeida, Chem. Eng. J., 2016, 286, 476 CrossRef CAS.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998 CrossRef CAS.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: One table and seventeen (17) additional figures; TGA curves, XRD patterns, and CO2 uptake isotherms. See DOI: 10.1039/d0ma00449a |
| This journal is © The Royal Society of Chemistry 2020 |