
Stabilization of Cu+ by tuning a CuO–CeO2 interface for selective electrochemical CO2 reduction to ethylene†
Senlin
Chu‡
a,
Xupeng
Yan‡
b,
Changhyeok
Choi‡
 c,
Song
Hong
a,
Alex W.
Robertson
d,
Justus
Masa§
e,
Buxing
Han
b,
Yousung
Jung
*c and
Zhenyu
Sun
*af
c,
Song
Hong
a,
Alex W.
Robertson
d,
Justus
Masa§
e,
Buxing
Han
b,
Yousung
Jung
*c and
Zhenyu
Sun
*af
aState Key Laboratory of Organic-Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: sunzy@mail.buct.edu.cn
bBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid and Interface and Thermodynamics, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
cDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: ysjn@kaist.ac.kr
dDepartment of Materials, University of Oxford, Oxford, OX1 3PH, UK
eAnalytische Chemie-Elektroanalytik & Sensorik, Ruhr University Bochum, D-44780 Bochum, Germany
fKey Laboratory of Low-Carbon Conversion Science & Engineering, Shanghai Advanced Research Institute, Chinese Academy of Sciences, China
First published on 1st September 2020
Abstract
Electrochemical conversion of carbon dioxide (CO2) into multi-carbon fuels and chemical feedstocks is important but remains challenging. Here, we report the stabilization of Cu+ within a CuO–CeO2 interface for efficient and selective electrocatalytic CO2 reduction to ethylene under ambient conditions. Tuning the CuO/CeO2 interfacial interaction permits dramatic suppression of proton reduction and enhancement of CO2 reduction, with an ethylene faradaic efficiency (FE) as high as 50.0% at −1.1 V (vs. the reversible hydrogen electrode) in 0.1 M KHCO3, in stark contrast to 22.6% over pure CuO immobilized on carbon black (CB). The composite catalyst presents a 2.6-fold improvement in ethylene current compared to that of CuO/CB at similar overpotentials, which also exceeds many recently reported Cu-based materials. The FE of C2H4 remained at over 48.0% even after 9 h of continuous polarization. The Cu+ species are believed to be the adsorption as well as active sites for the activation of CO2 molecules, which remain almost unchanged after 1 h of electrolysis. Further density functional theory calculations demonstrate the preferred formation of Cu+ at the CuO–CeO2 interface. This work provides a simple avenue to convert CO2 into high-value hydrocarbons by rational stabilization of Cu+ species.
Introduction
Electrochemical carbon dioxide (CO2) reduction (ECR) shows promise in reducing greenhouse gas emissions, storing intermittent renewable electricity, as well as attaining energy security and sustainability.1,2 Although this energy conversion process can be conducted under mild temperatures and atmospheric pressure, there are still many challenges, such as low conversion efficiency and poor product selectivity, which have to be overcome.3,4 To enable progress towards this goal, the development of catalysts with high efficiency, sufficient selectivity, and low cost is necessary.5,6 The synthesis of valuable hydrocarbons and other chemicals through ECR has drawn significant attention as a potential scheme for recycling CO2.7–18 In particular, C2+ (containing two or more carbon atoms) compounds such as ethylene have high energy densities and enjoy global demand in comparison to C1 products.19,20 For instance, ethylene is widely used as an industrial feedstock for manufacturing plastics and diesel, and its selective production in lieu of methane is important.Copper, with its unique electronic properties, has been shown to stabilize CO intermediates (*CO) and enable them to be further reduced to multi-carbon products via CO dimerization to yield an *OCCO adsorbate and subsequent hydrogenations.19 However, Cu is intrinsically limited by the scaling relations between the binding energies of various reaction intermediates on the metallic surfaces, which leads to wide product distributions and undesirable hydrogen evolution, thus hampering large-scale practical implementation.21 Selective reduction of CO2 into industrially important C2+ species remains an ongoing challenge. Recent investigations indicate that preferential conversion of CO2 to C2+ products can be achieved using Cu-based materials doped with foreign atoms,22 Cu alloys,23,24 or through control of exposed crystal lattice,25,26 oxidation state,27,28 and surface morphology.25,29–31 For example, single-crystal Cu(100) was demonstrated to display good selectivity for ethylene evolution with a faradaic efficiency (FE) of about 40.0%, which can be further improved to 50.0% over Cu(711) at 5.0 mA cm−2 in 0.1 M KHCO3.25 A recent study showed that CuAg bimetallic catalysts have enhanced selectivity to C2+ products, which was attributed to the suppression of the hydrogen evolution reaction (HER) due to the formation of compressively strained CuAg surface alloys.32 In addition, Cu/oxide interfaces are regarded to be critical to inhibit the parasitic HER during electrocatalytic CO2 reduction.33,34 Oxides of copper exhibit enhanced ECR activity and increased selectivity towards multi-carbon products. The selectivity of these catalysts is dependent on the copper oxidation state.31 Some computational studies have suggested that the coexistence of a Cu+/Cu0 mixture synergistically promotes CO2 reduction to C2+ products due to improved CO2 activation and CO dimerization.35,36 Experimentally, however, evidence for the stability of the active Cu+ species during CO2 reduction remains unclarified thus far.
Herein, we report on the stabilization of Cu+ by controlling the interplay between lattice-mismatched CuO and CeO2. This scheme allows one to design an efficient and selective catalyst for electrocatalytic CO2 reduction to produce ethylene, among other products (methane, carbon monoxide, formic acid, and ethanol). Catalytic selectivity can be greatly improved by taking advantage of the CuO–CeO2 interactions in different composition regimes and interfacial structures. A remarkable FE for ethylene production of up to 50.0% was obtained at mild overpotentials, outperforming many previously reported Cu-based electrocatalysts. Furthermore, density functional theory (DFT) calculations revealed that CeO2 changes the oxidation state of Cu atoms to Cu+ at the CuO–CeO2 interface.
Results and discussion
The X-ray diffraction (XRD) patterns of CuO–CeO2/CB together with individual CuO/CB and CeO2/CB are shown in Fig. 1a. Apart from a broad peak at 22.2° originating from carbon black with low crystallinity, diffraction peaks at about 28.1, 47.6, and 56.0° were observed in both CeO2/CB and CuO–CeO2/CB, corresponding to the (111), (220), and (311) planes of CeO2 (PDF# 34-0394). These indicate the formation of fluorite (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) CeO2 with a face-centered cubic (fcc) structure in the composites. Unlike the bare CuO/CB that displayed representative monoclinic CuO peaks (PDF# 44-0706), no diffraction peaks of any Cu compounds were discernible in the XRD pattern of CuO–CeO2/CB, likely due to the low loading and/or small size of CuO in the composite.
m) CeO2 with a face-centered cubic (fcc) structure in the composites. Unlike the bare CuO/CB that displayed representative monoclinic CuO peaks (PDF# 44-0706), no diffraction peaks of any Cu compounds were discernible in the XRD pattern of CuO–CeO2/CB, likely due to the low loading and/or small size of CuO in the composite.
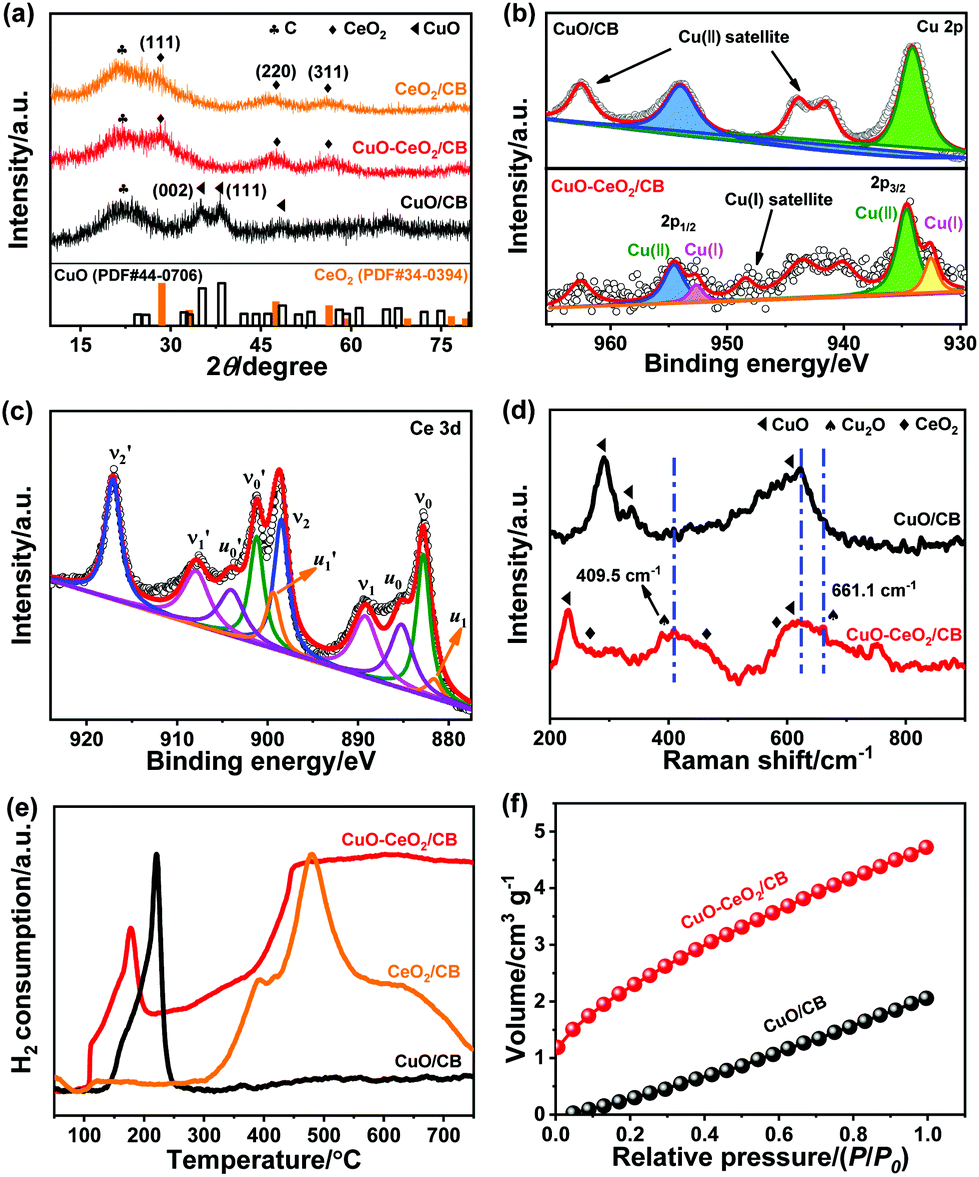 | ||
| Fig. 1 (a) XRD patterns of CeO2/CB, CuO/CB, and CuO–CeO2/CB. (b) Cu 2p XPS spectra of CuO/CB and CuO–CeO2/CB. (c) Ce 3d XPS spectrum of CuO–CeO2/CB. (d) Raman spectra of CuO/CB and CuO–CeO2/CB. (e) H2-TPR profiles of CeO2/CB, CuO/CB, and CuO–CeO2/CB. (f) CO2 adsorption isotherms of CuO/CB and CuO–CeO2/CB. | ||
X-ray photoelectron spectroscopy (XPS) was used to acquire information about the surface composition and chemical state of the Cu species, as well as possible interactions between copper and cerium oxides. Fig. 1b shows the Cu 2p signal of CuO/CB and CuO–CeO2/CB. The Cu 2p core-level spectrum of CuO/CB reveals pronounced CuO features, that is, Cu 2p1/2 and 2p3/2 peaks with binding energies (BEs) at 954.0 and 934.0 eV, respectively. Strong Cu2+ satellites at 962.6, 944.0, and 941.6 eV were also clearly observed.37 Nevertheless, no apparent peaks assigned to Cu+ can be identified. In contrast, two peaks located at 954.3 and 951.9 eV were observed in CuO–CeO2/CB, which can be attributed to Cu2+ 2p1/2 and Cu+ 2p3/2, respectively.10 This unambiguously verifies the formation and stabilization of Cu+, likely owing to electron transfer from Ce3+ to Cu2+. The relative Cu+ percentage was determined to be 23.4% in CuO–CeO2/CB, based on the peak area ratio of all copper oxidation states in the Cu 2p regions. Fig. 1c depicts the Ce 3d signals of CuO–CeO2/CB having a satellite structure due to the hybridization of Ce 3d orbitals with O 2p orbitals and partial occupation of the 4f levels.38 The 3d5/2 and 3d3/2 spin–orbit components (spin–orbit splitting, ∼18.5 eV) are denoted as ν and ν′, respectively, which is in line with the previous literature on Ce(IV).39 The peaks of ν0 and ν1 were attributed to a mixing configuration of the 3d9 4f2 (O 2p4) and 3d9 4f1 (O 2p5) Ce4+ states and ν2 to the 3d9 4f0 (O 2p6) Ce4+ state.38 The same assignment could be applied to the ν′ structures, which correspond to the Ce 3d3/2 level. This illustrates the major valence of Ce(IV) in the sample, consistent with the XRD result. Four peaks u0 (BE ≈ 885.8 eV), u1 (BE ≈ 880.6 eV), u0′ (BE ≈ 904.1 eV), and u1′ (BE ≈ 899.5 eV) associated with Ce3+ were identified, indicating the presence of Ce2O3 in the sample.11 The well-defined peak ν2′ typical of Ce4+ can be used to estimate the fraction of Ce4+.40 Given that the area of the ν2′ component comprises 14.0% of the overall area of the Ce 3d region, the Ce3+ percentage was estimated to be 27.0%. The deconvoluted O 1s XPS spectrum of CuO–CeO2/CB (Fig. S1a†) displays a predominant peak at 529.7 eV arising from the lattice oxygens in the metal oxides, and two less intense peaks at 531.2 and 532.6 eV that can be assigned to defective sites (surface oxygen vacancies) and physisorbed water, respectively.
The presence of Cu+ in CuO/CeO2 heterostructures was also evidenced by Raman scattering experiments. The bands centered at about 258.0, 462.9, and 595.0 cm−1 were identified as shown in Fig. 1d, which can be well assigned to the F2g mode, second-order transverse acoustic (2TA) mode, and defect-induced (D) mode of fluorite CeO2, respectively.41 It is worth noting that the three peaks at 290.4, 337.0, and 622.8 cm−1 that appeared for CuO/CB are attributed to the respective single Ag mode and two Bg optical modes of cupric oxide.42 However, for CuO–CeO2/CB, the feature around 290.4 cm−1 disappeared and a new band at 230.4 cm−1 was observed being tentatively assigned to one-magnon scattering, which arose from the antiferromagnetic ordering of the Cu2+ ions.42 Two additional distinct peaks at 409.5 and 661.1 cm−1 were observed that were typical of Cu+ Raman fingerprints.42 This further confirms that CuO was partially converted to Cu2O, possibly induced by adjacent CeO2 nanoparticles (NPs). These results are consistent with the XPS data in Fig. 1b.
Temperature-programmed reduction by hydrogen (H2-TPR, Fig. 1e) manifested two marked H2 consumption peaks at 110.0 and 178.0 °C for CuO–CeO2/CB, which were ascribed to the reduction of the subsurface Cuδ+ to Cu+ and further to Cu0, respectively, by consuming reducible oxygen from the CuOx species. Notably, the TPR reduction peaks shifted to lower temperatures relative to CuO/CB, which is likely a result of hydrogen spillover to CuO at the CuO/CeO2 interface. Furthermore, CuO–CeO2/CB exhibited a CO2 uptake capacity of 4.7 cm3 g−1 (Fig. 1f), 2.3 fold as large as that of CuO/CB. This could lead to enriched CO2 on the local surface of the working electrode, thus boosting *CO coverage and dimerization. The significant enhancement in CO2 capture ability is due to the introduction of CeO2, which can effectively adsorb CO2, forming carbonates and hydrogen carbonates.43 It can be envisioned that the as-made hybrid catalyst may facilitate multiple interesting functionalities such as adsorption, electronics, activation, and catalysis, among others, based on the synergistic interaction between CuO and CeO2.
To decipher the morphological features of our catalyst, aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was performed on CuO–CeO2/CB. Fig. 2a and b show the formation of many reticular NPs homogeneously distributed on carbon black. The (111) and (100) planes of CeO2 were indexed with the aid of fast Fourier transformation (FFT) (inset of Fig. 2b). Furthermore, the energy-dispersive X-ray spectroscopy (EDS) maps (Fig. 2c–g and Fig. S2b–f†) along with the EDS spectrum (Fig. 2h) confirmed that the NPs were composed of CuO and CeO2 crystallites. EDS elemental mapping revealed an almost full overlap of the Cu-rich and Ce-rich domains, indicating large interfaces between the two metal oxides. The crystallite sizes of CuO and CeO2 were found to be similar with the sizes being less than 5 nm as found using high-resolution TEM (Fig. 2i and j). By FFT, the fcc CuO and CeO2 NPs were discerned (Fig. 2k and l). Most of the CuO crystals exhibited a faceted cuboidal morphology and were surrounded by CeO2 NPs (Fig. 2i and j).
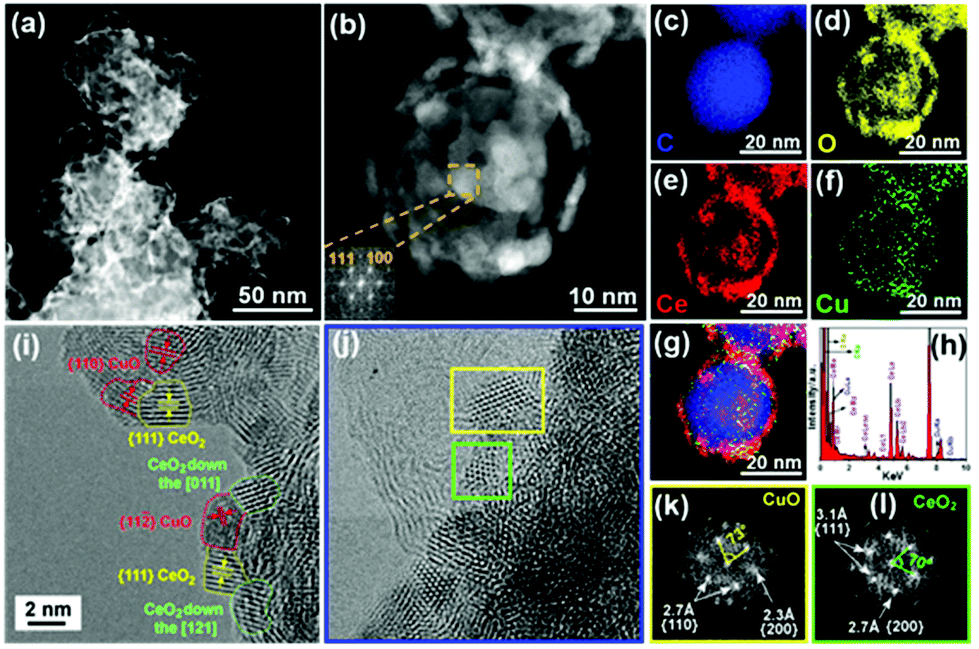 | ||
| Fig. 2 (a and b) HAADF-STEM images of CuO–CeO2/CB. The inset in image (b) shows the corresponding FFT of CuO–CeO2/CB. EDS elemental maps of (c) C, (d) O, (e) Ce, (f) Cu, and (g) overlay of C (blue), Ce (red), and Cu (green) over the region shown in image (b), along with corresponding EDS spectrum (h). (i) HRTEM image of CuO–CeO2/CB. (j–l) Magnified image of the blue rectangle in image (i) and FFTs of the regions encased by the yellow and green rectangles in image (j). | ||
ECR is very sensitive to operating conditions, such as the nature and properties of the electrocatalyst, electrolyte composition, and electrochemical cell type. To evaluate the intrinsic catalytic properties of the as-prepared hybrids, we conducted the ECR in CO2-saturated 0.1 M KHCO3 aqueous electrolyte (pH 6.8) using a reported design of liquid H-type cell with continuous CO2 bubbling.44 The potential-dependent geometric current densities of CuO–CeO2/CB within the potential range of 0.0 to −1.4 V (vs. RHE) were recorded by linear sweep voltammetry (LSV), as shown in Fig. 3a. Significantly higher cathodic currents were observed in a CO2 environment than in an Ar environment within the entire potential region. CO, H2, CH4, HCOOH, C2H4, and C2H5OH were detected at applied potentials ranging from −0.9 to −1.3 V (vs. RHE) in a CO2-saturated 0.1 M KHCO3 solution. The ECR preferably occurred over HER at potentials ranging from −0.9 to −1.2 V (vs. RHE), while HER dominated at more negative potentials (Fig. 3b and c). As demonstrated in Fig. 3d, CeO2/CB generated exclusively H2 with a very small amount of ECR products (FE < 3.0%). Commercial Cu2O decorated on CB exhibited an FE of 31.7% for ECR (vs. RHE) (Fig. S3†), whereas the FE for C2H4 formation was as low as 8.5%. Although CuO/CB can reduce CO2 at overpotentials larger than 0.97 V, the highest FE for ECR was below 32.0%, with the selectivity for C2+ products being less than 23.0%. It is worth noting that the CuO–CeO2/CB nanocomposites substantially promoted the activity toward CO2 reduction with an FE > 63.0%.
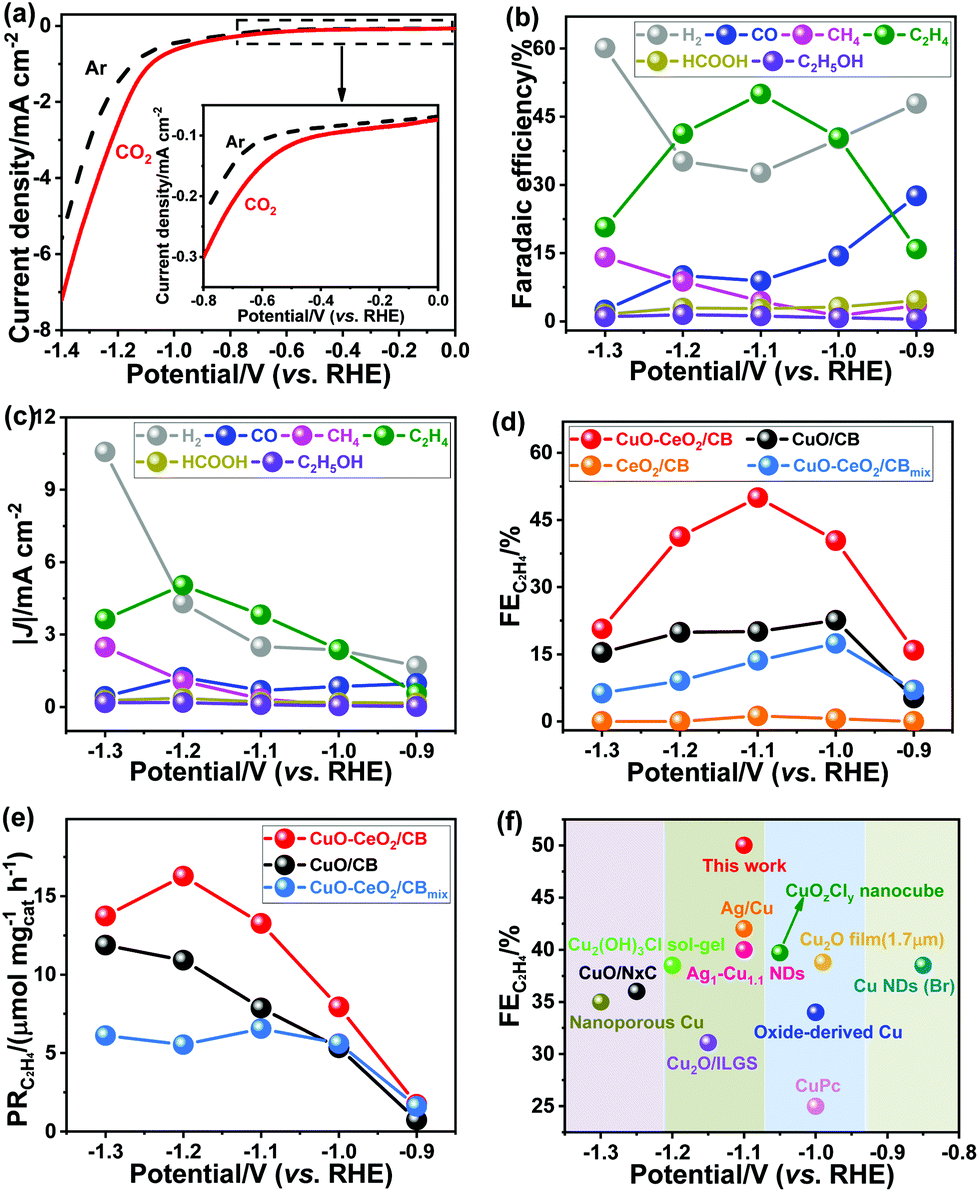 | ||
| Fig. 3 (a) LSV results of CuO–CeO2/CB on a glassy carbon electrode in Ar- (dashed black line) or CO2 (solid red line)-saturated 0.1 M KHCO3 solutions at a scan rate of 5 mV s−1. (b) Faradaic efficiencies and (c) partial current densities for ECR products over CuO–CeO2/CB at various applied potentials. (d) C2H4 FEs of CuO–CeO2/CB, CuO/CB, CeO2/CB, and CuO–CeO2/CBmix in the potential range from −0.9 to −1.3 V. (e) Production rates of C2H4 at different potentials over CuO–CeO2/CB, CuO/CB, and CuO–CeO2/CBmix. (f) C2H4 FEs of CuO–CeO2/CB and other reported Cu-based electrocatalysts. | ||
The C1 products were obtained at similar yields for CuO/CB and CuO–CeO2/CB, but C2+ product selectivity and C2H4 FE and production rate (Fig. 3e) were remarkably boosted for the latter sample. C2H4 emerged at an onset potential of −0.7 V (vs. RHE) over CuO–CeO2/CB and rose to a maximum with FE up to 50.0% at −1.1 V (vs. RHE) in contrast to that of 22.6% and 1.2% for CuO/CB and CeO2/CB, respectively (Fig. 3b and d). The C2H4 selectivity even outperforms many recently reported Cu-based electrocatalysts under similar overpotentials (Fig. 3f), such as the state-of-the-art Cu nanocubes with exposed (100) facets (maximum C2H4 FE is 32.0%)45,46 and Ag–Cu nanodimers (maximum C2H4 FE is 40.0%).24
The ECR activity was tunable by adjusting the amounts of CeO2 and CuO (Tables S1 and S2†). As seen in Fig. 4a, incorporation of CeO2 at various amounts was found to thwart hydrogen evolution and facilitate C2H4 generation. The optimal loading of CeO2 was 30.0 wt%. A continuous increase in CeO2 loading led to a slight decrease in ECR activity, probably owing to the reduction in electrical conductivity. Likewise, the ECR activity to yield C2H4 increased with the mass percentage of CuO in the range of 0.75–1.5 wt%. The C2H4 FE tended to diminish upon a further increase in CuO loading (Fig. 4b). This may be due to a combination of the less extended interface and formation of larger CuO particles, resulting in weakened binding of the reactants and intermediates. Furthermore, it was found that there is an optimum in the particle size of CuO, which maximized C2H4 generation (Table S3†), in line with the results observed for Cu in the literature.24 The effect of electrolytic temperature on ECR was also explored. The FE for ECR was found to be maximized at 3 ± 3 °C (Fig. S4†), indicating that the HER tends to be inhibited at low reaction temperatures.
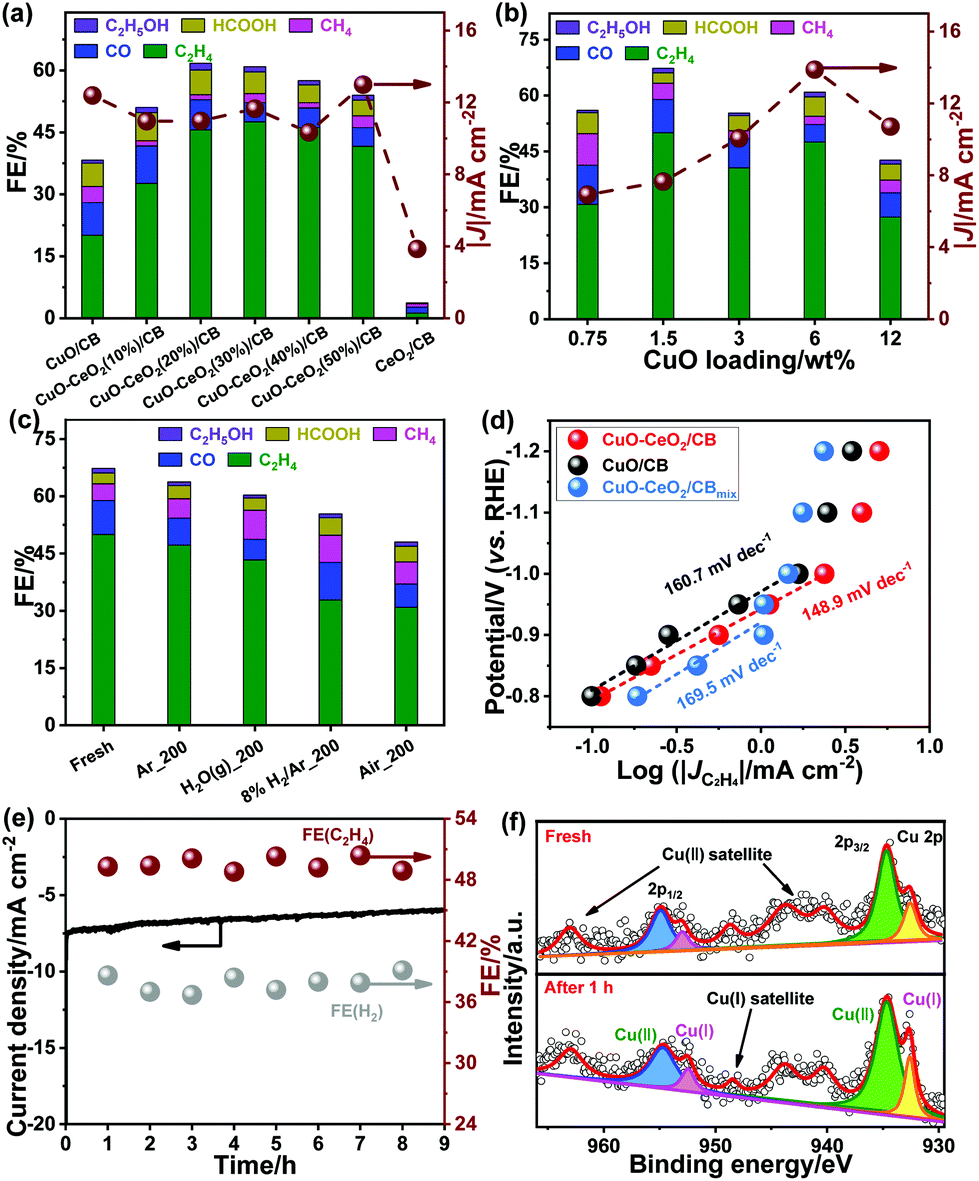 | ||
| Fig. 4 FE and geometric current density at −1.1 V as a function of (a) CeO2 loading at a fixed CuO content of 6.0 wt% and (b) CuO loading at a constant CeO2 loading of 30.0 wt%. (c) FEs at −1.1 V over fresh and treated CuO–CeO2/CB under Ar, H2O, 8.0% H2/Ar, and air. (d) Tafel plots of the partial geometric current density for C2H4 production over CuO–CeO2/CB, CuO/CB, and CuO–CeO2/CBmix. (e) Geometric current-, C2H4 FE-, and H2 FE-time responses of CuO–CeO2/CB at −1.1 V. (f) Cu 2p XPS spectra of CuO–CeO2/CB before and after 1 h of electrolysis. | ||
To check whether Ce3+ impacted the ECR, the synthesis of catalysts was performed in an air-free glove-box under otherwise similar conditions. The resulting CuO–Ce2O3/CB provided a much lower C2H4 FE (22.4% at −1.1 V vs. RHE) compared to CuO–CeO2/CB. This indicates that Ce3+ is unlikely to contribute to the enhanced ECR.
The role of Cu+ during ECR was investigated by treating CuO–CeO2/CB at 200 °C under different atmospheres. The relative fractions of Cu0, Cu+, and Cu2+ in the treated CuO–CeO2/CB samples were probed using XPS (Table S4 and Fig. S5†). As observed in Fig. 4c, the CO2 reduction activity and selectivity dropped slightly in an Ar environment, which may favor the transformation of a small fraction of Cu2+ to Cu+ and Cu+ to Cu0 (Table S4 and Fig. S5a†). The C2H4 FE mildly decreased with a simultaneous increase in CH4 FE, probably resulting from the aggregation of metal oxide NPs and the presence of Cu0. This phenomenon became a little more pronounced after being subjected to water vapor, which may be due to the promotion of CH4 formation by the adsorbed surface water molecules, in addition to the transformation of Cu+ (Table S4 and Fig. S5b†). However, annealing of the catalyst in 8% H2/Ar led to increased HER with a distinct drop in ECR performance. Despite CO FE being improved to 33.7%, the C2H4 FE dropped to 33.8%. This suggests that the reduction of Cu+/Cu0 ratio (Table S4 and Fig. S5c†) is detrimental to CO–CO coupling. In addition, the decrease in Cu+/Cu2+ (Table S4 and Fig. S5d†) upon exposure of the sample to air at elevated temperatures degraded C2H4 production, accompanied by substantially more H2 evolution.
To probe the role of the CuO–CeO2 interface, we made efforts to tailor the interfacial structure by fine-tuning synthetic parameters such as the feeding sequence of metal precursors. When the precursor Cu(Ac)2 was first added followed by the addition of Ce(NO3)3 to prepare the catalyst, only 22.0% of C2H4 FE was attained (Table S5†). Alternatively, a cascade addition of cerium precursor and copper precursor in sequence also increased C2H4 FE only to about 37.8%. In both cases, the accessible CuO–CeO2 interfaces with exposed copper domains were markedly reduced, which accounted for the declined ECR performance. A physical mixture of CuO/CB and CeO2/CB (CuO–CeO2/CBmix) with equivalent metal oxide loadings was also evaluated for ECR. It showed even worse CO2 reduction activity than that of CuO/CB (Fig. 3d and e), most likely due to poor mass transport. Taken together, we conclude that an intelligent design of CuO–CeO2 interfaces to yield and stabilize Cu+ is essential to facilitate the CO2-to-C2H4 conversion.
The interfacial reaction kinetics was explored by Tafel analysis. A Tafel slope of 148.9 mV dec−1 was observed for CuO–CeO2/CB, much lower than 160.7 mV dec−1 for CuO/CB and 169.5 mV dec−1 for CuO–CeO2/CBmix (Fig. 4d). This indicates that CuO–CeO2/CB has a comparatively faster kinetics for CO2 reduction. The formation of the *CO intermediate for tandem catalysis on the surface of the catalysts determines the reaction rate.
The long-term performances of the catalysts were examined by chronoamperometric measurements. The results (Fig. 4e) showed that the FE for C2H4 remained steady, exceeding 48.0% even after 9 h of continuous polarization at −1.1 V (vs. RHE). XPS analysis (Fig. 4f and S1b†) indicated that the surface concentration of Cu+ was preserved after 1 h of polarization at −1.1 V (vs. RHE), reflecting its good stability owing to the strong interplay between ceria and copper oxide.
To further investigate the role of CeO2 in stabilizing Cu+, we performed density functional theory (DFT) calculations (Fig. 5). We modeled the interface between CuO and CeO2 (denoted as CuO–CeO2) by constructing a small CeO2 cluster (Ce3O6) on the CuO(100) surface.47,48 The (100) facet, which has been known as the active site for C2 production in electrochemical CO2 reduction, is considered.49 We also considered the Cu-terminated surface of CuO since the surface O species would be reduced at the experimental electrode potential range (−0.9 ∼ −1.3 V vs. RHE). Previous studies have shown that subsurface oxygen in copper oxides plays an important role in facilitating the CO2 reduction,35,36 and hence, we focus on subsurface O rather than surface O.
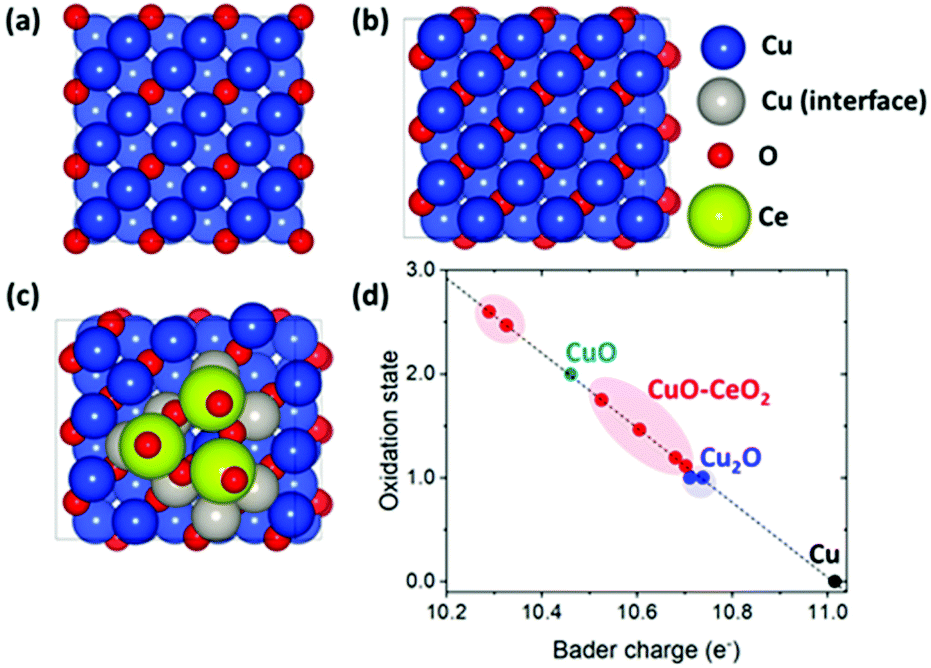 | ||
| Fig. 5 Top view of the optimized geometries of (a) Cu2O, (b) CuO, and (c) CuO–CeO2. (d) Oxidation states of surface Cu atoms obtained by Bader charge analysis. Only the surface Cu atoms adjacent to CeO2 (denoted as grey balls in (c)) are considered for the Bader charge analysis of CuO–CeO2. In (d), black, blue, cyan, and red colors represent Cu, Cu2O, CuO, and CuO–CeO2, respectively. | ||
We focused on the change/trend of the Bader charges50 of the surface Cu atoms with and without CeO2 clusters since the Bader charge agrees with the oxidation state qualitatively (albeit not quantitatively). Assuming that the Bader charges of surface Cu atoms in Cu(100), Cu2O(100), and CuO(100) correspond to the oxidation states of 0, +1, and +2, respectively, we obtained a linear relationship between the Bader charge and oxidation state (Fig. 5d), and from the latter correlation, we obtained the oxidation state of Cu atoms in CuO–CeO2.
This Bader charge analysis shows that the oxidation states of several Cu atoms at the CuO–CeO2 interface lie between that of Cu2O and CuO (Fig. 5d), indicating that the interfacial CeO2 cluster changes the oxidation state of neighboring Cu atoms in CuO toward that of Cu2O. More specifically, the oxidation states of two Cu atoms at the CuO–CeO2 interface are highly similar to those of surface Cu atoms in Cu2O (i.e. Cu+). This result agrees with the presence of Cu+ in the XPS characterization of CuO–CeO2/CB (Fig. 1b), and indicates that CeO2 plays an important role in stabilizing Cu+.
Conclusions
In summary, we present CuO–CeO2/CB as a highly promising electrocatalyst for enhancing the selective reduction of CO2 to ethylene. By utilizing the strong synergistic interaction between CuO and CeO2, stabilization of the Cu+ species at the metal–oxide interface is realized, while H2 production is simultaneously considerably suppressed, resulting in boosted ethylene production with a high FE of up to 50.0%. The existence of Cu+ species was confirmed by XPS, Raman spectroscopy, as well as TPR; Cu+ species are believed to be the adsorption as well as active sites for the activation of CO2 molecules. This work provides a simple way to enhance the conversion of CO2 into ethylene, and it is hoped that the findings will inspire the rational design of active copper domains for efficient electroreduction of CO2.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21972010); the State Key Laboratory of Organic-Inorganic Composites (No. oic-201901001); the Beijing Natural Science Foundation (No. 2192039); the Beijing University of Chemical Technology (XK180301 and XK1804-2); the Foundation of Key Laboratory of Low-Carbon Conversion Science & Engineering, Shanghai Advanced Research Institute, the Chinese Academy of Sciences (No. KLLCCSE-201901, SARI, CAS); the DCCEM at the Department of Materials, Oxford University, and the the Henry Royce Institute (EP/R010145/1). Y. J. acknowledges the National Research Foundation of Korea (NRF-2019M3D1A1079303 and NRF-2019M3D3A1A01069099).Notes and references
- J. Gu, C. Hsu, L. Bai, H. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS
.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS
- T. Ma, Q. Fan, X. Li, J. Qiu, T. Wu and Z. Sun, J. CO2 Util., 2019, 30, 168–182 CrossRef CAS
- H. Tao, Q. Fan, T. Ma, S. Liu, H. Gysling, J. Texter, F. Guo and Z. Sun, Prog. Mater. Sci., 2020, 100637 CrossRef CAS
- Q. Fan, P. Hou, C. Choi, T. S. Wu, S. Hong, F. Li, Y. L. Soo, P. Kang, Y. Jung and Z. Sun, Adv. Energy Mater., 2020, 10, 1903068 CrossRef CAS
- A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang, T.-K. Sham, C. Yang, Y. Ji, L. Qian and X. Xu, ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS
- Y. Pi, J. Guo, Q. Shao and X. Huang, Nano Energy, 2019, 62, 861–868 CrossRef CAS
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS
- J. Lv, M. Jouny, W. Luc, W. Zhu, J. Zhu and F. Jiao, Adv. Mater., 2018, 30, 1803111 CrossRef
- M. Jia, C. Choi, T. Wu, C. Ma, P. Kang, H. Tao, Q. Fan, S. Hong, S. Liu, Y. Soo and Z. Sun, Chem. Sci., 2018, 9, 8775–8780 RSC
- M. Jia, Q. Fan, S. Liu, J. Qiu and Z. Sun, Curr. Opin. Green Sustainable Chem., 2019, 16, 1–6 CrossRef
- S. Liu, X. Lu, J. Xiao, X. Wang and X. Lou, Angew. Chem. Int. Ed., 2019, 58, 13828–13833 CrossRef CAS
- Q. Gong, P. Ding, M. Xu, X. Zhu, M. Wang, J. Deng, Q. Ma, N. Han, Y. Zhu and J. Lu, Nat. Commun., 2019, 10, 1–10 CrossRef CAS
- C. Xia, P. Zhu, Q. Jiang, Y. Pan, W. t. Liang, E. Stavitsk, H. N. Alshareef and H. Wang, Nat. Energy, 2019, 4, 776–785 CrossRef CAS
- D. Gao, H. Zhou, F. Cai, J. Wang, G. Wang and X. Bao, ACS Catal., 2018, 8, 1510–1519 CrossRef CAS
- Z. Liu, Acta Phys.-Chim. Sin., 2019, 35, 1307–1308 Search PubMed
- Y. Yang, Y. Zhang, J. Hu and L. Wan, Acta Phys.-Chim. Sin., 2020, 36, 1906085–1906080 Search PubMed
- Q. Fan, M. Zhang, M. Jia, S. Liu, J. Qiu and Z. Sun, Mater. Today Energy, 2018, 10, 280–301 CrossRef
- Y. Gao, S. Liu, Z. Zhao, H. Tao and Z. Sun, Acta Phys.-Chim. Sin., 2018, 34, 858–872 CAS
- T. Ma, Q. Fan, H. Tao, Z. Han, M. Jia, Y. Gao, W. Ma and Z. Sun, Nanotechnology, 2017, 28, 472001–472019 CrossRef
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang and H. Xie, Nat. Chem., 2018, 10, 974–980 CrossRef CAS
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem, 2018, 4, 1809–1831 CAS
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS
- Y. A. Wu, I. McNulty, C. Liu, K. C. Lau, Q. Liu, A. P. Paulikas, C.-J. Sun, Z. h. Cai, J. R. Guest and Y. Ren, Nat. Energy, 2019, 4, 957–968 CrossRef CAS
- S. Y. Lee, H. Jung, N. K. Kim, H. S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2018, 140, 8681–8689 CrossRef CAS
- H. Jung, S. Y. Lee, C. W. Lee, M. K. Cho, D. H. Won, C. Kim, H.-S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2019, 141, 4624–4633 CrossRef CAS
- W. Luc, X. Fu, J. Shi, J.-J. Lv, M. Jouny, B. H. Ko, Y. Xu, Q. Tu, X. Hu and J. Wu, Nat. Catal., 2019, 2, 423–430 CrossRef CAS
- D.-H. Nam, O. S. Bushuyev, J. Li, P. De Luna, A. Seifitokaldani, C.-T. Dinh, F. P. García de Arquer, Y. h. Wang, Z. q. Liang and A. H. Proppe, J. Am. Chem. Soc., 2018, 140, 11378–11386 CrossRef CAS
- P. De Luna, R. Quintero-Bermudez, C. T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. d. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef CAS
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS
- S. Chu, S. Hong, J. Masa, X. Li and Z. Sun, Chem. Commun., 2019, 55, 12380–12383 RSC
- Y. Li, S. Chu, H. Shen, Q. Xia, A. W. Robertson, J. Masa, U. Siddiqui and Z. Sun, ACS Sustainable Chem. Eng., 2020, 8, 4948–4954 CrossRef CAS
- M. Favaro, H. Xiao, T. Cheng, W. A. Goddard, J. Yano and E. J. Crumlin, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6706–6711 CAS
- H. Xiao, W. A. Goddard, T. Cheng and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6685–6688 CrossRef CAS
- Q. Lei, H. Zhu, K. Song, N. Wei, L. Liu, D. Zhang, J. Yin, X. Dong, K. Yao and N. Wang, J. Am. Chem. Soc., 2020, 142, 4213 CrossRef CAS
- P. Burroughs, A. Hamnett, A. F. Orchard and G. Thornton, J. Chem. Soc., Dalton Trans., 1976, 1686–1698 RSC
- Z. Sun, X. Wang, Z. Liu, H. Zhang, P. Yu and L. Mao, Langmuir, 2010, 26, 12383–12389 CrossRef CAS
- J. Shyu, K. Otto, W. Watkins, G. Graham, R. Belitz and H. Gandhi, J. Catal., 1988, 114, 23–33 CrossRef CAS
- J. E. Spanier, R. D. Robinson, F. Zhang, S.-W. Chan and I. P. Herman, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 245407 CrossRef
- H. Hagemann, H. Bill, E. Walker and M. François, Solid State Commun., 1990, 73, 447–451 CrossRef CAS
- M. Li, U. Tumuluri, Z. Wu and S. Dai, ChemSusChem, 2015, 8, 3651–3660 CrossRef CAS
- M. Zhang, T. Wu, S. Hong, Q. Fan, Y. Soo, J. Masa, J. Qiu and Z. Sun, ACS Sustainable Chem. Eng., 2019, 7, 15030–15035 CrossRef CAS
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem. Int. Ed., 2016, 55, 5789–5792 CrossRef CAS
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Nat. Catal., 2018, 1, 111–119 CrossRef CAS
- C. W. Lee, S.-J. Shin, H. Jung, D. L. T. Nguyen, S. Y. Lee, W. H. Lee, D. H. Won, M. G. Kim, H.-S. Oh and T. Jang, ACS Energy Lett., 2019, 4, 2241–2248 CrossRef CAS
- D. Gao, Y. Zhang, Z. Zhou, F. Cai, X. Zhao, W. Huang, Y. s. Li, J. Zhu, P. Liu and F. Yang, J. Am. Chem. Soc., 2017, 139, 5652–5655 CrossRef CAS
- K. J. P. Schouten, Z. Qin, E. P. Gallent and M. T. M. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter., 2009, 21, 084204 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, XPS spectra, STEM images and EDS elemental maps, and FE vs. electrolysis temperature. See DOI: 10.1039/d0gc02279a |
| ‡ These authors contributed equally to this work. |
| § Current address: Max Planck Institute for Chemical Energy Conversion, Stiftstrasse 34–36, 45470 Mülheim an der Ruhr, Germany. |
| This journal is © The Royal Society of Chemistry 2020 |