
Exploration of a KI-catalyzed oxidation system for direct construction of bispyrrolidino[2,3-b]indolines and the total synthesis of (+)-WIN 64821†
Si-Kai
Chen‡
a,
Ju-Song
Yang‡
a,
Kun-Long
Dai
a,
Fu-Min
Zhang
 *a,
Xiao-Ming
Zhang
*a and
Yong-Qiang
Tu
*ab
*a,
Xiao-Ming
Zhang
*a and
Yong-Qiang
Tu
*ab
aState Key Laboratory of Applied Organic Chemistry and College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, P. R. China. E-mail: tuyq@lzu.edu.cn; zhangfm@lzu.edu.cn; zhangxiaom@lzu.edu.cn
bSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, P. R. China
First published on 26th November 2019
Abstract
A facile and environmentally benign KI(cat.)/NaBO3·4H2O oxidation system has been developed for the tandem oxidative aminocyclization/coupling of tryptamines, affording a series of 3a,3a′-bispyrrolidino[2,3-b]indolines with high efficiency (up to 94% yield). This reaction features an electrophilic “I+” mechanism, which is importantly quite different from and milder than the typical radical-involving process, and can be readily amplified for the total synthesis of (+)-WIN 64821.
Oxidation is one of the most important transformations for organisms to produce functional molecules. In vivo, the oxidation of tryptophan or tryptamine can result in a big family of structurally complex and biologically important 3a,3a′-bispyrrolidino[2,3-b]indoline alkaloids (Fig. 1), which show antifungal, antiviral and cytostatic acivities.1 Despite its significance for biological and medicinal chemistry, realizing this biotransformation by means of organic chemistry, especially with a catalytic amount of assistant oxidation reagent, is still rare and synthetically challenging.2 To date, several groups have devoted much pioneering effort to this aspect.3,4 However, most of the current solutions require either equivalent amounts of transition metals or excess of strong acid or base. Furthermore, these methodologies generally give insufficient yields. Therefore, the development of a catalytic and transition-metal-free oxidative system for the efficient synthesis of 3a,3a′-bispyrrolidino[2,3-b]indolines under mild condition is in demand.
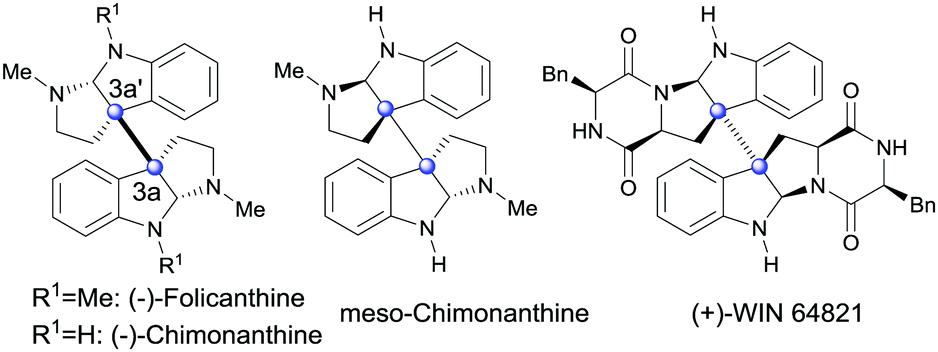 | ||
| Fig. 1 Representative bispyrrolidino[2,3-b]indoline alkaloids. | ||
During the past fifteen years, the iodide-catalyzed oxidative reaction has received widespread attention because of its versatile reactivity and environmentally benign property.5,6 In general, however, these systems are mostly limited to the application of synthesis of relatively simple organic compounds, and only a few can give access to sterically complex frameworks, such as those with the vicinal all-carbon quaternary centers.7 As indicated in Scheme 1, the oxidation systems mediated by a catalytic amount of iodide have not been expanded to effect the tandem coupling/cyclizations of tryptamine or tryptophan to construct the more complex 3a,3a′-bispyrrolidino[2,3-b]indoline 6, 8 and 11 with vicinal all-carbon quaternary centers. Instead, they are only applicable to mediate the simple cyclization to produce the monomer products 1–4 (Scheme 1(a)).8 Due to our continuing interest in the synthesis of 3a,3a′-bispyrrolidino[2,3-b]indoline alkaloids,9 we thus try to develop an alternative effective iodide-catalyzed oxidative system, which we expect will enable the coupling/cyclizations as shown in Scheme 1(b). Herein, we describe our research results.
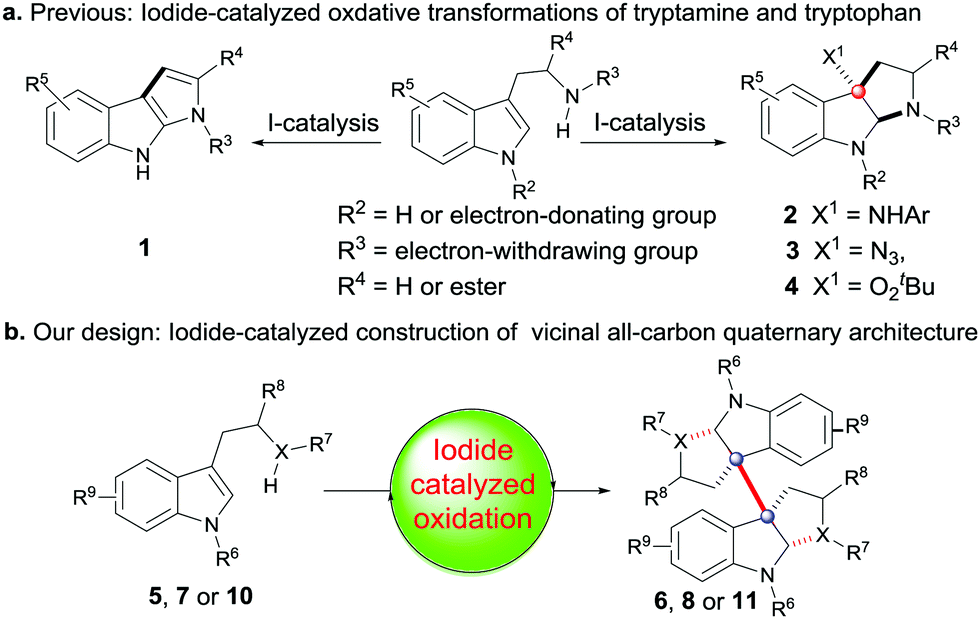 | ||
| Scheme 1 (a) Previous I-catalyzed oxidation of tryptamine or tryptophan; (b) our design. | ||
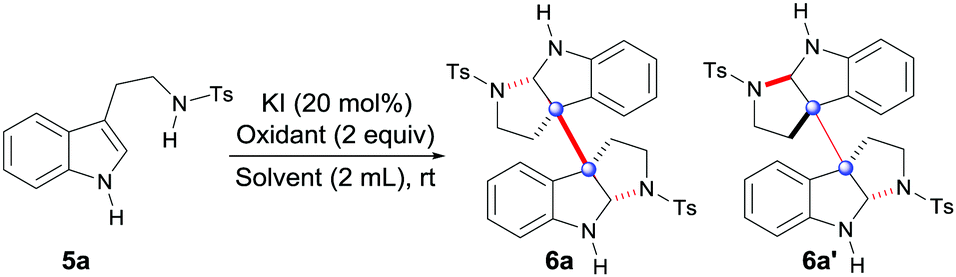
To achieve the hypothesis above, our initial optimization started toward the oxidation of tryptamine 5a with screening the oxidants and solvents using KI as a catalyst (Table 1). Fortunately, after testing H2O2 in several solvents, only TFE (trifluoroethanol) could generate the desired dimeric products (6a and 6a′) with a moderate 44% yield (entry 5), while the other solvents gave inferior results (entries 1–4). Subsequently, both organic (entries 6 and 7) and inorganic oxidants were screened (entries 8 and 9), among which, NaBO3·4H2O gave the best yield (64%, entry 9). In addition, other conditions which involved varying the iodide catalysts, additives, concentrations and component equivalents were widely investigated,10 with the conditions in entry 10 giving the best result (68% yield).
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Oxidant | Time | Yieldb (%) |
a Unless otherwise noted, reactions were carried out with 5a (0.2 mmol), KI (20 mol%) and oxidant (2.0 equiv.) in 2 mL solvent. The dr was 1.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 determined by 1H NMR.
b Determined by 1H NMR.
c 50 wt% in water.
d No reaction was observed.
e 5.5 M in decane.
f Not detected.
g 1 atm.
h Reaction was carried out with 5a (0.2 mmol), KI (10 mol%) and NaBO3·4H2O (1.2 equiv.) in 2 mL TFE. :1 determined by 1H NMR.
b Determined by 1H NMR.
c 50 wt% in water.
d No reaction was observed.
e 5.5 M in decane.
f Not detected.
g 1 atm.
h Reaction was carried out with 5a (0.2 mmol), KI (10 mol%) and NaBO3·4H2O (1.2 equiv.) in 2 mL TFE.
|
||||
| 1 | CH3CN | H2O2c | 24 h | NRd |
| 2 | Toluene | H2O2 | 24 h | NR |
| 3 | THF | H2O2 | 24 h | NR |
| 4 | CH2Cl2 | H2O2 | 24 h | NR |
| 5 | TFE | H2O2 | 4 h | 44 |
| 6 | TFE | t BuO2He | 10 h | 53 |
| 7 | TFE | m-CPBA | 10 min | NDf |
| 8 | TFE | O2g | 24 h | NR |
| 9 | TFE | NaBO3·4H2O | 1.5 h | 64 |
| 10h | TFE | NaBO3·4H2O | 2 h | 68 |
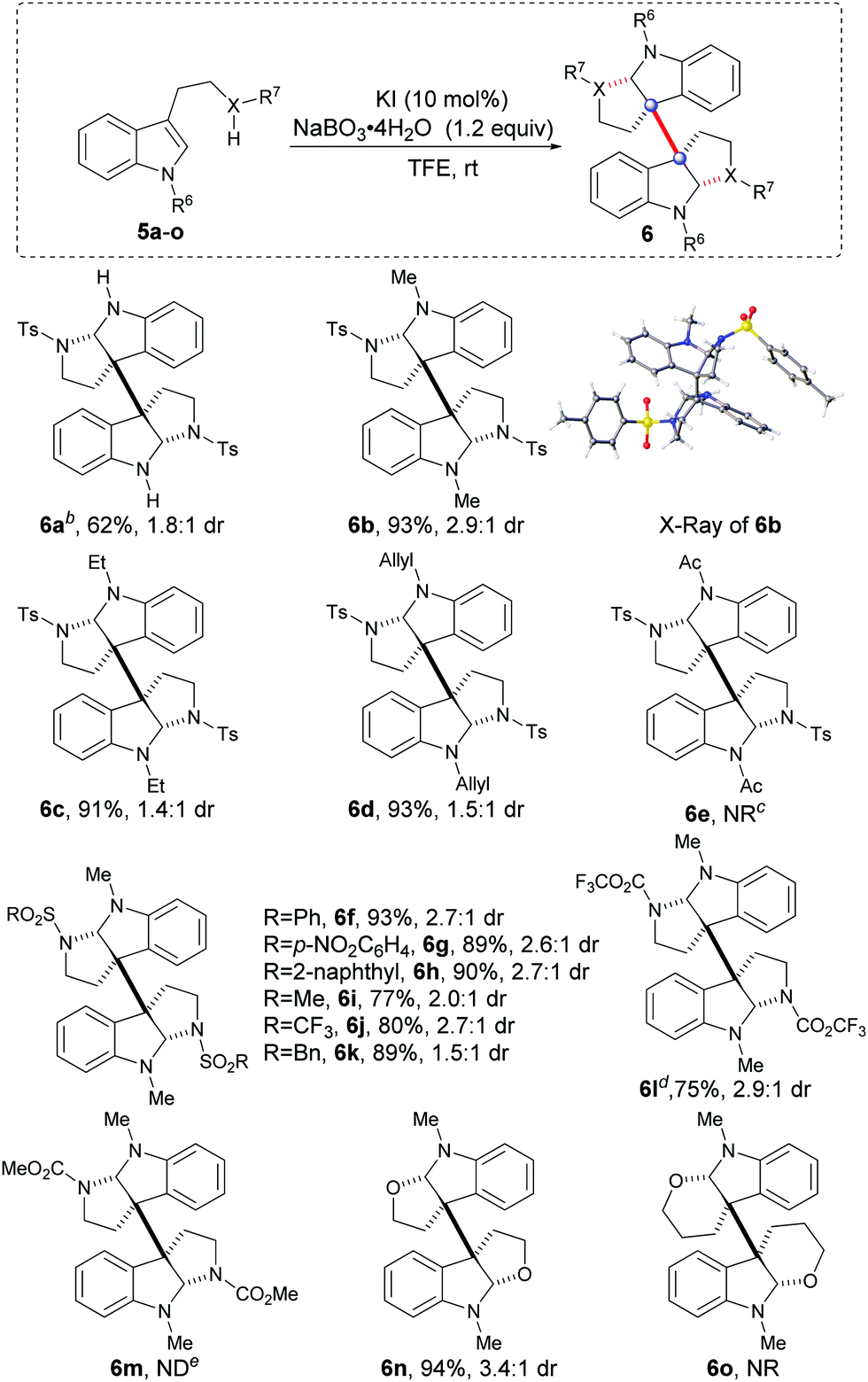
With the optimal catalytic oxidative system in hand, we then expanded the tryptamine substrate scope by varying the substituents R6 and X–R7 (Table 2, 5a–m). Initially, when X–R7 was selected as N-tosyl, varying the substituents R6 (5b–d) with the EDGs (electron-donating groups, e.g., methyl, ethyl and allyl) generally led to excellent reaction results (6b–d, 91–93% yields, 1.4:1 to 2.9:1 dr), while the EWG (electron-withdrawing group, e.g., Ac) substituted substrate (5e) remained inactive to give product 6e. Subsequently, when the optimal methyl was selected as R6 (as indicated in 6f–l), varying substitution of R7 (X = N) with the strong EWG sulfonyl (5f–k) and trifluoroacetyl (5l) could drive the reactions with satisfactory results (6f–l, 75–93% yields, 1.5:1 to 2.9:1 dr). Exceptionally, when a carbamate was introduced to tryptamine (5m, X–R7 = N-CO2Me), the reaction could not give the desired product 6m, but gave a complex mixture accompanied by a partial starting material. It was also important that when tryptophol 5n and 3-indolepropanol 5o were separately subjected to the catalytic oxidative systems, interestingly, the former could react efficiently to give the tetrahydrofuran 6n with an excellent yield of 94%, while the latter gave a mixture without hexahydropyran 6o detected.
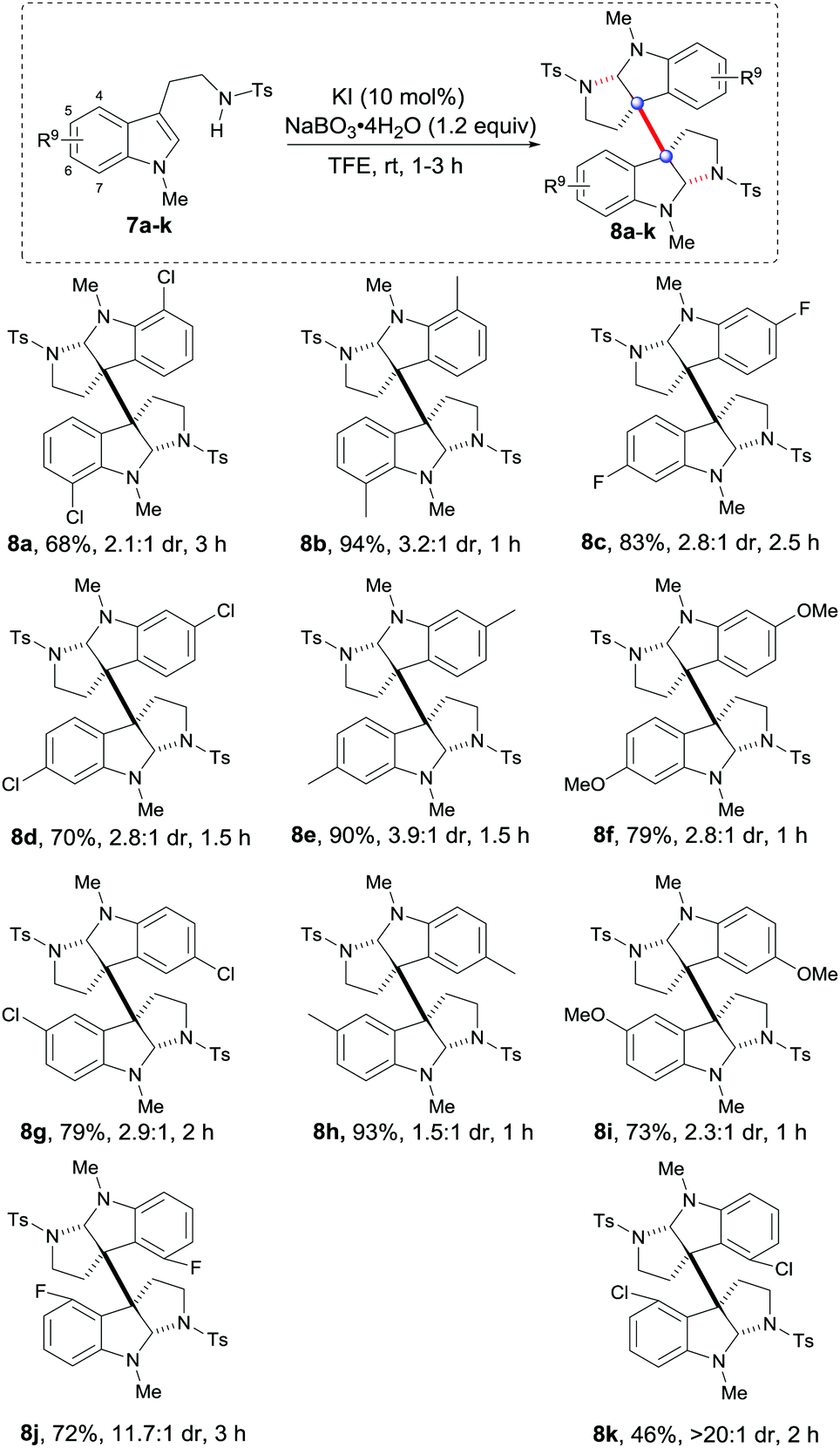
Next, a wide range of tryptamines with different substituents on the benzene rings were tested. As shown in Table 3, both EWG and EDG substituents at C7, C6 and C5 of the benzene rings were effective for the expected oxidative dimerization reactions and generally gave satisfactory results (8a–i, 68–94% yield, 1.5:1 to 3.9:1 dr). The differences of these examples in the reaction time revealed that the EDG substituents were more favorable for reaction rates than the EWGs (8f, 8ivs.8a, 8c, 8j, and 8k). Notably, the C4–F and C4–Cl substituted tryptamines 7j and 7k could give 8j and 8k with excellent diastereoselectivities (11.7:1 and >20:1 dr) and acceptable yields (72% and 46%), probably due to the steric interaction between F or Cl and C3a or C3a′.
| a Unless otherwise noted, all reactions were carried out with 5 (0.2 mmol), KI (10 mol%) and NaBO3·4H2O (1.2 equiv.) in 2 mL TFE and reacted for 1 h. Isolated yields and dr were determined by 1H NMR. |
|---|
|
In order to elucidate the mechanism of this catalytic oxidative coupling/cyclization reaction, we also conducted some additional control experiments (Scheme S1, ESI†).10 According to the experimental results, addition of the radical scavengers such as TEMPO (2,2,6,6-tetramethylpiperidinooxy) or DMPO (5,5-dimethyl-1-pyrroline N-oxide) was found to have little influence on the reaction, while use of the electrophilic iodide reagent NIS (N-iodosuccinimide) could efficiently promote the desired reaction with similar results (77% yield, 2.8:1 dr) compared to the standard reaction (Table 2, 6b). Therefore, an electrophilic “I+” mechanism (Scheme 2) rather than the classical radical process was proposed.6i As shown in Scheme 2, the reaction would begin with the oxidation of “I−”; thereafter, the resulting electrophilic “I+” species could readily interact with the nucleophilic tryptamine (5 or 7) to form a cyclic iodonium ion intermediate A.3g,8b,11 Subsequently, the active intermediate A underwent an intramolecular cyclization to generate the 3-iodo-hexahydropyrroloindole compound B, which was then transferred to indolium C and released iodide for further catalytic cycles. Finally, the indolium C could couple with another tryptamine (5 or 7) through an electrophilic addition/cyclization process via either favorable model M-1 or a sterically hindered model M-2 to give the major product 6 or 8 and the minor one 6′ or 8′, respectively. It is worth noting that this stereo-control model was also consistent with the diastereoselectivities obtained from our reactions.10
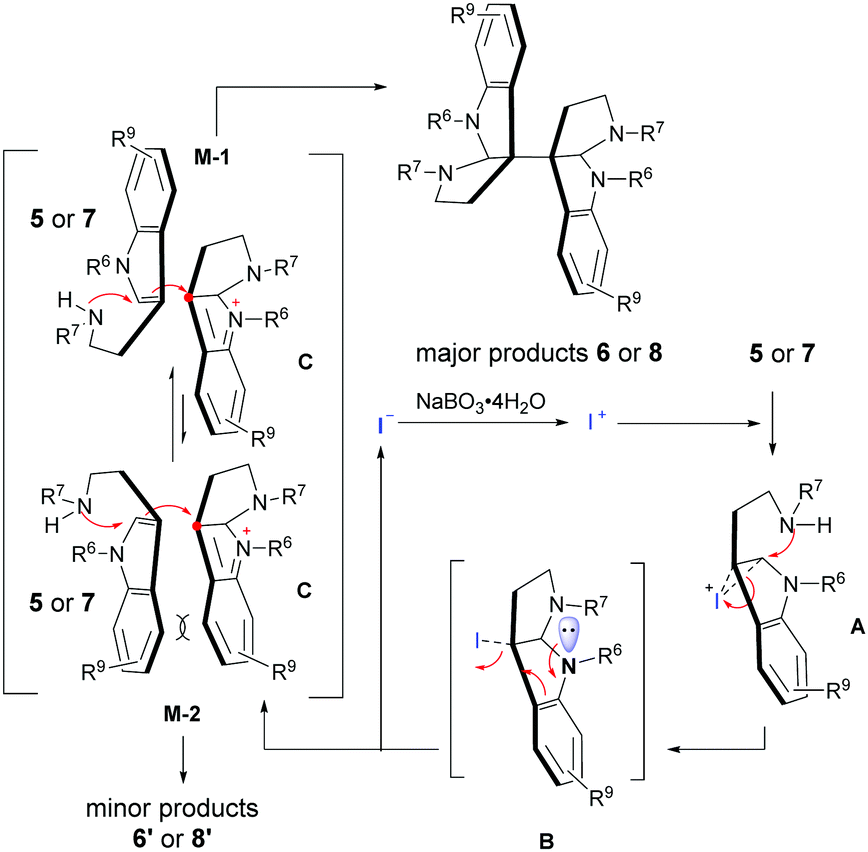 | ||
| Scheme 2 The plausible mechanism. | ||
To verify the utility of this methodology, the pharmacologically promising agent (+)-WIN 64821 was chosen as a synthetic target (Scheme 3).12 The efficient construction of the 3a,3a′-bispyrrolidino[2,3-b]indoline motif was the key to approach a concise total synthesis of (+)-WIN 64821. That could be conveniently realized by using our newly developed catalytic oxidative methodology. Initially, we synthesized the precursor 10 for the key reaction from commercially available (L)-tryptophan 9. Then, compound 10 was successfully applied, at the gram-scale, to the KI-catalyzed oxidative coupling/cyclization reaction to give the 3a,3a′-bispyrrolidino[2,3-b]indoline 11 with satisfactory results (79% yield, 1.2:1 dr). Subsequent deprotections of p-nitrobenzene sulfonyl and allyl of 11 successively afforded the dimeric diamine 13, which could be easily condensed with N-Cbz-L-phenylalanine over three steps4d,13 to finally give (+)-WIN 64821.
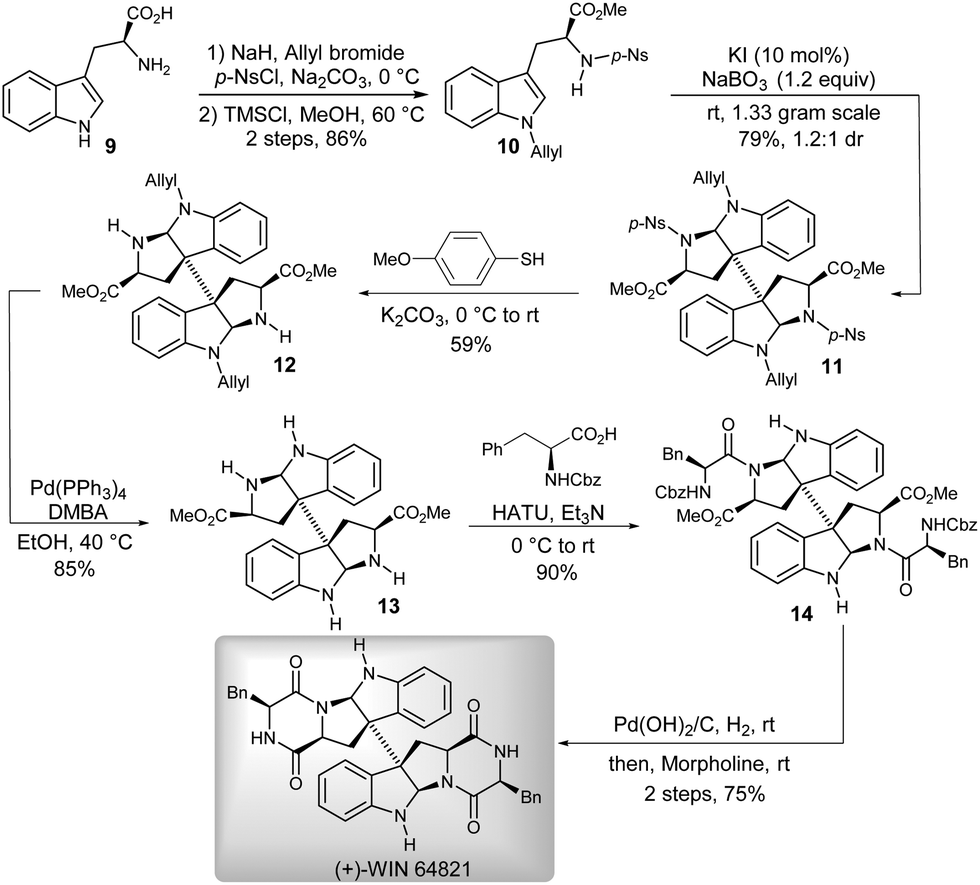 | ||
| Scheme 3 Asymmetric total synthesis of (+)-WIN 64821. | ||
In conclusion, we have successfully established an iodide-catalyzed oxidative coupling/cyclization approach for the dimerization of tryptamine, tryptophol and tryptophan analogues. This protocol features a plausible catalytic cycle comprising iodide and hypoiodite catalyst states, the use of inexpensive and readily available as well as an environmentally benign system (KI and NaBO3·4H2O) and mild reaction conditions. Particularly, it provides a practical solution for the construction of synthetically challenging vicinal all-carbon quaternary motifs. Although it gives moderate diastereoselectivities in some cases, we have optimized it up to >20:1 dr by adjusting the substituent at C4 of the substrate. Furthermore, the synthetic utility of this methodology has been verified by the asymmetric total synthesis of bioactive natural product (+)-WIN 64821.
We thank the NSFC (No. 21502080, 21772071, 21871117, 21772076, 21971095), STCSM (19JC1430100) and the “111” Program of MOE for their financial support. We also thank Prof. Wei Yu (Lanzhou University) for his kind mechanism discussion.
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. Ruiz-Sanchis, S. A. Savina, F. Albericio and M. Álvarez, Chem. – Eur. J., 2011, 17, 1388–1408 CrossRef CAS PubMed.
- For reviews of synthesis, see: (a) A. Steven and L. E. Overman, Angew. Chem., Int. Ed., 2007, 46, 5488–5508 CrossRef CAS PubMed; (b) M. A. Schmidt and M. Movassaghi, Synlett, 2008, 313–324 CAS; (c) J. Kim and M. Movassaghi, Chem. Soc. Rev., 2009, 38, 3035–3050 RSC; (d) L. M. Repka and S. E. Reisman, J. Org. Chem., 2013, 78, 12314–12320 CrossRef CAS; (e) J. Kim and M. Movassaghi, Acc. Chem. Res., 2015, 48, 1159–1171 CrossRef CAS; (f) J. Song, D.-F. Chen and L.-Z. Gong, Natl. Sci. Rev., 2017, 4, 381–396 CrossRef CAS.
- For representative pioneer examples, see: (a) A. I. Scott, F. McCapra and E. S. Hall, J. Am. Chem. Soc., 1964, 86, 302–303 CrossRef CAS; (b) T. Hino, S. Kodato, K. Takahashi, H. Yamaguchi and M. Nakagawa, Tetrahedron Lett., 1978, 19, 4913–4916 CrossRef; (c) H. Ishikawa, H. Takayama and N. Aimi, Tetrahedron Lett., 2002, 43, 5637–5639 CrossRef CAS; (d) T. Newhouse and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 10886–10887 CrossRef CAS PubMed; (e) M. Movassaghi and M. A. Schmidt, Angew. Chem., Int. Ed., 2007, 46, 3725–3728 CrossRef CAS PubMed; (f) J. Kim and M. Movassaghi, J. Am. Chem. Soc., 2010, 132, 14376–14378 CrossRef CAS; (g) Y.-X. Li, H.-X. Wang, S. Ali, X.-F. Xia and Y.-M. Liang, Chem. Commun., 2012, 48, 2343–2345 RSC; (h) S. Tadano, Y. Mukaeda and H. Ishikawa, Angew. Chem., Int. Ed., 2013, 52, 7990–7994 CrossRef CAS PubMed.
- For recent progress, see: (a) M. Tayu, K. Higuchi, T. Ishizaki and T. Kawasaki, Org. Lett., 2014, 16, 3613–3615 CrossRef CAS PubMed; (b) D. Sun, C. Xing, X. Wang, Z. Su and C. Li, Org. Chem. Front., 2014, 1, 956–960 RSC; (c) M. Ding, K. Liang, R. Pan, H. Zhang and C. Xia, J. Org. Chem., 2015, 80, 10309–10316 CrossRef CAS PubMed; (d) K. Liang, X. Deng, X. Tong, D. Li, M. Ding, A. Zhou and C. Xia, Org. Lett., 2015, 17, 206–209 CrossRef CAS PubMed; (e) M. Tayu, Y. Suzuki, K. Higuchi and T. Kawasaki, Synlett, 2016, 941–945 CAS; (f) S. Tadano, Y. Sugimachi, M. Sumimoto, S. Tsukamoto and H. Ishikawa, Chem. – Eur. J., 2016, 22, 1277–1291 CrossRef CAS PubMed.
- For representative pioneer examples, see: (a) T. Dohi, A. Maruyama, M. Yoshimura, K. Morimoto, H. Tohma and Y. Kita, Angew. Chem., Int. Ed., 2005, 44, 6193–6196 CrossRef CAS PubMed; (b) M. Ochiai, Y. Takeuchi, T. Katayama, T. Sueda and K. Miyamoto, J. Am. Chem. Soc., 2005, 127, 12244–12245 CrossRef CAS PubMed; (c) R. D. Richardson, T. K. Page, S. Altermann, S. M. Paradine, A. N. French and T. Wirth, Synlett, 2007, 538–542 CAS; (d) T. Dohi, A. Maruyama, N. Takenaga, K. Senami, Y. Minamitsuji, H. Fujioka, S. B. Caemmerer and Y. Kita, Angew. Chem., Int. Ed., 2008, 47, 3787–3790 CrossRef CAS PubMed; (e) M. Uyanik, M. Akakura and K. Ishihara, J. Am. Chem. Soc., 2009, 131, 251–262 CrossRef CAS PubMed; (f) K. Miyamoto, Y. Sei, K. Yamaguchi and M. Ochiai, J. Am. Chem. Soc., 2009, 131, 1382–1383 CrossRef CAS PubMed; (g) M. Uyanik, H. Okamoto, T. Yasui and K. Ishihara, Science, 2010, 328, 1376–1379 CrossRef CAS PubMed; (h) M. Uyanik, T. Yasui and K. Ishihara, Angew. Chem., Int. Ed., 2010, 49, 2175–2177 CrossRef CAS PubMed; (i) J. Zhang, D. Zhu, C. Yu, C. Wan and Z. Wang, Org. Lett., 2010, 12, 2841–2843 CrossRef CAS PubMed.
- For selected reviews, see: (a) M. Uyanik and K. Ishihara, ChemCatChem, 2012, 4, 177–185 CrossRef CAS; (b) P. Finkbeiner and B. J. Nachtsheim, Synthesis, 2013, 979–999 CAS; (c) F. V. Singh and T. Wirth, Chem. – Asian. J., 2014, 9, 950–971 CrossRef CAS PubMed; (d) X.-F. Wu, J.-L. Gong and X. Qi, Org. Biomol. Chem., 2014, 12, 5807–5817 RSC; (e) F. Berthiol, Synthesis, 2015, 587–603 CrossRef CAS; (f) D. Liu and A. Lei, Chem. – Asian. J., 2015, 10, 806–823 CrossRef CAS PubMed; (g) M. S. Yusubov and V. V. Zhdankin, Resour.-Effic. Technol., 2015, 1, 49–67 Search PubMed; (h) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed; (i) R. Chen, J. Chen, J. Zhang and X. Wan, Chem. Rec., 2018, 18, 1292–1305 CrossRef CAS PubMed; (j) A. Claraz and G. Masson, Org. Biomol. Chem., 2018, 16, 5386–5402 RSC; (k) A. Flores, E. Cots, J. Bergès and K. Muñiz, Adv. Synth. Catal., 2019, 361, 2–25 CrossRef CAS.
- For selected examples, see: (a) M. Uyanik, T. Yasui and K. Ishihara, Angew. Chem., Int. Ed., 2013, 52, 9215–9218 CrossRef CAS PubMed; (b) M. Ngatimin, R. Frey, A. Levens, Y. Nakano, M. Kowalczyk, K. Konstas, O. E. Hutt and D. W. Lupton, Org. Lett., 2013, 15, 5858–5861 CrossRef CAS PubMed; (c) H. Wu, Y.-P. He, L. Xu, D.-Y. Zhang and L.-Z. Gong, Angew. Chem., Int. Ed., 2014, 53, 3466–3469 CrossRef CAS PubMed; (d) B. Liu, J. Cheng, Y. Li and J.-H. Li, Chem. Commun., 2019, 55, 667–670 RSC.
- (a) Z.-J. Cai, S.-Y. Wang and S.-J. Ji, Org. Lett., 2013, 15, 5226–5229 CrossRef CAS PubMed; (b) Z.-Y. Yang, T. Tian, Y.-F. Du, S.-Y. Li, C.-C. Chu, L.-Y. Chen, D. Li, J.-Y. Liu and B. Wang, Chem. Commun., 2017, 53, 8050–8053 RSC; (c) J. Guo, S. Chen, J. Liu, J. Guo, W. Chen, Q. Cai, P. Liu and P. Sun, Eur. J. Org. Chem., 2017, 4773–4777 CrossRef CAS; (d) X. Lu, Y. Bai, Y. Li, Y. Shi, L. Li, Y. Wu and F. Zhong, Org. Lett., 2018, 20, 7937–7941 CrossRef CAS PubMed; (e) Y. Li, L. Li, X. Lu, Y. Bai, Y. Wang, Y. Wu and F. Zhong, Chem. Commun., 2019, 55, 63–66 RSC.
- S.-K. Chen, W.-Q. Ma, Z.-B. Yan, F.-M. Zhang, S.-H. Wang, Y.-Q. Tu, X.-M. Zhang and J.-M. Tian, J. Am. Chem. Soc., 2018, 140, 10099–10103 CrossRef CAS PubMed.
- For details, see ESI†.
- (a) D. Beukeaw, K. Udomsasporn and S. Yotphan, J. Org. Chem., 2015, 80, 3447–3454 CrossRef CAS PubMed; (b) J. Dhineshkumar, K. Gadde and K. R. Prabhu, J. Org. Chem., 2018, 83, 228–235 CrossRef CAS PubMed.
- (a) C. J. Barrow, P. Cai, J. K. Snyder, D. M. Sedlock, H. H. Sun and R. Cooper, J. Org. Chem., 1993, 58, 6016–6021 CrossRef CAS; (b) M. Wada, H. Suzuki, M. Kato, H. Oikawa, A. Tsubouchi and H. Oguri, ChemBioChem, 2019, 20, 1273–1281 CrossRef CAS PubMed.
- C. Pérez-Balado and Á. R. de Lera, Org. Lett., 2008, 10, 3701–3704 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1940250. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc08646c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |