
Fluorinated carboxylic acids as powerful building blocks for the formation of bimolecular monolayers†
Harry
Pinfold
 ,
Christopher
Greenland
,
Graham
Pattison‡
and
Giovanni
Costantini
*
,
Christopher
Greenland
,
Graham
Pattison‡
and
Giovanni
Costantini
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: g.costantini@warwick.ac.uk
First published on 20th November 2019
Abstract
We compare the ability of a prototypical dicarboxylic acid and its fluorinated analogue to act as molecular building blocks for the formation of self-assembled monolayers. Whilst fluorination is found to prevent homomolecular self-assembly, it greatly increases the ability of the carboxylic acid to act as a hydrogen bond donor for the formation of bimolecular networks.
Lateral intermolecular interactions between surface-adsorbed molecules play an essential role in driving the formation of self-assembled monolayers. A range of different interactions have been exploited for this purpose, including hydrogen bonds,1–4 van der Waals interactions5 and metal–organic coordination.6 Of these, hydrogen bonds are perhaps the most extensively utilised. Their strength and directional nature can be used for the formation of stable networks with relatively predictable morphologies.
Carboxylic acids are widely employed as building blocks for the fabrication of hydrogen-bond-driven self-assembled monolayers.3,4 A significant proportion of such networks are homomolecular systems in which the carboxyl groups of adjacent molecules interact via R22(8) hydrogen bonds. This simple, robust motif has been exhaustively studied in surface-confined supramolecular chemistry. Comparatively little attention has been directed towards to the use of carboxylic acid building blocks as hydrogen bond donors for the formation of bimolecular networks. By pairing a carboxylic acid with an appropriate hydrogen bond acceptor, it is possible to form bimolecular systems based on straightforward hydrogen bond donor–acceptor interactions. Although such systems have been reported,7–13 the number of studies is sparse. This is likely because their reliable fabrication requires that the stability of the two-component assembly exceeds that of the homomolecular assembly of either of the individual components. This criterion can be difficult to fulfil when using carboxylic acids as hydrogen bond donors due to their propensity to self-assemble into homomolecular networks via R22(8) hydrogen bonds. Furthermore, additional challenges are encountered when experiments are conducted at the solid–liquid interface. Since the solvents used are often carboxylic acids themselves, the solvent molecules can compete for hydrogen bond acceptor sites and prevent the formation of the desired bimolecular network. An ideal carboxylic acid building block should thus be a strong enough hydrogen bond donor to form stable networks and outcompete solvent molecules whilst not being inclined towards stable homomolecular self-assembly.
Here we explore fluorination as a potential route towards circumventing the issues associated with using carboxylic acids to form bimolecular networks. Peripheral fluorination of planar molecules has previously been shown to disfavour on-surface homomolecular assembly due to the electrostatic repulsion between the fluorine atoms.14,15 At the same time, fluorine atoms are highly electron withdrawing and, as such, can increase the hydrogen bond donor strength of neighbouring functional groups. It has previously been demonstrated that fluorinated compounds, including alcohols16–20 and carboxylic acids,20,21 can form particularly strong hydrogen bonds. These two complementary properties could allow fluorinated carboxylic acids to act as strong hydrogen bond donors which are less likely to form homomolecular networks than their unfluorinated counterparts. Furthermore, the electron-withdrawing influence of the fluorine atoms may also decrease the hydrogen bond acceptor ability of carboxylic acids. This should make their carboxyl groups less competitive as hydrogen bond acceptor sites, thereby favouring the formation of bimolecular networks when a fluorinated carboxylic acid is paired with a potential hydrogen bond acceptor.
In order to test these ideas, we have performed a comparative study on the self-assembly behaviour of a simple aromatic dicarboxylic acid (TPA, Fig. 1) and its fluorinated analogue (F4TPA, Fig. 1) using scanning tunnelling microscopy (STM) operated at the solid–liquid interface. Our results show that while TPA readily self-assembles into extended 2D networks, fluorination prevents the homomolecular self-assembly of F4TPA. Moreover, we demonstrate that F4TPA is a superior building block to TPA for the formation of bimolecular networks when coupled with a range of hydrogen bond acceptors.
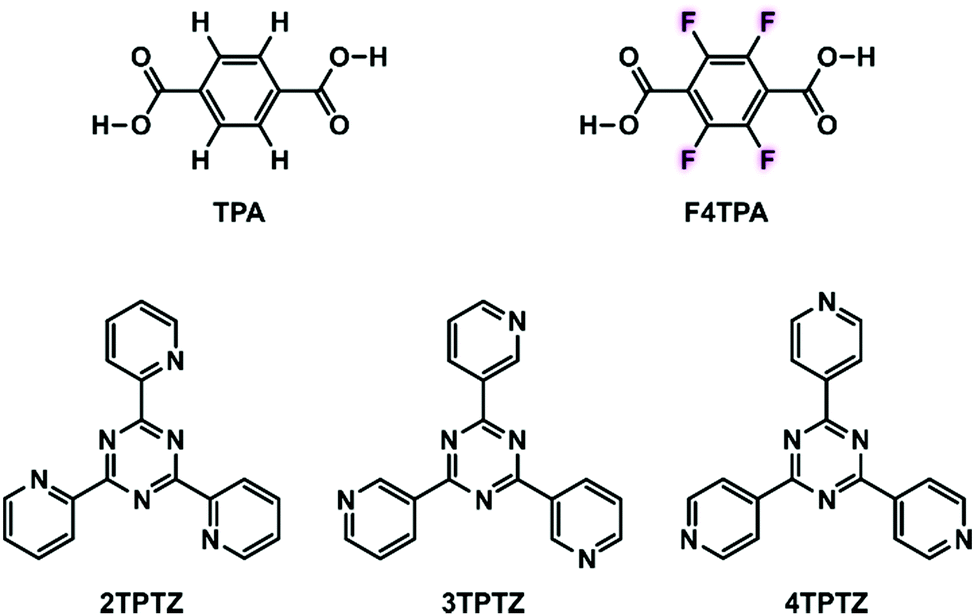 | ||
| Fig. 1 Structures of the molecular building blocks used within this study: terephthalic acid (TPA), tetrafluoroterephthalic acid (F4TPA), 2,4,6-tri(2-pyridyl)-s-triazine (2TPTZ), 2,4,6-tri(3-pyridyl)-s-triazine (3TPTZ) and 2,4,6-tri(4-pyridyl)-s-triazine (4TPTZ). | ||
TPA is a prototypical molecule used to study hydrogen-bond-controlled self-assembly, and its ability to form ordered homomolecular monolayers is well known.22–33 As the homomolecular self-assembly of TPA (Fig. S1 and S2, ESI†) has been extensively studied at the heptanoic acid/highly oriented pyrolytic graphite (HOPG) interface,22–25 we decided to investigate the assembly of F4TPA at the same interface. However, despite multiple attempts performed with a range of different solution concentrations, we never observed any evidence for the homomolecular self-assembly of F4TPA (see ESI†). We ascribe this to electrostatic repulsion between the fluorine atoms preventing close packing of the F4TPA molecules. This effect has previously been reported for other surface-adsorbed fluorinated molecules.14,15
We then proceeded to test the relative ability of TPA and F4TPA to act as hydrogen bond donors in bimolecular systems. Both molecules were mixed with a range of hydrogen bond acceptors and the formation of bimolecular networks was examined via STM. The three tripyridyltriazine isomers shown in Fig. 1 (2TPTZ, 3TPTZ and 4TPTZ) were employed as acceptors as their pyridyl nitrogen atoms are expected to be strong hydrogen bond acceptor sites. As is shown in the ESI,† in the absence of either F4TPA or TPA, both 2TPTZ (Fig. S3 and S4, ESI†) and 3TPTZ (Fig. S6 and S7, ESI†) self-assemble at the heptanoic acid/HOPG interface, whereas there was no evidence for the self-assembly of 4TPTZ.
Of the three acceptors tested, 4TPTZ is unique as it is the only one able to form bimolecular networks with TPA. As is described in the ESI,† when solutions containing both TPA and either 2TPTZ or 3TPTZ were used, the only ordered networks that could be observed were those corresponding to the homomolecular assemblies of the individual components. Kampschulte et al. previously demonstrated that TPA and 4TPTZ coassemble at the heptanoic acid/HOPG interface.10 Our measurements confirm this result, revealing the formation of extended domains of a bimolecular network (Fig. S14, ESI†). High-resolution STM images, such as Fig. 2a, can be used to elucidate the structure of the assembly (the proposed model is given in Fig. 2b). One of the three pyridyl nitrogen atoms in each 4TPTZ molecule interacts with a TPA molecule via an O–H⋯N(pyridyl) hydrogen bond, and the TPA molecules bridge two 4TPTZ molecules via these interactions. Secondary C–H⋯O and C–H⋯N(pyridyl) interactions may also contribute towards stabilising the assembly.
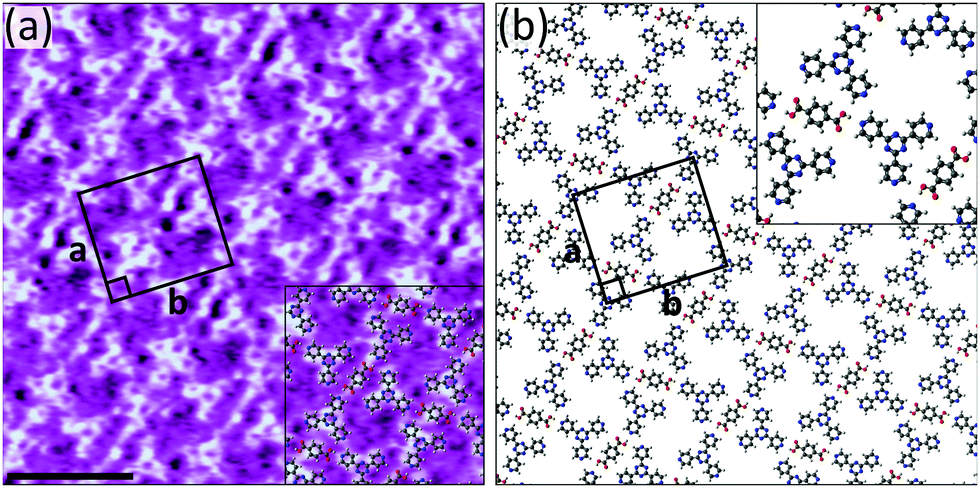 | ||
Fig. 2 (a) STM image showing the bimolecular assembly of TPA and 4TPTZ at the heptanoic acid/HOPG interface. The assembly contains a TPA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4TPTZ ratio of 1:2. Tunnelling parameters: Vbias = −1.2 V, Iset = 50 pA. Unit cell parameters: a = 2.7 ± 0.2 nm, b = 3.0 ± 0.2 nm, angle 90 ± 3°. Scale bar = 3 nm. (b) Proposed model for the assembly. This assembly was first reported in ref. 10. :4TPTZ ratio of 1:2. Tunnelling parameters: Vbias = −1.2 V, Iset = 50 pA. Unit cell parameters: a = 2.7 ± 0.2 nm, b = 3.0 ± 0.2 nm, angle 90 ± 3°. Scale bar = 3 nm. (b) Proposed model for the assembly. This assembly was first reported in ref. 10. | ||
Given the failure of TPA to act as a reliable hydrogen bond donor with two of the three acceptors, we tested the performance of F4TPA. Unlike with TPA, we were able to fabricate bimolecular networks by combining 2TPTZ with F4TPA (Fig. S15, ESI†). The structural details of the assembly are revealed by high-resolution STM images such as Fig. 3a. The proposed model is given in Fig. 3b. It should be noted that the 2TPTZ molecules can adopt two distinct conformations, one in which all three pyridyl rings are orientated in the same direction and another in which one of the pyridyl rings is flipped such that it is orientated in the opposite way relative to the other two (see Fig. S12, ESI†). The conformation of the individual molecules cannot be identified in the STM images, but DFT calculations (see ESI†) show that the threefold-symmetric conformation is more stable than the alternate conformation by 12.2 kJ mol−1. Therefore, we expect that the 2TPTZ molecules within the assembly adopt the high-symmetry conformation, which is consistent with previous studies.34 The F4TPA molecules are positioned such that they can interact with the 2TPTZ molecules via O–H⋯N(pyridyl) hydrogen bonds. Two of the three pyridyl nitrogen atoms in each 2TPTZ molecule partake in these interactions. Additional C–H⋯O and C–H⋯F interactions also likely stabilise the assembly.
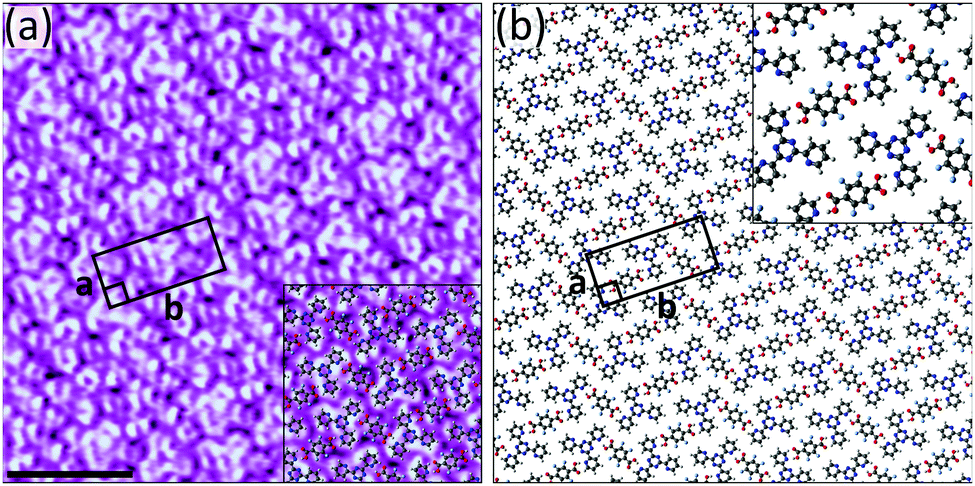 | ||
| Fig. 3 (a) STM image showing the bimolecular assembly of F4TPA and 2TPTZ at the heptanoic acid/HOPG interface. The assembly contains a F4TPA:2TPTZ ratio of 1:1. Tunnelling parameters: Vbias = −1.1 V, Iset = 60 pA. Unit cell parameters: a = 1.3 ± 0.1 nm, b = 2.9 ± 0.2 nm, angle 90 ± 3°. Scale bar = 3 nm. (b) Proposed model for the assembly. | ||
The results obtained when using 3TPTZ as a hydrogen bond acceptor are comparable to those obtained with 2TPTZ: again, F4TPA succeeds at forming bimolecular networks (Fig. S16, ESI†) where TPA fails. Fig. 4a shows a high-resolution STM image of the assembly in which both species can be clearly resolved. The F4TPA molecules are positioned such that each can interact with two 3TPTZ molecules via O–H⋯N(pyridyl) hydrogen bonds. Similarly to 2TPTZ, 3TPTZ can also adopt two distinct planar conformations (Fig. S13, ESI†). In this case, the threefold-symmetric conformation was found to be only 0.9 kJ mol−1 more stable than the alternate conformation (see ESI†). As this energy difference is smaller than thermal energy, either conformation could be present. However, in order to maximise the number of favourable O–H⋯N(pyridyl) interactions, the 3TPTZ molecules are required to be in their non-threefold-symmetric conformation; therefore, we expect that this is the conformation of the 3TPTZ molecules within the assembly. Two of the pyridyl nitrogen atoms in each 3TPTZ molecule partake in these interactions. C–H⋯F, C–H⋯O and C–H⋯N(pyridyl) interactions may also contribute towards stabilising the assembly. The proposed model is given in Fig. 4b.
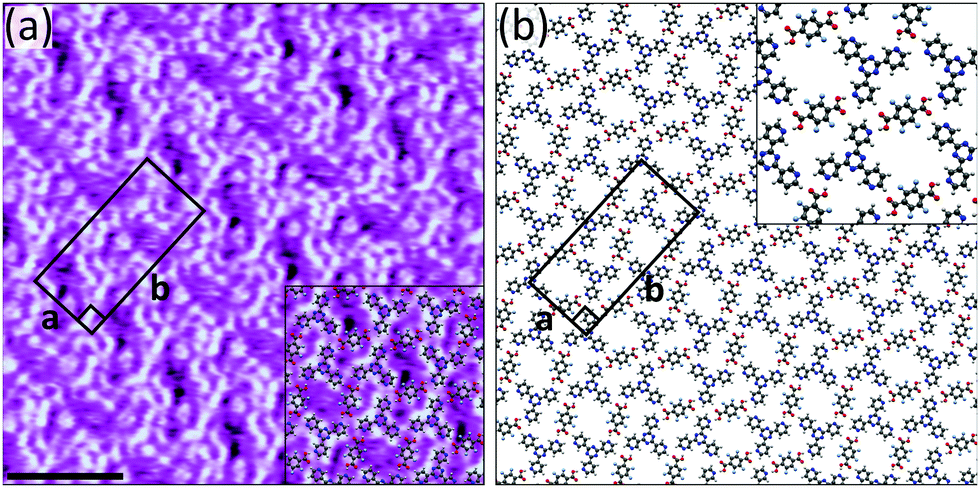 | ||
| Fig. 4 (a) STM image showing the bimolecular assembly of F4TPA and 3TPTZ at the heptanoic acid/HOPG interface. The assembly contains a F4TPA:3TPTZ ratio of 1:1. Tunnelling parameters: Vbias = −1.2 V, Iset = 50 pA. Unit cell parameters: a = 2.0 ± 0.2 nm, b = 4.3 ± 0.3 nm, angle 90 ± 3°. Scale bar = 3 nm. (b) Proposed model for the assembly. | ||
Finally, although neither of the individual components form self-assembled monolayers in isolation, bimolecular networks of F4TPA and 4TPTZ could be formed (see Fig. S17 (ESI†) and Fig. 5). Strikingly, the resulting coassembly is characterised by lattice parameters that are identical to those of the network formed between TPA and 4TPTZ. In fact, high-resolution STM images reveal that the two bimolecular networks are isostructural (compare Fig. 2 and 5). The interactions between the molecules within the two assemblies are analogous. However, in the F4TPA/4TPTZ case there may also be an additional stabilising contribution from C–H⋯F interactions. The fact that isostructural networks can be formed when either TPA or F4TPA are employed as hydrogen bond donors reflects the minimal steric impact associated with exchanging hydrogen for fluorine. This well-known property, extensively utilised in bioorganic chemistry,35 has also previously been observed in self-assembled monolayers.36
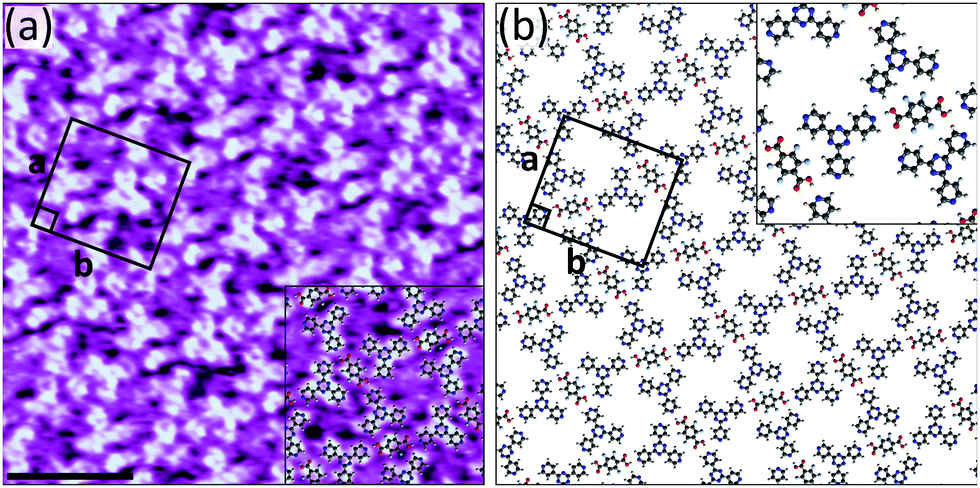 | ||
| Fig. 5 (a) STM image showing the bimolecular assembly of F4TPA and 4TPTZ at the heptanoic acid/HOPG interface. The assembly contains a F4TPA:4TPTZ ratio of 1:2. Tunnelling parameters: Vbias = −1.1 V, Iset = 80 pA. Unit cell parameters: a = 2.7 ± 0.2 nm, b = 3.0 ± 0.2 nm, angle 90 ± 3°. Scale bar = 3 nm. (b) Proposed model for the assembly. | ||
Of the three potential hydrogen bond acceptors, only 4TPTZ was observed to coassemble with TPA, whilst all three form bimolecular networks with F4TPA. This clearly demonstrates that F4TPA is the superior building block in these bimolecular systems. However, precisely understanding the mechanism for this remains challenging. The difference in the adsorption energies of these two similarly sized small molecules is expected to be minimal. Additionally, no significant difference in the solubility of the two molecules was observed. Hence, these factors are unlikely to be the origin of the superior performance of F4TPA. The clearest difference between the two molecules appears to be the intermolecular interactions in which they can partake. We expect that the electron-withdrawing influence of the fluorine atoms may allow F4TPA to form stronger O–H⋯N(pyridyl) hydrogen bonds with the acceptors when compared to TPA. Furthermore, F4TPA can engage in additional C–H⋯F interactions which may also stabilise the assemblies. Although C–H⋯F interactions are typically quite weak, they have been shown to play a significant role in other monolayer systems.37–40 Additionally, we cannot eliminate the possibility that there may be a significant ionic contribution stabilising the assemblies formed with F4TPA. Due to the acidity of F4TPA, proton transfer to the acceptors may occur to some extent. This could result in the acceptors interacting with the F4TPA molecules via O−⋯H–N+(pyridinium) salt bridges rather than simple O–H⋯N(pyridyl) hydrogen bonds. As the acidity of TPA is expected to be significantly lower than that of F4TPA, TPA may be unable to form such salt bridges. Although the relative importance of these different factors remains unclear, it is evident that the net interaction between the acceptors and F4TPA likely exceeds that of TPA. Further theoretical exploration is required to fully understand the improved performance of F4TPA.
In conclusion, we have demonstrated that fluorination can significantly modify the on-surface self-assembly behaviour of carboxylic acids. Whilst peripheral fluorination was found to prevent homomolecular self-assembly, it was shown to be advantageous in the construction of bimolecular networks. We expect that this approach can be used for the successful fabrication of stable bimolecular networks in instances where unfluorinated carboxylic acids are insufficient. This might be particularly relevant in cases where the homomolecular self-assembly of the carboxylic acid is found to outcompete the formation of bimolecular networks.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. G. Slater, L. M. A. Perdigão, P. H. Beton and N. R. Champness, Acc. Chem. Res., 2014, 47, 3417–3427 CrossRef CAS.
- S. De Feyter and F. C. De Schryver, Chem. Soc. Rev., 2003, 32, 139–150 RSC.
- M. Lackinger and W. M. Heckl, Langmuir, 2009, 25, 11307–11321 CrossRef CAS.
- O. Ivasenko and D. F. Perepichka, Chem. Soc. Rev., 2011, 40, 191–206 RSC.
- K. Tahara, S. Lei, J. Adisoejoso, S. De Feyter and Y. Tobe, Chem. Commun., 2010, 46, 8507–8525 RSC.
- L. Dong, Z. Gao and N. Lin, Prog. Surf. Sci., 2016, 91, 101–135 CrossRef CAS.
- K. Eichhorst-Gerner, A. Stabel, G. Moessner, D. Declerq, S. Valiyaveettil, V. Enkelmann, K. Müllen and J. P. Rabe, Angew. Chem., Int. Ed. Engl., 1996, 35, 1492–1495 CrossRef CAS.
- X. Peng, L. Cheng, X. Zhu, Y. Geng, F. Zhao, K. Hu, X. Guo, K. Deng and Q. Zeng, Nano Res., 2018, 11, 5823–5834 CrossRef CAS.
- X. Zhang, S. Wang, Y. Shen, Y. Guo, Q. Zeng and C. Wang, Nanoscale, 2012, 4, 5039–5042 RSC.
- L. Kampschulte, S. Griessl, W. M. Heckl and M. Lackinger, J. Phys. Chem. B, 2005, 109, 14074–14078 CrossRef CAS.
- F. Cometto, K. Frank, B. Stel, N. Arisnabarreta, K. Kern and M. Lingenfelder, Chem. Commun., 2017, 53, 11430–11432 RSC.
- M. Li, P. Xie, K. Deng, Y.-L. Yang, S.-B. Lei, Z.-Q. Wei, Q.-D. Zeng and C. Wang, Phys. Chem. Chem. Phys., 2014, 16, 8778–8782 RSC.
- M. Li, Y.-L. Yang, K.-Q. Zhao, Q.-D. Zeng and C. Wang, J. Phys. Chem. C, 2008, 112, 10141–10144 CrossRef CAS.
- S. Barja, M. Garnica, J. J. Hinarejos, A. L. Vázquez de Parga, N. Martín and R. Miranda, Chem. Commun., 2010, 46, 8198–8200 RSC.
- L. Scudiero, K. W. Hipps and D. E. Barlow, J. Phys. Chem. B, 2003, 107, 2903–2909 CrossRef CAS.
- W. J. Middleton and R. V. Lindsey, J. Am. Chem. Soc., 1964, 86, 4948–4952 CrossRef CAS.
- M. Heger, T. Scharge and M. A. Suhm, Phys. Chem. Chem. Phys., 2013, 15, 16065–16073 RSC.
- A. S. Hansen, L. Du and H. G. Kjaergaard, Phys. Chem. Chem. Phys., 2014, 16, 22882–22891 RSC.
- B. Juršić, M. Ladika and D. E. Sunko, Tetrahedron Lett., 1985, 26, 5323–5324 CrossRef.
- M. H. Abraham, P. L. Grellier, D. V. Prior, P. P. Duce, J. J. Morris and P. J. Taylor, J. Chem. Soc., Perkin Trans. 2, 1989, 699–711 RSC.
- L. W. Reeves, Can. J. Chem., 1961, 39, 1711–1720 CrossRef CAS.
- M. Lackinger, S. Griessl, T. Markert, F. Jamitzky and W. M. Heckl, J. Phys. Chem. B, 2004, 108, 13652–13655 CrossRef CAS.
- A. Della Pia, D. Luo, R. Blackwell, G. Costantini and N. Martsinovich, Faraday Discuss., 2017, 204, 191–213 RSC.
- M. Lackinger, S. Griessl, L. Kampschulte, F. Jamitzky and W. M. Heckl, Small, 2005, 1, 532–539 CrossRef CAS.
- S. Yasuda, A. Furuya and K. Murakoshi, RSC Adv., 2014, 4, 58567–58572 RSC.
- Y.-G. Kim, S.-L. Yau and K. Itaya, Langmuir, 1999, 15, 7810–7815 CrossRef CAS.
- T. Suzuki, T. Lutz, D. Payer, N. Lin, S. L. Tait, G. Costantini and K. Kern, Phys. Chem. Chem. Phys., 2009, 11, 6498–6504 RSC.
- R. Addou and M. Batzill, Langmuir, 2013, 29, 6354–6360 CrossRef CAS PubMed.
- A. Carrera, L. J. Cristina, S. Bengió, A. Cossaro, A. Verdini, L. Floreano, J. D. Fuhr, J. E. Gayone and H. Ascolani, J. Phys. Chem. C, 2013, 117, 17058–17065 CrossRef CAS.
- W. Song, N. Martsinovich, W. M. Heckl and M. Lackinger, J. Am. Chem. Soc., 2013, 135, 14854–14862 CrossRef CAS.
- Z. P. L. Laker, A. J. Marsden, O. De Luca, A. D. Pia, L. M. A. Perdigão, G. Costantini and N. R. Wilson, Nanoscale, 2017, 9, 11959–11968 RSC.
- T. W. White, N. Martsinovich, A. Troisi and G. Costantini, J. Phys. Chem. C, 2018, 122, 17954–17962 CrossRef CAS.
- S. Clair, S. Pons, A. P. Seitsonen, H. Brune, K. Kern and J. V. Barth, J. Phys. Chem. B, 2004, 108, 14585–14590 CrossRef CAS.
- J. Zhang, B. Li, X. Cui, B. Wang, J. Yang and J. G. Hou, J. Am. Chem. Soc., 2009, 131, 5885–5890 CrossRef CAS PubMed.
- D. O’Hagan and H. S. Rzepa, Chem. Commun., 1997, 645–652 RSC.
- A. Stabel, L. Dasaradhi, D. O’Hagan and J. P. Rabe, Langmuir, 1995, 11, 1427–1430 CrossRef CAS.
- A. Y. Brewer, M. Sacchi, J. E. Parker, C. L. Truscott, S. J. Jenkins and S. M. Clarke, Phys. Chem. Chem. Phys., 2014, 16, 19608–19617 RSC.
- J. Niederhausen, Y. Zhang, F. Cheenicode Kabeer, Y. Garmshausen, B. M. Schmidt, Y. Li, K.-F. Braun, S. Hecht, A. Tkatchenko, N. Koch and S.-W. Hla, J. Phys. Chem. C, 2018, 122, 18902–18911 CrossRef CAS.
- Z. Mu, L. Shu, H. Fuchs, M. Mayor and L. Chi, J. Am. Chem. Soc., 2008, 130, 10840–10841 CrossRef CAS.
- V. Oison, M. Koudia, M. Abel and L. Porte, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 035428 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, additional STM images and calculations. See DOI: 10.1039/c9cc08361h |
| ‡ Present address: Chemistry Research Group, School of Pharmacy and Biomolecular Sciences, University of Brighton, Brighton, BN2 4GJ, UK. |
| This journal is © The Royal Society of Chemistry 2020 |