
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA conceptual framework for the development of iridium(III) complex-based electrogenerated chemiluminescence labels†
Lifen
Chen‡
a,
David J.
Hayne‡
 *a,
Egan H.
Doeven
a,
Johnny
Agugiaro
b,
David J. D.
Wilson
b,
Luke C.
Henderson
a,
Timothy U.
Connell§
c,
Yi Heng
Nai
a,
Richard
Alexander
a,
Serena
Carrara
b,
Conor F.
Hogan
b,
Paul S.
Donnelly
d and
Paul S.
Francis
*a
*a,
Egan H.
Doeven
a,
Johnny
Agugiaro
b,
David J. D.
Wilson
b,
Luke C.
Henderson
a,
Timothy U.
Connell§
c,
Yi Heng
Nai
a,
Richard
Alexander
a,
Serena
Carrara
b,
Conor F.
Hogan
b,
Paul S.
Donnelly
d and
Paul S.
Francis
*a
aDeakin University, School of Life and Environmental Sciences, Centre for Regional and Rural Futures (CeRRF), Institute for Frontier Materials (IFM), Geelong, Victoria 3220, Australia. E-mail: paul.francis@deakin.edu.au; d.hayne@deakin.edu.au
bDepartment of Chemistry and Physics, La Trobe Institute for Molecular Sciences (LIMS), La Trobe University, Melbourne, Victoria 3086, Australia
cCommonwealth Scientific and Industrial Research Organisation (CSIRO), Clayton, Victoria 3168, Australia
dSchool of Chemistry, Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Victoria 3010, Australia
First published on 31st July 2019
Abstract
Translation of the highly promising electrogenerated chemiluminescence (ECL) properties of Ir(III) complexes (with tri-n-propylamine (TPrA) as a co-reactant) into a new generation of ECL labels for ligand binding assays necessitates the introduction of functionality suitable for bioconjugation. Modification of the ligands, however, can affect not only the photophysical and electrochemical properties of the complex, but also the reaction pathways available to generate light. Through a combined theoretical and experimental study, we reveal the limitations of conventional approaches to the design of electrochemiluminophores and introduce a new class of ECL label, [Ir(C^N)2(pt-TOxT-Sq)]+ (where C^N is a range of possible cyclometalating ligands, and pt-TOxT-Sq is a pyridyltriazole ligand with trioxatridecane chain and squarate amide ethyl ester), which outperformed commercial Ir(III) complex labels in two commonly used assay formats. Predicted limits on the redox potentials and emission wavelengths of Ir(III) complexes capable of generating ECL via the dominant pathway applicable in microbead supported ECL assays were experimentally verified by measuring the ECL intensities of the parent luminophores at different applied potentials, and comparing the ECL responses for the corresponding labels under assay conditions. This study provides a framework to tailor ECL labels for specific assay conditions and a fundamental understanding of the ECL pathways that will underpin exploration of new luminophores and co-reactants.
Introduction
Electrogenerated chemiluminescence (ECL) has been widely adopted over the past few decades for affinity/ligand binding assays,1,2 with ∼2 billion tests now performed each year on commercial ECL instrumentation for clinical diagnostics, life science research, food testing and biodefense applications.3 Remarkably, despite the extensive on-going research into new approaches and applications, the vast majority of ECL-based assays published in the open literature and all commercial systems still rely on a single orange luminophore: tris(2,2′-bipyridine)ruthenium(II) ([Ru(bpy)3]2+; Fig. 1).1,2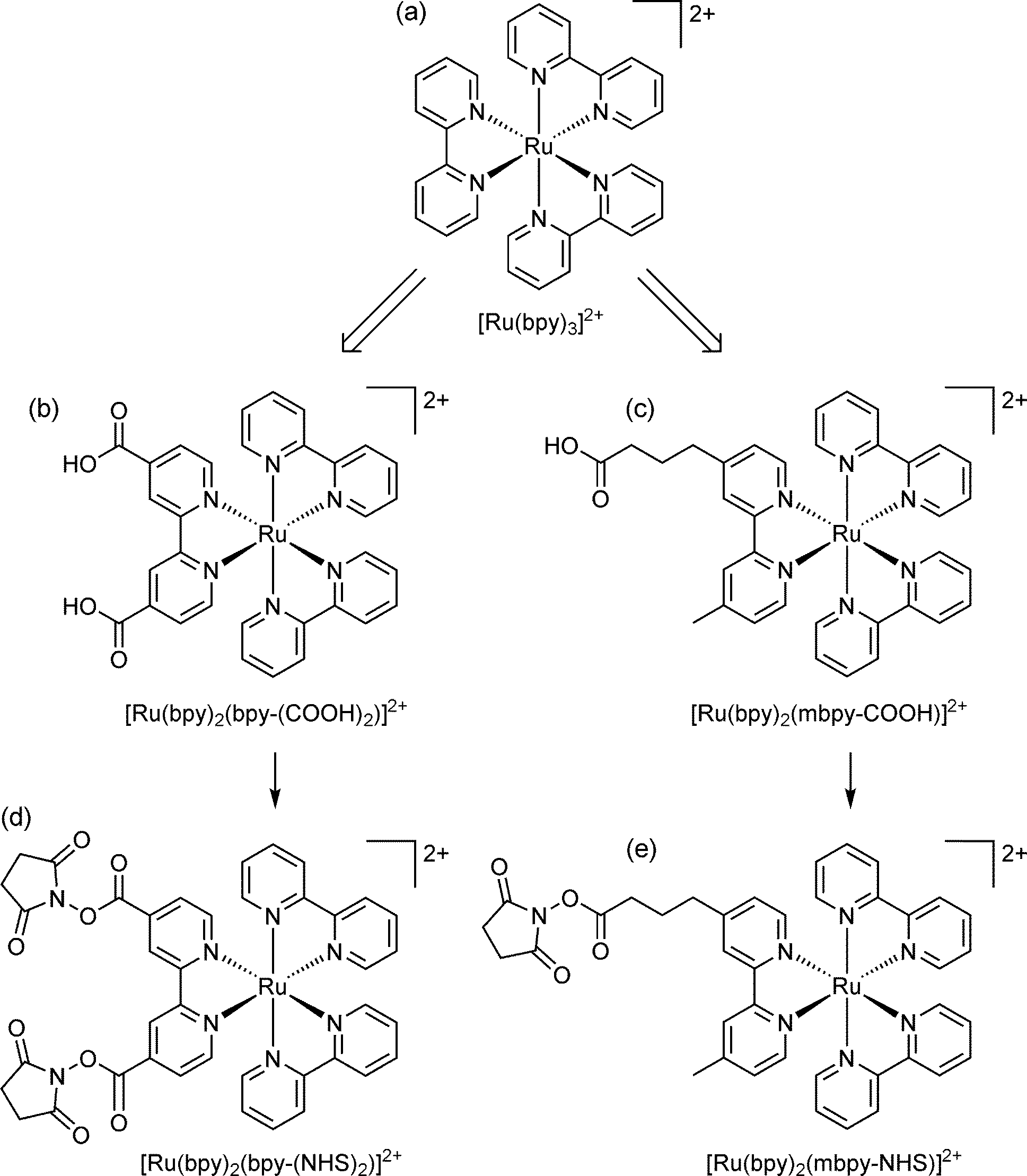 | ||
| Fig. 1 (a) The parent luminophore, tris(2,2′-bipyridine)ruthenium(II); and examples of (b) disubstituted33,34 and (c) monosubstituted6,8,35,36 derivatives suitable for bioconjugation. The single binding group separated from the luminophore by an alkyl chain is the approach that has been adopted in commercial ECL systems. Carboxylic acid functionality is common, but other groups, such as amines, maleimides, hydrazides, and phosphoramidites have also been used.37 (d, e) The carboxylic acids are converted to more reactive N-hydroxysuccinimide (NHS) esters for binding to amines (such as lysine units of proteins).35 | ||
The underlying principles of this detection chemistry were established through an extensive series of investigations that included the inception of ‘co-reactant’ ECL,4 in which the oxidation of a sacrificial species such as tri-n-propylamine (TPrA) forms the chemi-excitation source (Scheme 1a), and the elucidation of an alternative pathway to the excited luminophore (Scheme 1b)5 that has been shown to be the dominant light-producing mechanism within typical ECL-based assays immobilised on microbeads.6,7 ECL labels for bioconjugation were created by adding suitable functional groups to one or both pyridine rings of a single ligand (see Fig. 1), which exert, in this case, only minor influence on the electrochemical and photophysical properties of the luminophore.8
 | ||
| Scheme 1 Co-reactant ECL mechanisms involving (a) electrochemical oxidation the tri-n-propylamine co-reactant (TPrA) and metal complex (M; [Ru(bpy)3]2+), or (b) oxidation of the co-reactant only,5,21 where TPrA+˙ is an aminium radical cation, TPrA˙ is a neural α-amino alkyl radical, and P is its subsequent products. The additional ‘catalytic route’ involving oxidation of TPrA by M+, and an ‘annihilation’ pathway, in which the excited state is generated from the reaction of M+ (from a) with M− (from b), are shown in Scheme S1 in the ESI.† | ||
Cyclometallated iridium(III) complexes (e.g., Fig. 2a–c) have emerged as promising candidates for a new generation of ECL labels.2,9,10 Compared to the traditional Ru(II) polypyridine chelates, the Ir(III) complexes exhibit much greater quantum yields (offering enhanced analytical performance) and their emission wavelengths and electrochemical potentials can be readily manipulated through changes in ligand structure,11 creating exciting opportunities for multi-colour and/or potential-resolved multiplexed ECL systems.12–15 However, despite more than a decade of exploration of co-reactant ECL of Ir(III) complexes with these goals in mind, the properties of the most promising luminophores have not been effectively translated into ECL labels for the extensive range of possible assays, which we attribute to several major limitations:
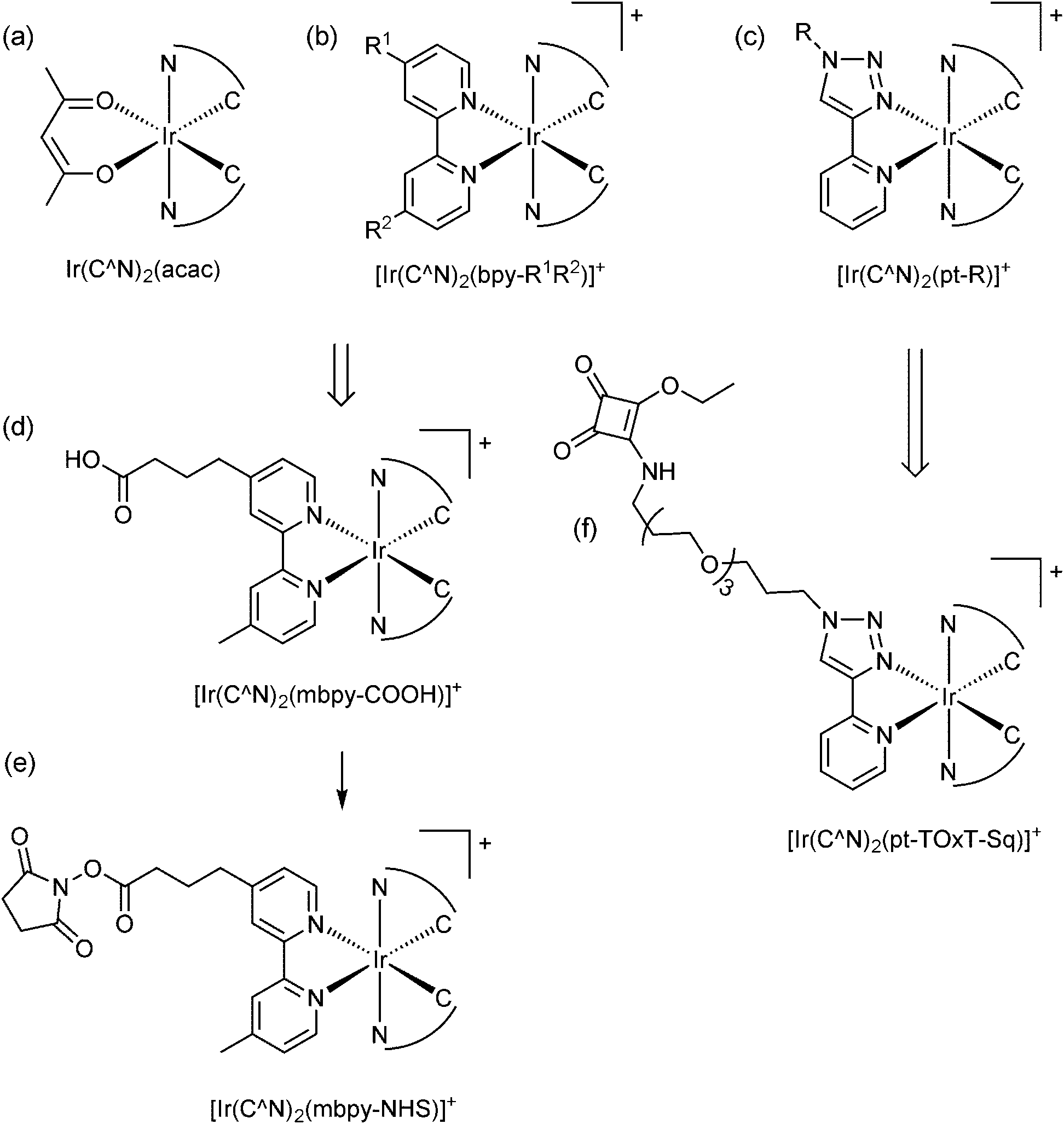 | ||
| Fig. 2 The conceptual development of [Ir(C^N)2(L)]+ complex ECL labels from the early promising examinations of (a) neutral heteroleptic Ir(III) complexes such as Ir(C^N)2(acac), where C^N = 2-phenylpyridine (ppy), 2-phenylbenzo[d]thiazole (bt), 2-phenylisoquinoline (piq) or various other ligands.9,16,17 (b) A representative example of the wider class of cationic [Ir(C^N)2(N^N)]+ complexes, the co-reactant ECL of many of which has also been examined.40,41 (c) More recently reported analogues incorporating a 1-substituted-4-(2-pyridyl)-1,2,3-triazole ligand, which show promising properties for ECL.22,23,29 (d) The most common approach to convert neutral and cationic Ir(III) complexes into ECL labels has been to substitute one ligand for the same bipyridine derivative as that used in Ru(II) labels (see Fig. 1).15,18,19 (e) As with the Ru(II)-based labels shown in Fig. 1, the carboxylic acids are converted to the corresponding NHS esters for binding to amines. (f) The alternative design for ECL labels described herein. | ||
(1) The available Ir(III) complexes are generally far less soluble in water than [Ru(bpy)3]2+, and so their ECL properties have almost exclusively been evaluated under conditions that are not compatible with common assay formats.9,16,17
(2) Most ligands utilised in Ir(III) complexes do not have readily available derivatives with functional groups suitable for bioconjugation and consequently, very few of the promising Ir(III) complexes have been adapted into labels. Of the few that have been created, the predominant approach has been to replace one ligand with the same bipyridine derivatives that have been used in the [Ru(bpy)3]2+-based labels (e.g., Fig. 2d).15,18,19 However, this appears to limit the range of electrochemical potentials, emission wavelengths and ECL intensities of the complexes.
(3) The two dominant ECL reaction pathways (Scheme 1a and b) elucidated for [Ru(bpy)3]2+ (with TPrA as co-reactant)5 are not necessarily both feasible for all Ir(III) complexes,20 which has implications for their effectiveness under assay conditions. This has not been considered in the previous development of Ir(III)-based ECL labels.
We sought to devise a framework for the development of ECL labels from Ir(III) complex luminophores in which each of the above challenges are addressed. With this in mind, we selected a pyridyltriazole ancillary ligand (Fig. 2c) as the scaffold for the novel ECL labelling complexes. This ligand class exhibits several favourable properties for ECL detection,22–24 including simple ‘click chemistry’ preparation25 that provides a versatile point for derivatisation or attachment.26
De Cola and co-workers have previously explored various [Ir(C^N)2(pt-R)]+ complexes (Fig. 2c; where C^N = ppy or df-ppy, and R = methyl, phenyl, benzyl, adamantyl, β-cyclodextrin and other groups) for photoluminescence,27 light emitting electrochemical cells28 and ECL22 applications. The [Ir(df-ppy)2(pt-R)]+ species exhibited a deeper blue emission than most charged Ir(III) complexes, and intense co-reactant ECL under aprotic and aqueous conditions. Similarly, we have shown that [Ir(df-ppy)2(ptb)]+ (Fig. 2c; C^N = df-ppy; R = Bn) exhibits more intense co-reactant ECL than related blue luminophores Ir(df-ppy)3 and [Ir(df-ppy)2(ptp)]+ (where ptp = 3-phenyl-1,2,4-triazol-5-ylpyridinato) in acetonitrile,23 which we exploited in foundational investigations of multi-colour annihilation ECL.29,30 Using an analogous synthetic strategy, we prepared a highly water-soluble derivative [Ir(df-ppy)2(pt-TEG)]+ (Fig. 2c; C^N = df-ppy; R = tetraethylene glycol (TEG)) that provided more intense co-reactant ECL24,31 and chemiluminescence32 than related Ir(III) complexes in buffered aqueous solution.
Although most prior studies of the ECL of [Ir(C^N)2(pt-R)]+ complexes have focused on developing blue luminophores (such as [Ir(df-ppy)2(ptb)]+; λem = 453, 481 nm),22–24 we have previously observed efficient red photoluminescence (λem = 592, 632 nm) from [Ir(piq)2(ptb)]+ (where piq = 1-phenyl-isoquinoline),38 indicating that the emission of these complexes can be tuned over a wide range via simple modifications to the C^N ligands. Moreover, in our previous development of luminescent Ir(III) complexes for live cell imaging,39 we explored several strategies for their covalent attachment to biomolecules, involving the introduction of maleimide, N-hydroxysuccinimide activated ester, or squarate ethyl ester (Fig. 2f; C^N = ppy or 2-phenylquinoline (pq)) functional groups.
We now draw together these advances to overcome several barriers to the adoption of promising Ir(III)-complex electrochemiluminophores to labelling in binding assays. This includes an in-depth examination of the influence of bioconjugation ligands on Ir(III) complex luminophores, a simple synthetic approach to prepare analogues suitable for organic solvents or aqueous conditions, the creation of novel ECL labels, and the evaluation of the new labels within two common ECL-based assay formats.
Results and discussion
Comparison of luminophores
The ECL efficiency (ϕECL) of a luminophore is dependent on both its redox potentials and excited state character. In the case of heteroleptic Ir(III) complexes, these parameters can be readily tuned through minor modifications of ligand structure, to stabilise or destabilise the frontier molecular orbitals with some degree of selectivity.9,10,42 Early exploration of Ir(C^N)2(acac) complexes showed high ECL efficiencies with a wide range of emission colours, in reactions with radical anions of aromatic nitriles.43 Efficient ECL from Ir(C^N)2(acac) complexes with TPrA as a co-reactant has also been demonstrated,40,44 but little attention has been paid to the influence of the different redox potentials of the complexes on the competing light-producing reaction pathways (Scheme 1). Moreover, the effects of replacing ancillary ligands such as acac with those suitable for bioconjugation (ECL-labelling) must also be considered.To understand these effects, we initially conducted an experimental and theoretical study of the relevant properties of twelve Ir(III) complexes. This included: (i) four commercially available Ir(C^N)2(acac) complexes (Fig. 2a), where C^N = piq, bt, ppy and df-ppy, which exhibit red, yellow, green and blue luminescence, respectively; (ii) four [Ir(C^N)2(dm-bpy)]+ complexes (Fig. 2b; R1 and R2 = Me), with the same C^N ligands, as the parent luminophores of complexes with bpy-based ligands for bioconjugation (Fig. 2e), and (iii) four [Ir(C^N)2(ptb)]+ complexes (Fig. 2c; R = Bn) containing the parent luminophore of the proposed [Ir(C^N)2(pt-TOxT-Sq)]+ labelling complexes. The model luminophores were employed because they were commercially available or readily synthesised and they avoided complications from the reactive peripheral functionality of their labelling derivatives8 when assessing the properties of the luminophore. The [Ir(C^N)2(dm-bpy)]+ and [Ir(C^N)2(ptb)]+ complexes containing piq, ppy and df-ppy ligands have previously been reported,23,28,38,45 but the two bt analogues were prepared in this study for the first time.
The properties of the Ir(C^N)2(acac) complexes (Table S1† and Fig. 3, 4, S1–S6†) were in good agreement with those reported across prior studies.17,46 DFT calculations on Ir(C^N)2(acac) complexes indicate that the HOMO is predominantly localised on the phenyl ring of the C^N ligands and the metal centre, the LUMO is on the C^N ligand (Fig. S7†), and the observed luminescence (Fig. 3) originates from mixed metal-to-ligand and intra-ligand charge transfer (3MLCT/3ILCT) excited states.47 As the acac ligand has minimal direct involvement in the frontier molecular orbitals, trends in spectroscopic and electrochemical properties across the four complexes mirror those of their homoleptic Ir(C^N)3 counterparts.17,23,48
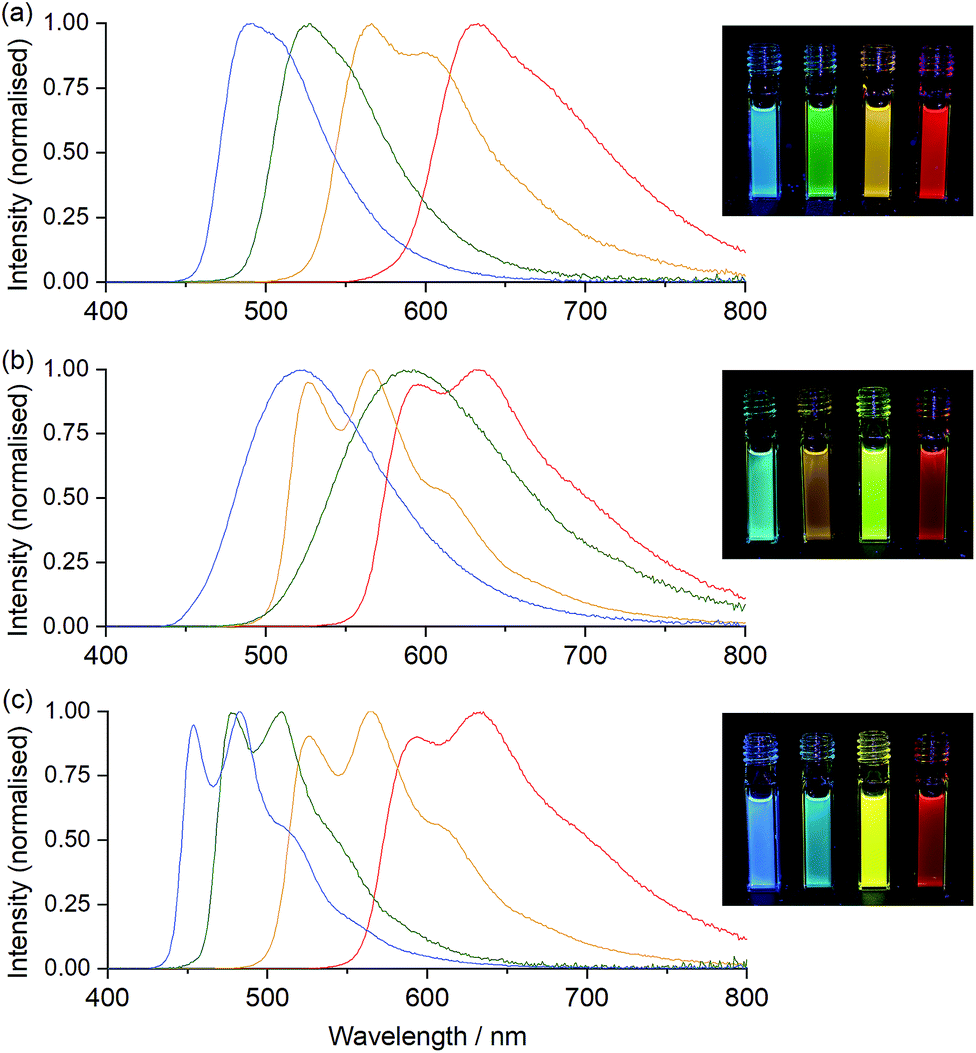 | ||
| Fig. 3 Photoluminescence emission spectra (corrected) of (a) Ir(C^N)2(acac), (b) [Ir(C^N)2(dm-bpy)]+, and (c) [Ir(C^N)2(ptb)]+ complexes, where C^N = df-ppy (blue lines), ppy (green lines), bt (yellow lines), or piq (red lines), at a concentration of 10 μM in acetonitrile. The inset photos show the emissions under UV light, with the complexes (100 μM in acetonitrile) containing df-ppy, ppy, bt, and then piq ligands in cuvettes from left to right. | ||
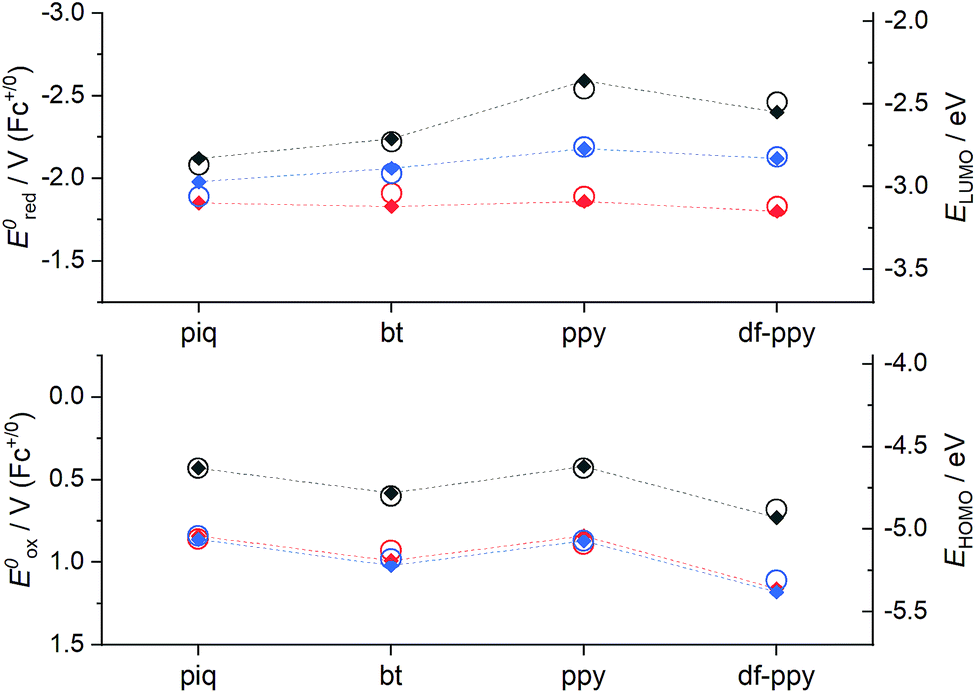 | ||
| Fig. 4 Effects of ligand structure on electrochemical properties (solid diamonds; left axes) and calculated MO energies (open circles; right axes; BP86/def2-TZVP calculations), for Ir(C^N)2(acac) (grey symbols), [Ir(C^N)2(dm-bpy)]+ (red symbols), and [Ir(C^N)2(ptb)]+ (blue symbols) complexes, where C^N = piq, bt, ppy, or df-ppy. The upper graph shows reduction potentials and LUMO energies, and the lower graph shows oxidation potentials and HOMO energies. | ||
Ir(ppy)2(acac) exhibits green luminescence (Fig. 3a) and the highest LUMO energy of the four complexes, which is reflected in it having the most negative reduction potential (Fig. 4). The electron-withdrawing fluoro substituents of Ir(df-ppy)2(acac) strongly stabilise the HOMO and to a lesser extent the LUMO, with corresponding positive shifts in associated electrochemical potentials and a hypsochromic shift to give its characteristic blue emission. The bt ligand also lowers the energy of the frontier orbitals compared to ppy, but with a slightly lesser effect on the HOMO, resulting in a yellow emission. The resolved band structure in the emission spectrum can be attributed to the greater proportion of the LUMO on the phenyl ring of the C^N ligands. The extended aromaticity of the piq ligand greatly stabilises the LUMO through its low lying π* orbital, but the HOMO energy is relatively unchanged, resulting in a large bathochromic shift in emission.
If the ancillary acac ligand is replaced by dm-bpy, the HOMO is stabilised by ∼0.4 eV (observed as commensurate increases in the oxidation potentials; Fig. 4) but remains located on the metal and C^N ligands (Fig. S7 and Table S6†). The low-lying π* orbital of the dm-bpy ligand, however, is now the dominant contributor to the LUMO of the df-ppy, ppy, and bt complexes, although both the dm-bpy and two piq ligands contribute to the LUMO of the [Ir(piq)2(dm-bpy)]+ complex. This results in a similar reduction potential for all four [Ir(C^N)2(dm-bpy)]+ complexes at −1.83 ± 0.03 V vs. Fc+/0 (Fig. 4). As Ir(ppy)2(acac) has the highest energy LUMO, the influence of the dm-bpy ligand on the emission spectra is most prominent for [Ir(ppy)2(dm-bpy)]+. This changes the order of emission energy from df-ppy > ppy > bt > piq in Ir(C^N)2(acac) complexes (Fig. 3a) to df-ppy > bt > ppy > piq in [Ir(C^N)2(dm-bpy)]+ complexes (Fig. 3b). Moreover, the near identical LUMO energies of the four complexes narrows the difference in their emission wavelengths, reducing the possible selectivity of multi-colour ECL systems.
The HOMOs calculated for the [Ir(C^N)2(ptb)]+ complexes had similar energies to those of their [Ir(C^N)2(dm-bpy)]+ counterparts (Fig. 4), and were again distributed on the metal centre and C^N ligands. The LUMOs, however, were intermediate in energy between those of the Ir(C^N)2(acac) and [Ir(C^N)2(dm-bpy)]+ complexes. The order of emission energies matched that of the Ir(C^N)2(acac) complexes (Fig. 3c). The difference in the λmax between the blue and red emitters is 151 nm (or 140 nm if the highest energy peaks are compared), which is much greater than that of the [Ir(C^N)2(dm-bpy)]+ complexes (71 nm), and similar to that of the Ir(C^N)2(acac) complexes (142 nm).
The red and yellow luminophore Ir(C^N)2(acac) complexes exhibited greater ECL intensities (2.05 and 0.81, vs. [Ru(bpy)3]2+ = 1, see Table S1†) than the green and blue emitters (0.015 and 0.17), but it should be noted that the CCD detector provides a fairly even response across the wavelength range and other commonly used photodetectors (such as photomultiplier tubes) exhibit much greater sensitivity in the blue and green regions. The ECL intensities of the [Ir(C^N)2(dm-bpy)]+ and [Ir(C^N)2(ptb)]+ complexes were distributed over narrower ranges (0.27–0.89 and 0.10–0.43, respectively).
The above characterisations allow us to assess the feasibility of the ECL reaction pathways shown in Scheme 1a and b for these Ir(III) complexes. This is critical, because studies of the ECL of [Ru(bpy)3]2+ with TPrA co-reactant have shown that the conditions of some assays heavily favour one ECL pathway over others.5–7,49 The requirements can be visualised using the graphs shown in Fig. 5a–d,20 in which the electrochemical potential for oxidation and reduction of the metal complex is plotted against the excited state energy (shown as the λmax from low temperature emission spectra).
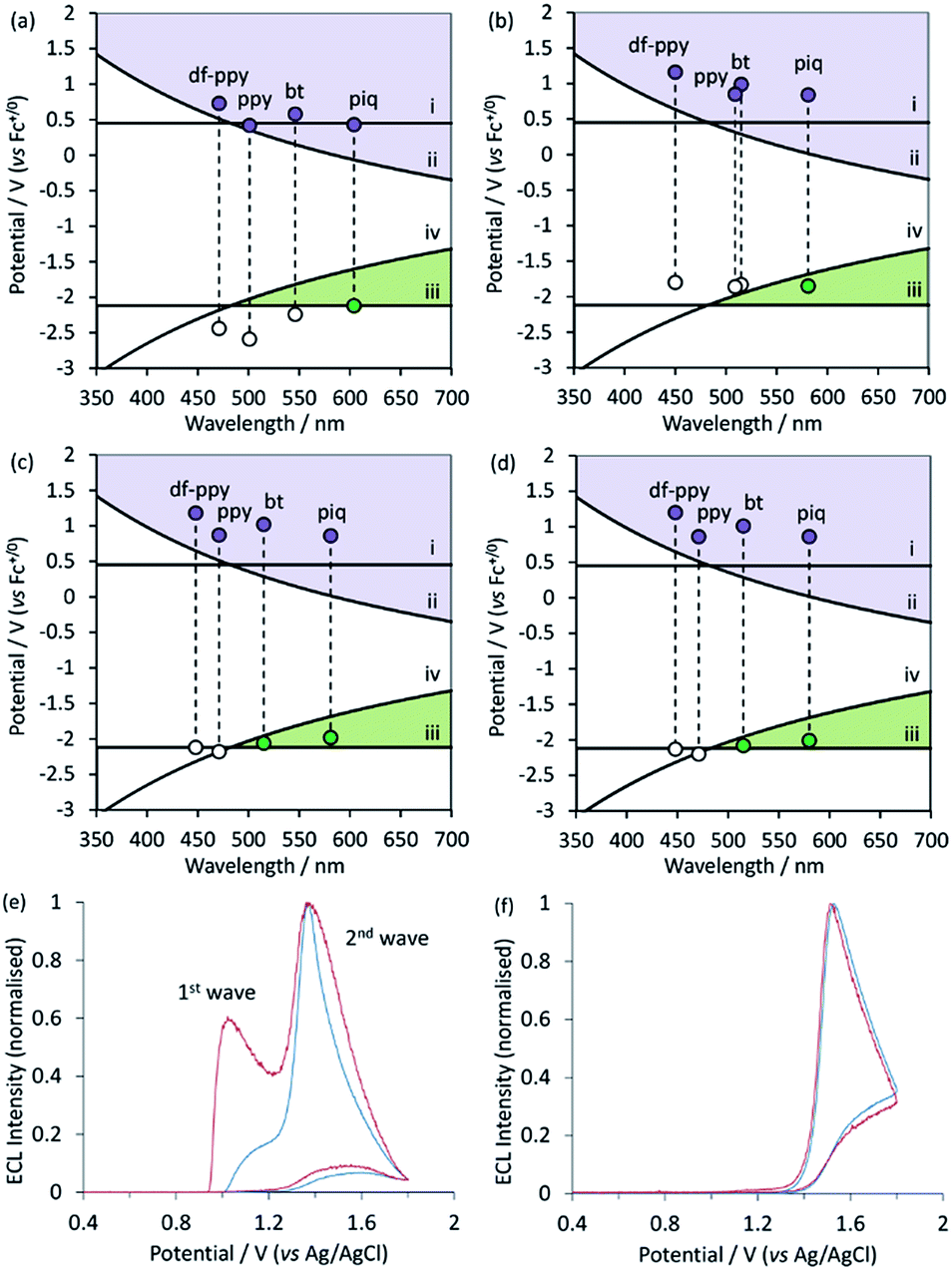 | ||
| Fig. 5 (a–d) Redox potentials vs. low-temperature emission wavelengths (λmax) for (a) Ir(C^N)2(acac), (b) [Ir(C^N)2(dm-bpy)]+, (c) [Ir(C^N)2(ptb)]+, and (d) [Ir(C^N)2(pt-TEG)]+, on graphs depicting the redox potential requirements of the ECL mechanisms shown in Scheme 1a and b, indicating which ECL pathway(s) are feasible for each complex. Lines (i)–(iv) show the redox potential requirements for reactions (1)–(4), respectively. (e and f) Normalised ECL intensity during an applied potential sweep from 0 V to 1.8 V and back to 0 V (vs. Ag|AgCl) for (e) [Ir(bt)2(pt-TEG)]+ and (f) [Ir(df-ppy)2(pt-TEG)]+ at 1 μM (blue lines) and 0.1 μM (red lines) in buffer (ProCell) solution containing TPrA as a co-reactant (plots for [Ir(piq)2(pt-TEG)]+ and [Ir(ppy)2(pt-TEG)]+ are shown in ESI†), providing experimental confirmation of the predictions made in (d) under the aqueous conditions commonly used in ECL assays. The concentration of TPrA in the Procell solution is higher than that typically used in comparisons of ECL intensities in buffered aqueous solutions (for example, see Table 1), which will favour the ‘first wave’ of ECL.5 The absence of the first wave in (f) therefore indicates that it will not be observed for this complex with any relevant concentration of TPrA. | ||
Overlaid on the graphs in Fig. 5a–d are the redox potential requirements of the two ECL pathways. Line (i) is the oxidation potential of TPrA. For complexes with an oxidation potential above this line, reaction (1) is favourable. This ‘catalytic route’5 provides an efficient means to generate TPrA+˙,50 but it is not essential for ECL, because TPrA is also electrochemically oxidised. Line (ii) represents the oxidation potential that the metal complex requires to attain its electronically excited state via reaction (2) (from Scheme 1a). As can be seen in the curve of this line, the energy demands become greater as the emission wavelength becomes shorter (i.e., blue-shifted luminophores must possess higher oxidation potentials to generate ECL viaScheme 1a). This requirement for Scheme 1a, which we referred to in our previous work as the ‘ECL wall of energy sufficiency’,51 was met by all of the metal complexes under investigation.
| M+ + TPrA → M + TPrA+˙ | (1) |
| M+ + TPrA˙ → M* + P | (2) |
To generate ECL viaScheme 1b, the metal complex must first be able to be reduced by TPrA˙ (reaction (3)) and then react with TPrA+˙ with sufficient excess energy to generate the electronically excited luminophore (reaction (4)). These requirements are met in complexes with a reduction potential above line (iii) and below line (iv) in Fig. 5a–c (i.e., the green coloured region of the graph).
| M + TPrA˙ → M− + P | (3) |
| M− + TPrA+˙ → M* + TPrA | (4) |
Of the Ir(C^N)2(acac) and [Ir(C^N)2(dm-bpy)]+ complexes (Fig. 5a and b), only the red emitters with piq ligands clearly satisfy the criteria for generating ECL with TPrA viaScheme 1b. It should be noted, however, that there are numerous sources of error in these predictions20 and borderline cases (such as [Ir(bt)2(dm-bpy)]+ and [Ir(ppy)2(dm-bpy)]+) should be treated with caution. Of the [Ir(C^N)2(ptb)]+ complexes, both [Ir(piq)2(ptb)]+ and [Ir(bt)2(ptb)]+ satisfy the criteria for generating ECL viaScheme 1b. Moreover, whilst the data depicted in Fig. 5a–d enable evaluation of which mechanisms are thermodynamically feasible, the intensity of ECL generated through the available pathways is dependent on the efficiency of both excitation (determined by the relative rates of ground and excited state product formation52) and emission (which is equivalent to the photoluminescence quantum yield).
Water soluble [Ir(C^N)2(pt-TEG)]+ complexes
The click chemistry synthesis of pyridyltriazole ligands provides a synthetically robust approach to introduce functional groups that improve the solubility of the Ir(III) complex.22,27,32,53 Using this approach, we prepared [Ir(C^N)2(pt-TEG)]+ (Fig. 2c, C^N = piq, bt, ppy, df-ppy; R = TEG), with chloride counter ions instead of hexafluorophosphate, for evaluation under conditions akin to those of typical ECL assays.The electrochemical potentials of the [Ir(C^N)2(pt-TEG)]+ complexes matched those of [Ir(C^N)2(ptb)]+ in acetonitrile (Fig. S6†) and the oxidation potentials of the [Ir(C^N)2(pt-TEG)]+ complexes in buffered aqueous solution (vs. Ag|AgCl) exhibited a similar trend (Table 1). The luminescence λmax of [Ir(C^N)2(pt-TEG)]+ in water and [Ir(C^N)2(ptb)]+ in acetonitrile at ambient temperature were near identical, as was their λmax at low temperature in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethanol:methanol (Fig. S4 and S5,†Tables 1 and S1†). The ECL intensities of the [Ir(C^N)2(pt-TEG)]+ complexes in buffered aqueous solution, however, were greater than those of [Ir(C^N)2(ptb)]+ in acetonitrile, relative to that of [Ru(bpy)3]2+ under each set of conditions.
:1 ethanol:methanol (Fig. S4 and S5,†Tables 1 and S1†). The ECL intensities of the [Ir(C^N)2(pt-TEG)]+ complexes in buffered aqueous solution, however, were greater than those of [Ir(C^N)2(ptb)]+ in acetonitrile, relative to that of [Ru(bpy)3]2+ under each set of conditions.
| Photoluminescence | Electrochemical potentials | ECL | ||||||
|---|---|---|---|---|---|---|---|---|
| λ max , /nm | λ max , (85 K)/nm | E 0–0 /eV | ϕ PL (%) | E ox /V (vs. Ag|AgCl) | E ox /V (vs. Fc+/0) | E red /V (vs. Fc+/0) | I ECL | |
|
a Metal complexes at 10 μM in water at ambient temperature.
b Corrected for the change in instrument sensitivity over the wavelength range.
c Metal complexes at 5 μM in ethanol:methanol (4:1) at 85 K.
d Energy gap between the zeroth vibrational levels of the ground and excited states, estimated from the highest energy peak of the low-temperature emission spectrum.
e Photoluminescence quantum yield in ‘ProCell’ phosphate buffer used in commercial ECL instruments.
f Metal complexes at 0.5 mM in 0.1 M phosphate buffer solution (pH 7.5); squarewave voltammetry; 5 mV step, 0.02 amplitude, 25 Hz.
g Metal complexes at 0.25 mM in acetonitrile with 0.1 M TBAPF6; cyclic voltammetry; scan rate: 0.1 V s−1.
h ECL intensities relative to [Ru(bpy)3]2+ (10 μM metal complex in phosphate buffer (pH 7.5) with 10 mM TPrA; 10 s pulse, 10 Hz).
|
||||||||
| [Ru(bpy)3]2+ | 625 | 581, 629 | 2.13 | 3 | 1.09 | 0.89 | −1.73, −1.92, −2.15 | 1 |
| [Ir(piq)2(pt-TEG)]+ | 596, 628 | 580, 629, 684 | 2.14 | 9 | 1.15 | 0.86 | −2.01, −2.23 | 0.86 |
| [Ir(bt)2(pt-TEG)]+ | 526, 562 | 515, 557, 604, 659 | 2.41 | 23 | 1.28 | 1.01 | −2.08 | 2.76 |
| [Ir(ppy)2(pt-TEG)]+ | 475, 505 | 471, 506, 536 | 2.63 | 14 | 1.08 | 0.86 | −2.20 | 1.57 |
| [Ir(df-ppy)2(pt-TEG)]+ | 452, 481 | 448, 480, 507 | 2.77 | 19 | 1.44 | 1.20 | −2.13 | 0.26 |
Due to the similar properties of the [Ir(C^N)2(ptb)]+ and [Ir(C^N)2(pt-TEG)]+ complexes, the same predictions (Fig. 5d) were made for the feasibility of the two ECL reaction mechanisms (Scheme 1a and b), where [Ir(piq)2(pt-TEG)]+ and [Ir(bt)2(pt-TEG)]+ are anticipated to generate ECL (with TPrA co-reactant) via both pathways, whereas [Ir(ppy)2(pt-TEG)]+ and [Ir(df-ppy)2(pt-TEG)]+ are limited to Scheme 1a. We sought validation for these predictions using the ‘two-wave’ ECL experiment utilised by Bard and co-workers5 in their elucidation of Scheme 1b for [Ru(bpy)3]2+, in which the ECL intensity is monitored during a voltammetric sweep from low to high anodic potentials. Scheme 1b is initiated at the oxidation potential for TPrA, whereas Scheme 1a also requires electrochemical oxidation of the metal complex, which in the case of the [Ir(C^N)2(pt-TEG)]+ complexes, occurs at considerably higher potentials (Fig. 5d).
The two ‘waves’ of ECL intensity corresponding to the oxidation of TPrA and the metal complex (Table 1) for [Ir(bt)2(pt-TEG)]+ (Fig. 5e) and [Ir(piq)2(pt-TEG)]+ (Fig. S8b†), and the single wave of ECL associated with the oxidation of [Ir(df-ppy)2(pt-TEG)]+ (Fig. 5f) and [Ir(ppy)2(pt-TEG)]+ (Fig. S8a†) in aqueous buffered solution support the prediction made for each complex based on electrochemical potentials and emission energies. Bard and co-workers observed that a lower concentration of [Ru(bpy)3]2+ favoured the first wave of ECL (viaScheme 1b), which we also observed for [Ir(bt)2(pt-TEG)]+ (Fig. 5e) adding further evidence of the competing ECL pathways of this complex.
We note here that interpretation of ECL intensity profiles during voltammetric experiments without considering the energy requirements of the competing ECL pathways (as depicted in Fig. 5a–d) can be misleading, as other factors can contribute. The ECL of Ir(ppy)3 with TPrA, for example, is strongly inhibited at high over-potentials,13,14 which has been attributed to oxidative quenching of the excited state Ir(ppy)3* by TPrA˙+. Moreover, De Cola and co-workers16 observed more than two maxima in the ECL of Ir(pph)2(pic) (where pph = phenylphenanthridine; pic = picolinate) and TPrA, which is yet to be understood.
Incorporation of the new ECL labels in binding assays
Using the synthetic strategies outlined in our preparation of [Ir(ppy)2(pt-TOxT-Sq)]+ for live cell photoluminescence imaging39 (details in ESI†), we adapted the promising [Ir(C^N)2(pt-TEG)]+ electrochemiluminophores (Fig. 2c, R = TEG) for ECL labelling. Conventional ECL labels with carboxylic acid functionality (e.g., Fig. 1b, c and 2d) for attachment to amine groups require initial conversion to the NHS esters, which can only be stored for short periods of time at low temperature. In contrast, the squarate ethyl ester functionality of the novel [Ir(C^N)2(pt-TOxT-Sq)]+ ECL labels (Fig. 2f) does not require further activation, and the labels can be stored at room temperature for extended periods of time. The emission spectra and electrochemical properties of the [Ir(C^N)2(pt-TOxT-Sq)]+ and commercial [Ir(C^N)2(mbpy-COOH)]+ labels (Table 2 and Fig. S9†) were closely aligned to those of their parent luminophores, [Ir(C^N)2(pt-TEG)]+ (Table 1) and [Ir(C^N)2(dm-bpy)]+ (Table S1†), respectively. The photoluminescence quantum yields (ϕPL) and emission lifetimes (τ) for the [Ir(C^N)2(pt-TOxT-Sq)]+ labels were greater than those of their commercial analogues (Table 2).| ECL labela | Photoluminescence | Electrochemical potentials | |||
|---|---|---|---|---|---|
| λ max , /nm | ϕ PL , (%) | τ , /ns | E ox /V (vs. Fc+/0) | E red /V (vs. Fc+/0) | |
| a The chemical structure of [Ru(bpy)2(mbpy-COOH)]2+ is shown in Fig. 1c; the [Ir(C^N)2(mbpy-COOH)]+ labels are depicted by Fig. 2d, except that the piq complex does not contain the 4-methyl group, and the bt complex contains a 4′-carboxy instead of 4′-carboxypropyl group on the bpy-based ligand (due to the availability of the different commercial labels at the time of the study); and the [Ir(C^N)2(pt-TOxT-Sq)]+ labels are depicted by Fig. 2f. b The ECL labels were dissolved in DMF (1 mM) and diluted to 10 μM in a 0.1 M PBS solution (pH 7.4). c Corrected for the change in instrument sensitivity over the wavelength range. d Photoluminescence quantum yield. e Emission lifetime; excitation wavelength at 344 nm or 451 nm using Nanoled light sources. f ECL labels at 0.25 mM in acetonitrile with 0.1 M TBAPF6; cyclic voltammetry; scan rate: 0.1 V s−1. Peaks associated with the counter ion or labelling functional group not listed. | |||||
| [Ru(bpy)2(mbpy-COOH)]2+ | 632 | 4.9 | 311 | 0.84 | −1.78, −1.96, −2.24 |
| [Ir(piq)2(bpy-COOH)]+ | 592(sh), 640 | 10.4 | 378 | 0.85 | −1.90, −2.17 |
| [Ir(bt)2(mbpy-COOH)]+ | 530(sh), 572, 615(sh) | 5.9 | 174 | 1.04 | −1.87, −2.28 |
| [Ir(ppy)2(mbpy-COOH)]+ | 606 | 2.0 | 398 | 0.84 | −1.87 |
| [Ir(df-ppy)2(mbpy-COOH)]+ | 533 | 2.7 | 392 | 1.16 | −1.84, −2.49 |
| [Ir(piq)2(pt-TOxT-Sq)]+ | 590(sh), 632 | 11.8 | 927 | 0.87 | −2.01, −2.22, −2.56 |
| [Ir(bt)2(pt-TOxT-Sq)]+ | 526, 564 | 26.7 | 1051 | 1.01 | −2.11, −2.35, −2.56 |
| [Ir(ppy)2(pt-TOxT-Sq)]+ | 476, 503 | 8.8 | 308 | 0.86 | −2.22 |
| [Ir(df-ppy)2(pt-TOxT-Sq)]+ | 453, 483, 515 | 15.7 | 559 | 1.16 | −2.14 |
As a proof-of-concept demonstration of the influence of the electrochemical and spectroscopic properties on the ECL of Ir(III) complexes in the context of the reaction pathways available to each luminophore, we compared the relative ECL intensities of the bioconjugated [Ir(C^N)2(pt-TOxT-Sq)]+ and [Ir(C^N)2(mbpy-COOH)]+ labels in two different modes of binding assay (Fig. 6). The first was a sandwich hybridisation RNA assay on magnetic bead support, and the second was a C-reactive protein (CRP) sandwich immunoassay with the capture monoclonal antibody (mAb) immobilised on a gold electrode.
 | ||
| Fig. 6 Schematic representation of (a) the RNA assay on a magnetic-bead support held at a glassy carbon working electrode, and (b) the sandwich immunoassay for CRP where the capture antibody was immobilised on a gold electrode. | ||
In the sandwich RNA hybridisation assay, the target was mixed with capture probe-functionalised magnetic beads (2.8 μm diameter, streptavidin coated) and a detection probe with ECL label attached, and heated to 45 °C for 15 min. The beads were then washed, resuspended in ProCell solution (a phosphate buffer enriched with TPrA and various surfactants with a confidential specific chemical composition, which was specially designed and optimised for ECL assays in commercial systems), and dispersed above a screen-printed electrode in a custom-made holder (Fig. S10†) containing a magnet to move the beads to the electrode surface. The ECL was initiated by applying 1.4 V vs. Ag|AgCl at the working electrode for 10 s and measured using a silicon photomultiplier detector (Fig. S11†).
Bard and co-workers' elucidation of an alternative ECL reaction pathway of the [Ru(bpy)3]2+ luminophore with TPrA co-reactant5 revealed Scheme 1b as the dominant light-producing pathway of the magnetic bead-supported assays used in commercial ECL instruments. In these assays, only an infinitesimal fraction of ECL-labels are held within the nanometric electron tunnelling distance from the electrode surface required for their direct oxidation (required for Scheme 1a).5,7,54 Diffusion of the TPrA radicals, however, allows excitation via homogeneous electron transfer to the [Ru(bpy)3]2+ luminophores (Scheme 1b) at much greater (micrometric) distances from the electrode surface.
Based on the above considerations of the parent luminophores of the ECL labels (Fig. 5), the reaction pathway depicted in Scheme 1b should only be feasible for the novel red and yellow emitters ([Ir(piq)2(pt-TOxT-Sq)]+ and [Ir(bt)2(pt-TOxT-Sq)]+) and the commercial red emitter ([Ir(piq)2(bpy-COOH)]+).
As shown in Fig. 7a, comparison of the ECL signal-to-blank (S/B) ratios for the different labels for the same target RNA concentration and assay conditions shows that these labels gave the greatest response. The commercial bt analogue, for which the parent luminophore was considered a borderline case (Fig. 5b), also showed a minor response. The two [Ir(C^N)2(pt-TOxT-Sq)]+ labels gave superior S/B ratios than their commercial counterparts, but the response with [Ir(piq)2(pt-TOxT-Sq)]+ was still 3-fold poorer than that of [Ru(bpy)2(mbpy-COOH)]2+.
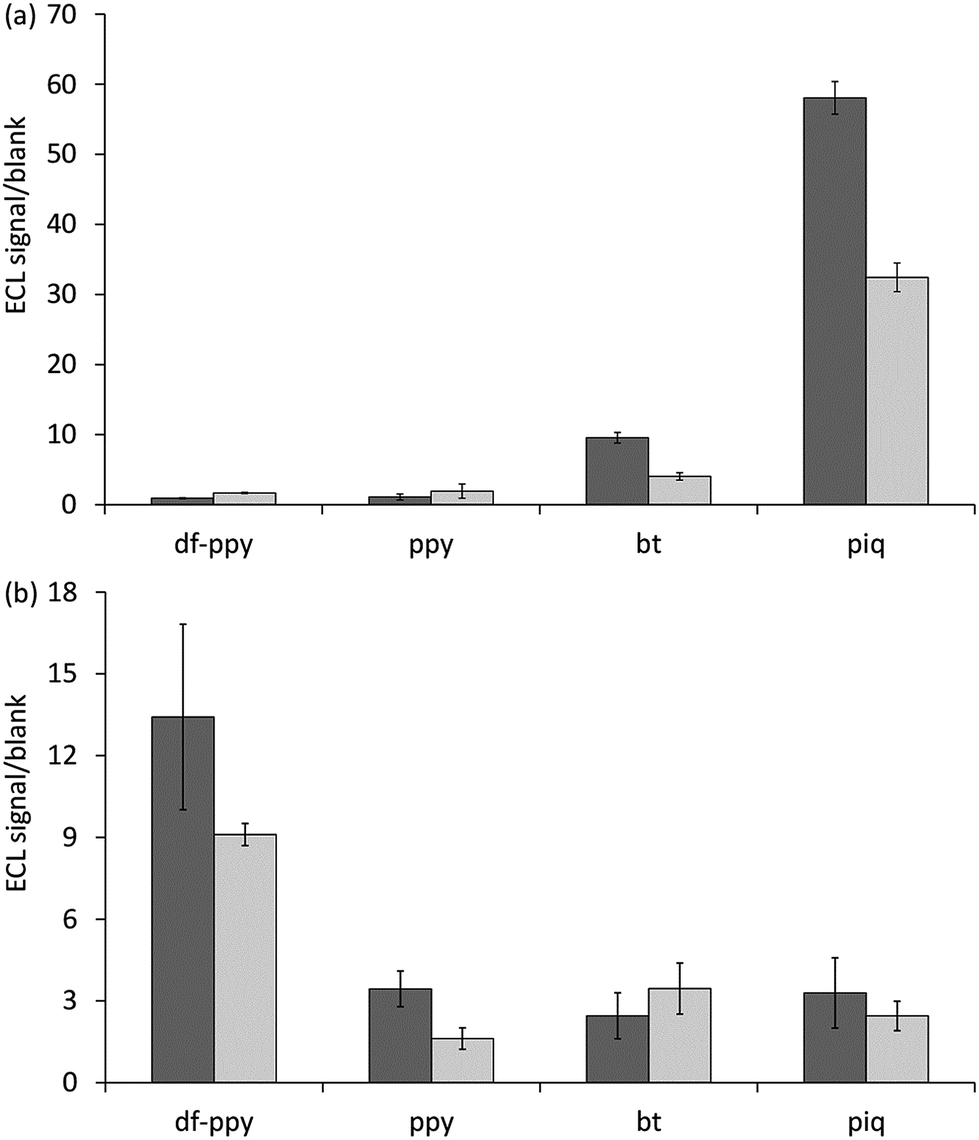 | ||
| Fig. 7 ECL signal/blank ratio for (a) the detection of target RNA by sandwich hybridisation assay on a magnetic bead support, and (b) the detection of C-reactive protein by sandwich immunoassay with the capture monoclonal antibody immobilised on a gold electrode, using [Ir(C^N)2(pt-TOxT-Sq)]+ (black columns) or [Ir(C^N)2(mbpy-COOH)]+ (grey columns) ECL labels. | ||
For the [Ir(piq)2(pt-TOxT-Sq)]+, [Ir(bt)2(pt-TOxT-Sq)]+, [Ir(piq)2(bpy-COOH)]+ and [Ru(bpy)2(mbpy-COOH)]2+ labels, the reactions depicted in Scheme 1b are exergonic, but considerable variation in ECL responses were observed in the RNA assay due to differences in the efficiencies of (i) excitation (dependent on the relative rate of reactions leading to the excited state in addition to various ‘dark’ reactions), and (ii) emission (which can be estimated from the photoluminescence quantum yield, although the excited state may also be vulnerable to quenching from reactive oxidation products of the co-reactant in some cases13).
As the quantum yield of the labels decreased in the order: [Ir(bt)2(pt-TOxT-Sq)]+ > [Ir(piq)2(pt-TOxT-Sq)]+ > [Ir(piq)2(bpy-COOH)]+ > [Ru(bpy)2(mbpy-COOH)]2+ (Table 2), the excitation efficiency appears to be the dominant factor. It is therefore not surprising that most Ir(III) complexes reported to exhibit the greatest ECL intensities to date have exhibited red luminescence and reduction potentials that would place them in the same regions of Fig. 5 as the [Ru(bpy)3]2+ complex, even though they were not evaluated under bead-based assay conditions that would limit the ECL pathway to that depicted in Scheme 1b. Examples include Ir(pq)2(acac) (λem = 609 nm, E0′ = 0.57 V and −2.05 V vs. Fc+/0),40 Ir(pph)2(pic) (λem = 649 nm, E0′ = 0.61 V and −1.94 V vs. Fc+/0),16 and [Ir(dmpq)2(mbpy-COOH)]+ (λem = 590 nm, E0′ = 0.78 V and −1.66 V vs. Fc+/0),19 where dmpq = 2-(3,5-dimethylphenyl)quinoline.
For assays in which the Ru(II)/Ir(III) complex luminophore can diffuse to the electrode or is immobilised in very close proximity to the electrode surface, co-reactant ECL with TPrA is feasible viaScheme 1a (with possible involvement of Scheme S1a†), but only if a sufficient potential is applied to oxidise the metal complex, and the reaction between the oxidised complex and TPrA˙ is sufficiently exergonic to populate the excited state responsible for emission (reaction (2)). As illustrated by Fig. 5a–d, all metal complexes examined in this study meet this requirement. The potentials required to oxidise these metal complexes are generally greater than that for TPrA, and so the pathway depicted in Scheme 1b (and Scheme S1b†) may also contribute to the overall ECL intensity for complexes meeting its requirements (described above).
In our second assay, the capture antibody was covalently immobilised on a monolayer of 16-mercaptohexadecanoic acid on a gold electrode, which was incubated with the target CRP and then the detection antibody. The electrode was then introduced to an electrochemical cell containing ProCell solution and the ECL was initiated using a voltammetric sweep (0.05 V s−1) from +0.5 V to +1.5/1.6 V (vs. Ag|AgCl) and measured using a photomultiplier tube (S20 multi-alkali photocathode) module. The ECL labels in this assay are much closer to the electrode than the vast majority of those in the bead-based approach, but still outside the typical electron tunnelling region.5,54 O'Reilly et al.,55 however, showed that the dominant ECL mechanism of a similar assay (CRP by sandwich immunoassay; capture antibody absorbed on a Pt electrode; detection antibody with an [Ru(bpy)2(N^N)]2+-type ECL label) involved oxidation of the label (Scheme 1a) and the catalytic oxidation of TPrA (Scheme S1a†). As noted by the authors,55 electron hopping between ECL luminophores49,54 is unlikely in this system due to their relatively low concentration, but multi-step electron transfer through proteins via their redox active side chains is well known.56
As shown in Fig. 7b, comparison of the ECL signal-to-blank (S/B) ratios for the different labels under the same CRP assay conditions shows that the two blue emitter labels ([Ir(df-ppy)2(pt-TOxT-Sq)]+ and [Ir(df-ppy)2(mbpy-COOH)]+), which both require oxidation to generate ECL, gave the greatest response. Moreover, the S/B ratio for the novel [Ir(df-ppy)2(pt-TOxT-Sq)]+ ECL label was only 15% lower than that of [Ru(bpy)2(mbpy-COOH)]2+.
Experimental
Chemicals
Syntheses of the pt-TEG and pt-TOxT-Sq ligands, the [Ir(C^N)2(dm-bpy)](PF6), [Ir(C^N)2(ptb)](PF6) and [Ir(C^N)2(pt-TEG)]Cl complexes, and the [Ir(C^N)2(pt-TOxT-Sq)]+ ECL labels (where C^N = piq, bt, ppy, or df-ppy) are described in the ESI.† The solubility of the [Ir(C^N)2(pt-TEG)]+ complexes was approximately 1 mM [Ir(bt)2(pt-TEG)]+ and [Ir(ppy)2(pt-TEG)]+ and 0.5 mM [Ir(df-ppy)2(pt-TEG)]+ in water, and 0.1 mM [Ir(piq)2(pt-TEG)]+ in water with 10% acetonitrile. Stock [Ir(C^N)2(pt-TEG)]+ solutions were subsequently prepared at 0.1 mM. Acetonitrile (Scharlau, Spain) was distilled over calcium hydride under a nitrogen atmosphere and collected as needed. All other chemicals were obtained from commercial sources and used as received.¶Photophysical measurements
For the characterisation of Ir(C^N)2(acac), [Ir(C^N)2(dm-bpy)](PF6), [Ir(C^N)2(ptb)](PF6) and [Ir(C^N)2(pt-TEG)]Cl complexes, absorption spectra were obtained with a Cary 300 Bio UV/Vis spectrophotometer (Varian Australia, Vic., Australia) with 1 cm pathlength quartz cuvettes. Emission spectra were measured on a Cary Eclipse fluorescence spectrometer (Varian Australia; 5 nm band pass, 1 nm data interval, PMT voltage: 800 V). Metal complexes were prepared at a concentration of 10 μM in deionised water or freshly distilled acetonitrile. For the low temperature emission spectra, the complexes were prepared at 5 μM in ethanol:methanol (4:1) and cooled to 85 K using an OptistatDN Variable Temperature Liquid Nitrogen Cryostat equipped with custom-made quartz sample holder. The low temperature spectra were collected at 85 K to avoid damage to the spectroscopic cuvettes near 77 K observed during our previous study17 and by others.57 No significant difference in the λmax for metal complexes such as [Ru(bpy)3]2+ and Ir(ppy)3 between 77 K and 85 K was observed under these instrumental conditions.30 All emission spectra were corrected by standard correction curves established using a quartz halogen tungsten lamp.
For the characterisation of [Ir(C^N)2(pt-TOxT-Sq)]+ and other ECL labels, the complexes were dissolved in DMF (1 mM) and diluted to 10 μM in a 0.1 M PBS solution (pH = 7.4). Steady-state emission spectra were collected on a Nanolog (HORIBA Jobin Yvon IBH) spectrofluorometer. A 450 W xenon-arc lamp was used to excite the complexes using a 1200 g mm−1 grating blazed at 330 nm excitation monochromators, a 1200 g mm−1 grating blazed at a 500 nm emission monochromator, and a thermoelectrically cooled TBX picosecond single-photon detector. Emission and excitation spectra were corrected for source intensity, gratings, and detector response. Lifetimes were measured using the time correlated single photon counting (TCSPC) option on the spectrometer and correlated by a time-to-amplitude converter (TAC) in forward TAC mode. A Nanoled laser (λex = 344 nm or 451 nm) was pulsed at 100 kHz, signals were collected using a FluoroHub counter and the data was analysed using DAS6 software (HORIBA Jobin Yvon IBH). Spectra for absolute quantum yields were measured at room temperature (22 ± 2 °C) with a Quanta-phi HORIBA Scientific 6 in. diameter integrating sphere connected to the Nanolog via optical fibres. The complexes were excited using a 450 W xenon lamp and detected with a liquid nitrogen cooled Symphony II (Model SII-1LS-256−06) CCD.
Electrochemistry
An Autolab PGSTAT204 potentiostat (Metrohm Autolab B.V., Netherlands) with a conventional three-electrode system housed in a custom-made light-tight Faraday cage was used. The electrochemical cell contained a glassy carbon working electrode (3 mm diameter), platinum counter electrode, and a low leakage Ag|AgCl (3.4 M) (Innovative Instruments, Fl, USA) or silver wire reference electrode. Cyclic voltammograms were performed at a scan rate of 0.1 V s−1 and metal complex concentration of 0.25 mM with a supporting electrolyte of 0.1 M TBAPF6 in freshly distilled acetonitrile. The glassy carbon electrode was polished using 0.3 and 0.05 μm alumina powder, sonicated in water and then distilled acetonitrile, and dried before use. Prior to analysis, solutions were deoxygenated for 15 min (using Argon). Potentials were referenced to the ferrocenium/ferrocene redox couple. All electrochemical experiments were carried out at room temperature. Square wave voltammetric measurements were performed using 0.5 mM metal complex in 0.1 M phosphate buffer (pH 7.5), with 5 mV step, 25 Hz frequency, 0.02 V amplitude and 100 mV s−1 scan rate.Electrogenerated chemiluminescence
ECL experiments were performed using the same electrochemical cell configuration as described above, and the light emitted near the working electrode surface was detected using a photomultiplier tube (PMT; Electron Tubes model 9124B; ETP, NSW, Australia) positioned under the cell, or a CCD spectrometer (QE65Pro, Ocean Optics, FL, USA) interfaced with the cell using a collimating lens (74-UV, Ocean Optics) and optic fibre (1.0 m, 1.0 mm core diameter; Ocean Optics). Acquisition was synchronised with the electrochemical experiment by sending a trigger from the potentiostat to the HR4000 (Ocean Optics) break out box. Comparisons of co-reactant ECL intensities (integrated peak area) between Ir(III) complexes and [Ru(bpy)3]2+ were performed using chronoamperometry (potential of Epa + 0.1 V, 10 s pulse time, 10 Hz), with the CCD spectrometer, and metal complex concentration of 10 μM. The ‘two-wave’ ECL experiments involved a voltammetric sweep from 0 V to 1.8 V vs. Ag|AgCl followed by the reverse sweep back to 0 V, with the resulting ECL measured by PMT. The ECL detection for each assay is described in later sections.Computational methods
DFT calculations were carried out within the Gaussian 09 suite of programs.58 Ground state geometries were optimised in the absence of solvent with the mPW1PW91 functional59 in conjunction with the def2-SVP basis set and associated effective core potential.60 Stationary points were characterised as minima by calculating the Hessian matrix analytically at the same level of theory. All structures are minima with no imaginary frequencies. Molecular orbital (MO) analysis was carried out with the def2-TZVP basis set and core potential60 together with the BP86 functional,61 with solvent effects included for all single-point energy calculations for consistency with the experimental system (water for [Ir(C^N)2(pt-TEG)]+ complexes, and acetonitrile for all other Ir(III) complexes). The polarisable continuum model (PCM)62 self-consistent reaction field (SCRF) was used together with Truhlar's SMD solvent model.63 TD-DFT calculations were performed at the CAM-B3LYP/def2-SVP level of theory. MO analysis was carried out with the QMForge program.64Conversion of carboxylic acid ECL labels to N-hydroxy-succinimide (NHS) esters
N,N′-Dicyclohexylcarbodiimide (DCC, 146 mmol; Sigma Aldrich, >99%) and NHS (146 mmol; Sigma Aldrich, 98%) were dissolved in 1.5 mL chilled (water ice-bath), dried (molecular sieves) DMF (Sigma Aldrich, molecular biology grade) with stirring. To this solution, 28 mmol of [Ru(bpy)2(mbpy-COOH)]2+ (Fig. 1c) or [Ir(C^N)2(mbpy-COOH)]+ (Fig. 2d) dissolved in 0.5 mL dry, chilled DMF was added. The mixture was stirred on ice for 30 min, before returning to room temperature (22 °C) and stirring was continued for 5 h. The reaction mixture was then chilled (−18 °C), and the solids were removed by centrifugation. The [Ru(bpy)2(mbpy-NHS)]2+ and [Ir(C^N)2(mbpy-NHS)]+ solutions were stored at −20 °C in a desiccator. The [Ir(C^N)2(pt-TOxT-Sq)]+ labels (Fig. 2e) did not require this step.Assay 1: sandwich hybridisation RNA assay on magnetic bead support
Attachment of the capture probe to the magnetic beads
40 μL (400 μg) of Dynabeads M-280 Streptavidin (MB) were washed with binding buffer (20 mM Tris HCl, pH 8, 0.5 M NaCl) by vortexing and magnetic separation, then resuspended in 100 μL of binding buffer at 4 mg mL−1, the capture probe (CP) was bound to the MB by adding 20 μL of CP solution (10 μM) to the bead solution and incubating for 20 min at room temperature with gentle mixing. Excess CP was removed by washing the beads three times in binding buffer, followed by resuspension in 200 μL binding buffer (2 mg mL−1). The CP@MB solution was stored at 4 °C and was stable for several weeks.Conjugation of ECL labels with the detection probe
The detection probe (as purchased) was resuspended to a concentration of 500 μM in 100 mM sodium borate buffer (pH 8). 100 μL (50 nmol) of this solution was combined with 1000 nmol of the complex (20-fold excess, 80 μL at 12.5 mM) [Ru(bpy)2(mbpy-NHS)]2+ or [Ir(C^N)2(mbpy-NHS)]+ in DMF and 620 μL 100 mM borate buffer. The solution was shielded from ambient light with aluminium foil and reacted at room temperature for 24 h on a rotating mixer. The labelled oligo was washed and purified as described by Zhou et al.,34 before being resuspended in nuclease free water at 20 μM. The concentration of the labelled oligo was measured using a Nanodrop 2000 UV-Vis spectrophotometer and the purity was checked by RP-HPLC. When necessary, the labelled oligo was further purified by collecting the appropriate fraction eluted from the column and precipitating the labelled oligo by solvent evaporation and salt precipitation before re-suspension. The [Ir(C^N)2(pt-TOxT-Sq)]+ labels were attached to the oligo and purified in a similar manner, except the complex concentration was 5 mM in acetonitrile, and 700 μL additional borate buffer was added. If the solution turned cloudy on the combination of the complex and oligo or borate buffer, acetonitrile (<100 μL) was added dropwise until the solution turned clear. The analytical and semi-preparative HPLC was performed using an Agilent Technologies 1260 Infinity LC system (CA, USA) with a Phenomenex Luna 5μ C18(2) 100 Å column (150 × 4.6 mm) (CA, USA). The mobile phase was a solvent gradient using solvent A (0.1% ammonium acetate in deionised water) and solvent B (acetonitrile) at a total flow rate of 1 mL min−1. The oligo labelled with the Ru complex was examined and purified using a gradient of 5% → 100% solvent B over 40 min. The oligos labelled with Ir(III) complexes were examined and purified using a gradient of 5% → 100% solvent B over 20 min. For semi-preparative RP-HPLC, samples were made up to approximately 20 μM in acetonitrile and loaded onto the column at a maximum injection volume of 100 μL.Assay procedure
7 μL of CP@MB (2 mg mL−1), 1 μL of detection probe (20 μM, 20 pmol) and differing amounts of target RNA solution were mixed in a PCR tube and made up to 20 μL final volume using binding buffer. The sample was heated to 45 °C for 15 min to allow RNA hybridisation to occur. The beads were then washed two times using binding buffer with 5% T-20 and 0.1% T-100 detergent, removing excess detection probe. The beads were then resuspended in ProCell solution (Roche Diagnostics Australia), before dispersing above the working electrode of a Zensor screen printed electrode, mounted in a custom-made holder (Fig. S10†) pre-filled with 80 μL ProCell solution. The magnet (3× 4 mm diameter rod shaped N42 rare earth; Aussie Magnets, Australia) positioned behind the electrode ensured the beads were rapidly collected at the surface of the working electrode for analysis. The ECL was measured using a 3 × 3 mm silicon photomultiplier detector (SiPM; ASD-RGB3S-P; AdvanSiD, Italy) interfaced with an ASD-EP-EB-N amplifier board (AdvanSiD; Fig. S11†). The SPE holder and photodetector were housed in a light-tight Faraday cage. An Autolab PGSTAT 101 (Metrohm Autolab B.V.) potentiostat with NOVA software was used to apply a single-step chronoamperometry experiment (1.4 V vs. Ag|AgCl for 10 s) and record the electrochemical signals. Data from the SiPM was recorded and processed using an eDAQ401 (eDAQ, Australia) data recording unit using the supplied eDAQ Chart software.Assay 2: C-reactive protein sandwich immunoassay on a gold electrode
Conjugation of ECL labels with the detection antibody
The Ir(III) complex was dissolved in DMF (0.01 M) and 10 μL was added to an Eppendorf tube containing 1 mL 0.1 M PBS solution (pH = 7.4) of the monoclonal antibody mAbTJ330 (100 μg mL−1) and slowly stirred at 4 °C for 4 h. The reaction mixture was purified by different cycles of centrifugation using an ultra-centrifuge tube with a cut-off of 30 kDa.Assay procedure
The Au/MHA electrodes with immobilised capture antibody were incubated with 10 ng mL−1 CRP solution for 1 h at 37 °C. The functionalised electrodes were then immersed in a PBS solution containing the soluble labelled detection antibody (100 μg mL−1 stock) for 1 h at 37 °C and again washed. A custom system was used for ECL characterisation, consisting in an electrochemical cell based on modified gold-disk working electrode shrouded in Teflon (CH Instruments, Austin, TX, USA, 3 mm diameter), which were closely facing (a few millimetres) the photomultiplier tube (PMT) module (Sens-Tech model P30A-05, ETP, NSW, Australia). The PMT signal was amplified by TA-GI-74 Ames Photonics Inc. amplifier (Model D7280) and controlled by a CHI660C Electrochemical Workstation (CH Instruments, Inc., Austin, TX, USA). The reference electrode employed was an Ag|AgCl (1 M KCl) from CHI-Instruments and was separated from the catholyte by a glass frit. A platinum wire served as the counter electrode. ProCell solution (Roche Diagnostics Australia) was used as the aqueous solvent and source of TPrA. The solutions were scanned at 0.05 V s−1 from +0.5 V to +1.5/+1.6 V (according to the oxidation potential of the different complexes).Conclusions
This exploration of spectroscopic, electrochemical and ECL properties of analogous series of Ir(C^N)2(acac), [Ir(C^N)2(dm-bpy)]+, [Ir(C^N)2(ptb)]+, [Ir(C^N)2(pt-TEG)]+, [Ir(C^N)2(mbpy-COOH)]+, and [Ir(C^N)2(pt-TOxT-Sq)]+ complexes has provided a new understanding of the translation of promising Ir(III) complexes to ECL labelling, whilst at the same time introducing a new class of ECL label. As the possibility of multi-coloured or potential-resolved ECL systems has been the main driver for the exploration of Ir(III) ECL systems, we selected a group of common C^N ligands (df-ppy, ppy, bt and piq) that would impart a wide range of properties. This highlighted the effects of modifying the ancillary ligand for ECL labelling purposes, the availability of ECL reaction pathways, and the implications on the performance of ECL labels in different assay formats.Adaption of promising Ir(C^N)2(acac) complexes for ECL labelling through the common approach of replacing the ancillary ligand with 2,2′-bipyridine derivatives introduces a low-lying π* LUMO that contracts the spread of emission colours over a series of complexes, which will be detrimental for multi-colour applications. The change is most prominent in complexes with the furthest negative reduction potentials (i.e., the highest LUMO energies). Of the Ir(C^N)2(acac) complexes examined in this study, this was the ppy complex, and the change in ancillary ligand to dm-bpy visibly switched the order of emission energies with the Ir(bt)2(acac) complex. These effects can be largely ameliorated with alternative ancillary ligands such as pyridyltriazole ligands, which are prepared by simple click chemistry procedures to access more water-soluble analogues and functionality suitable for labelling, without significant modification to the properties of the luminophore.
Graphical representations of the key energy requirements of the competing ECL reactions elucidated for the [Ru(bpy)3]2+ complex with TPrA as a co-reactant show fundamental limits on the redox potentials and emission wavelengths of complexes that can generate ECL through a mechanism involving oxidation of only the TPrA co-reactant (Scheme 1b). Most importantly in the context of developing multi-colour ECL systems, the ‘window’ of reduction potentials enabling this pathway becomes narrower as emission energy increases, and it does not extend across the entire visible region. Plotting the redox potentials and (low-temperature) emissions of the Ir(III) complexes on these graphs enables simple prediction of the feasible ECL pathways, which for the [Ir(C^N)2(pt-TEG)]+ complexes was supported by the relative ECL intensity at different applied potentials under aqueous conditions relevant to binding assays.
The practical outcome of this limitation is seen in the bead-based assay, in which only some of the Ir(III) complexes (those for which Scheme 1b is feasible) result in a significant ECL signal for the target analyte. This provides the simplest experimental verification of Bard and co-workers' reasoning5 (supported by a range of experiments and simulations by other groups7,54,67) of the dominant ECL reaction pathway for bead-based assays using [Ru(bpy)3]2+ with TPrA as co-reactant. Examination of a greater range of electrochemiluminophores with this approach will enable the energy boundaries (carrying considerable error due to the difficulty in establishing the redox potentials of TPrA and TPrA˙) to be clarified. Moreover, this approach will be valuable for the evaluation of alternative co-reactants in conjunction with various metal complexes (which may provide an analogous pathway to Scheme 1b for ECL in the blue region of spectrum), as previous studies68 have predominantly focused on [Ru(bpy)3]2+, and not under bioconjugated assay conditions. Assays in which the metal complex can diffuse to the electrode surface, or is immobilised within a few nanometres of the electrode, or where electrons can be transferred over greater distances from the metal complex to the electrode, are not subject to the above limitations. In these cases, the effectiveness of the label is defined only by the efficiency of its excitation and emission under the specific assay conditions, and the relative sensitivity of the photodetector towards that luminophore.
The novel [Ir(C^N)2(pt-TOxT-Sq)]+ ECL labels were obtained through a more convenient and versatile synthetic approach and provided superior ECL responses to the commercial [Ir(C^N)2(mbpy-COOH)]+ analogues in both assays. In both assays, the ECL was less intense than that of the conventional [Ru(bpy)2(mbpy-COOH)]2+ label, but assay conditions have been optimised specifically for the [Ru(bpy)3]2+-based labels since the inception of the technique, and there is considerable scope to improve the relative performance of the Ir(III) complex labels, in areas such as TPrA concentration and electrochemical pulse time.17 Despite a few alternative co-reactants providing greater ECL intensities with [Ru(bpy)3]2+ under specific conditions, TPrA remains the ‘gold standard’ co-reactant for ECL assays. Other co-reactants, however, may provide superior ECL performance from various Ir(III) complexes. Finally, the ability through DFT calculations to predict the influence of changes in ligand structure on the redox and luminescence character of the complex, and consider these changes with respect to the energy requirements of various ECL pathways, will enable the design of superior Ir(III) complex ECL labels with specific emission colours and targeting different reaction pathways.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Australian Research Council (DP160103046). LCH thanks the ARC Training Centre for Lightweight Automotive Structures (IC160100032) and the ARC Research Hub for Future Fibres (IH140100018) funded by the Australian Government. LC was supported by a Deakin University International Postgraduate Scholarship. We acknowledge generous allocations of computing from La Trobe University, Intersect, and NCI. We thank Linxi Shi (Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, China) for assistance in obtaining the quantum yields of the [Ir(C^N)2(pt-TOxT-Sq)]+ complexes, and Tien T. Pham (Deakin University, Australia) for assistance in collecting low-temperature emission spectra.Notes and references
- L. Li, Y. Chen and J.-J. Zhu, Anal. Chem., 2017, 89, 358–371 CrossRef CAS PubMed; Y. N. Khonsari and S. Sun, Chem. Commun., 2017, 53, 9042–9054 RSC; K. Muzyka, Biosens. Bioelectron., 2014, 54, 393–407 CrossRef PubMed; L. Hu and G. Xu, Chem. Soc. Rev., 2010, 39, 3275–3304 RSC; W. Miao, Chem. Rev., 2008, 108, 2506–2553 CrossRef PubMed.
- Z. Liu, W. Qi and G. Xu, Chem. Soc. Rev., 2015, 44, 3117–3142 RSC.
- Roche Diagnostics, Products and Solutions, 2017, http://www.cobas.com/content/dam/cobas_com/pdf/lists/products-and-solutions.pdf, Meso Scale Diagnostics, https://www.mesoscale.com/en/technical_resources/our_technology/multi-array.
- I. Rubinstein and A. J. Bard, J. Am. Chem. Soc., 1981, 103, 512–516 CrossRef CAS; J. K. Leland and M. J. Powell, J. Electrochem. Soc., 1990, 137, 3127–3131 CrossRef.
- W. Miao, J.-P. Choi and A. J. Bard, J. Am. Chem. Soc., 2002, 124, 14478–14485 CrossRef CAS PubMed.
- K. Komori, K. Takada, O. Hatozaki and N. Oyama, Langmuir, 2007, 23, 6446–6452 CrossRef CAS PubMed.
- M. Sentic, M. Milutinovic, F. Kanoufi, D. Manojlovic, S. Arbault and N. Sojic, Chem. Sci., 2014, 5, 2568–2572 RSC.
- G. J. Barbante, C. F. Hogan, D. J. D. Wilson, N. A. Lewcenko, F. M. Pfeffer, N. W. Barnett and P. S. Francis, Analyst, 2011, 136, 1329–1338 RSC.
- A. Kapturkiewicz, Anal. Bioanal. Chem., 2016, 408, 7013–7033 CrossRef CAS PubMed.
- S. Laird and C. F. Hogan, in Iridium(III) in Optoelectronic and Photonics Applications, ed. E. Zysman-Colman, John Wiley & Sons, Inc., Chichester, UK, 2017, pp. 359–414 Search PubMed.
- M. S. Lowry and S. Bernhard, Chem.–Eur. J., 2006, 12, 7970–7977 CrossRef CAS PubMed; S. Ladouceur and E. Zysman-Colman, Eur. J. Inorg. Chem., 2013, 2985–3007 CrossRef; C. E. Housecroft and E. C. Constable, Coord. Chem. Rev., 2017, 350, 155–177 CrossRef.
- D. Bruce and M. M. Richter, Anal. Chem., 2002, 74, 1340–1342 CrossRef CAS PubMed; B. D. Muegge and M. M. Richter, Anal. Chem., 2004, 76, 73–77 CrossRef; E. H. Doeven, E. M. Zammit, G. J. Barbante, C. F. Hogan, N. W. Barnett and P. S. Francis, Angew. Chem., Int. Ed., 2012, 51, 4354–4357 CrossRef PubMed; E. H. Doeven, G. J. Barbante, C. F. Hogan and P. S. Francis, ChemPlusChem, 2015, 80, 456–470 CrossRef; H. Li, L. Bouffier, S. Arbault, A. Kuhn, C. F. Hogan and N. Sojic, Electrochem. Commun., 2017, 77, 10–13 CrossRef; Y.-Z. Wang, C.-H. Xu, W. Zhao, Q.-Y. Guan, H.-Y. Chen and J.-J. Xu, Anal. Chem., 2017, 89, 8050–8056 CrossRef PubMed; Y.-Z. Wang, S.-Y. Ji, H.-Y. Xu, W. Zhao, J.-J. Xu and H.-Y. Chen, Anal. Chem., 2018, 90, 3570–3575 CrossRef PubMed; W. Guo, H. Ding, C. Gu, Y. Liu, X. Jiang, B. Su and Y. Shao, J. Am. Chem. Soc., 2018, 140, 15904–15915 CrossRef PubMed.
- E. H. Doeven, E. M. Zammit, G. J. Barbante, P. S. Francis, N. W. Barnett and C. F. Hogan, Chem. Sci., 2013, 4, 977–982 RSC.
- E. H. Doeven, G. J. Barbante, E. Kerr, C. F. Hogan, J. A. Endler and P. S. Francis, Anal. Chem., 2014, 86, 2727–2732 CrossRef CAS PubMed.
- H. Gao, Q. Dang, S. Xia, Y. Zhao, H. Qi, Q. Gao and C. Zhang, Sens. Actuators, B, 2017, 253, 69–76 CrossRef CAS.
- J. M. Fernandez-Hernandez, E. Longhi, R. Cysewski, F. Polo, H.-P. Josel and L. De Cola, Anal. Chem., 2016, 88, 4174–4178 CrossRef CAS PubMed.
- L. Chen, E. H. Doeven, D. J. D. Wilson, E. Kerr, D. J. Hayne, C. F. Hogan, W. Yang, T. T. Pham and P. S. Francis, ChemElectroChem, 2017, 4, 1797–1808 CrossRef CAS.
- C. Li, J. Lin, Y. Guo and S. Zhang, Chem. Commun., 2011, 47, 4442–4444 RSC; Y. Zhao, Y. Luo, T. Li and Q. Song, RSC Adv., 2014, 4, 57709–57714 RSC.
- Y. Zhou, K. Xie, R. Leng, L. Kong, C. Liu, Q. Zhang and X. Wang, Dalton Trans., 2017, 46, 355–363 RSC.
- E. Kerr, E. H. Doeven, D. J. D. Wilson, C. F. Hogan and P. S. Francis, Analyst, 2016, 141, 62–69 RSC.
- G. Valenti, A. Fiorani, H. Li, N. Sojic and F. Paolucci, ChemElectroChem, 2016, 3, 1990–1997 CrossRef CAS.
- S. Zanarini, M. Felici, G. Valenti, M. Marcaccio, L. Prodi, S. Bonacchi, P. Contreras-Carballada, R. M. Williams, M. C. Feiters, R. J. M. Nolte, L. De Cola and F. Paolucci, Chem.–Eur. J., 2011, 17, 4640–4647 CrossRef CAS PubMed.
- G. J. Barbante, E. H. Doeven, E. Kerr, T. U. Connell, P. S. Donnelly, J. M. White, T. Lópes, S. Laird, C. F. Hogan, D. J. D. Wilson, P. J. Barnard and P. S. Francis, Chem.–Eur. J., 2014, 20, 3322–3332 CrossRef CAS PubMed.
- E. Kerr, E. H. Doeven, G. J. Barbante, T. U. Connell, P. S. Donnelly, D. J. D. Wilson, T. D. Ashton, F. M. Pfeffer and P. S. Francis, Chem.–Eur. J., 2015, 21, 14987–14995 CrossRef CAS PubMed.
- B. Beyer, C. Ulbricht, D. Escudero, C. Friebe, A. Winter, L. Gonzalez and U. S. Schubert, Organometallics, 2009, 28, 5478–5488 CrossRef CAS; P. A. Scattergood, A. Sinopoli and P. I. P. Elliott, Coord. Chem. Rev., 2017, 350, 136–154 CrossRef; T. Hosseinnejad, F. Ebrahimpour-Malmir and B. Fattahi, RSC Adv., 2018, 8, 12232–12259 RSC.
- T. U. Connell and P. S. Donnelly, Coord. Chem. Rev., 2018, 375, 267–284 CrossRef CAS.
- M. Felici, P. Contreras-Carballada, Y. Vida, J. M. Smits, R. J. Nolte, L. De Cola, R. M. Williams and M. C. Feiters, Chem.–Eur. J., 2009, 15, 13124–13134 CrossRef CAS PubMed.
- M. Mydlak, C. Bizzarri, D. Hartmann, W. Sarfert, G. Schmid and L. De Cola, Adv. Funct. Mater., 2010, 20, 1812–1820 CrossRef CAS.
- E. Kerr, E. H. Doeven, G. J. Barbante, C. F. Hogan, D. Bower, P. S. Donnelly, T. U. Connell and P. S. Francis, Chem. Sci., 2015, 6, 472–479 RSC.
- L. C. Soulsby, D. J. Hayne, E. H. Doeven, D. J. D. Wilson, J. Agugiaro, T. U. Connell, L. Chen, C. F. Hogan, E. Kerr, J. L. Adcock, P. S. Donnelly, J. M. White and P. S. Francis, Phys. Chem. Chem. Phys., 2018, 20, 18995–19006 RSC.
- E. H. Doeven, G. J. Barbante, A. J. Harsant, P. S. Donnelly, T. U. Connell, C. F. Hogan and P. S. Francis, Sens. Actuators, B, 2015, 216, 608–613 CrossRef CAS.
- Z. M. Smith, E. Kerr, E. H. Doeven, T. U. Connell, N. W. Barnett, P. S. Donnelly, S. J. Haswell and P. S. Francis, Analyst, 2016, 141, 2140–2144 RSC.
- WO Pat., 8602734, 1986 CrossRef CAS PubMed; L. Xu, Y. Li, S. Wu, X. Liu and B. Su, Angew. Chem., Int. Ed., 2012, 51, 8068–8072 CrossRef CAS PubMed; Y. Liao, X. Zhou, Y. Fu and D. Xing, Anal. Chem., 2017, 89, 13016–13023 CrossRef PubMed.
- X. Zhou, D. Zhu, Y. Liao, W. Liu, H. Liu, Z. Ma and D. Xing, Nat. Protoc., 2014, 9, 1146–1159 CrossRef CAS PubMed.
- G. F. Blackburn, H. P. Shah, J. H. Kenten, J. Leland, R. A. Kamin, J. Link, J. Peterman, M. J. Powell, A. Shah, D. B. Talley, S. K. Tyagi, E. Wilkins, T.-G. Wu and R. J. Massey, Clin. Chem., 1991, 37, 1534–1539 CAS.
- S. Carrara, F. Arcudi, M. Prato and L. De Cola, Angew. Chem., Int. Ed., 2017, 56, 4757–4761 CrossRef CAS PubMed; G. Valenti, S. Scarabino, B. Goudeau, A. Lesch, M. Jović, E. Villani, M. Sentic, S. Rapino, S. p. Arbault, F. Paolucci and N. Sojic, J. Am. Chem. Soc., 2017, 139, 16830–16837 CrossRef PubMed.
- J. D. Debad, E. N. Glezer, J. Wohlstadter, G. B. Sigal and J. K. Leland, in Electrogenerated Chemiluminescence, ed. A. J. Bard, Marcel Dekker, New York, 2004, pp. 359–396 RSC; B. A. Gorman, P. S. Francis and N. W. Barnett, Analyst, 2006, 131, 616–639 RSC.
- T. U. Connell, J. M. White, T. A. Smith and P. S. Donnelly, Inorg. Chem., 2016, 55, 2776–2790 CrossRef CAS PubMed.
- T. U. Connell, J. L. James, A. R. White and P. S. Donnelly, Chem.–Eur. J., 2015, 21, 14146–14155 CrossRef CAS PubMed.
- J. I. Kim, I.-S. Shin, H. Kim and J.-K. Lee, J. Am. Chem. Soc., 2005, 127, 1614–1615 CrossRef CAS PubMed.
- L. Yu, Z. Huang, Y. Liu and M. Zhou, J. Organomet. Chem., 2012, 718, 14–21 CrossRef CAS; S. Ladouceur, K. N. Swanick, S. Gallagher-Duval, Z. Ding and E. Zysman-Colman, Eur. J. Inorg. Chem., 2013, 2013, 5329–5343 CrossRef.
- M. A. Haghighatbin, S. E. Laird and C. F. Hogan, Curr. Opin. Electrochem., 2018, 8, 52–59 CrossRef.
- A. Kapturkiewicz, T.-M. Chen, I. R. Laskar and J. Nowacki, Electrochem. Commun., 2004, 6, 827–831 CrossRef CAS; A. Kapturkiewicz, J. Nowacki and P. Borowicz, Electrochim. Acta, 2005, 50, 3395–3400 CrossRef.
- C. Li, J. Lin, X. Yang and J. Wan, J. Organomet. Chem., 2011, 696, 2445–2450 CrossRef CAS; Y. Zhou, W. Li, L. Yu, Y. Liu, X. Wang and M. Zhou, Dalton Trans., 2015, 44, 1858–1865 RSC; Y. Zhou, H. Gao, X. Wang and H. Qi, Inorg. Chem., 2015, 54, 1446–1453 CrossRef PubMed.
- M. S. Lowry, W. R. Hudson, R. A. Pascal Jr and S. Bernhard, J. Am. Chem. Soc., 2004, 126, 14129–14135 CrossRef CAS PubMed; S.-J. Liu, Q. Zhao, Q.-L. Fan and W. Huang, Eur. J. Inorg. Chem., 2008, 2177–2185 CrossRef; K. P. S. Zanoni, B. K. Kariyazaki, A. Ito, M. K. Brennaman, T. J. Meyer and N. Y. Murakami Iha, Inorg. Chem., 2014, 53, 4089–4099 CrossRef PubMed; Y. Kuramochi and O. Ishitani, Inorg. Chem., 2016, 55, 5702–5709 CrossRef PubMed; T. K. Kang, C. H. Kang, J. Lee, S. H. Kim, B. H. Kim and W.-Y. Lee, J. Electroanal. Chem., 2016, 775, 83–90 CrossRef.
- H. A. Bronstein, C. E. Finlayson, K. R. Kirov, R. H. Friend and C. K. Williams, Organometallics, 2008, 27, 2980–2989 CrossRef CAS; E. Baranoff, B. F. E. Curchod, J. Frey, R. Scopelliti, F. Kessler, I. Tavernelli, U. Rothlisberger, M. Gratzel and M. K. Nazeeruddin, Inorg. Chem., 2012, 51, 215–224 CrossRef PubMed; M. Lepeltier, F. Dumur, G. Wantz, N. Vila, I. Mbomekalle, D. Bertin, D. Gigmes and C. R. Mayer, J. Lumin., 2013, 143, 145–149 CrossRef; J. Frey, B. F. E. Curchod, R. Scopelliti, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and E. Baranoff, Dalton Trans., 2014, 43, 5667–5679 RSC.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, R. Kwong, I. Tsyba, M. Bortz, B. Mui, R. Bau and M. E. Thompson, Inorg. Chem., 2001, 40, 1704–1711 CrossRef CAS PubMed; T. Liu, B.-H. Xia, X. Zhou, Q.-C. Zheng, Q.-J. Pan and H.-X. Zhang, Theor. Chem. Acc., 2008, 121, 155–164 Search PubMed; X. Gu, T. Fei, H. Zhang, H. Xu, B. Yang, Y. Ma and X. Liu, Eur. J. Inorg. Chem., 2009, 2407–2414 CrossRef; J. M. Younker and K. D. Dobbs, J. Phys. Chem. C, 2013, 117, 25714–25723 CrossRef.
- J. C. Deaton, R. H. Young, J. R. Lenhard, M. Rajeswaran and S. Huo, Inorg. Chem., 2010, 49, 9151–9161 CrossRef CAS PubMed; R. Wang, D. Liu, H. Ren, T. Zhang, X. Wang and J. Li, J. Mater. Chem., 2011, 21, 15494–15500 RSC.
- G. Valenti, E. Rampazzo, S. Bonacchi, L. Petrizza, M. Marcaccio, M. Montalti, L. Prodi and F. Paolucci, J. Am. Chem. Soc., 2016, 138, 15935–15942 CrossRef CAS PubMed.
- F. Kanoufi, Y. Zu and A. J. Bard, J. Phys. Chem. B, 2001, 105, 210–216 CrossRef CAS.
- B. D. Stringer, L. M. Quan, P. J. Barnard, D. J. D. Wilson and C. F. Hogan, Organometallics, 2014, 33, 4860–4872 CrossRef CAS; L. M. Quan, B. D. Stringer, M. Haghighatbin, J. Agugiaro, G. J. Barbante, D. J. Wilson, C. F. Hogan and P. Barnard, Dalton Trans., 2018, 48, 653–663 RSC.
- A. Kapturkiewicz, Adv. Electrochem. Sci. Eng., 1997, 5, 1–60 CAS.
- J. Truong, K. B. Spilstead, G. J. Barbante, E. H. Doeven, D. J. D. Wilson, N. W. Barnett, L. C. Henderson, J. M. Altimari, S. C. Hockey, M. Zhou and P. S. Francis, Analyst, 2014, 139, 6028–6035 RSC.
- K. Imai, G. Valenti, E. Villani, S. Rapino, E. Rampazzo, M. Marcaccio, L. Prodi and F. Paolucci, J. Phys. Chem. C, 2015, 119, 26111–26118 CrossRef CAS.
- E. J. O'Reilly, P. J. Conroy, S. Hearty, T. E. Keyes, R. O'Kennedy, R. J. Forster and L. Dennany, RSC Adv., 2015, 5, 67874–67877 RSC.
- B. Giese, M. Graber and M. Cordes, Curr. Opin. Chem. Biol., 2008, 12, 755–759 CrossRef CAS PubMed; J. J. Warren, M. E. Ener, A. Vlcek, J. R. Winkler and H. B. Gray, Coord. Chem. Rev., 2012, 256, 2478–2487 CrossRef PubMed; X. Ru, P. Zhang and D. N. Beratan, J. Phys. Chem. B, 2019, 123, 5035–5047 CrossRef PubMed.
- C. Mallet, A. Bolduc, S. Bishop, Y. Gautier and W. G. Skene, Phys. Chem. Chem. Phys., 2014, 16, 24382–24390 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev E.01, 2009 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed; J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- A. L. Tenderholt, QMForge, Version 2.4, http://https://qmforge.net Search PubMed.
- Y. Wei, C. Yang, B. Wei, J. Huang, L. Wang, S. Meng, R. Zhang and J. Li, J. Clin. Microbiol., 2008, 46, 1734–1740 CrossRef CAS PubMed.
- E. Spain, S. Carrara, K. Adamson, H. Ma, R. O'Kennedy, L. De Cola and R. J. Forster, ACS Omega, 2018, 3, 17116–17124 CrossRef CAS.
- I. Svir, A. Oleinick, O. V. Klymenko and C. Amatore, ChemElectroChem, 2015, 2, 811–818 CrossRef CAS.
- X. Liu, L. Shi, W. Niu, H. Li and G. Xu, Angew. Chem., Int. Ed., 2007, 46, 421–424 CrossRef CAS PubMed; S. Han, W. Niu, H. Li, L. Hu, Y. Yuan and G. Xu, Talanta, 2010, 81, 44–47 CrossRef PubMed; N. Kebede, P. S. Francis, G. J. Barbante and C. F. Hogan, Analyst, 2015, 140, 7142–7145 RSC; S. A. Kitte, C. Wang, S. Li, Y. Zholudov, L. Qi, J. Li and G. Xu, Anal. Bioanal. Chem., 2016, 408, 7059–7065 CrossRef PubMed; A. Fiorani, Irkham, G. Valenti, F. Paolucci and Y. Einaga, Anal. Chem., 2018, 90, 12959–12963 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of ligands, Ir(III) complexes and ECL labels. Additional data: absorption and emission spectra; cyclic voltammograms; tabulated spectroscopic and electrochemical properties in acetonitrile; calculated MO energies, contributions to the respective MOs, and contour plots; primers and NASBA amplicon fragment sequences. See DOI: 10.1039/c9sc01391a |
| ‡ These authors contributed equally. |
| § Present address: RMIT University, Melbourne, Victoria 3001 Australia. |
| ¶ Commercial sources of chemicals: tris(2,2′-bipyridine)ruthenium(II) hexafluorophosphate ([Ru(bpy)3](PF6)2) and tetrabutylammonium hexafluorophosphate (TBAPF6; electrochemical grade) were purchased from Sigma-Aldrich (NSW, Australia). Tris(2,2′-bipyridine)ruthenium(II) chloride hexahydrate ([Ru(bpy)3]Cl2·6H2O) and bis(cyclopentadienyl)iron (ferrocene; Fc) were purchased from Strem Chemicals (MA, USA). The four Ir(C^N)(acac) complexes and five ECL labels (bis(2,2′-bipyridyl)(4-methyl-4′-carboxypropyl-2,2′-bipyridyl)ruthenium(II) hexafluorophosphate ([Ru(bpy)2(mbpy-COOH)](PF6)2), bis(4,6-difluoro-2-(2-pyridyl)phenyl-C2,N)(4-methyl-4′-carboxypropyl-2,2′-bipyridyl)iridium(III) chloride ([Ir(df-ppy)2(mbpy-COOH)]Cl), bis(2-phenylpyridine-C2,N)(4-carboxypropyl-2,2′-bipyridyl)iridium(III) hexafluorophosphate ([Ir(ppy)2(mbpy-COOH)](PF6)), bis(2-phenylbenzothiazole-C2,N)(4-methyl-4′-carboxy-2,2′-bipyridyl)iridium(III) chloride ([Ir(bt)2(mbpy-COOH)]Cl), bis(1-phenylisoquinoline)(4-methyl-4′-carboxypropyl-2,2′-bipyridyl)iridium(III) chloride ([Ir(piq)2(bpy-COOH)]Cl)) were purchased from SunaTech (Jiangsu, China). |
| This journal is © The Royal Society of Chemistry 2019 |