
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCanonical DNA minor groove insertion of bisbenzamidine–Ru(II) complexes with chiral selectivity†
Mateo I.
Sánchez
 a,
Gustavo
Rama
b,
Renata
Calo-Lapido
a,
Kübra
Ucar
c,
Per
Lincoln
c,
Miguel Vázquez
López
b,
Manuel
Melle-Franco
d,
José L.
Mascareñas
*a and
M. Eugenio
Vázquez
*a
a,
Gustavo
Rama
b,
Renata
Calo-Lapido
a,
Kübra
Ucar
c,
Per
Lincoln
c,
Miguel Vázquez
López
b,
Manuel
Melle-Franco
d,
José L.
Mascareñas
*a and
M. Eugenio
Vázquez
*a
aCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS), Departamento de Química Orgánica, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain. E-mail: eugenio.vazquez@usc.es
bCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS), Departamento de Química Inorgánica, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain
cDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, SE 412 96 Gothenburg, Sweden
dCiceco - Aveiro Institute of Materials, University of Aveiro Campus Universitario de Santiago, Aveiro, 3810-193, Portugal
First published on 1st August 2019
Abstract
We report the first Ru(II) coordination compounds that interact with DNA through a canonical minor groove insertion mode and with selectivity for A/T rich sites. This was made possible by integrating a bis-benzamidine minor groove DNA-binding agent with a ruthenium(II) complex. Importantly, one of the enantiomers (Δ-[Ru(bpy)2b4bpy]2+, Δ-4Ru) shows a considerably higher DNA affinity than the parent organic ligand and the other enantiomer, particularly for the AATT sequence, while the other enantiomer preferentially targets long AAATTT sites with overall lower affinity. Finally, we demonstrate that the photophysical properties of these new binders can be exploited for DNA cleavage using visible light.
Introduction
Over the past few decades there has been great interest in the development of transition metal complexes that target DNA as antitumor agents.1 In this context, intercalative ruthenium(II) complexes are particularly attractive due to their good kinetic stability, as well as their rich redox and photochemistry.2 These complexes typically contain two bipyridine ligands, and one large heteroaromatic unit that penetrates into DNA through the major groove and stacks between consecutive base pairs.3 Unfortunately, just like polycyclic organic intercalators,4 these complexes display poor sequence selectivity with preference for G/C-rich sequences. In contrast, organic minor groove DNA-binding agents typically show good discrimination between binding sites.5 Although minor groove contacts have been proposed for a number of coordination compounds,6 to our knowledge, a canonical minor groove binding complex that inserts one of its ligands into the DNA minor groove has not yet been demonstrated.7We reasoned that in the same way polyaromatic ligands, such as dppn, dppz or dpq, define the intercalative binding mode of traditional DNA-binding metal complexes,8 engineering an organic minor groove binder as a metal ligand could yield complexes capable of inserting into the minor groove of DNA, and thus display new DNA binding properties not observed with traditional metallointercalators.9,10 More specifically, we considered the use of aza-bisbenzamidines as model minor groove binders, owing to their synthetic accessibility and their well-established fluorogenic properties.11 This type of compound tends to insert into the minor groove of A/T rich sequences with dissociation constants in the low μM range.12 Herein we describe the synthesis of several ruthenium(II) complexes incorporating bis-(methylamino-benzamidine)-2,2′-bipyridine ligands, and demonstrate that they bind to A/T-rich DNA sequences by insertion of such a benzamidine ligand into the minor groove. Importantly, we also found that the DNA binding profile of these complexes is heavily dependent on their chirality, which not only affects their overall binding affinity, but also determines their preferred binding sequence.
Results and discussion
Synthesis and characterization of the bisbenzamidine complexes
The aza-bisbenzamidine ligands and their corresponding complexes were synthesized as shown in Scheme 1.11 Thus, reductive amination of 2,2′-bipyridine-4,4′-dicarbaldehyde (1a, Scheme 1), with commercially available 4-aminobenzene carboximidamide dihydrochloride, afforded the desired bis-benzamidine-bipyridine ligand b4bpy in good yield.22 The reaction of this ligand with each of the enantiopure Hua and von Zelewsky's reagents, Λ- and Δ-cis-[Ru(bpy)2(py)2]2+,13 afforded the enantiomeric complexes Λ-4Ru (Λ-[Ru(bpy)2b4bpy]2+) and Δ-4Ru (Δ-[Ru(bpy)2b4bpy]2+), respectively. The same sequence of transformations starting with 2,2′-bipyridine-5,5′-dicarbaldehyde (1b, Scheme 1) leads to the regioisomeric complexes Λ-5Ru (Λ-[Ru(bpy)2b5bpy]2+) and Δ-5Ru (Δ-[Ru(bpy)2b5bpy]2+), also in good yields (Scheme 1).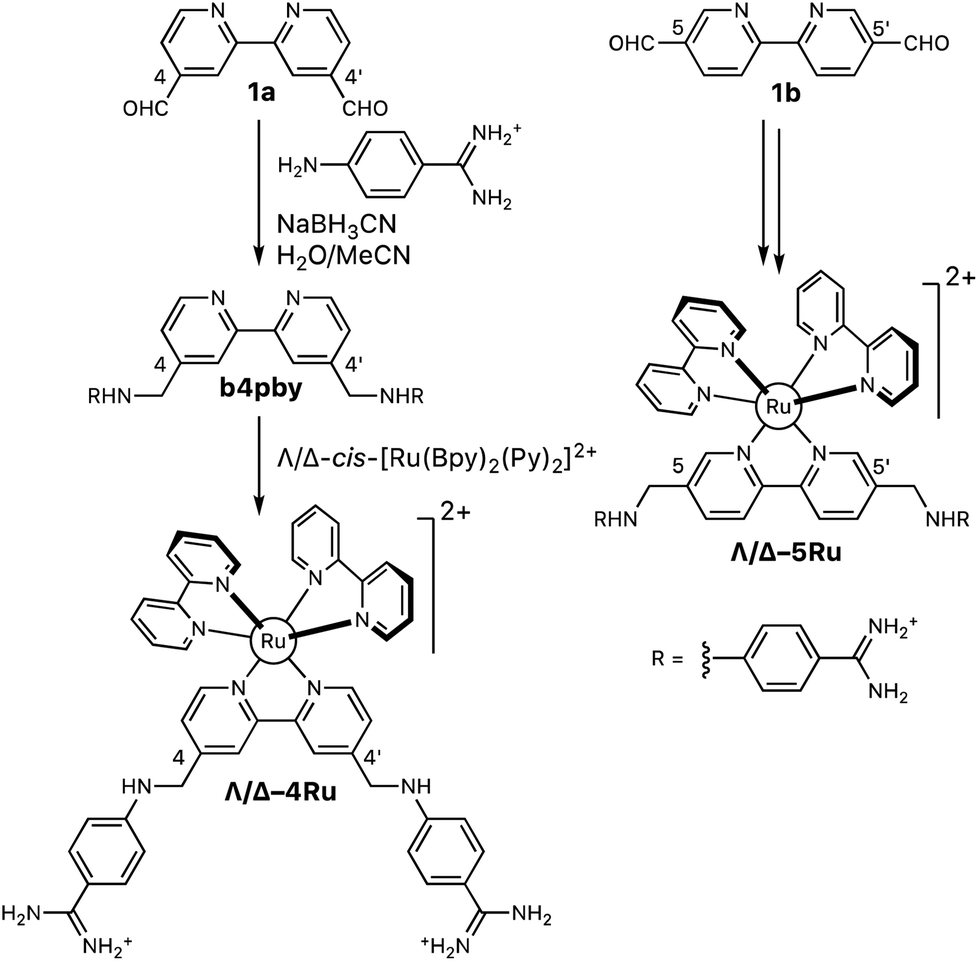 | ||
| Scheme 1 Synthesis of the set of Λ- and Δ-bisbipyridyl ruthenium(II) complexes containing the ligands b4bpy and b5bpy. | ||
The CD spectra of both Λ-complexes (Λ-4Ru and Λ-5Ru) are dominated by a large LC transition band with a positive Cotton effect at 285–310 nm and a broad and less intense metal-to-ligand charge transfer (MLCT) band centered at ca. 460 nm, also displaying positive Cotton effects. These features are consistent with the Λ-configuration around the metal center in octahedral coordination compounds.14 Likewise, their enantiomeric complexes (Δ-4Ru and Δ-5Ru) display mirror image CD spectra with negative Cotton effects (see the ESI†).
DNA binding of the bisbenzamidine–Ru(II) complexes
Having the two pairs of enantiomeric complexes at hand, we studied their DNA binding properties by taking advantage of the intrinsic fluorogenic properties of the ruthenium(II) polypyridyl complexes.15 Thus, successive aliquots of a 250 μM solution of a short double stranded hairpin oligonucleotide containing an extended six-base-pair A/T-rich binding site (A3T3) were added to a 0.5 μM solution of Λ-4Ru in Tris–HCl buffer and the luminescence emission spectra upon irradiation at the benzamidine excitation wavelength (329 nm) were recorded after each addition. This resulted in a series of spectra displaying a progressive increase in the emission intensity of the Λ-4Ru3MLCT band at 605 nm, which could be fitted to a one to one binding model including contribution from non-specific binding, with a dissociation constant of KD ≈ 0.62 μM (Fig. 1a and Table 1, see the ESI† for details about curve fitting analysis).16 Titrations with other DNA oligonucleotides exhibiting five (A2T3) and four (A2T2) consecutive A/T base pairs resulted in progressively weaker binding. Not surprisingly, Λ-4Ru displayed considerably lower affinity for a G/C-rich oligo (G2C3). Similar experiments were carried out with the enantiomeric complex Δ-4Ru, the isomeric Λ-5Ru and Δ-5Ru complexes, and the parent bis-benzamidine ligands (b4bpy and b5bpy, λexc = 329 nm; λem = 389 nm). The resulting apparent dissociation constants are summarized in Table 1 and graphically compared in Fig. 1b.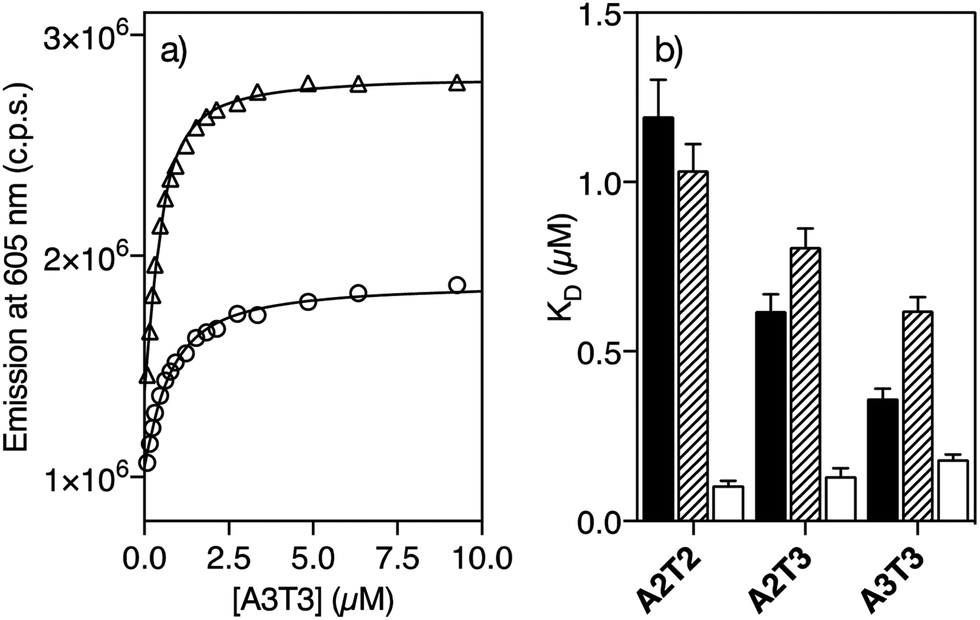 | ||
| Fig. 1 (a) Representative titration profiles of 0.5 μM solutions of Λ-4Ru (○) and Δ-4Ru (△) complexes with increasing concentration of the oligo A3T3. (b) Dissociation constants of b4bpy (black), Λ-4Ru (striped), and Δ-4Ru (white) with a set of A/T-rich oligonucleotides. Hairpin sequences (binding sites in italics): A3T3: 5′-GGC AAATTT CAG T5 CTG AAATTT GCC-3′; A2T3: 5′-GGCG AATTT CGC T5 GCG AAATT CGCC-3′; A2T2: 5′-GGCG AATT CAGC T5 GCTG AATT CGCC-3′A/T-rich binding sites, and the central hairpin loop (T5) is shown in italics. All titrations were performed in 20 mM Tris–HCl buffer with 100 mM NaCl, pH 7.5, at 298 K. | ||
| A2T2 | A2T3 | A3T3 | G2C3 | |
|---|---|---|---|---|
| a K D (μM) was measured in 20 mM Tris–HCl buffer with 100 mM NaCl, pH 7.5, at 298 K and calculated from three independent titrations. The estimated KD error is shown in brackets. n.b. indicates that no binding was observed under the experimental conditions used in the titrations. Hairpin oligonucleotide sequence of G2C3: 5′-GGCA GGCC CAGC T5 GCTG GGCC TGCC-3′. | ||||
| b4bpy | 1.19 (0.11) | 0.62 (0.05) | 0.36 (0.03) | n.b. |
| b5bpy | 2.58 (0.58) | 1.29 (0.15) | 0.84 (0.07) | n.b. |
| Δ-4Ru | 0.11 (0.01) | 0.13 (0.02) | 0.18 (0.01) | 1.69 (0.09) |
| Λ-4Ru | 1.04 (0.08) | 0.81 (0.06) | 0.62 (0.04) | 2.64 (0.28) |
| Δ-5Ru | 4.24 (0.49) | 3.64 (0.26) | 3.08 (0.16) | 4.90 (0.05) |
| Λ-5Ru | 4.04 (0.76) | 3.81 (0.76) | 3.24 (0.28) | 3.63 (0.39) |
The ligand b5bpy and its derivatives Λ-5Ru and Δ-5Ru display lower affinity than its b4bpy counterparts. The reduced binding of b5bpy probably arises from its linear structure, which cannot simultaneously optimize the interactions of the two amidinium groups with the bottom of the DNA minor groove.12,17,18 Moreover, Λ-5Ru and Δ-5Ru showed weaker DNA binding than b5bpy, regardless of the chirality of the metal center. On the other hand, the ligand b4bpy and Λ-4Ru display comparable binding affinities and a clear preference for those DNAs featuring longer A/T sites, so titrations with oligos exhibiting six (A3T3), five (A2T3), and four (A2T2) consecutive A/T base pairs resulted in progressively weaker dissociation constants (Table 1 and Fig. 1b). Remarkably, Δ-4Ru displays much higher affinity for all the above A/T-rich DNAs than b4bpy or Λ-4Ru, particularly for A2T2 (KD = 0.11 μM, Table 1). Indeed, its interaction with the A2T2 oligo is over 10 times stronger than that of its enantiomer Λ-4Ru, or that of the parent organic ligand b4bpy. Hence, proper ligand engineering allowed transforming a weak and nonspecific DNA binder, such as [Ru(bpy)3]2+19 into complexes capable of inserting into the minor groove of specific DNA sequences with high affinity. Curiously, in the case of the isomer b5bpy, the metal coordination has a negative effect on the affinity (e.g., Λ/Δ-5Ruvs.b5bpy).
DNA binding occurs through insertion into the DNA minor groove
In order to confirm that the interaction of Δ-4Ru with the target DNA occurs by insertion into the minor groove, we measured the circular dichroism spectra of the oligonucleotide A3T3 in the presence of increasing concentrations of the ligand b4bpy, Λ-4Ru, and Δ-4Ru. Incubation with b4bpy resulted in the appearance of a positive induced CD band at ca. 345 nm, consistent with its insertion into the DNA minor groove (Fig. 2a).20,21 Importantly, mixing the DNA with either Λ-4Ru or Δ-4Ru produced a similar induced CD band, although with reduced intensity compared to that of the free ligand b4bpy, perhaps due to the conformational restrictions imposed by coordination to the metal ion (Fig. 2b). The observation of this induced CD band, which arises from a chiral twisting of the bis-benzamidine ligand, supports the formation of DNA complexes of similar nature to those formed with b4bpy.22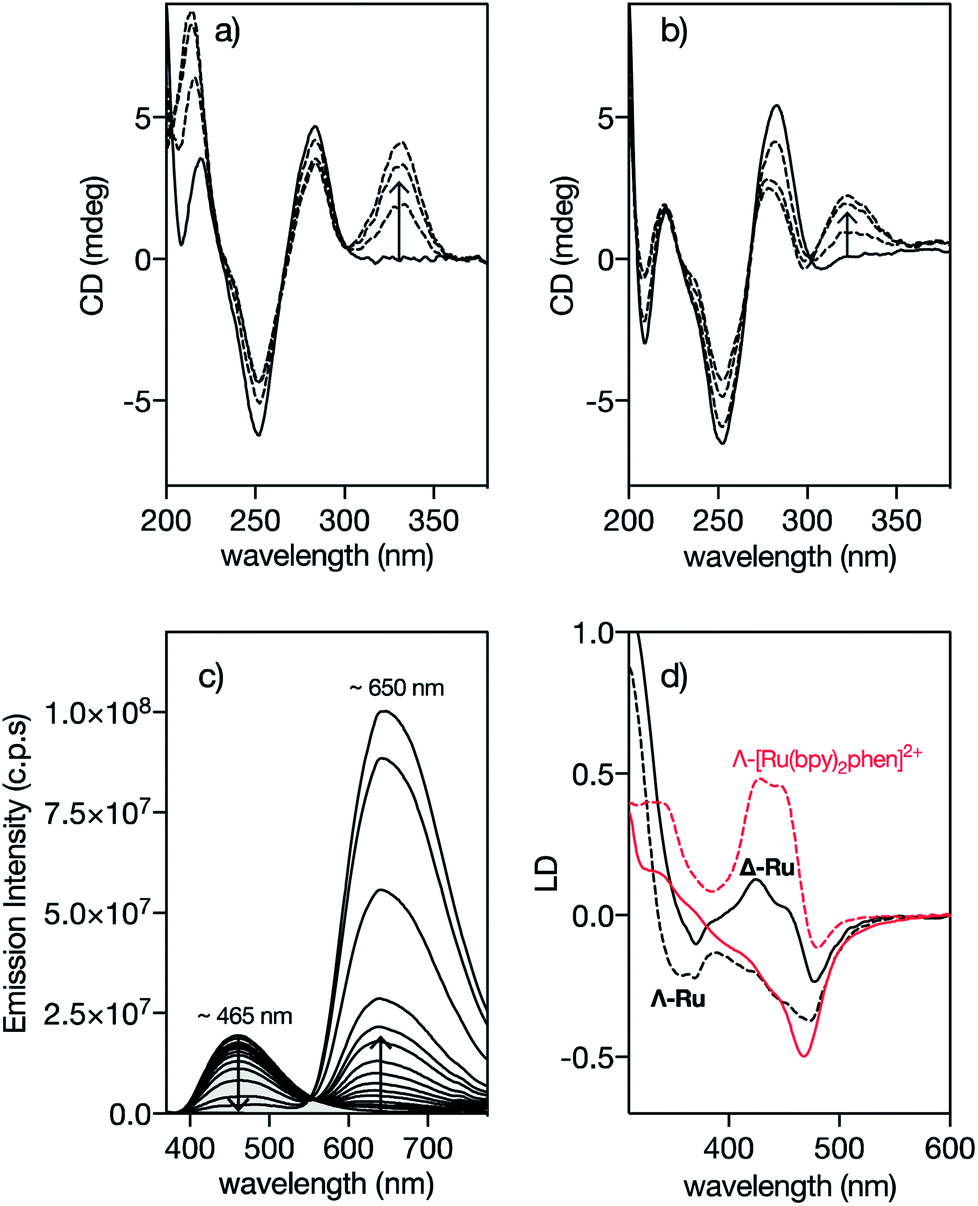 | ||
| Fig. 2 Circular Dichroism spectra of 5 μM solutions of the A3T3 oligo in 20 mM Tris–HCl buffer with 100 mM NaCl, pH 7.5 (solid lines) in the presence of 1, 3 and 5 eq. of (a) 4bpy and (b) Λ-4Ru showing the induced CD band at ca. 330 nm corresponding to the benzamidine chromophore. A3T3: 5′-GGC AAATTT CAG T5 CTG AAATTT GCC-3′; A/T-rich binding sites and the central hairpin loop (T5) are shown in italics. The CD spectra obtained upon incubation of the Δ-4Ru isomer with the A3T3 DNA are qualitatively the same as those for the enantiomeric Λ-4Ru; (c) DAPI displacement assay showing a series of emission spectra of a mixture of 0.25 μM DAPI and 0.5 μM A2T2 in the presence of increasing concentration of Δ-4Ru; (d) Linear dichroism (LD) spectra of flow-oriented calf thymus DNA with the two enantiomers of 4Ru (black lines, P/Ru = 20) and [Ru(bpy)2phen]2+ (red lines, P/Ru = 30) in 10 mM NaCl.29 Λ-enantiomers as dashed lines and Δ-enantiomers as solid lines in both cases. Spectra are normalized to A = 1 for the long wavelength absorption maximum of the free complex. The LD spectra are further normalized to perfect orientation (S = 1) by setting the LDr value at the DNA band at 260 nm to −1.5.29 | ||
Fluorescence competition assays show that Δ-4Ru displaces DAPI (4,6-diamidino-2-phenylindole) and Hoechst 33258, typical A/T-rich minor groove binders, very efficiently,23 thus reinforcing the hypothesis that these complexes insert into the DNA minor groove (Fig. 2c and S10 in the ESI†).24 Linear Dichroism (LD) experiments, which provide information about the orientation of the bound complexes with respect to the DNA,25 were also consistent with minor groove insertion. Thus, the LD spectra of a reference intercalative complex Λ-[Ru(bpy)2phen]2+ show a strong positive LD signal in the B(E)-polarized MLCT band at 440 nm, arising from the placement of the phen ligand almost coplanar to the DNA base pairs and the complex two-fold symmetry axis slightly rotated clockwise (by about 10°) from the ideal intercalation geometry. This arrangement results in a decreased LD at 440 nm for its enantiomer Δ-[Ru(bpy)2phen]2+ (Fig. 2d).26,29 By substituting the intercalative phen ligand with the minor groove binder 4bpy, the Λ-4Ru enantiomer now shows a more negative LD band compared to Δ-4Ru, which is unprecedented among mono-nuclear ruthenium polypyridyl complexes. This inversion in the relative intensities is consistent with the alignment of the substituted 4bpy ligand in the direction of the DNA minor groove, and a counter-clockwise rotation of the complex from the ideal intercalation geometry by about 45°, moving the B(E) transition away from the helix axis and consequently giving rise to a decreased, or even negative, LD at 440 nm.
Computational modeling of the interaction
To gain some structural insight into the interaction of our molecules with the DNA, we performed modelling studies using previously tested docking procedures27 and taking as reference the high-resolution crystal structure of the Dickerson–Drew dodecamer, which features a short A/T-rich binding site in the middle of its sequence (5′-CGCGAATTCGCG-3′, xA2T2).28 The lowest energy docking poses of the complexes Λ-Ru and Δ-4Ru on this DNA present the bisbenzamidine ligand inserted into the minor groove in the A/T-rich region of the oligonucleotide.More importantly, the bulkier bipyridine ligands match the indentations between the T7–T8/T19–T20 phosphate groups in the DNA backbone (Fig. 3), allowing the benzamidine ligand to reach the bottom of the DNA minor groove. Docking studies with a different model DNA based on fiber diffraction data (fA2T2)30 resulted in most populated poses qualitatively similar to those observed with xA2T2. Importantly, the binding energies resulting from the docking experiments are highly dependent on the DNA model used in the calculations. Thus, the intermolecular binding energies obtained with the DNA fA2T2 were −16.5 and −15.6 kcal mol−1 for Δ-4Ru and Λ-Ru respectively, in line with the observed experimental difference in the binding affinity. However, the interaction energy with xA2T2 turned out to be about −20 kcal mol−1 for both isomers. Summing up, these docking studies support the minor groove binding mode for both Δ-4Ru and Λ-Ru and also suggest that the strength and sequence selectivity are highly dependent on the microstructure of the DNA substrates, as previously reported for related minor groove binders.31 The shape complementarity with the DNA minor groove plays a key role in the strength of the binding, as well as in the sequence discrimination between both enantiomeric forms, which ultimately appears to be related to the way in which the accessory bipyridine ligands match the DNA backbone.32 Taken together, these experimental and computational data demonstrate that through proper ligand engineering it is possible to obtain metal complexes that selectively recognize DNA through a canonical minor groove insertion mechanism.
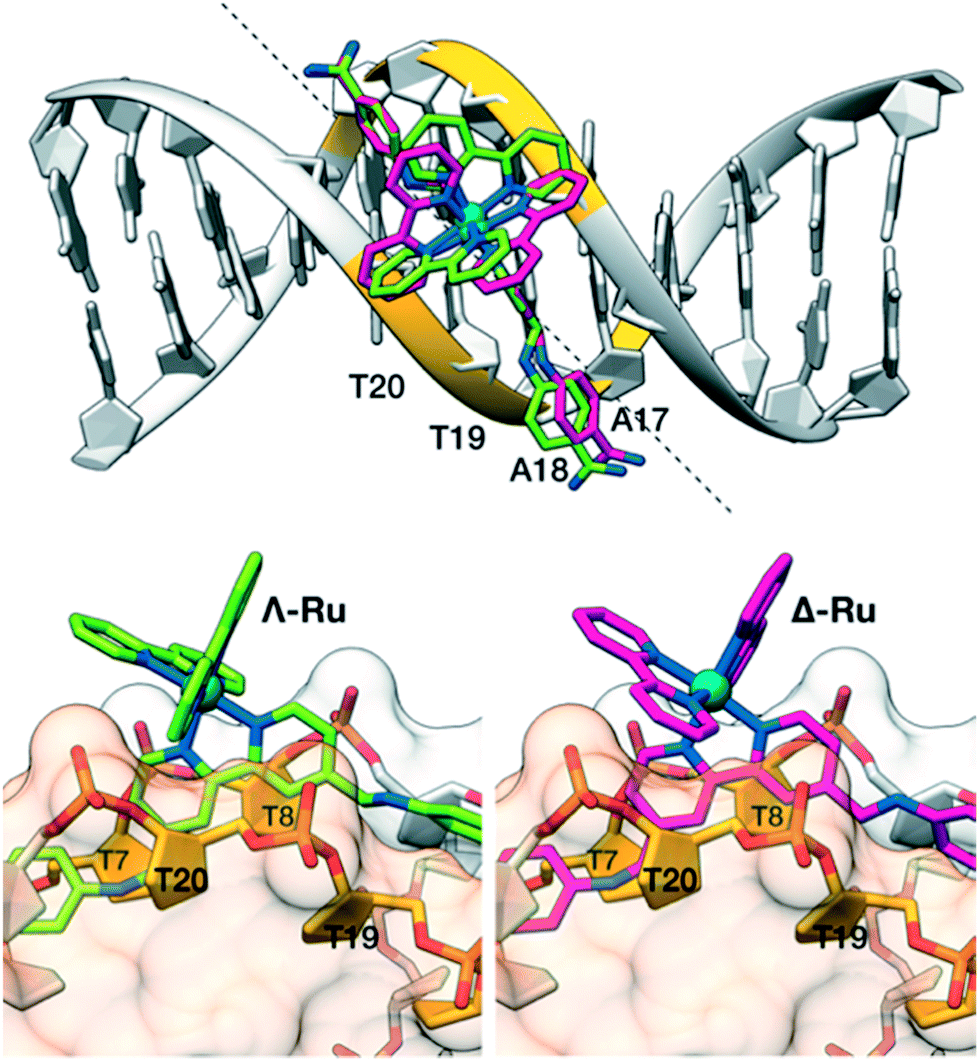 | ||
| Fig. 3 Docking structures of Λ-Ru (green) and Δ-4Ru (magenta). Top view: alignment of the b4bpy ligands with the minor groove in the Dickerson–Drew dodecamer (xA2T2). The dashed line forms a 45° angle with the DNA axis and matches the approximate orientation of the b4bpy ligand in the complexes. Bottom left: side view of Λ-Ru showing the bipyridine ligand between the phosphate groups flanking T20 (front, light orange surface) and T8 (back, light grey surface). Bottom right: the same view of Λ-Ru.33 | ||
Study of the bisbenzamidine–Ir(III) analogs Δ-4Ir and Λ-4Ir
We also studied the DNA binding of the 2-phenylpyridine cyclometalated iridium(III) complexes containing the minor groove binding ligand b4bpy. These monocationic complexes have roughly the same geometry as dicationic ruthenium(II) complexes, but lower charge (+1). Thus we synthesized the complexes Δ- and Λ-[Ir(ppy)2b4bpy]+ (Δ-4Ir and Λ-4Ir respectively; ppy = 2-phenylpyridine), by reaction of b4bpy with a dimeric bis(2-phenylpyridinato) iridium chloride precursor [(ppy)2Ir(μ-Cl)]2, followed by HPLC resolution of the resulting enantiomeric mixture (see the ESI†).34The interaction of these complexes with DNA was studied by steady-state luminescence titrations monitoring the emission from the complexes at 575 nm upon irradiation of the benzamidine fluorophore at 329 nm (ESI†). As shown in Table 2, both Δ-4Ir and Λ-4Ir display similar trends to Δ-4Ru and Λ-4Ru, although with slightly reduced affinities, which might be likely related to the lower charge of these complexes. Thus, for example, Δ-4Ir also displays higher affinity for A2T2 than Λ-4Ir. Interestingly, the two iridium isomers show different sequence selectivity, so while the affinity of Δ-4Ir for DNA is higher for shorter A/T-tracts, Λ-4Ir shows a marked preference for longer A/T-rich sites. Curiously, the affinity for G/C-rich oligos is higher than for the ruthenium analogs.
| A2T2 | A2T3 | A3T3 | G2C3 | |
|---|---|---|---|---|
| a K D (μM) was measured in 20 mM Tris–HCl buffer with 100 mM NaCl, pH 7.5, at 298 K and calculated from three independent titrations. The estimated KD error is shown in brackets. n.b. if no significant binding is observed. | ||||
| b4bpy | 1.19 (0.11) | 0.62 (0.05) | 0.36 (0.03) | n.b. |
| Δ-4Ir | 0.27 (0.09) | 0.49 (0.03) | 1.01 (0.12) | 1.44 (0.09) |
| Λ-4Ir | 1.15 (0.13) | 0.62 (0.04) | 0.47 (0.03) | 0.70 (0.05) |
Photo-endonuclease activity of Δ-4Ru
Finally, we studied the potential application of these newly developed DNA minor groove binders as photo-endonucleases. It is known that irradiation of trisbipyridyl Ru(II) complexes gives rise to a 3MLCT (metal-to-ligand charge transfer) excited state that can act as a photosensitizer to generate singlet oxygen (1O2),35 which ultimately leads to DNA strand breaks.36 Thus, a 50 ng μL−1 solution of the pCDNA3.1(+) plasmid was incubated with increasing concentration of the complex Δ-4Ru (20 and 100 μM) and irradiated with a 455 nm LED source for either 5 or 10 min, and the resulting mixtures were analyzed by agarose electrophoresis. As expected, in the absence of light, Δ-4Ru was inert, and no new bands indicating the degradation of the pCDNA3.1(+) plasmid were observed. However, the irradiated solutions displayed new bands in the agarose gel consistent with the illumination time and a concentration-dependent scission of the plasmid (Fig. 4). It is important to note that the parent ruthenium complex [Ru(bpy)3]2+ does not induce the light-promoted DNA cleavage, as it is not capable of interacting with DNA.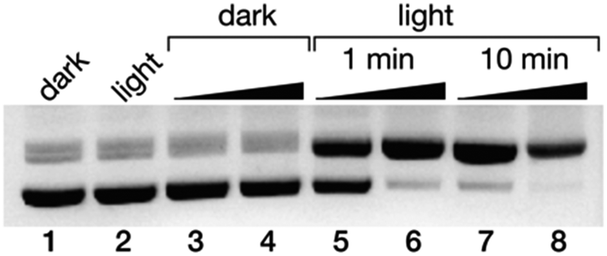 | ||
| Fig. 4 Ethidium bromide-stained agarose gels (0.7%) of the photocleavage of 25 ng μL−1 pCDNA3.1(+) in 20 mM Tris–HCl buffer with 100 mM NaCl, pH 7.5, with Δ-4Ru. All lanes contain the plasmid pCDNA3.1(+); lane 1: plasmid in the absence of Δ-4Ru in the dark; lane 2, the same plasmid after 10 min of irradiation in the absence of the Δ-4Ru complex; lanes 3 and 4: 20 and 100 μM Δ-4Ru in the dark; lanes 5 and 6: 20 and 100 μM Δ-4Ru irradiated for 1 min; lanes 7 and 8: 20 and 100 μM Δ-4Ru irradiated for 10 min. Irradiation was performed with a Thorlabs M455L3 Royal Blue (455 nm) Mounted High-Power LED, 1000 mA, using a custom made setup (see the ESI†). | ||
Conclusions
In summary, ruthenium(II) coordination complexes containing a designed bis-benzamidine ligand selectively bind to A/T-rich sequences in DNA by means of a classic minor groove insertion mechanism. To our knowledge, this type of interaction has not been demonstrated for metal-based DNA-binding agents. Importantly, the two enantiomers display markedly different DNA binding properties, so Δ-4Ru binds more strongly than Λ-4Ru to all the studied DNA sites and preferentially to those with a short A/T site (AATT) with ∼10-fold higher affinity than Λ-4Ru. In contrast the Λ-4Ru isomer preferentially binds to DNA with longer A/T sites (A3T3) and shows only residual binding affinity for the shorter DNA A2T2 preferred by its enantiomer. Finally, Δ-4Ru exhibited efficient nuclease activity upon irradiation, which might find applications in photodynamic therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish grants CTQ2015-70698-R, SAF2016-76689-R and Orfeo-cinqa network CTQ2016-81797-REDC; the Ministerio de Ciencia, Innovación y Universidades of Spain, RTI2018-099877-B-I00; the Xunta de Galicia (2015-CP082, ED431C 2017/19, Centro singular de investigación de Galicia accreditation 2016–2019, ED431G/09), the European Union (European Regional Development Fund – ERDF), and the European Research Council (Advanced Grant No. 340055) are gratefully acknowledged. We also acknowledge the support from the Portuguese Foundation for Science and Technology, FCT, (IF/00894/2015) and CICECO – Aveiro Institute of Materials, FCT Ref. UID/CTM/50011/2019, financed by national funds through the FCT/MCTES. M. E. V. also acknowledges the support of the Fundación Asociación Española Contra el Cáncer (AECC) (IDEAS197VAZQ grant). We also thank A. Arda and J. Jiménez-Barbero (CIC bioGUNE) for carrying out the preliminary spectroscopic characterization.References
- (a) K. L. Haas and K. J. Franz, Chem. Rev., 2009, 109, 4921–4960 CrossRef CAS PubMed; (b) P. C. A. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS; (c) M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 183–194 RSC; (d) J. K. Barton, Science, 1986, 233, 727–734 CrossRef CAS PubMed.
- (a) B. M. Zeglis, V. C. Pierre and J. K. Barton, Chem. Commun., 2007, 4565–4579 RSC; (b) M. R. Gill, J. Garcia-Lara, S. J. Foster, C. Smythe, G. Battaglia and J. A. Thomas, Nat. Chem., 2009, 1, 662–667 CrossRef CAS PubMed; (c) M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 RSC; (d) J. Rodríguez, J. Mosquera, J. R. Couceiro, M. E. Vázquez and J. L. Mascareñas, Angew. Chem., Int. Ed., 2016, 55, 15615–15618 CrossRef PubMed; (e) B. C. Poulsen, S. Estalayo-Adrián, S. Blasco, S. A. Bright, J. M. Kelly, D. Clive Williams and T. Gunnlaugsson, Dalton Trans., 2016, 45, 18208–18220 RSC.
- (a) C. L. Kielkopf, K. E. Erkkila, B. P. Hudson, J. K. Barton and D. C. Rees, Nat. Struct. Biol., 2000, 7, 117–121 CrossRef CAS PubMed; (b) H. Song, J. T. Kaiser and J. K. Barton, Nat. Chem., 2012, 4, 615–620 CrossRef CAS PubMed.
- (a) A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960–4962 CrossRef CAS; (b) H.-K. Liu and P. J. Sadler, Acc. Chem. Res., 2011, 44, 349–359 CrossRef CAS PubMed.
- (a) P. Guo, A. A. Farahat, A. Paul, N. K. Harika, D. W. Boykin and W. D. Wilson, J. Am. Chem. Soc., 2018, 140, 14761–14769 CrossRef CAS PubMed; (b) S. Neidle, Nat. Prod. Rep., 2001, 18, 291–309 RSC; (c) M. E. Vazquez, A. M. Caamaño, J. Martínez-Costas, L. Castedo and J. L. Mascareñas, Angew. Chem., Int. Ed., 2001, 40, 4723–4725 CrossRef CAS; (d) O. Vázquez, M. E. Vázquez, J. B. Blanco, L. Castedo and J. L. Mascareñas, Angew. Chem., Int. Ed., 2007, 46, 6886–6890 CrossRef PubMed.
- (a) J. K. Barton, J. M. Goldberg, C. V. Kumar and N. J. Turro, J. Am. Chem. Soc., 1986, 108, 2081–2088 CrossRef CAS; (b) C. V. Kumar, J. K. Barton and N. J. Turro, J. Am. Chem. Soc., 1985, 107, 5518–5523 CrossRef CAS; (c) A. Papakyriakou, G. Malandrinos and A. Garoufis, J. Inorg. Biochem., 2006, 100, 1842–1848 CrossRef CAS PubMed; (d) H. Ahmad, A. Wragg, W. Cullen, C. Wombwell, A. J. H. M. Meijer and J. A. Thomas, Chem.–Eur. J., 2014, 20, 3089–3096 CrossRef CAS PubMed; (e) I. Gamba, I. Salvadó, G. Rama, M. Bertazzon, M. I. Sánchez, V. M. Sánchez-Pedregal, J. Martínez-Costas, R. F. Brissos, P. Gamez, J. L. Mascareñas, M. Vázquez López and M. E. Vázquez, Chem.–Eur. J., 2013, 19, 13369–13375 CrossRef CAS PubMed.
- (a) A. Greguric, I. D. Greguric, T. W. Hambley, J. R. Aldrich-Wright and J. G. Collins, J. Chem. Soc., Dalton Trans., 2002, 849–855 RSC; (b) H. Niyazi, J. P. Hall, K. O'Sullivan, G. Winter, T. Sorensen, J. M. Kelly and C. J. Cardin, Nat. Chem., 2012, 4, 621–628 CrossRef CAS; (c) J. P. Hall, P. M. Keane, H. Beer, K. Buchner, G. Winter, T. L. Sorensen, D. J. Cardin, J. A. Brazier and C. J. Cardin, Nucleic Acids Res., 2016, 44, 9472–9482 CrossRef CAS PubMed; (d) H. Song, J. T. Kaiser and J. K. Barton, Nat. Chem., 2012, 4, 615–620 CrossRef CAS; (e) A. N. Boynton, L. Marcélis and J. K. Barton, J. Am. Chem. Soc., 2016, 138, 5020–5023 CrossRef CAS PubMed.
- A. C. Komor and J. K. Barton, Chem. Commun., 2013, 49, 3617–3630 RSC.
- S. Neidle, Nat. Prod. Rep., 2001, 18, 291–309 RSC.
- (a) C. Kaes, A. Katz and M. W. Hosseini, Chem. Rev., 2000, 100, 3553–3590 CrossRef CAS; (b) T. Wang, N. Zabarska, Y. Wu, M. Lamla, S. Fischer, K. Monczak, D. Y. W. Ng, S. Rau and T. Weil, Chem. Commun., 2015, 51, 12552–12555 RSC; (c) E. Wachter, D. K. Heidary, B. S. Howerton, S. Parkin and E. C. Glazer, Chem. Commun., 2012, 48, 9649–9651 RSC.
- (a) O. Vázquez, M. I. Sánchez, J. Martínez-Costas, M. E. Vázquez and J. L. Mascareñas, Org. Lett., 2010, 12, 216–219 CrossRef PubMed; (b) O. Vázquez, M. I. Sánchez, J. L. Mascareñas and M. E. Vázquez, Chem. Commun., 2010, 46, 5518–5520 RSC; (c) M. I. Sánchez, O. Vázquez, J. Martínez-Costas, M. Eugenio Vázquez and J. L. Mascareñas, Chem. Sci., 2012, 3, 2383–2387 RSC; (d) R. Nanjunda and W. D. Wilson, Curr. Protoc. Nucleic Acid Chem., 2012, http://https://currentprotocols.onlinelibrary.wiley.com/doi/abs/10.1002/0471142700.nc0808s51 Search PubMed; (e) E. Pazos, J. Mosquera, M. E. Vázquez and J. L. Mascareñas, ChemBioChem, 2011, 12, 1958–1973 CrossRef CAS.
- (a) A. Paul, A. Kumar, R. Nanjunda, A. A. Farahat, D. W. Boykin and W. D. Wilson, Org. Biomol. Chem., 2017, 15, 827–835 RSC; (b) P. Guo, A. A. Farahat, A. Paul, N. K. Harika, D. W. Boykin and W. D. Wilson, J. Am. Chem. Soc., 2018, 140, 14761–14769 CrossRef CAS PubMed.
- (a) X. Hua and A. Von Zelewsky, Inorg. Chem., 1991, 30, 3796–3798 CrossRef CAS; (b) X. Hua and A. von Zelewsky, Inorg. Chem., 1995, 34, 5791–5797 CrossRef CAS; (c) M. Brissard, O. Convert, M. Gruselle, C. Guyard-Duhayon and R. Thouvenot, Inorg. Chem., 2003, 42, 1378–1385 CrossRef CAS.
- (a) C. Fu, M. Wenzel, E. Treutlein, K. Harms and E. Meggers, Inorg. Chem., 2012, 51, 10004–10011 CrossRef CAS PubMed; (b) S. G. Telfer, T. M. McLean and M. R. Waterland, Dalton Trans., 2011, 40, 3097–3108 RSC.
- (a) C. V. Kumar, J. K. Barton and N. J. Turro, J. Am. Chem. Soc., 1985, 107, 5518–5523 CrossRef CAS; (b) M. R. Gill, H. Derrat, C. G. W. Smythe, G. Battaglia and J. A. Thomas, ChemBioChem, 2011, 12, 877–880 CrossRef CAS PubMed; (c) M.-J. Han, L.-H. Gao, Y.-Y. Lü and K.-Z. Wang, J. Phys. Chem. B, 2006, 110, 2364–2371 CrossRef CAS; (d) S. Arounaguiri and B. G. Maiya, Inorg. Chem., 1999, 38, 842–843 CrossRef CAS PubMed; (e) C. Hiort, P. Lincoln and B. Norden, J. Am. Chem. Soc., 1993, 115, 3448–3454 CrossRef CAS.
- P. Thordarson, Chem. Soc. Rev., 2010, 40, 1305–1323 RSC.
- (a) D. Roeland Boer, A. Canals and M. Coll, Dalton Trans., 2008, 399–414 Search PubMed; (b) C. Bailly and J. B. Chaires, Bioconjugate Chem., 1998, 9, 513–538 CrossRef CAS; (c) B. S. Reddy, S. M. Sondhi and J. W. Lown, Pharmacol. Ther., 1999, 84, 1–111 CrossRef CASM. A. Ismail, R. K. Arafa, R. Brun, T. Wenzler, Y. Miao, W. D. Wilson, C. Generaux, A. Bridges, J. E. Hal and D. W. Boykin, J. Med. Chem., 2006, 49, 5324–5332 CrossRef CAS.
- Although it has been reported that water-mediated contacts can compensate for poor shape complementarity, the length of the bipyridine ligand (more than 20 Å between the aminidium carbon atoms) would likely not allow the mechanism to operate. (a) B. Nguyen, S. Neidle and W. D. Wilson, Acc. Chem. Res., 2009, 42, 11–21 CrossRef CAS PubMed; (b) B. Nguyen, M. P. H. Lee, D. Hamelberg, A. Joubert, C. Bailly, R. Brun, S. Neidle and W. D. Wilson, J. Am. Chem. Soc., 2002, 124, 13680–13681 CrossRef CAS PubMed.
- A. B. Tossi and J. M. Kelly, Photochem. Photobiol., 1989, 49, 545–556 CrossRef CAS PubMed.
- N. Holmgaard List, J. Knoops, J. Rubio-Magnieto, J. Idé, D. Beljonne, P. Norman, M. Surin and M. Linares, J. Am. Chem. Soc., 2017, 139, 14947–14953 CrossRef CAS PubMed.
- The changes in the CD spectra are different to those observed with intercalative complexes: S. D. Choi, M. S. Kim, S. K. Kim, P. Lincoln, E. Tuite and B. Nordén, Biochemistry, 1997, 36, 214–223 CrossRef CAS PubMed.
- Circular dichroism experiments require high concentrations (5–10 times the KDs), which is why no differences are observed between Δ-4Ru and Λ-4Ru using this technique.
- T. A. Larsen, D. S. Goodsell, D. Cascio, K. Grzeskowiak and R. E. Dickerson, J. Biomol. Struct. Dyn., 1989, 7, 477–491 CrossRef CAS PubMed.
- Earlier studies have shown that DAPI is particularly suited for the study of DNA-binding complexes through competitive titrations: C. B. Spillane, J. A. Smith, J. L. Morgan and F. R. Keene, J. Biol. Inorg Chem., 2007, 12, 819–824 CrossRef CAS PubMed.
- A. Rodger, G. Dorrington and D. L. Ang, Analyst, 2016, 141, 6490–6498 RSC.
- P. Lincoln, A. Broo and B. Nordén, J. Am. Chem. Soc., 1996, 118, 2644–2653 CrossRef CAS.
- D. Bouzada, I. Salvadó, G. Barka, G. Rama, J. Martínez-Costas, R. Lorca, Á. Somoza, M. Melle-Franco, M. E. Vázquez and M. Vázquez López, Chem. Commun., 2018, 54, 658–661 RSC.
- L. Lercher, M. A. McDonough, A. H. El-Sagheer, A. Thalhammer, S. Kriaucionis, T. Brown and C. J. Schofield, Chem. Commun., 2014, 50, 1794–1796 RSC (PDB ID: 4C64).
- Data for [Ru(bpy)2phen]2+ from: P. Lincoln and B. Nordén, J. Phys. Chem. B, 1998, 102, 9583–9594 CrossRef CAS.
- Model built with 3dna software: A. V. Colasanti, X.-J. Lu and W. K. Olson, J. Visualized Exp., 2013, e4401 Search PubMed.
- (a) B. J. Pages, D. L. Ang, E. P. Wright and J. R. Aldrich-Wright, Dalton Trans., 2015, 44, 3505–3526 RSC; (b) C. Oguey, N. Foloppe and B. Hartmann, PLoS One, 2010, 5, e15931 CrossRef CAS PubMed; (c) S. Laughlin-Toth, E. K. Carter, I. Ivanov and W. D. Wilson, Nucleic Acids Res., 2017, 45, 1297–1306 CrossRef CAS PubMed; (d) P. M. Keane, F. E. Poynton, J. P. Hall, I. V. Sazanovich, M. Towrie, T. Gunnlaugsson, S. J. Quinn, C. J. Cardin and J. M. Kelly, Angew. Chem., Int. Ed., 2015, 54, 8364–8368 CrossRef CAS PubMed; (e) P. M. Keane, J. P. Hall, F. E. Poynton, B. C. Poulsen, S. P. Gurung, I. P. Clark, I. V. Sazanovich, M. Towrie, T. Gunnlaugsson, S. J. Quinn, C. J. Cardin and J. M. Kelly, Chemistry, 2017, 23, 10344–10351 CrossRef CAS PubMed; (f) J. P. Hall, F. E. Poynton, P. M. Keane, S. P. Gurung, J. A. Brazier, D. J. Cardin, G. Winter, T. Gunnlaugsson, I. V. Sazanovich, M. Towrie, C. J. Cardin, J. M. Kelly and S. J. Quinn, Nat. Chem., 2015, 7, 961–967 CrossRef CAS PubMed.
- C. Laughton and B. Luisi, J. Mol. Biol., 1999, 288, 953–963 CrossRef CAS PubMed.
- Molecular graphics obtained with UCSF Chimera, by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311. E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- S. Sprouse, K. A. King, P. J. Spellane and R. J. Watts, J. Am. Chem. Soc., 1984, 106, 6647–6653 CrossRef CAS.
- J. N. Demas, E. W. Harris and R. P. McBride, J. Am. Chem. Soc., 1977, 99, 3547–3551 CrossRef CAS.
- M. B. Fleisher, K. C. Waterman, N. J. Turro and J. K. Barton, Inorg. Chem., 1986, 25, 3549–3551 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03053k |
| This journal is © The Royal Society of Chemistry 2019 |