
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The chemical reactions in electrosprays of water do not always correspond to those at the pristine air–water interface†
Adair
Gallo
Jr
 abc,
Andreia S. F.
Farinha
abc,
Miguel
Dinis
ad,
Abdul-Hamid
Emwas
ae,
Adriano
Santana
abc,
Robert J.
Nielsen
f,
William A.
Goddard
III
f and
Himanshu
Mishra
*abc
abc,
Andreia S. F.
Farinha
abc,
Miguel
Dinis
ad,
Abdul-Hamid
Emwas
ae,
Adriano
Santana
abc,
Robert J.
Nielsen
f,
William A.
Goddard
III
f and
Himanshu
Mishra
*abc
aKing Abdullah University of Science and Technology (KAUST), Saudi Arabia. E-mail: himanshu.mishra@kaust.edu.sa
bWater Desalination and Reuse Center (WDRC), Saudi Arabia
cDivision of Biological and Environmental Sciences (BESE), Saudi Arabia
dKAUST Catalysis Center (KCC), Saudi Arabia
eImaging and Characterization Core Laboratory, Thuwal 23955-6900, Saudi Arabia
fMaterials and Process Simulation Center, California Institute of Technology, Pasadena, CA 91125, USA
First published on 21st December 2018
Abstract
The recent application of electrosprays to characterize the air–water interface, along with the reports on dramatically accelerated chemical reactions in aqueous electrosprays, have sparked a broad interest. Herein, we report on complementary laboratory and in silico experiments tracking the oligomerization of isoprene, an important biogenic gas, in electrosprays and isoprene–water emulsions to differentiate the contributions of interfacial effects from those of high voltages leading to charge-separation and concentration of reactants in the electrosprays. To this end, we employed electrospray ionization mass spectrometry, proton nuclear magnetic resonance, ab initio calculations and molecular dynamics simulations. We found that the oligomerization of isoprene in aqueous electrosprays involved minimally hydrated and highly reactive hydronium ions. Those conditions, however, are non-existent at pristine air–water interfaces and oil–water emulsions under normal temperature and pressure. Thus, electrosprays should be complemented with surface-specific platforms and theoretical methods to reliably investigate chemistries at the pristine air–water interface.
The air–water interface plays a critical role in numerous natural and applied contexts, such as atmospheric chemistries,1–3 precipitation,4 spray coatings,5 and materials synthesis.6,7 Indeed, it has been hypothesized that microdroplets generated during the splashing of waves in oceans could have been the chemical reactors leading to the origin of life.8–10 Despite its ubiquity and importance, a variety of fundamental phenomena at the air–water interface remain incompletely understood, such as the specific adsorption of ions11–13 and chemistries therein.10,14–20 The interfacial region, with a typical thickness δo ≈ 0.5 nm, separates the gas-phase (vapor) from the condensed phase (water), two drastically different regions in terms of hydration – reactions spontaneous in one phase are forbidden in the other.21 In fact, the chemical activities of species at the air–water interface can depart significantly from those in the bulk, as has been demonstrated by surface-specific techniques, including vibrational second harmonic generation and sum frequency generation,20,22,23 and polarization-modulated infrared absorption reflection spectroscopy,10,13 and indirect approaches, including NMR24 and confocal fluorescence microscopy.25 Even though vibrational spectroscopy-based techniques report directly on thermodynamic properties of the air–water interface, they suffer from interpretational ambiguities and limitations due to low signal-to-noise ratios.26–32 Thus, new techniques with higher sensitivity and unambiguous response are needed to help resolve the poorly understood features of the air–water interface while providing benchmarks to judge previous interpretations.33 In this work, we assess the application of electrospray ionization mass spectrometry (ESIMS) to unravel the thermodynamic properties of pristine air–water interface (Henceforth, we will use the qualifier ‘pristine’ to refer to the air–water interface that is not under the influence of any external sources/agents, such as electrical voltage or a drying gas).
In the recent years, ESIMS, which has been widely used to characterize ionic/molecular species in polar/apolar solvents,34 has been adapted to investigate the pristine air–water interface. In the standard configuration, ESIMS experiments entail the formation of electrosprays by the application of electrical potential and/or pneumatic pressure leading microscale droplets with excess electrical charge; those microdroplets pass through a glass/metallic capillary maintained at elevated temperature (∼473 K) to evaporate the solvent and facilitate the mass spectrometric detection of analytes downstream.34–38 In the experiments designed to investigate chemistries at the air–water interface, electrosprays containing one or more reactant(s) are intersected with gases or other electrosprays containing other reactant(s) followed by mass spectrometric detection. For instance, using this platform, thermodynamic properties of the pristine air–water interface have been explored under ambient conditions, including the relative concentrations of interfacial hydronium and hydroxide ions and their activities39–43 leading to interpretations that have elicited scientific debate.11,12,17–19,44,45 Further, by intersecting electrosprays of pH-adjusted water with gaseous organic acids,40 isoprene,41 and terpenes,40,41,46,47 researchers observed instantaneous protonation (<1 ms), and in some cases oligomerization of organics, which led them to conclude that as the bulk acidity of water approaches pH ≤ 3.6, the pristine air–water interface behaves as a superacid. While a clear understanding of the emergence of the putative superacidity at the air–water interface is unavailable, we note that in the condensed phase proton-catalyzed oligomerization of isoprene (or olefins in general) requires 60–80% concentrated H2SO4 solutions (pH < −0.5).48 Similar rate enhancements in aqueous electrosprays have also been observed for the syntheses of abiotic sugar phosphates,15,49 the Pomeranz–Fritsch synthesis of isoquinoline,50 the reaction between o-phthalaldehyde and alanine,51 and the ozonation of oleic acid,52 among others.16,53 Herein, we assess the relationships between the chemistries observed in aqueous electrosprays to those at pristine air–water interfaces; we also seek to decouple the factors that contribute to the mechanisms underlying reported dramatic rate enhancements by addressing the following questions:
(i) Do aqueous electrospray-based platforms report on thermodynamic properties of the pristine air–water interface?
(ii) Do accelerated reactions in aqueous electrosprays arise only from the significant enhancement of the hydrophobe–water (air–water) interfacial area? If yes, the mechanisms underlying the dramatic rate enhancements therein should be insightful in explaining the accelerated organic reactions in oil–water emulsions also referred to as ‘on-water’ catalysis.54–57
(iii) Are the rate accelerations in aqueous electrosprays driven solely by the non-equilibrium conditions therein, especially the enhanced concentration of reactants in the micro-/nano-droplets due to the evaporation of water?58–61
(iv) Are gas-phase reactions implicated in the acceleration of chemical reactions in aqueous electrosprays?36,44,50,62–64
To address those questions, we investigated the oligomerization of isoprene by proton nuclear magnetic resonance (1H-NMR), a non-invasive technique, as a complementary platform to the ESIMS. Questions (i and ii) were addressed by comparing the effects of enhancing the water–hydrophobe interfacial area in both liquid–vapor and liquid–liquid systems; questions (iii and iv) were addressed by varying the capillary voltages, ionic strengths of the aqueous solutions electrosprayed and intersected with gas-phase isoprene, and 1H-NMR analysis of condensed vapor from the electrosprays. To highlight the role of hydration in electrosprays, we performed quantum mechanical calculations employing density functional theory (M06 flavor).
Materials and methods
In our experiments, we used isoprene (99% purity from Sigma-Aldrich), Mili-Q deionized water (18 MΩ m resistivity), D2O (99.9% purity from Sigma Aldrich), ethanol (absolute from Merck Millipore), acetone (HPLC standard from VWR Chemicals), NaCl (>99% purity from Sigma Aldrich), HCl (36.9% concentration from Fisher Scientific), DCl (35% concentration 99% deuterium purity from Sigma Aldrich), and NaOH (>97% purity from Sigma Aldrich) to adjust the pH and ionic strengths.ESIMS
All experiments were conducted in a commercial Thermo Scientific – LCQ Fleet electrospray ionization mass spectrometer in the positive ion mode, where a DC potential of 6–8 kV was applied to the needle, the tube lens voltage was 75 V, the sheath gas flow rate was 10 arb, the pressure was 1.2 torr at the convection gauge and 0.8 × 10−5 torr at the ion-gauge, the flow rates of analytes were controlled by a calibrated syringe pump and ranged between 1–10 μL min−1, the distances from the ion source and the inlet to the mass spectrometer were ∼2 cm, and the distance between the electrospray and the tube ejecting isoprene was 1 cm.1H-NMR
All NMR spectra were acquired using a Bruker 700 AVANAC III spectrometer equipped with a Bruker CP TCI multinuclear CryoProbe (BrukerBioSpin, Rheinstetten, Germany); Bruker Topspin 2.1 software was used to collect and analyze the data. We transferred 100 μL of the (A1) samples into 5 mm NMR tubes, followed by 600 μL of deuterated chloroform (CDCl3). The 1H-NMR spectra were recorded at 298 K by collecting 32 scans with a recycle delay of 5 s, using a standard 1D 90° pulse sequence and standard (zg) program from the Bruker pulse library. The chemical shifts were adjusted using tetramethylsilane (TMS) as an internal chemical shift reference. The (A) samples and a sample of as-purchased isoprene (B) were prepared by transferring 100 μL of each to 5 mm NMR tubes, and then adding 550 μL of deuterated water D2O to the NMR tubes. The 1H-NMR spectra were recorded by collecting 512 scans with a recycle delay time of 5 s, using an excitation sculpting pulse sequence (zgesgp) program from the Bruker pulse library. The chemical shifts were adjusted using 3-trimethylsilylpropane sulfonic acid (DSS) as an internal chemical shift reference. The free induction decay (FID) data were collected at a spectral width of 16 ppm into 64 k data points. The FID signals were amplified by an exponential line-broadening factor of 1 Hz before Fourier transformation.Computational methods
To gain molecular-level insights into the protonation and oligomerization of the isoprene in our experiments, we performed (and benchmarked) DFT calculations at the following levels: (A) geometry and transition state optimization with M06/6-311G* followed by single point calculations with a larger basis set (6-311++G**);65 (B) geometry and transition state optimization with M06/6-311++G**; (C) geometry and transition state optimization with M06/6-311++G**, followed by single point calculations with CCSD(T).66 We calculated the internal reaction coordinate (IRC) along the potential energy surface connecting adduct (A), transition state (TS), and product (P). Frequency calculations of the optimized geometries yielded Hessians with zero and one imaginary frequency, respectively, for the minima and the transition state structures. The force constants from the frequency calculations on the TS structures were subsequently used to calculate the IRC path along the forward and backward direction of the transition vector, connecting transition state to the minima.67,68 The vibrational frequencies from the Hessians were also used to provide the zero-point energies and vibrational contributions to the enthalpies and entropies. The free energies of isoprene at 1 atm were calculated using statistical mechanics for ideal gases.Molecular simulations
Born–Oppenheimer molecular dynamics (BOMD)66 simulations were carried out with the M06 functional and 6-311G(d,p) basis set in the NVT ensemble coupled with a Nose–Hoover thermostat69 at 298 K. The total simulation times were 5 ps with 1 fs timesteps.We used the Gaussian 09 software package70 to perform these calculations and simulations.
Results
We investigated chemical reactions between pH-adjusted water and isoprene (C5H8, 2-methyl-l,3-butadiene, MW = 68 amu, and solubility in water, S = 0.7 g L−1 at normal temperature and pressure (NTP): 293 K and 1 atm). We chose to examine reactions of isoprene because (i) we wanted to reproduce previous experimental results to ensure a clear comparison, (ii) isoprene is an important biogenic gas whose fate in the atmosphere is not completely understood,1,41,71,72 and (iii) we could investigate chemistries in electrosprays and emulsions by taking advantage of the low boiling point of isoprene (Tb = 307 K) and the high vapor pressure at NTP (p = 61 kPa).73As delineated in Fig. 1 and summarized in Table 1, we report on the following sets of ESIMS (detection limit = ∼1 nM) and 1H-NMR (detection limit = ∼10 μM) experiments:
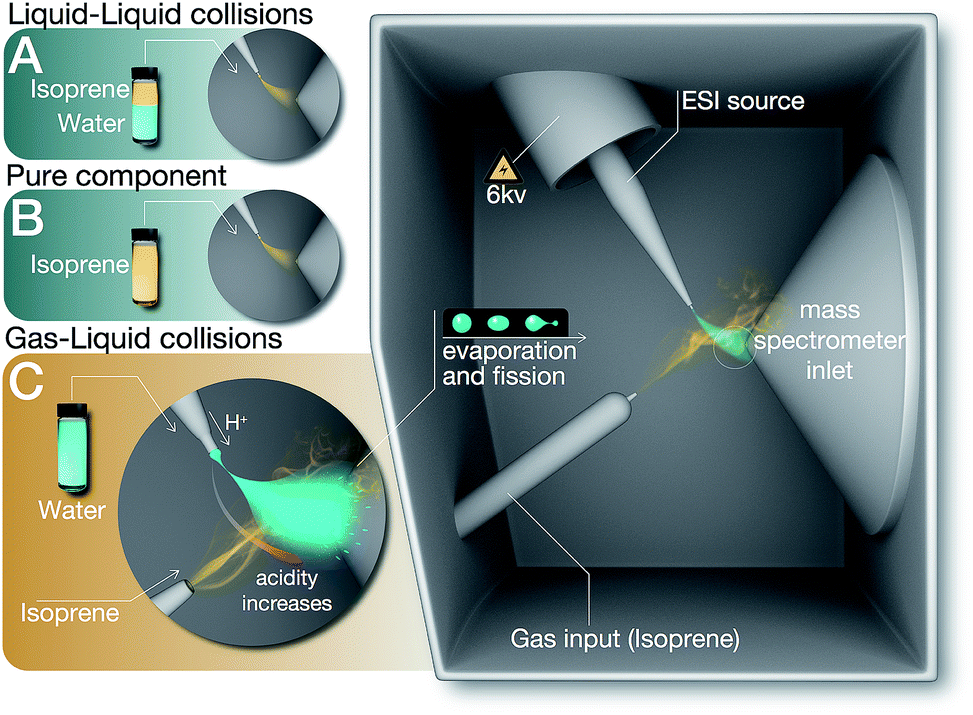 | ||
Fig. 1 Summary of the experiments (A), (B), and (C) reported in this work. (A) Liquid–liquid collisions: mixtures of isoprene, pH-adjusted water, and air in the volumetric ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:3 was agitated at 1200 rpm (for 6, 60, 360 minutes) followed by ESIMS analysis of the organic phase. (B) As-purchased liquid isoprene was injected directly in the ESIMS. (C) Gas–liquid collisions: electrosprays of water (pH range 1–13) were collided with a stream of air carrying isoprene gas, followed by mass spectrometric detection (Methods). :6:3 was agitated at 1200 rpm (for 6, 60, 360 minutes) followed by ESIMS analysis of the organic phase. (B) As-purchased liquid isoprene was injected directly in the ESIMS. (C) Gas–liquid collisions: electrosprays of water (pH range 1–13) were collided with a stream of air carrying isoprene gas, followed by mass spectrometric detection (Methods). | ||
| (A) Liquid–liquid collisions | (B) Pure components | (C) Gas–liquid collisions | |
|---|---|---|---|
| Water(L)–isoprene(L) | Isoprene(L), acetone(L), ethanol(L) | Isoprene(G)–water(L) | |
| ESIMS | Organics injected | Components injected | Water injected |
| pH | 1–13 | — | 1–13 |
| pNaCl | — | — | 1–9 |
| Shaking time | 6, 60, 360 min | — | — |
| Voltage | 6 kV | 6 kV | 6–8 kV |
| Capillary temperature | 150 °C | 30–330 °C | 150 °C |
| 1 H-NMR | Organics from (A) and condensed vapors (A1) | Isoprene(L) | — |
| pH | 1.5 | — | — |
| Shaking time | 6, 60, 360 min | — | — |
| Aqueous phase | D2O, H2O | — | — |
(A) Liquid–liquid collisions
At NTP, we combined liquid isoprene with pH-adjusted H2O or D2O, 1 ≤ pH ≤ 13, in a volumetric ratio 1:6:3 (isoprene:water:air), agitated the emulsions at 1200 rpm in a vortexer for 6, 60, or 360 minutes, and analyzed the organic phases after phase-separation by ESIMS and 1H-NMR. Since the air in the reaction vessels was saturated with isoprene, those experiments also ensured the presence of the products of reactions between the gas-phase isoprene and pH-adjusted water in the organic phase.
(B) Pure components
We analyzed as-purchased isoprene, acetone, and ethanol by ESIMS and 1H-NMR.(C) Gas–liquid collisions
We created electrosprays of aqueous solutions with varying ionic strengths and pH, and intersected them with a stream of gas-phase isoprene (0.48 g min−1 carried by N2 gas flowing at 600 mL min−1, i.e. isoprene gas concentration was 800 mg L−1) followed by mass spectrometric detection (Methods).Hereafter, throughout the paper, we will refer to our experiments on the liquid–liquid collisions as (A), condensed vapors from the electrosprays (A1), pure isoprene as (B), and gas–liquid collisions in the ESIMS as (C) (Fig. 1, Table 1).
Intriguingly, the ESIMS spectra from the above-mentioned experiments (A) at pH = 1, (B), and (C) at pH = 1 were nearly identical after normalizing with the maximum intensity (Fig. 2A–C and Section Sa†). The positions of the main peaks in the mass spectra fitted the general formula, [(Isop)n·H]+, which corresponded to covalently bonded oligomers of isoprene with one excess proton. In Section Sb and Fig. S1† we present the evidence proving that the peaks did not correspond to physisorbed clusters. In experiments (A), the mass spectra remained the same as the duration of mixing varied from 6 min to 6 h (Fig. S2†). We also observed numerous secondary peaks between the primary [(Isop)n·H]+ peaks. While a detailed characterization of those peaks falls beyond the scope of this work, we speculate that they are composed of C1–C4 fragments covalently bonded with the [(Isop)n·H]+ oligomers, for example m/z = 233 (Fig. 2C and S3†).
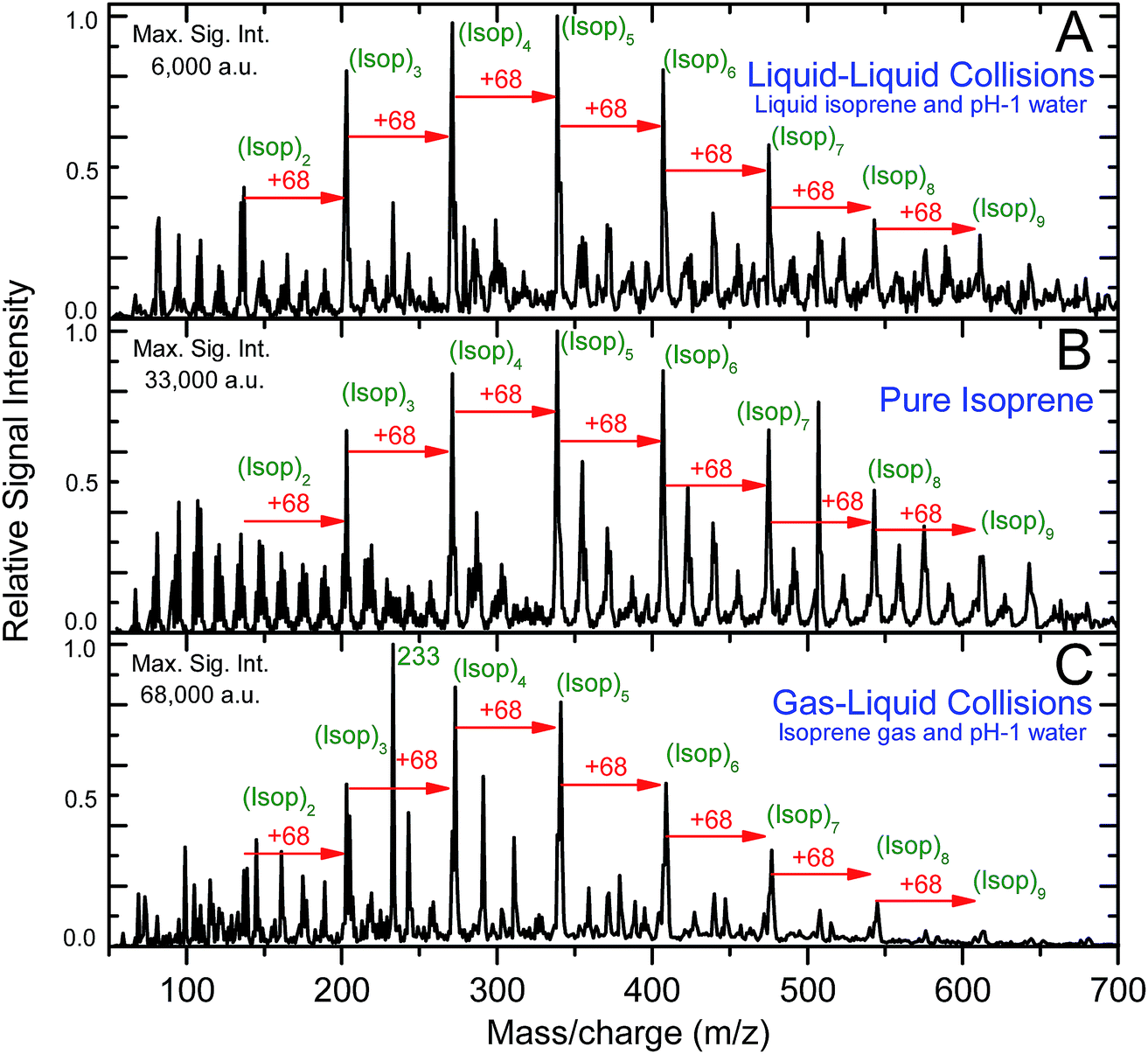 | ||
| Fig. 2 ESIMS spectra for sets of experiments A, B, and C: the dominant peaks correspond to protonated oligomers of isoprene, [(Isop)n·H]+, and the secondary peaks correspond to fragments of the isoprene molecules bonded to the primary oligomers. (A) ESIMS spectra of the organic phase from the emulsion of liquid isoprene in water at pH = 1 and air (1:6:3 v/v/v) that was agitated at 1200 rpm for 360 minutes. (B) ESIMS spectra of as-purchased liquid isoprene. (C) ESIMS spectra of products of gas–liquid collisions between water (pH = 1) and gas-phase isoprene (Methods). | ||
Next, we investigated the role of water pH on the oligomerization of isoprene in experiments (A) and (C). When the products were characterized by ESIMS, we noticed that the oligomers [(Isop)n·H]+ appeared when the aqueous phase had pH ≤ 3.6 (Fig. 3). Those observations have been reported previously18,40,41,46,47 and ascribed to the superacidity of the air–water interface at pH ≤ 3.6. However, we also found that the ESIMS spectra from both experiments, (A) and (C), yielded oligomers [(Isop)n·H]+ for the acidic, basic, and pH-neutral salty solutions (Fig. 3; compare Fig. 2A and C with Fig. S3† panels C1, C2, and C3). We note that for electrosprays produced from pH > 7 water and salty water, counterions, such as Na+, could influence the fate of reactions, but we have not investigated those factors.
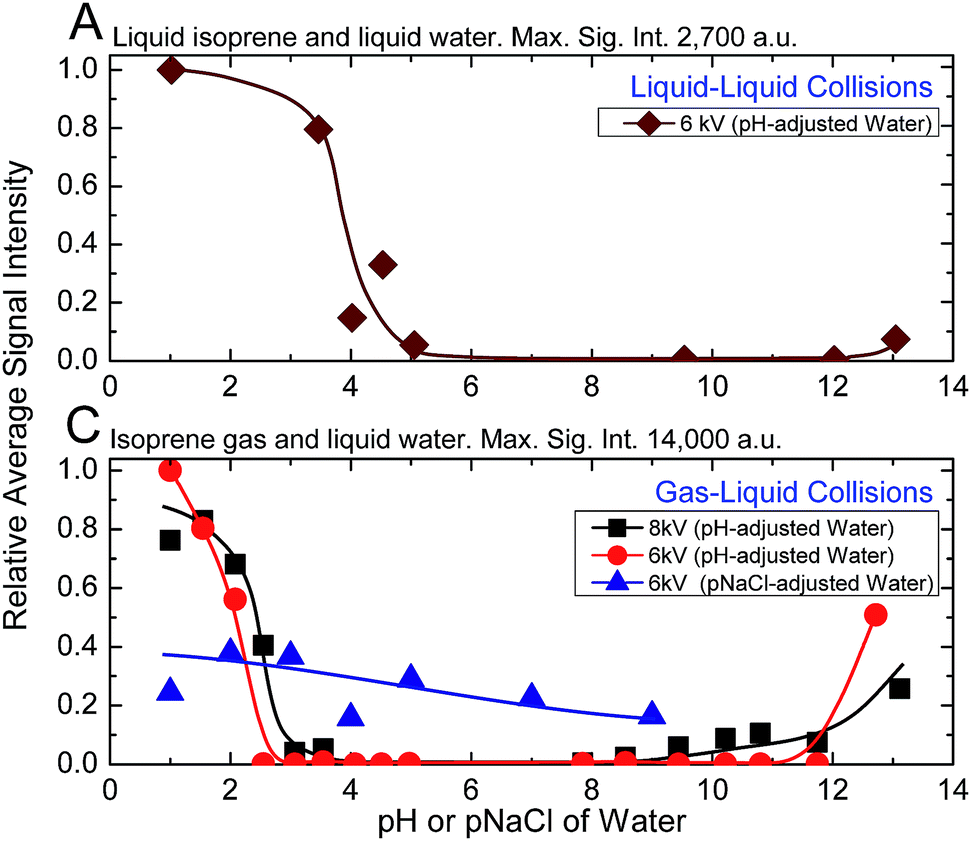 | ||
Fig. 3 Influence of the ionic strength and ESI voltage on the ESIMS spectra of experimental sets (A) and (C). On the y-axis, we plot the average mass spectral intensity of all the oligomeric peaks [(Isop)n·H]+, given by 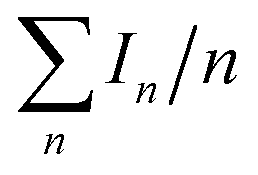 , normalized by the highest datum in each plot. (A) Liquid–liquid collisions: the ESIMS spectra demonstrated protonation and oligomerization of isoprene after emulsions of isoprene in water with pH ≤ 3.6 and pH > 12 and air in a 1:6:3 ratio (v/v/v) were agitated at 1200 rpm for 360 minutes. (C) Gas–liquid collisions: the ESIMS spectra demonstrated protonation and oligomerization of isoprene gas after collision with electrosprays of water with pH ≤ 3.6 and pH > 12, and pH-neutral salty solutions. Curves are added to the plots to aid visualization. , normalized by the highest datum in each plot. (A) Liquid–liquid collisions: the ESIMS spectra demonstrated protonation and oligomerization of isoprene after emulsions of isoprene in water with pH ≤ 3.6 and pH > 12 and air in a 1:6:3 ratio (v/v/v) were agitated at 1200 rpm for 360 minutes. (C) Gas–liquid collisions: the ESIMS spectra demonstrated protonation and oligomerization of isoprene gas after collision with electrosprays of water with pH ≤ 3.6 and pH > 12, and pH-neutral salty solutions. Curves are added to the plots to aid visualization. | ||
In experiments (A), after the emulsions comprising liquid isoprene, liquid water at pH = 1.5, and air (containing saturated gaseous isoprene) were vigorously mixed (for 6 min, 60 min, and 360 min) we compared the organic layers after phase separation by 1H-NMR. We also recorded the 1H-NMR spectra of pure, as-purchased isoprene (B). To our surprise, the 1H-NMR spectra from all of the set (A) samples were identical to those of set (B), indicating that the effect of the duration of shaking (6–360 min), the pH (1–13), the isotope (H2O versus D2O) and the presence of gaseous isoprene colliding with pH-adjusted water did not lead to any oligomers within the detection limit of 10 μM (Fig. 4A and B). To investigate further, we condensed the vapors from the ESIMS exhaust (A1) after injecting the set (A) samples (1 ≤ pH ≤ 13), and obtained their 1H-NMR spectra. All of (A1) samples showed spectra similar to each other (Fig. 4A1). The 1H-NMR spectra of (A) and (B) showed no sign of oligomers in the products: they contained a singlet at 1.87 ppm due to the resonance of the 3 protons in CH3; three dublets at 5.02 ppm due to the resonance of the two protons H5b and H5b (coupling constant, J = 13.2 Hz), 5.09 ppm due to the resonance of H1a with a cis-coupling constant, J = 10.8 Hz; a dublet at 5.20 ppm due to the resonance of H1b with a trans-coupling constant of J = 17.5 Hz; and two dublets at 6.47 ppm due to the resonance of the protons H2a and H2b with the corresponding trans- and cis-coupling constants, J = 17.5 Hz and J = 10.8 Hz. In contrast, the 1H-NMR spectra of the condensed vapors from the electrosprays (A1) of the organic phase after the liquid–liquid collisions (A) demonstrated a dramatic increase in the complexity of the spectrum,74 indicating that the protonation and oligomerization of isoprene took place exclusively in the electrosprays.
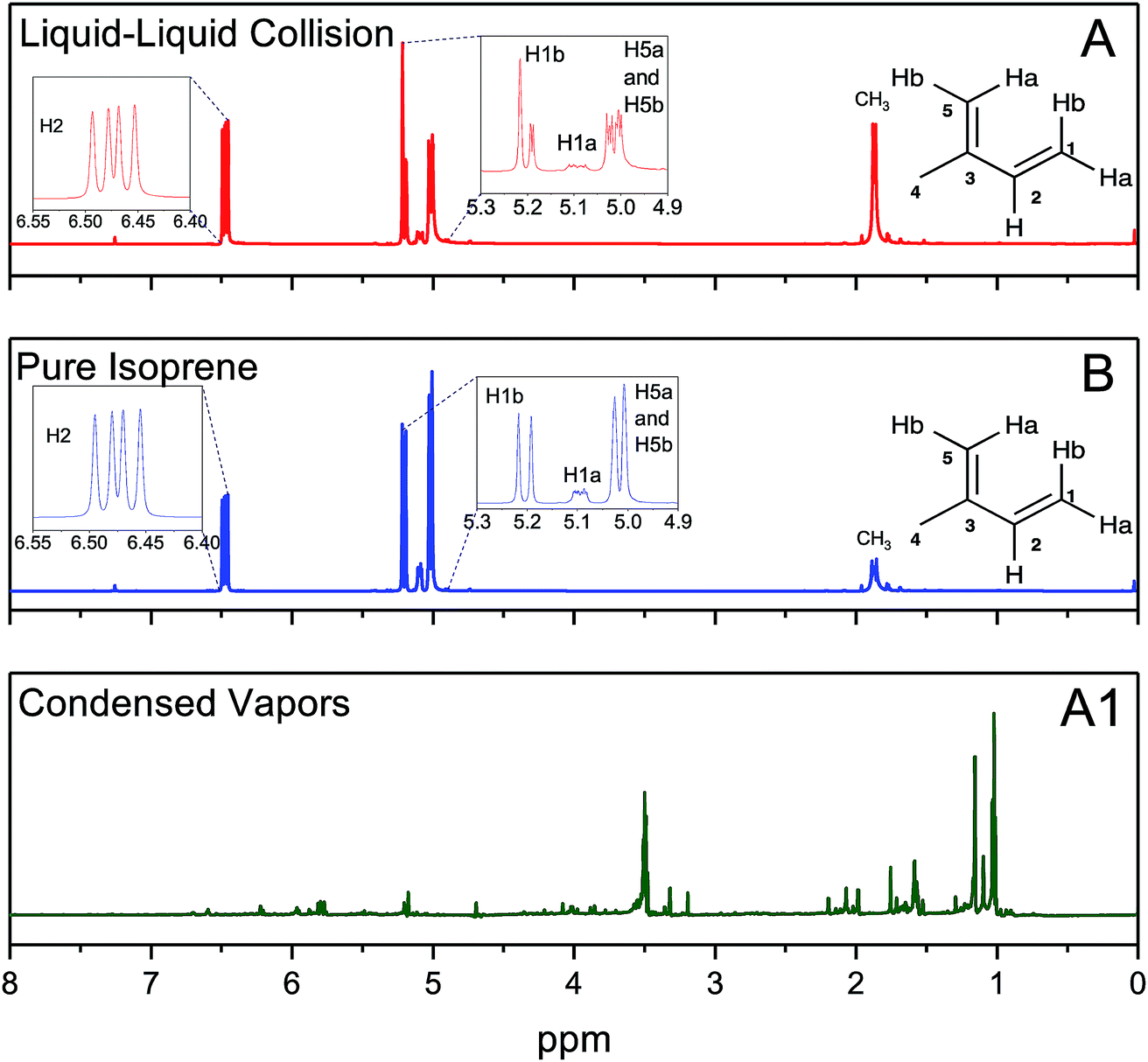 | ||
| Fig. 4 (A) 1H-NMR spectra of the organic phase after shaking liquid isoprene with pH 1.5 water and air in a volumetric ratio of 1:6:3 for 60 minutes. (B) 1H-NMR spectra of as-purchased isoprene. (A1) 1H-NMR spectra of the condensed exhaust from the electrosprays of the organic phase after the liquid–liquid collision (A) experiments. The nearly identical spectra for experimental sets (A) and (B) demonstrate that there was no detectable oligomerization of isoprene during the vigorous shaking of emulsions comprised of liquid isoprene with pH 1.5 water and air in a volumetric ratio of 1:6:3 (NMR resolution ∼10 μM). | ||
Discussion
Our investigation of experiments (A) with 1H-NMR revealed that a significant enhancement in the hydrophobe–water surface area was not sufficient for observable rate accelerations in emulsions of isoprene (gas and liquid) with pH-adjusted water at NTP conditions. On the other hand, analysis of experiments (A), (B), and (C) by ESIMS and experiments (A1) with 1H-NMR unambiguously demonstrated that the chemical reactions took place exclusively in aqueous electrosprays – the acidity, basicity, and saltiness of water all promoted the reactions. Further, as the capillary voltage was increased from 6 kV to 8 kV, the inflection points in experiments (A) and (C) shifted such that the oligomers [(Isop)n·H]+ were detected at lower ionic strengths (Fig. 3C). Collectively, these findings contradict previous claims of ‘superacidity’ of pristine air–water interfaces at pH ≤ 3.6.Next, we sought to identify the mechanisms underlying the protonation and oligomerization of isoprene in electrosprays of mildly acidic water (experiments C). As discussed above, a variety of parameters could influence reactions therein, including electrical voltage, salts, pH, electrochemical reactions, concentration of reactants in rapidly evaporating drops, and gas-phase reactions.34–36,44,50,63,64,75–79 Interestingly, by monitoring the changes in the surface tension of pendant water drops exposed to isoprene gas, we found that gas-phase isoprene molecules could adsorb at the air–water interface under NTP conditions (Fig. S4†). While the adsorption of non-polar molecules at the air–water interface might appear unexpected, similar phenomenon at the macroscale, entailing the adsorption of hydrophobic particles onto water drops of size 10−3 m in air forming ‘liquid marbles’ is well known.80 Thus, gas-phase isoprene molecules (partial pressure in our chamber: 0.28 atm) may adsorb onto the positively charged aqueous electrosprays comprising excess protons.62 From this stance, three potential mechanisms for the oligomerization of isoprene emerge, which we discuss and evaluate based on our experimental results and quantum mechanical predictions: mechanism M1 – the adsorption of isoprene molecules onto the electrosprays increases their concentration at the interface, leading to reactions under the influence of high electric fields, similarly to the oligomerization of pure liquid isoprene on injection into ESIMS (Fig. 2B); mechanism M2 – continuous evaporation of positively charged electrosprays renders them increasingly acidic, akin to 50% H2SO4 solutions,48 which drives the liquid-phase oligomerization of the adsorbed isoprene molecules (Section Sc and Fig. S5†); mechanism M3 – high capillary voltages electrolyze mildly acidic water and the positive charge ensures that the electrosprayed drops contain excess protons that, during Coulomb explosions, eject highly reactive water clusters that protonate and oligomerize isoprene molecules in the gas phase. In the gas phase, strictly, protonation takes place if the proton affinity of the donor, e.g., H2O (165 kcal mol−1 at NTP), is lower than that of the acceptor, e.g., Isop (197 kcal mol−1 at NTP).81 This observation has been extensively discussed by Enke and co-workers in the context of water-alcohol mixtures,82 among others;64,77 these proton–transfer reactions also underlie the basis of the Proton Transfer Reaction Mass Spectrometry (PTRMS) that has been used to detect trace gases in the atmosphere.77,83
In all those mechanisms, the initial ionic strength of water (acidic, basic, or salty) and electrical voltage were crucial for the formation of a stable stream of charged microdroplets – the higher the ionic strength of solutions, the lower the requirement for the electrical voltage.35,36,84–86 In fact, due to the electrochemical reactions at the electrospray needle under the influence of high electric fields, the electrosprayed droplets from a positively charged capillary should contain more positive ions than in the bulk58,63,85,87,88 (Section Sc, Fig. S5†). Interestingly, for pH-adjusted water electrosprayed at 6 kV, we detected oligomers (Fig. 3C) when pH ≤ 3.6 or pH > 12, whereas for the NaCl solutions, we observed oligomers at concentrations as low as 10−9 M (pNa = 9). Yet, the higher intensities of the [(Isop)n·H]+ at pH ≤ 3.6 in comparison to the salty solutions (Fig. 3C) indicate that the proposed mechanism M1 is unlikely to play a crucial role in the case of gaseous isoprene interacting with electrosprays of water.
Following our logical exclusion of mechanism M1, we are left with mechanisms M2 and M3, i.e. did the reactions take place on the surface of electrosprayed water droplets or in the gas-phase? Whether or not the electrospray spectra represent the solvent- or gas-phase chemistries/characteristics is a much-debated matter and case-specific.36,38,76,89,90 Obviously, the answer would have a bearing on the questions (ii–iv) outlined above, because the kinetics and thermodynamics of reactions in bulk and gas-phase differ dramatically.21 Recently, Silveira and co-workers employed cryogenic ion mobility mass spectrometry to demonstrate the effects of rapid dehydration on the structures of undecapeptite substance during the final stages of electrospray ionization.90 To further clarify the role of hydration on the protonation and oligomerization of isoprene in our experiments with mildly acidic water (electrosprays and emulsions), we carried out quantum mechanics calculations.
Computational calculations
To gain qualitative molecular-scale insights into our experiments with mildly acidic water, we carried out density functional theory (DFT) calculations (Computational methods). M06, a hybrid meta-generalized gradient approximation (meta-GGA) functional, is known to provide an accurate description of the ground-state thermochemistry, thermochemical kinetics, and transition state structures and energies for a wide series of organic reactions.91–96 We have previously confirmed that the binding energies of water clusters, (H2O)n (range n = 2–8, 20), along with the hydration and neutralization energies of hydroxide and hydronium ions using DFT functionals (M06, M06-2X, M06-L, B3LYP, X3LYP) are in excellent agreement with the high-level theory (CCSD(T)/aug-cc-p VDZ level), both with and without the basis set superposition error correction.91 For instance, our ab initio predictions of the proton transfer thermodynamics between H3O+(g) and Isop(g) was ΔG0 = −30.7 kcal mol−1, in accordance with the difference in the experimental gas-phase basicity (GB) of H2O (GBH2O = −157.7 kcal mol−1) and Isop (GBISO = −190.6 kcal mol−1), ΔGB = −32.9 kcal mol−1 (Fig. S6†).97 Furthermore, the trans- or cis-Isop(g) spontaneously added onto (Isop·H+ + H2O), leading to cyclic (ΔG0 = −40 kcal mol−1) or acyclic monoterpenes (ΔG0 = −9 kcal mol−1), (Fig. S7 and S8†), as also noted by other researchers.48,98 Next, we calculated the kinetic barriers for protonation and oligomerization of Isop on a small water cluster containing an extra proton, (H2O)3·H+, as representative of our electrospray experiments and a larger cluster, (H2O)36·H+, representing the pristine air–water interface. The sizes of the clusters were guided, in principle, by previous experimental and theoretical work on the hydration of protons,99–101 showing the asymptotic stabilization of a proton with increasing cluster size, and limited by computational expense. The initial geometry of the smaller cluster was obtained from the Cambridge Cluster Database,102 while the larger cluster geometry was obtained from an SPC/E bulk water box equilibrated at 298 K and at 1 atm pressure yielding the bulk density of 1 g cm−3. We took a cluster of 36 water molecules from the equilibrated water box as a surrogate for the air–water interface. We added a proton to this cluster and applied Born–Oppenheimer Molecular Dynamics (BOMD) simulations (Computational methods) to obtain various low-energy conformers (Table S2, Section Sd†). We chose a cluster with the proton on the surface that facilitated the subsequent study of the activation barriers through DFT calculations (Computational methods).In the case of (H2O)3·H+, representative of electrosprays, the proton was extremely reactive/acidic due to insufficient hydration – the incipient isoprene molecule (Isop) fell into a shallow potential well forming an adduct (Fig. 5). The free energy barrier for proton transfer from (H2O)3·H+ to Isop(g) was ΔG‡ = 6.9 kcal mol−1, and the barrier to the oligomerization with another free Isop(g) was ΔG‡ = 2.1 kcal mol−1 (Fig. 5). On the other hand, the predictions for those barriers for protonation and oligomerization of isoprene with the larger water cluster, (H2O)36·H+, representative of the air–water interface, were ΔG‡ = 25.5 kcal mol−1 and ΔG‡ = 40.2 kcal mol−1, respectively, which are insurmountable under ambient conditions within a 1 ms time-frame (Fig. 6). The stark differences in the free energy landscapes for the interactions of Isop(g) with minimally hydrated hydronium ions, available in electrosprays, and larger water clusters can simply be understood in terms of the enthalpies of hydration of gas-phase protons that decrease monotonically with the addition of water molecules;99–101 the proton activity also decreases commensurately and, in fact, is expected to be even lower for the air–water interface than in the (H2O)36·H+ cluster.
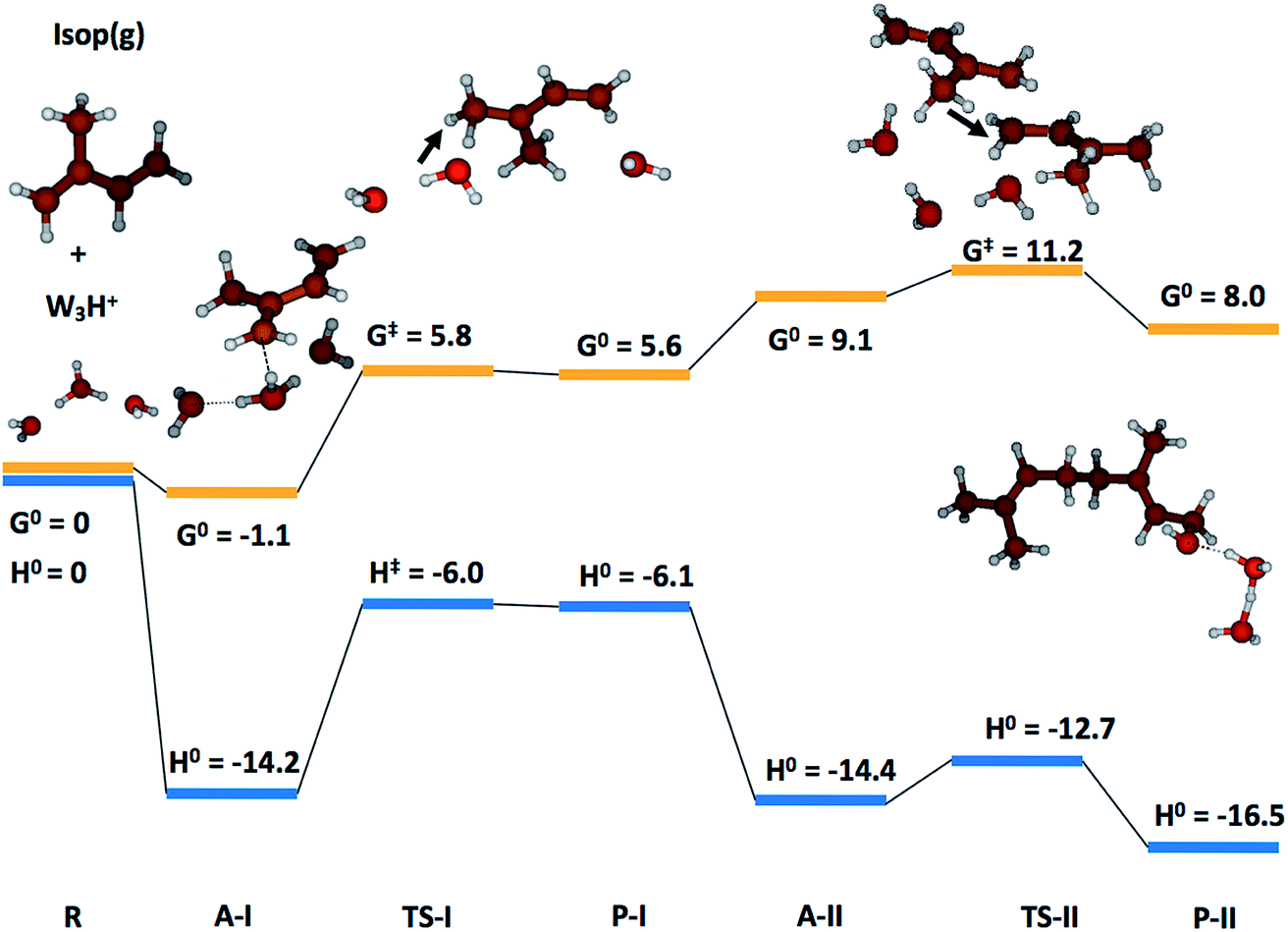 | ||
| Fig. 5 Quantum mechanics-based free energy and enthalpy landscapes for protonation and oligomerization of isoprene while interacting with a cluster comprising three water molecules and one excess proton, (H2O)3·H+. Due to its incomplete hydration (compared to bulk), the proton exhibited extreme acidity. The free energy barrier for the proton transfer from (H2O)3·H+ to isoprene(g) was ΔG‡ = 6.9 kcal mol−1 and the barrier to subsequent oligomerization with another free isoprene(g) was ΔG‡ = 2.1 kcal mol−1, which are easily surmountable under ambient NTP conditions within the timescale of our experiments (∼1 ms). These model predictions support mechanism M3. | ||
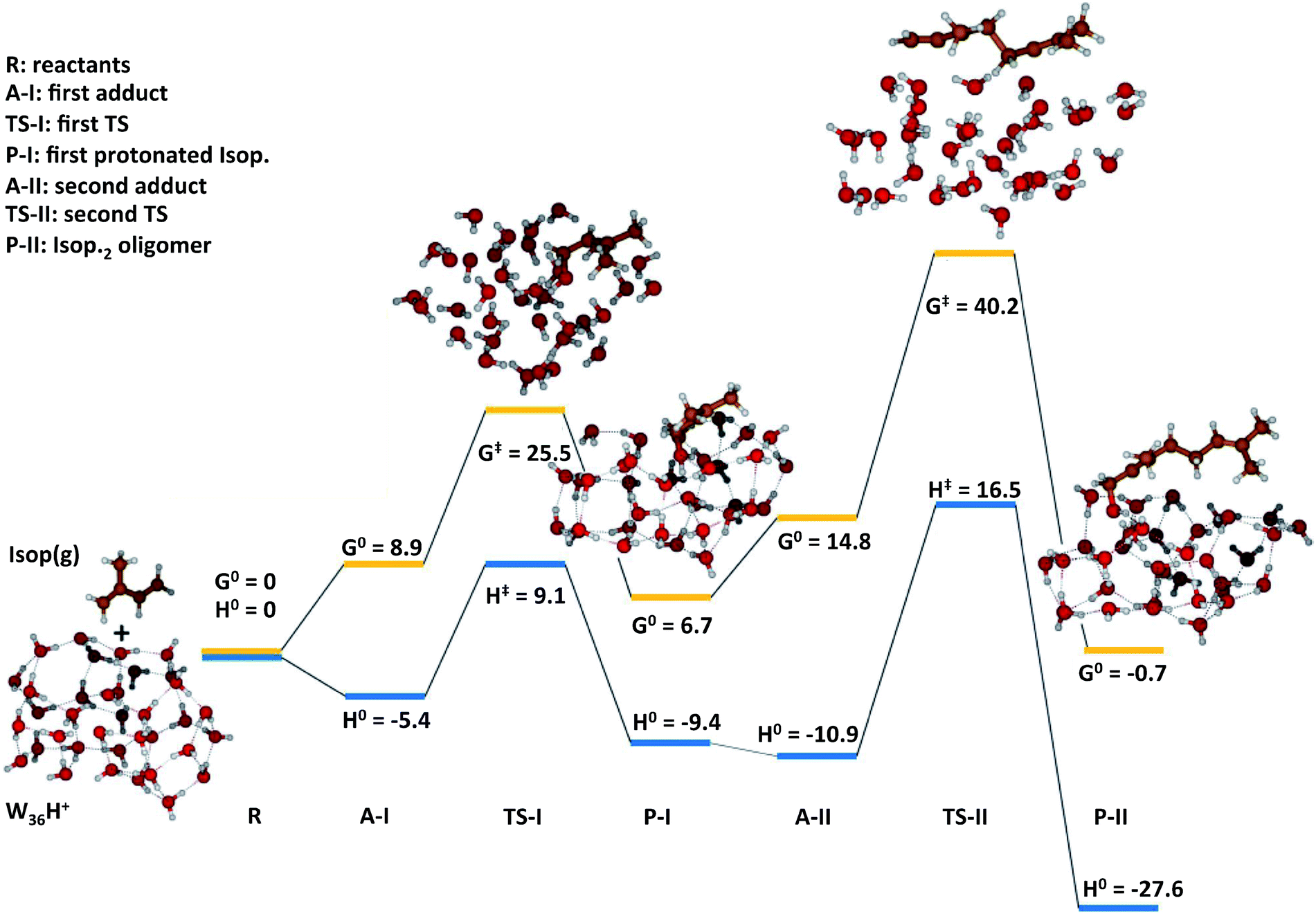 | ||
| Fig. 6 Quantum mechanics-based free energy and enthalpy landscapes for protonation and oligomerization of isoprene on a cluster consisting of 36 water molecules and an excess proton, (H2O)36·H+, representative of very small water droplets. The kinetic barriers preventing proton transfer to isoprene and its subsequent oligomerization were ΔG‡ = 25.5 kcal mol−1 and ΔG‡ = 40.2 kcal mol−1 respectively, which were insurmountable under ambient NTP conditions within the timescale of our experiments (∼1 ms). The predictions of this model suggest that the reactions of isoprene in electrosprays cannot involve liquid-phase drops. These predictions also support the proposed mechanisms M3 by ruling out the possible reactions through M2. | ||
To test the effects of larger basis-sets on these predictions, we compared the results of our M06/6-311+G*/6-311++G** calculations with M06/6-311++G** for the smaller cluster, and found very similar results (Section Sd, Table S1†). Towards further benchmarking, we carried out single point energy calculations on the structures optimized at the M06/6-311++G**-level with the CCSD(T)-level theory,66 and found that the qualitative trends in the reaction kinetics remained unchanged (Table S1†). We note that we could only perform the benchmarking for the smaller system due to the computational expense. Thus, we expect that the relative differences in the barrier heights for the reactions are unlikely to change significantly between DFT/M06 and CC for our systems. Besides, our 1H-NMR experiments present unambiguous evidence for these predictions. To summarize, our calculations demonstrate that the kinetic barriers for the protonation and oligomerization of isoprene are easily surmountable in the smaller acidic cluster (available in electrosprays only) and prohibited in larger acidic clusters at 298 K and 1 atm (a surrogate for the pristine air–water interface).
Conclusion and outlook
Based on our experimental investigation of oil–water and air–water interfaces of isoprene with pH-adjusted water, analyzed by ESIMS and 1H-NMR along with quantum mechanical predictions, we address the questions outlined in the introduction as follows:(i) Aqueous electrosprays do not always report on the thermodynamic properties of pristine air–water interfaces.
(ii) The observed chemical reactions of isoprene in aqueous electrosprays were not driven by the enhancement in the hydrophobe–water interfacial area, as evidenced by the lack thereof in vigorously mixed emulsions of isoprene and pH-adjusted water. Thus, the mechanisms underlying the ‘on-water’ catalysis54–57 must be different from those leading to rate accelerations in the aqueous electrosprays.103 Electrosprays of water must facilitate additional chemical pathways, such as reactions with partially hydrated (gas-phase) hydroniums, which are not accessible in vigorously mixed oil–water emulsions or pristine aqueous interfaces.
(iii) Reactions of isoprene in aqueous electrosprays were driven by non-equilibrium conditions therein – most importantly, due to the rapid evaporation of water leading to highly concentrated droplets and partially hydrated hydronium ions (mechanism M3). It is, thus, crucial to distinguish their contribution from purely ‘interfacial effects’ towards dramatic rate enhancements in aqueous electrosprays.50,53,64,78,104,105
(iv) Gas-phase reactions could play a significant role in the electrosprays – in our experiments, reactions between partially hydrated protons and isoprene led to its protonation and oligomerization, as recently suggested by Yan & co-workers53 and demonstrated by Jacobs & co-workers.106
Our experimental and theoretical results demonstrate that chemistries in aqueous electrosprays do not necessarily correspond to those at the pristine air–water interface and oil–water emulsions at NTP. These findings also contradict the previous claims of the superacidity of the pristine air–water interface as the bulk acidity approaches pH ≤ 3.6;40 the proposal for the mildly acidic environmental surfaces to act as the primary sink for the atmospheric isoprene/terpenes should also be reevaluated.41,46,47 While the potential of aqueous electrosprays to produce high-value products appears promising, those reactions are unlikely to be realized at pristine aqueous interfaces because of the seminal role of the non-equilibrium effects, such as the formation of water clusters with minimally hydrated hydronium ions.49,78,106 We do note that air–water interfaces could, perhaps, be investigated semi-quantitatively through electrosprays, if the reactants do not participate in gas-phase, acid catalyzed, or redox reactions therein51,107–109 and/or the gas-phase reactants do not dissolve in the droplets to re-emerge as interfacial species; a careful case-by-case assessment is needed. The mechanisms underlying the protonation and oligomerization of isoprene gas on electrosprays of pH > 7 water also warrant further investigation. We conclude by stressing on the importance of combining complementary experimental techniques, ab initio calculations and molecular dynamics simulations in the quest to unravel phenomena occurring at the pristine air–water interface.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research reported in this publication was supported by funding from King Abdullah University of Science and Technology (#OSR-2016-CRG5-2992). The authors thank Mr Ivan Gromicho, Scientific Illustrator at KAUST, for preparing Fig. 1. The authors also thank Professor Richard Saykally and Professor Evan Williams (University of California Berkeley), and Dr Manuel Monge Palacios (KAUST) for fruitful discussions. This research used the resources of the Supercomputing Laboratory at King Abdullah University of Science & Technology (KAUST) in Thuwal, Saudi Arabia.References
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, Wiley-Interscience, 2edn, 2006 Search PubMed.
- J. Zhong, M. Kumar, J. S. Francisco and X. C. Zeng, Insight into Chemistry on Cloud/Aerosol Water Surfaces, Acc. Chem. Res., 2018, 51(5), 1229–1237 CrossRef CAS PubMed.
- M. Kumar and J. S. Francisco, Ion pair particles at the air–water interface, Proc. Natl. Acad. Sci., India, 2017, 114(47), 12401–12406 CAS.
- A. V. Shavlov and V. A. Zdzhumandzhi, Metastable states and coalescence of charged water drops inside clouds and fog, J. Aerosol Sci., 2016, 91, 54–61 CrossRef CAS.
- L. Pawlowski, The Science and Engineering of Thermal Spray Coatings, John Wiley & Sons Ltd., Britain, 2008 Search PubMed.
- J. H. Bang and K. S. Suslick, Applications of Ultrasound to the Synthesis of Nanostructured Materials, Adv. Mater., 2010, 22(10), 1039–1059 CrossRef CAS PubMed.
- A. Y. Li, Q. J. Luo, S. J. Park and R. G. Cooks, Synthesis and Catalytic Reactions of Nanoparticles formed by Electrospray Ionization of Coinage Metals, Angew. Chem., Int. Ed., 2014, 53(12), 3147–3150 CrossRef CAS PubMed.
- C. M. Dobson, G. B. Ellison, A. F. Tuck and V. Vaida, Atmospheric aerosols as prebiotic chemical reactors, Proc. Natl. Acad. Sci., India, 2000, 97(22), 11864–11868 CAS.
- K. Ariga and J. P. Hill, Monolayers at Air–Water Interfaces: From Origins-of-Life to Nanotechnology, Chem. Rec., 2011, 11(4), 199–211 CrossRef CAS PubMed.
- E. C. Griffith and V. Vaida, In situ observation of peptide bond formation at the water–air interface, Proc. Natl. Acad. Sci., India, 2012, 109(39), 15697–15701 CAS.
- N. Agmon, H. J. Bakker, R. K. Campen, R. H. Henchman, P. Pohl, S. Roke, M. Thämer and A. Hassanali, Protons and Hydroxide Ions in Aqueous Systems, Chem. Rev., 2016, 116(13), 7642–7672 CrossRef CAS PubMed.
- R. J. Saykally, Air/water interface: Two sides of the acid–base story, Nat. Chem., 2013, 5(2), 82–84 CrossRef CAS PubMed.
- E. C. Griffith and V. Vaida, Ionization state of L-phenylalanine at the air–water interface, J. Am. Chem. Soc., 2013, 135(2), 710–716 CrossRef CAS PubMed.
- National Academies of Sciences, Engineering, and Medicine, The Future of Atmospheric Chemistry Research: Remembering Yesterday, Understanding Today, Anticipating Tomorrow, The National Academies Press, Washington, DC, 2016, DOI:10.17226/23573.
- V. Vaida, Prebiotic phosphorylation enabled by microdroplets, Proc. Natl. Acad. Sci., India, 2017, 114(47), 12359–12361 CAS.
- S. Banerjee, E. Gnanamani, X. Yan and R. N. Zare, Can all bulk-phase reactions be accelerated in microdroplets?, Analyst, 2017, 142(9), 1399–1402 RSC.
- Y. Tabe, N. Kikkawa, H. Takahashi and A. Morita, Surface Acidity of Water Probed by Free Energy Calculation for Trimethylamine Protonation, J. Phys. Chem. A, 2013, 118(2), 977–988 Search PubMed.
- A. J. Colussi and S. Enami, Comment on Surface Acidity of Water Probed by Free Energy Calculation for Trimethylamine Protonation, J. Phys. Chem. C, 2014, 118(5), 2894 CrossRef CAS.
- Y. Tabe, N. Kikkawa, H. Takahashi and A. Morita, Reply to Comment on Surface Acidity of Water Probed by Free Energy Calculation for Trimethylamine Protonation, J. Phys. Chem. C, 2014, 118(5), 2895 CrossRef CAS.
- C. Macias-Romero, I. Nahalka, H. I. Okur and S. Roke, Optical imaging of surface chemistry and dynamics in confinement, Science, 2017, 357(6353), 784–787 CrossRef CAS PubMed.
- E. M. Arnett, Gas-phase proton transfer- Breakthrough for solution chemistry, Acc. Chem. Res., 1973, 6(12), 404–409 CrossRef CAS.
- K. B. Eisenthal, Liquid interfaces probed by second-harmonic and sum-frequency spectroscopy, Chem. Rev., 1996, 96(4), 1343–1360 CrossRef CAS PubMed.
- R. K. Lam, S. L. Raj, T. A. Pascal, C. D. Pemmaraju, L. Foglia, A. Simoncig, N. Fabris, P. Miotti, C. J. Hull, A. M. Rizzuto, J. W. Smith, R. Mincigrucci, C. Masciovecchio, A. Gessini, E. Allaria, G. De Ninno, B. Diviacco, E. Roussel, S. Spampinati, G. Penco, S. Di Mitri, M. Trovo, M. Danailov, S. T. Christensen, D. Sokaras, T. C. Weng, M. Coreno, L. Poletto, W. S. Drisdell, D. Prendergast, L. Giannessi, E. Principi, D. Nordlund, R. J. Saykally and C. P. Schwartz, Soft X-Ray Second Harmonic Generation as an Interfacial Probe, Phys. Rev. Lett., 2018, 120, 023901 CrossRef CAS PubMed.
- K. Ariga, T. Nakanishi, J. P. Hill, M. Shirai, M. Okuno, T. Abe and J. Kikuchi, Tunable pK of amino acid residues at the air–water interface gives an L-zyme (Langmuir enzyme), J. Am. Chem. Soc., 2005, 127(34), 12074–12080 CrossRef CAS PubMed.
- H. Y. Yang, Y. Imanishi and A. Harata, Estimating pH at the Air/Water Interface with a Confocal Fluorescence Microscope, Anal. Sci., 2015, 31(10), 1005–1010 CrossRef CAS PubMed.
- J. S. Samson, R. Scheu, N. Smolentsev, S. W. Rick and S. Roke, Sum frequency spectroscopy of the hydrophobic nanodroplet/water interface: Absence of hydroxyl ion and dangling OH bond signatures, Chem. Phys. Lett., 2014, 615, 124–131 CrossRef CAS.
- C. S. Tian and Y. R. Shen, Structure and charging of hydrophobic material/water interfaces studied by phase-sensitive sum-frequency vibrational spectroscopy, Proc. Natl. Acad. Sci., India, 2009, 106(36), 15148–15153 CAS.
- S. Nihonyanagi, R. Kusaka, K.-i. Inoue, A. Adhikari, S. Yamaguchi and T. Tahara, Accurate determination of complex χ(2) spectrum of the air/water interface, J. Chem. Phys., 2015, 143(12), 124707 CrossRef PubMed.
- S. J. Byrnes, P. L. Geissler and Y. R. Shen, Ambiguities in surface nonlinear spectroscopy calculations, Chem. Phys. Lett., 2011, 516(4–6), 115–124 CrossRef CAS.
- O. Carrier, E. H. G. Backus, N. Shahidzadeh, J. Franz, M. Wagner, Y. Nagata, M. Bonn and D. Bonn, Oppositely Charged Ions at Water–Air and Water–Oil Interfaces: Contrasting the Molecular Picture with Thermodynamics, J. Phys. Chem. Lett., 2016, 7(5), 825–830 CrossRef CAS PubMed.
- R. K. Lam, J. W. Smith, A. M. Rizzuto, O. Karslıoğlu, H. Bluhm and R. J. Saykally, Reversed interfacial fractionation of carbonate and bicarbonate evidenced by X-ray photoemission spectroscopy, J. Chem. Phys., 2017, 146(9), 094703 CrossRef.
- A. M. Jubb, W. Hua and H. C. Allen, Organization of Water and Atmospherically Relevant Ions and Solutes: Vibrational Sum Frequency Spectroscopy at the Vapor/Liquid and Liquid/Solid Interfaces, Acc. Chem. Res., 2012, 45(1), 110–119 CrossRef CAS PubMed.
- J. R. Platt, Strong Inference – Certain Systematic Methods of Scientific Thinking May Produce Much More Rapid Progress Than Others, Science, 1964, 146(364), 7 Search PubMed.
- E. De Hoffmann and V. Stroobant, Mass spectrometry: principles and applications, John Wiley & Sons, 2007 Search PubMed.
- P. Kebarle and M. Peschke, On the mechanisms by which the charged droplets produced by electrospray lead to gas phase ions, Anal. Chim. Acta, 2000, 406(1), 11–35 CrossRef CAS.
- P. Kebarle and U. H. Verkerk, Electrospray: From Ions in Solution to Ions in the Gas Phase, What We Know Now, Mass Spectrom. Rev., 2009, 28(6), 898–917 CrossRef CAS PubMed.
- S. Banerjee and S. Mazumdar, Electrospray Ionization Mass Spectrometry: A Technique to Access the Information beyond the Molecular Weight of the Analyte, Int. J. Anal. Chem., 2012, 2012, 40 Search PubMed.
- S. Nguyen and J. B. Fenn, Gas-phase ions of solute species from charged droplets of solutions, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(4), 1111–1117 CrossRef CAS PubMed.
- S. Enami, M. R. Hoffmann and A. J. Colussi, Proton Availability at the Air/Water Interface, J. Phys. Chem. Lett., 2010, 1(10), 1599–1604 CrossRef CAS.
- S. Enami, M. R. Hoffmann and A. J. Colussi, Superacid chemistry on mildly acidic water, J. Phys. Chem. Lett., 2010, 1(24), 3488–3493 CrossRef CAS.
- S. Enami, H. Mishra, M. R. Hoffmann and A. J. Colussi, Protonation and Oligomerization of Gaseous Isoprene on Mildly Acidic Surfaces: Implications for Atmospheric Chemistry, J. Phys. Chem. A, 2012, 116(24), 6027–6032 CrossRef CAS PubMed.
- H. Mishra, S. Enami, R. J. Nielsen, L. A. Stewart, M. R. Hoffmann, W. A. Goddard and A. J. Colussi, Bronsted basicity of the air–water interface, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(46), 18679–18683 CrossRef CAS PubMed.
- H. Mishra, S. Enami, R. J. Nielsen, M. R. Hoffmann, W. A. Goddard and A. J. Colussi, Anions dramatically enhance proton transfer through aqueous interfaces, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(26), 10228–10232 CrossRef CAS PubMed.
- D. Schroder, Ion clustering in electrospray mass spectrometry of brine and other electrolyte solutions, Phys. Chem. Chem. Phys., 2012, 14(18), 6382–6390 RSC.
- X. Yan, M. Delgado, J. Aubry, O. Gribelin, A. Stocco, F. Boisson-Da Cruz, J. Bernard and F. Ganachaud, Central Role of Bicarbonate Anions in Charging Water/Hydrophobic Interfaces, J. Phys. Chem. Lett., 2018, 9(1), 96–103 CrossRef CAS PubMed.
- K. Matsuoka, Y. Sakamoto, T. Hama, Y. Kajii and S. Enami, Reactive Uptake of Gaseous Sesquiterpenes on Aqueous Surfaces, J. Phys. Chem. A, 2017, 121(4), 810–818 CrossRef CAS PubMed.
- S. Enami, M. R. Hoffmann and A. J. Colussi, Dry deposition of biogenic terpenes via cationic oligomerization on environmental aqueous surfaces, J. Phys. Chem. Lett., 2012, 3, 3102–3108 CrossRef CAS PubMed.
- V. B. Kazansky, Solvation as a main factor that determines the strength of liquid superacids and the selectivity of the acid-catalyzed reactions of olefins, Catal. Today, 2002, 73(1–2), 127–137 CrossRef CAS.
- I. Nam, H. G. Nam and R. N. Zare, Abiotic synthesis of purine and pyrimidine ribonucleosides in aqueous microdroplets, Proc. Natl. Acad. Sci., India, 2018, 115(1), 36–40 CAS.
- J. K. Lee, S. Banerjee, H. G. Nam and R. N. Zare, Acceleration of reaction in charged microdroplets, Q. Rev. Biophys., 2015, 48(04), 437–444 CrossRef CAS PubMed.
- M. I. Jacobs, J. F. Davies, L. Lee, R. D. Davis, F. Houle and K. R. Wilson, Exploring Chemistry in Microcompartments Using Guided Droplet Collisions in a Branched Quadrupole Trap Coupled to a Single Droplet, Paper Spray Mass Spectrometer, Anal. Chem., 2017, 89(22), 12511–12519 CrossRef CAS PubMed.
- D. A. Thomas, L. T. Wang, B. Goh, E. S. Kim and J. L. Beauchamp, Mass Spectrometric Sampling of a Liquid Surface by Nanoliter Droplet Generation from Bursting Bubbles and Focused Acoustic Pulses: Application to Studies of Interfacial Chemistry, Anal. Chem., 2015, 87(6), 3336–3344 CrossRef CAS PubMed.
- X. Yan, R. M. Bain and R. G. Cooks, Organic Reactions in Microdroplets: Reaction Acceleration Revealed by Mass Spectrometry, Angew. Chem., Int. Ed., 2016, 55(42), 12960–12972 CrossRef CAS PubMed.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, On water: unique reactivity of organic compounds in aqueous suspension, Angew. Chem., Int. Ed., 2005, 44(21), 3275–3279 CrossRef CAS PubMed.
- Y. S. Jung and R. A. Marcus, On the theory of organic catalysis on water, J. Am. Chem. Soc., 2007, 129(17), 5492–5502 CrossRef CAS PubMed.
- J. K. Beattie, C. S. P. McErlean and C. B. W. Phippen, The Mechanism of On-Water Catalysis, Chem.–Eur. J., 2010, 16(30), 8972–8974 CrossRef CAS PubMed.
- L. L. Thomas, J. Tirado-Rives and W. L. Jorgensen, Quantum Mechanical/Molecular Mechanical Modeling Finds Diels-Alder Reactions Are Accelerated Less on the Surface of Water Than in Water, J. Am. Chem. Soc., 2010, 132(9), 3097–3104 CrossRef CAS PubMed.
- W. D. Luedtke, U. Landman, Y. H. Chiu, D. J. Levandier, R. A. Dressler, S. Sok and M. S. Gordon, Nanojets, electrospray, and ion field evaporation: Molecular dynamics simulations and laboratory experiments, J. Phys. Chem. A, 2008, 112(40), 9628–9649 CrossRef CAS PubMed.
- J. F. de la Mora, The fluid dynamics of Taylor cones, Annu. Rev. Fluid Mech., 2007, 39, 217–243 CrossRef.
- D. N. Mortensen and E. R. Williams, Ultrafast (1 μs) Mixing and Fast Protein Folding in Nanodrops Monitored by Mass Spectrometry, J. Am. Chem. Soc., 2016, 138(10), 3453–3460 CrossRef CAS PubMed.
- H. J. Sterling, C. A. Cassou, A. C. Susa and E. R. Williams, Electrothermal Supercharging of Proteins in Native Electrospray Ionization, Anal. Chem., 2012, 84(8), 3795–3801 CrossRef CAS PubMed.
- B. Yuan, A. R. Koss, C. Warneke, M. Coggon, K. Sekimoto and J. A. de Gouw, Proton-Transfer-Reaction Mass Spectrometry: Applications in Atmospheric Sciences, Chem. Rev., 2017, 117(21), 13187–13229 CrossRef CAS PubMed.
- S. Zhou, B. S. Prebyl and K. D. Cook, Profiling pH changes in the electrospray plume, Anal. Chem., 2002, 74(19), 4885–4888 CrossRef CAS PubMed.
- S. L. Zhou and K. D. Cook, Protonation in electrospray mass spectrometry: Wrong-way-round or right-way-round?, J. Am. Soc. Mass Spectrom., 2000, 11(11), 961–966 CrossRef CAS PubMed.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. Defrees and J. A. Pople, Self-consitent molecular-orbital methods. 23. A polarization-type basis set for 2nd -row elements, J. Chem. Phys., 1982, 77(7), 3654–3665 CrossRef CAS.
- C. J. Cramer, Essentials of computational chemistry: theories and models, John Wiley & Sons, 2013 Search PubMed.
- K. Fukui, The path of chemical reactions-the IRC approach, Acc. Chem. Res., 1981, 14(12), 363–368 CrossRef CAS.
- C. Dykstra, G. Frenking, K. Kim and G. Scuseria, Theory and Applications of Computational Chemistry: the first forty years, Elsevier, 2011 Search PubMed.
- S. Nosé, A molecular dynamics method for simulations in the canonical ensemble, Mol. Phys., 1984, 52(2), 255–268 CrossRef.
- M. J. Frisch, G. Trucks, H. B. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Gaussian 09, revision A. 1, Gaussian Inc., Wallingford CT, 2009, vol. 27, p. 34 Search PubMed.
- T. D. Sharkey, A. E. Wiberley and A. R. Donohue, Isoprene emission from plants: Why and how, Ann. Bot., 2008, 101(1), 5–18 CrossRef CAS PubMed.
- A. T. Archibald, J. G. Levine, N. L. Abraham, M. C. Cooke, P. M. Edwards, D. E. Heard, M. E. Jenkin, A. Karunaharan, R. C. Pike, P. S. Monks, D. E. Shallcross, P. J. Telford, L. K. Whalley and J. A. Pyle, Impacts of HO(x) regeneration and recycling in the oxidation of isoprene: Consequences for the composition of past, present and future atmospheres, Geophys. Res. Lett., 2011, 38(L05804), 6 Search PubMed.
- Neutral Thermochemical Data, in NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg MD, 2017, p. 20899, DOI:10.18434/T4D303.
- M. Deiana, F. Corongiu, M. Dessi, P. Scano, M. Casu and A. Lai, NMR determination of site-specific deuterium distribution (SNIF-NMR) in squalene from different sources, Magn. Reson. Chem., 2001, 39(1), 29–32 CrossRef CAS.
- R. M. Bain, C. J. Pulliam and R. G. Cooks, Accelerated Hantzsch electrospray synthesis with temporal control of reaction intermediates, Chem. Sci., 2015, 6(1), 397–401 RSC.
- D. Schroder, M. Budesinsky and J. Roithova, Deprotonation of p-Hydroxybenzoic Acid: Does Electrospray Ionization Sample Solution or Gas-Phase Structures?, J. Am. Chem. Soc., 2012, 134(38), 15897–15905 CrossRef CAS PubMed.
- J. F. d. l. Mora, G. J. Van Berkel, C. G. Enke, R. B. Cole, M. Martinez-Sanchez and J. B. Fenn, Electrochemical processes in electrospray ionization mass spectrometry, J. Mass Spectrom., 2000, 35(8), 939–952 CrossRef.
- A. J. Ingram, C. L. Boeser and R. N. Zare, Going beyond electrospray: mass spectrometric studies of chemical reactions in and on liquids, Chem. Sci., 2016, 7(1), 39–55 RSC.
- R. M. Bain, C. J. Pulliam, F. Thery and R. G. Cooks, Accelerated Chemical Reactions and Organic Synthesis in Leidenfrost Droplets, Angew. Chem., Int. Ed., 2016, 55(35), 10478–10482 CrossRef CAS PubMed.
- P. Aussillous and D. Quere, Liquid marbles, Nature, 2001, 411(6840), 924–927 CrossRef CAS PubMed.
- E. P. L. Hunter and S. G. Lias, Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update, J. Phys. Chem. Ref. Data, 1998, 27(3), 413–656 CrossRef CAS.
- M. a. H. Amad, N. B. Cech, G. S. Jackson and C. G. Enke, Importance of gas-phase proton affinities in determining the electrospray ionization response for analytes and solvents, J. Mass Spectrom., 2000, 35(7), 784–789 CrossRef CAS.
- R. S. Blake, P. S. Monks and A. M. Ellis, Proton-Transfer Reaction Mass Spectrometry, Chem. Rev., 2009, 109(3), 861–896 CrossRef CAS PubMed.
- R. T. Collins, J. J. Jones, M. T. Harris and O. A. Basaran, Electrohydrodynamic tip streaming and emission of charged drops from liquid cones, Nat. Phys., 2008, 4(2), 149–154 Search PubMed.
- J. N. Smith, R. C. Flagan and J. L. Beauchamp, Droplet evaporation and discharge dynamics in electrospray ionization, J. Phys. Chem. A, 2002, 106(42), 9957–9967 CrossRef CAS.
- Y. Wang, M. K. Tan, D. B. Go and H.-C. Chang, Electrospray cone-jet breakup and droplet production for electrolyte solutions, EPL, 2012, 99(6), 64003 CrossRef.
- L. W. Zilch, J. T. Maze, J. W. Smith, G. E. Ewing and M. F. Jarrold, Charge Separation in the Aerodynamic Breakup of Micrometer-Sized Water Droplets, J. Phys. Chem. A, 2008, 112(51), 13352–13363 CrossRef CAS PubMed.
- R. L. Grimm and J. L. Beauchamp, Dynamics of field-induced droplet ionization: Time-resolved studies of distortion, jetting, and progeny formation from charged and neutral methanol droplets exposed to strong electric fields, J. Phys. Chem. B, 2005, 109(16), 8244–8250 CrossRef CAS PubMed.
- K. Breuker and F. W. McLafferty, Stepwise evolution of protein native structure with electrospray into the gas phase, 10–12 to 102 s, Proc. Natl. Acad. Sci., India, 2008, 105(47), 18145–18152 CAS.
- J. A. Silveira, K. L. Fort, D. Kim, K. A. Servage, N. A. Pierson, D. E. Clemmer and D. H. Russell, From Solution to the Gas Phase: Stepwise Dehydration and Kinetic Trapping of Substance P Reveals the Origin of Peptide Conformations, J. Am. Chem. Soc., 2013, 135(51), 19147–19153 CrossRef CAS PubMed.
- V. S. Bryantsev, M. S. Diallo and W. A. Goddard, Computational Study of Copper(II) Complexation and Hydrolysis in Aqueous Solutions Using Mixed Cluster/Continuum Models, J. Phys. Chem. A, 2009, 113(34), 9559–9567 CrossRef CAS PubMed.
- A. M. Sarotti, A. G. Suarez and R. A. Spanevello, DFT calculations induced a regiochemical outcome revision of the Diels-Alder reaction between levoglucosenone and isoprene, Tetrahedron Lett., 2011, 52(24), 3116–3119 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120(1–3), 215–241 Search PubMed.
- C. P. Kelly, C. J. Cramer and D. G. Truhlar, Predicting Adsorption Coefficients at Air–Water Interfaces Using Universal Solvation and Surface Area Models, J. Phys. Chem. B, 2004, 108, 12882–12897 CrossRef CAS.
- M. Kumar, H. Li, X. Zhang, X. C. Zeng and J. S. Francisco, Nitric Acid–Amine Chemistry in the Gas Phase and at the Air–Water Interface, J. Am. Chem. Soc., 2018, 140(20), 6456–6466 CrossRef CAS PubMed.
- H. Mishra, R. J. Nielsen, S. Enami, M. R. Hoffmann, A. J. Colussi and W. A. Goddard, Quantum chemical insights into the dissociation of nitric acid on the surface of aqueous electrolytes, Int. J. Quantum Chem., 2013, 113(4), 413–417 CrossRef CAS.
- E. P. L. Hunter and S. G. Lias, Evaluated gas phase basicities and proton affinities of molecules: an update, J. Phys. Chem. Ref. Data, 1998, 27(3), 413–656 CrossRef CAS.
- V. B. Kazansky, Solvation effects in catalytic transformations of olefins in sulfuric acid, Catal. Rev., 2001, 43(3), 199–232 CrossRef CAS.
- C. E. Klots, Solubility of protons in water, J. Phys. Chem., 1981, 85(24), 3585–3588 CrossRef CAS.
- M. D. Tissandier, K. A. Cowen, W. Y. Feng, E. Gundlach, M. H. Cohen, A. D. Earhart, J. V. Coe and T. R. Tuttle, The Proton's Absolute Aqueous Enthalpy and Gibbs Free Energy of Solvation from Cluster-Ion Solvation Data, J. Phys. Chem. A, 1998, 102(40), 7787–7794 CrossRef CAS.
- C. P. Kelly, C. J. Cramer and D. G. Truhlar, Aqueous Solvation Free Energies of Ions and Ion-Water Clusters Based on an Accurate Value for the Absolute Aqueous Solvation Free Energy of the Proton, J. Phys. Chem. B, 2006, 110(32), 16066–16081 CrossRef CAS PubMed.
- M. P. Hodges and D. J. Wales, Global minima of protonated water clusters, Chem. Phys. Lett., 2000, 324(4), 279–288 CrossRef CAS.
- R. N. Butler and A. G. Coyne, Organic synthesis reactions on-water at the organic–liquid water interface, Org. Biomol. Chem., 2016, 14(42), 9945–9960 RSC.
- T. Muller, A. Badu-Tawiah and R. G. Cooks, Accelerated Carbon–Carbon Bond-Forming Reactions in Preparative Electrospray, Angew. Chem., Int. Ed., 2012, 51(47), 11832–11835 CrossRef PubMed.
- D. N. Mortensen and E. R. Williams, Theta-Glass Capillaries in Electrospray Ionization: Rapid Mixing and Short Droplet Lifetimes, Anal. Chem., 2014, 86(18), 9315–9321 CrossRef CAS PubMed.
- M. I. Jacobs, R. D. Davis, R. J. Rapf and K. R. Wilson, Studying Chemistry in Micro-compartments by Separating Droplet Generation from Ionization, J. Am. Soc. Mass Spectrom., 2018, 1–5 Search PubMed.
- R. L. Grimm, R. Hodyss and J. L. Beauchamp, Probing interfacial chemistry of single droplets with field-induced droplet ionization mass spectrometry: physical adsorption of polycyclic aromatic hydrocarbons and ozonolysis of oleic acid and related compounds, Anal. Chem., 2006, 78(11), 3800–3806 CrossRef CAS PubMed.
- H. I. Kim, H. J. Kim, Y. S. Shin, L. W. Beegle, S. S. Jang, E. L. Neidholdt, W. A. Goddard, J. R. Heath, I. Kanik and J. L. Beauchamp, Interfacial Reactions of Ozone with Surfactant Protein B in a Model Lung Surfactant System, J. Am. Chem. Soc., 2010, 132(7), 2254–2263 CrossRef CAS PubMed.
- Y. Zhao, M. C. Fairhurst, L. M. Wingen, V. Perraud, M. J. Ezell and B. J. Finlayson-Pitts, New insights into atmospherically relevant reaction systems using direct analysis in real-time mass spectrometry (DART-MS), Atmos. Meas. Tech., 2017, 10(4), 1373–1386 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05538f |
| This journal is © The Royal Society of Chemistry 2019 |