
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAchieving efficient and robust catalytic reforming on dual-sites of Cu species†
Kui
Ma
ab,
Ye
Tian
ab,
Zhi-Jian
Zhao
ac,
Qingpeng
Cheng
ab,
Tong
Ding
ab,
Jing
Zhang
d,
Lirong
Zheng
 d,
Zheng
Jiang
e,
Takayuki
Abe
f,
Noritatsu
Tsubaki
g,
Jinlong
Gong
*ac and
Xingang
Li
*ab
d,
Zheng
Jiang
e,
Takayuki
Abe
f,
Noritatsu
Tsubaki
g,
Jinlong
Gong
*ac and
Xingang
Li
*ab
aSchool of Chemical Engineering & Technology, Tianjin University, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, China. E-mail: jlgong@tju.edu.cn; xingang_li@tju.edu.cn
bTianjin Key Laboratory of Applied Catalysis Science & Engineering, Tianjin 300072, China
cKey Laboratory for Green Chemical Technology of Ministry of Education, Tianjin 300072, China
dBeijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China
eShanghai Synchrotron Radiation Facility, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201800, China
fHydrogen Isotope Research Center, University of Toyama, Gofuku 3190, Toyama 930-8555, Japan
gDepartment of Applied Chemistry, School of Engineering, University of Toyama, Gofuku 3190, Toyama 930-8555, Japan
First published on 23rd January 2019
Abstract
Catalytic reforming provides a practical technique for on-board hydrogen production in fuel cell applications. The high energy density, easy transportation and non-toxicity of biomass-derived dimethyl ether (bio-DME) offer potential to replace methanol for on-board steam reforming (SR). Presently, the reaction mechanism over conventional Cu-based SR catalysts remains elusive, limiting the rational design of highly efficient reforming systems. Herein, we build a catalytic system for bio-DME SR with dual-sites of Cu species, i.e., Cu+ and Cu0 sites, and achieve a record-high H2 production rate of 1145 mol kgcat−1 h−1. Via regulating the ratios of the dual-sites of Cu, we clearly describe molecular understandings on SR. And we discover that the substantially boosted activity is induced by a new Cu+-determined reaction path substituting the conventional Cu0-determined path. Intrinsically, Cu2O can act as a physical spacer and hydroxyl consumer to suppress the aggregation of metallic Cu species in SR. Due to the unique structure of metallic Cu surrounded by Cu2O, the catalyst exhibits robust catalytic performance even after severe thermal treatment. These findings open a new avenue for designing efficient catalytic reforming systems with commercial potential.
Introduction
Hydrogen, a renewable resource with a high energy density (142 MJ kg−1), is consumed today as a chemical raw material (about 5 × 1010 kg per year worldwide).1 As the core of the hydrogen economy, fuel cells running on hydrogen are attractive power supplies for versatile applications.2,3 The required hydrogen comes from a stable on-board production system, ensuring its safe storage and transportation.4,5 Recently, the development of technologies for converting biomass has resuscitated the surge of reforming renewable oxygenated hydrocarbons, e.g., methanol, ethanol, dimethyl ether (DME), and glycerol, with H2O for on-board hydrogen production.6–8 In view of the mild SR conditions, methanol and DME without C–C bonds are the primary choices.9 The usage of DME can release hydrogen with a higher gravimetric density of 26.1 wt% than methanol (18.8 wt%). As the European project BioDME has been industrially built and operated in Sweden, bio-DME is becoming a potential reforming feed to replace the traditional methanol.10,11 The non-toxicity of DME offers another important advantage. DME SR is an integrated technology consisting of hydrolysis of DME to methanol over solid acid catalysts, and subsequently methanol SR over metal catalysts. Currently, γ-Al2O3 is the commonly used acid catalyst for DME hydrolysis operated above 300 °C,12 requiring higher operating temperatures for the following SR. And copper-based catalysts have been intensively explored in reforming reactions with the inherent drawback of thermal deactivation by metal-particle growth.13It is commonly perceived that Cu0 is primary and indispensable for SR,14,15 bringing debate about the role of Cu+. Some researchers have proposed that Cu+ is active in suppressing the by-product of carbon monoxide (CO) from the reverse water–gas shift, the main side reaction in SR.16–18 However, others still suggest that Cu+ could directly catalyze the steam reforming, and speculate that there may exist an optimum cooperation between Cu0 and Cu+ to boost activities,19–21 but this lacks effective evidence. Currently, Cu+ species are usually introduced through addition of metal oxides to Cu-based catalysts, and self-redox reaction of copper species under feed and reaction conditions.22–25 The complexity of these systems impairs efforts to identify the exact role of copper. In this regard, copper phyllosilicates [Cu2Si2O5(OH)2], providing stable Cu+ species under hydrogen-rich conditions in catalytic hydrogenation,26–28 may be adopted as a controllable Cu+-supplier to investigate SR mechanisms. However, the tendency of Cu nanoparticles (NPs) to grow into larger crystallites is an impediment to the stable performance of Cu-based catalysts, especially for those without promoters. Intrinsically below the Tammann temperature of copper (407 °C), the aggregation of Cu NPs is mainly induced by Ostwald ripening occurring across the support surface through the functional groups (e.g., hydroxyl).29 It is another stumbling block in further design of Cu-based catalysts.
This paper describes the design and fabrication of a hierarchical Cu–Si system with dual-sites of Cu species (denoted as Cu-AE) via a vacuum-rotary ammonia evaporation method by employing SBA-15 as the silicon source. The ordered three-dimensional (3D) mesoporous channels of SBA-15 can improve molecular diffusion, and restrict the growth of copper NPs. As described in Fig. 1A, the well-ordered ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–OH sites on SBA-15 and vacuum conditions facilitate the dispersion and uniform arrangement of the Cu species.30 Consequently, CuO nanoparticles (NPs) dispersed into Cu2Si2O5(OH)2 nanotubes (NTs) are successfully developed on SBA-15. After reduction, metallic Cu NPs are generated from the CuO NPs, while the Cu2O NPs are from the surrounding Cu2Si2O5(OH)2 NTs. Based on this synthesis mechanism, we tune the dual-sites of copper to quantify their contributions to SR, and achieve a record-high H2 production rate of 1145 mol kgcat−1 h−1 with the structure of metallic Cu surrounded by Cu2O. We also discover that the formation of Cu2O consumes the hydroxyl groups on the support, which retards the Ostwald ripening of metallic Cu, thereby leading to a robust SR performance.
Si–OH sites on SBA-15 and vacuum conditions facilitate the dispersion and uniform arrangement of the Cu species.30 Consequently, CuO nanoparticles (NPs) dispersed into Cu2Si2O5(OH)2 nanotubes (NTs) are successfully developed on SBA-15. After reduction, metallic Cu NPs are generated from the CuO NPs, while the Cu2O NPs are from the surrounding Cu2Si2O5(OH)2 NTs. Based on this synthesis mechanism, we tune the dual-sites of copper to quantify their contributions to SR, and achieve a record-high H2 production rate of 1145 mol kgcat−1 h−1 with the structure of metallic Cu surrounded by Cu2O. We also discover that the formation of Cu2O consumes the hydroxyl groups on the support, which retards the Ostwald ripening of metallic Cu, thereby leading to a robust SR performance.
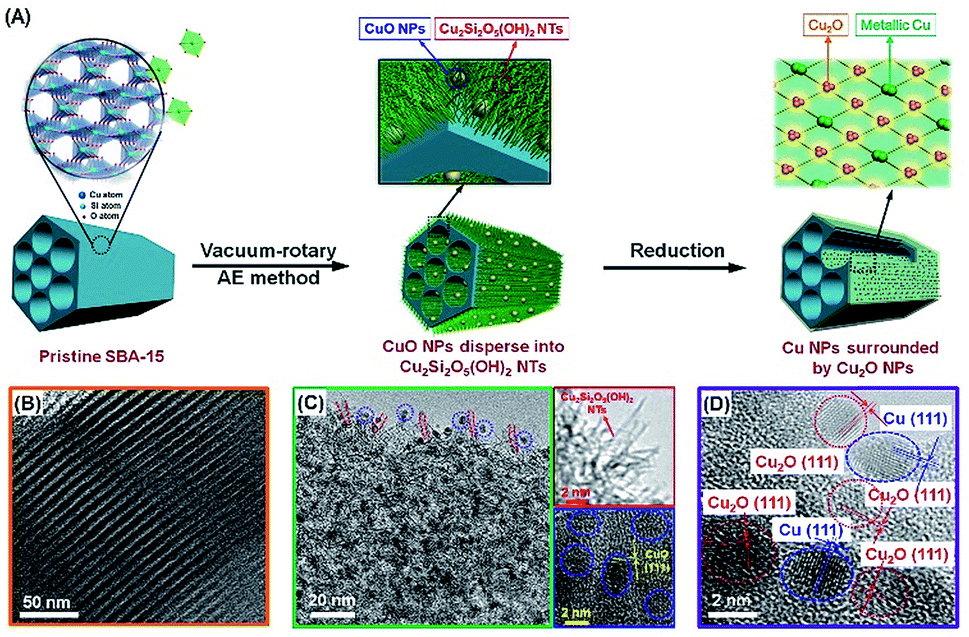 | ||
| Fig. 1 (A) Schematic procedure for the synthesis of the Cu-AE catalysts; TEM images of (B) the pristine SBA-15, and (C) calcined and (D) reduced 25Cu-AE catalyst. | ||
Results and discussion
The TEM images of 25Cu-AE in Fig. 1B–D are obtained to illustrate the synthetic procedure. As indicated by the FTIR and UV-Raman results (Fig. S1†), abundantSi–OH groups (the νSi–OH vibration at 958 cm−1)31 exist on the SiO4 tetrahedron-shaped matrix of the mesoporous SBA-15 (Fig. 1B). After ammonia evaporation, the disappearance of the νSi–OH vibration band in 25Cu-AE implies that the Si–OH groups are consumed during the Cu2Si2O5(OH)2 formation. On 25Cu-AE, the NPs are CuO, while the NTs lacking lattice fringes are Cu2Si2O5(OH)2 (Fig. 1C).32 This formation mechanism is also applied to the other Cu-AE catalysts with different Cu loadings. The XRD and X-ray absorption near-edge structure (XANES) results also verify the coexistence of Cu2Si2O5(OH)2 and CuO in Cu-AE catalysts (Fig. S2†).27
Semi-quantitatively, we adopted linear combination fitting (LCF) of the XANES spectra (Fig. S3†) to estimate the contents of Cu2Si2O5(OH)2 in the catalysts. It shows a volcano-type behavior following the increase of the Cu content and maximizes at 25Cu-AE (Table S1†). Obviously, in Fig. S4A,† CuO NPs are continuously enriched with the increase of the copper content in the catalysts, and eventually cap the Cu2Si2O5(OH)2 NTs. From the low-angle XRD patterns in Fig. S5,† all the samples present three well-resolved diffractions at a 2θ of 0.5–3.0, which can be indexed to the ordered hexagonal lattice (p6mm) of SBA-15.30 Noticeably, the diffraction peaks of the Cu-AE catalysts shift to higher angles compared with those of the pristine SBA-15, reflecting a slight contraction of the unit cell. This phenomenon is contributed by the partial destruction of the ordered pore structure of SBA-15 due to the dissolution of silica during the preparation process, as can be directly observed by TEM as shown above. Here, the existence of these characteristic low-angle diffraction peaks of Cu-AE suggests that most of the channels of SBA-15 are still preserved. Furthermore, the Cu-AE catalysts show weaker d100 intensity, indicating that the copper loading in the mesopore channels reduces X-ray diffraction.33,34 Based on the above results, the schematic illustrations of the Cu species distribution with the increased Cu loading are presented in Fig. S4C.† Summarily, the 25Cu-AE possesses the well-proportioned and uniformly arranged Cu2Si2O5(OH)2 NTs and CuO NPs.
In the reduced Cu-AE catalysts, both the preserved characteristic diffractions of SBA-15 in low-angle XRD patterns (Fig. S6A†) and the narrowed size distribution of 4.3 nm (Fig. S7†) demonstrate the existence of regular channels. We also observed Cu2O (JCPDS no. 05-0667) and metallic Cu (JCPDS no. 04-0836) from wide-angle XRD patterns in Fig. S6B.† Cu2O and metallic Cu originate from the reduction of Cu2Si2O5(OH)2 and CuO, respectively.27 In particular, the metallic Cu is surrounded by Cu2O on 25Cu-AE (Fig. 1D), which is due to the appropriate proportion of Cu species in the calcined catalysts (Fig. S4†). Table S2† gives the physicochemical properties of the catalysts, indicating that we can modulate the physicochemical properties of Cu-AE via tuning the Cu loading. Compared with other catalysts, the Cu-AE catalysts possess the smaller Cu crystal sizes and more accessible surface Cu atoms.
We employed H2-TPR (Fig. 2A) and in situ reduction QXAFS (Fig. 2B) to identify the reducibility and copper species of the catalysts. Generally, the reduction of copper phyllosilicates to Cu+ and highly dispersed CuO to Cu0 occurs at ca. 247–267 °C (ref. 28) and 237 °C,35 respectively. Thus, we attribute the single peak below 300 °C here to the overlap of these two reduction processes. Correspondingly in this range, we observe the co-existence of Cu2O with the first Cu–O coordination (ca. 0.15 nm) and metallic Cu with the first Cu–Cu coordination (ca. 0.20 nm). The existence of Cu+ in the reduced Cu-AE catalysts is also verified by the Cu+-carbonyl band (2122 cm−1) observed from the in situ DRIFTS spectrum of CO adsorption (Fig. S8†). Notably, the broadening peaks above 600 °C (inset in Fig. 2A) indicate the further reduction of Cu+ to metallic Cu.35 Simultaneously, the Cu–O coordination peaks decrease, also indicating the gradual reduction of the Cu species to Cu0 as shown in Fig. 2B. For 25Cu-IM, the main reduction peak at ca. 368 °C is assigned to the reduction of large CuO particles to metallic Cu.26
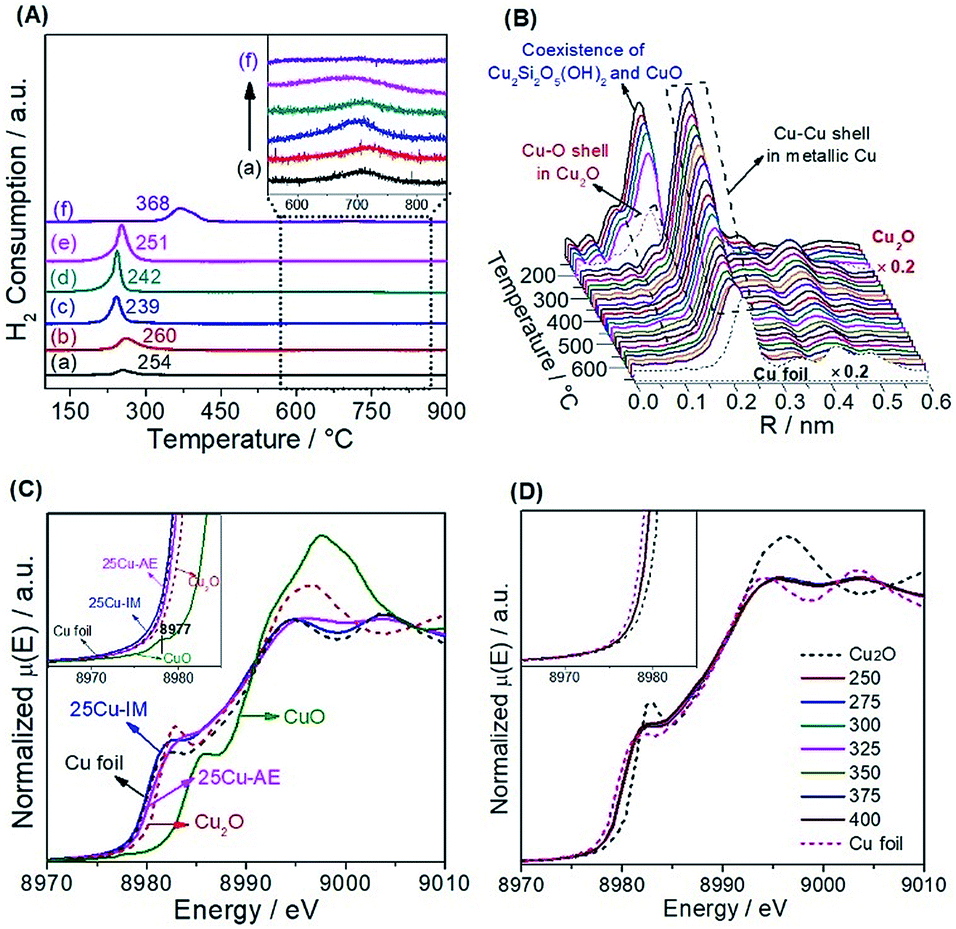 | ||
| Fig. 2 (A) H2-TPR profiles of (a) 10Cu-AE, (b) 20Cu-AE, (c) 25Cu-AE, (d) 30Cu-AE, (e) 40Cu-AE, and (f) 25Cu-IM; the inset shows the reduction of Cu+ in copper phyllosilicates; (B) temperature-resolved in situ RSFs of the Cu K-edge for 25Cu-AE in 5% H2/N2 from RT to 650 °C; (C) in situ steady-state XANES spectra of the Cu K-edge for the reduced 25Cu-AE and 25Cu-IM catalysts in 5% reactants (n(CH3OH)/n(H2O) = 1/2) in N2 at 400 °C; the inset shows the amplified curves; (D) in situ temperature-resolved XANES spectra of the Cu K-edge for the reduced 25Cu-AE catalyst under reaction conditions: 5% reactants (n(CH3OH)/n(H2O) = 1/2) in N2, from 250 to 400 °C with an interval of 25 °C; the inset shows the amplified curves. | ||
We also distinguished the superficial copper species of Cu+ and Cu0 using the Cu LMM Auger kinetic energy peaks36 at ca. 915.5 and 919.0 eV, respectively, for the reduced catalysts (Fig. S9†). The modified Auger parameter (A.P.) values, which represent the summation of the kinetic energy of the Cu LMM Auger electron and the binding energy of the Cu 2p3/2 photoelectron, are calculated and summarized in Table S3.† The smaller A.P. values of Cu+ here (ca. 1848.5 eV) than that of the Cu2O bulk (ca. 1849.0 eV)36 are attributed to the electron transfer from Cu+ to SBA-15, inducing a strong interaction to keep the Cu+ stable. Moreover, the Cu 2p XPS spectra (Fig. S10†) before and after a single reaction cycle indicates that the change in the chemical environment and content of surface Cu (Table S3†) are nearly negligible, also suggesting that the equilibrium between Cu+ and Cu0 over the reduced Cu-AE catalysts is very stable, and is broken with difficultly by the SR reaction. In addition, as summarized in Table S3,† the ratio of Cu+/(Cu0 + Cu+) decreases with the elevated Cu loading, coinciding with the tendency of calculated surface Cu atoms in Table S2.†
Generally, the chemical state of Cu species is sensitive to the environmental atmosphere.37 We performed in situ steady-state XANES to monitor the chemical state of Cu under reaction conditions. As calibrated by the first derivatives of the K-edge XANES spectra in Fig. S11,† the absorption edge at ca. 8980.4 eV of 25Cu-AE in Fig. 2C is located in the middle of those of Cu foil (8979.4 eV) and Cu2O (8980.9 eV). Combined with the absence of the pre-edge feature at ca. 8977 eV for Cu2+ (inset in Fig. 2C),38 we conclude that Cu+ species indeed exist in 25Cu-AE and remain stable during the operation. But for 25Cu-IM, its absorption edge location and white line feature are close to Cu foil, indicating the lack of Cu+ in the bulk of 25Cu-IM during the reaction. Fig. 2D shows the in situ temperature-resolved XANES spectra of 25Cu-AE. The curves are quite similar in shape at different reaction temperatures, and their absorption edges locate in the middle of those of Cu foil and Cu2O. These results demonstrate that the copper oxidation state has little change in dynamic reactive atmospheres at different reaction temperatures, in accordance with the XPS results in Fig. S9, S10 and Table S3.† The above comprehensive characterization results demonstrate a successful synthesis of the catalytic system with tuneable and stable dual-sites of Cu species.
We investigated the catalytic performance and stability of the catalysts after 12 h severe thermal treatment (GHSV = 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1, T = 450 °C) for DME SR. Table 1 summarizes the performance of the catalysts in a numerical form, and Table S4† lists the activity of the representative catalysts for DME SR and methanol SR. In Table 1, 25Cu-AE exhibits a higher catalytic activity in comparison with others. It achieves a DME conversion of 100%, CO selectivity of ca. 10%, and H2 yield exceeding 95%, leading to a record-high H2 production rate of 1145 molH2 kgcat−1 h−1 (Table S4†), 6-fold higher than that of commercial CuZnAl.39 The highest TOFH2 over 25Cu-AE reaches 0.147 s−1, about 2 and 10 times higher than those of the as-prepared Cu/ZnO/Al2O3 and 25Cu-IM, respectively (Table 1). Additionally, 25Cu-IM shows a lower H2 yield and higher CO selectivity as compared with the Cu-AE catalysts, indicating that the latter's excellent catalytic activities probably lie in the presence of Cu+ species.
000 h−1, T = 450 °C) for DME SR. Table 1 summarizes the performance of the catalysts in a numerical form, and Table S4† lists the activity of the representative catalysts for DME SR and methanol SR. In Table 1, 25Cu-AE exhibits a higher catalytic activity in comparison with others. It achieves a DME conversion of 100%, CO selectivity of ca. 10%, and H2 yield exceeding 95%, leading to a record-high H2 production rate of 1145 molH2 kgcat−1 h−1 (Table S4†), 6-fold higher than that of commercial CuZnAl.39 The highest TOFH2 over 25Cu-AE reaches 0.147 s−1, about 2 and 10 times higher than those of the as-prepared Cu/ZnO/Al2O3 and 25Cu-IM, respectively (Table 1). Additionally, 25Cu-IM shows a lower H2 yield and higher CO selectivity as compared with the Cu-AE catalysts, indicating that the latter's excellent catalytic activities probably lie in the presence of Cu+ species.
| Catalysts | Conv. (%) | Y (%) | Sel. (%) | TOFH2b (s−1 × 10−2) | TOFDMEc (s−1 × 10−3) | |||
|---|---|---|---|---|---|---|---|---|
| DME | H2 | CO | CO2 | CH4 | CH3OH | |||
|
a Reaction conditions: gas hourly space velocity (GHSV) = 18000 h−1, steam-to-carbon ratio (S/C) = 2/1, T = 400 °C.
b TOFH2 based on the Cu0 surface area and yield of H2 at 350 °C.
c TOFDME based on the total Cu surface areas and conversion of DME at 350 °C.
|
||||||||
| 10Cu-AEa | 58.3 ± 6.8 | 51.6 ± 9.3 | 12.5 ± 6.3 | 85.8 ± 7.9 | 1.7 ± 0.3 | — | 7.9 ± 0.4 | 3.0 ± 0.7 |
| 20Cu-AEa | 89.0 ± 0.6 | 80.9 ± 3.3 | 13.4 ± 3.8 | 86.3 ± 3.9 | 0.3 ± 0.1 | — | 12.1 ± 0.5 | 8.3 ± 0.8 |
| 25Cu-AEa | 100.0 ± 0.0 | 95.0 ± 1.5 | 10.4 ± 3.0 | 89.3 ± 2.9 | 0.3 ± 0.1 | — | 14.7 ± 0.9 | 12.2 ± 0.3 |
| 30Cu-AEa | 96.7 ± 0.7 | 85.4 ± 2.5 | 12.4 ± 0.8 | 87.4 ± 2.0 | 0.2 ± 0.1 | — | 11.3 ± 1.0 | 9.2 ± 0.4 |
| 40Cu-AEa | 77.5 ± 2.4 | 69.4 ± 4.0 | 31.5 ± 7.2 | 68.0 ± 7.5 | 0.5 ± 0.2 | — | 6.1 ± 1.1 | 6.0 ± 0.9 |
| 25Cu-IMa | 16.0 ± 0.6 | 12.5 ± 0.7 | 41.5 ± 5.9 | 57.9 ± 6.7 | 0.6 ± 0.2 | 5.5 × 10−2 | 1.5 ± 0.4 | 1.9 ± 0.6 |
| Cu/ZnO/Al2O3a | 92.6 ± 0.1 | 73.6 ± 2.3 | 20.3 ± 3.2 | 79.7 ± 4.0 | 0.2 ± 0.1 | — | 7.2 ± 0.7 | 4.4 ± 0.9 |
Fig. 3A displays the catalytic stability of 25Cu-AE, and Fig. 3B shows the TEM images at the corresponding stages. At stage I prior to the thermal treatment, the activity of 25Cu-AE shows no change while that of 25Cu-SiO2 drops rapidly as shown in Fig. S12.† Intrinsically, unlike the steady Cu NP distribution of ∼3.5 nm over 25Cu-AE (Fig. S13A†), the Cu NPs over 25Cu-SiO2 increase from ∼4.1 to ∼9.2 nm (Fig. S13B†) after stage I.
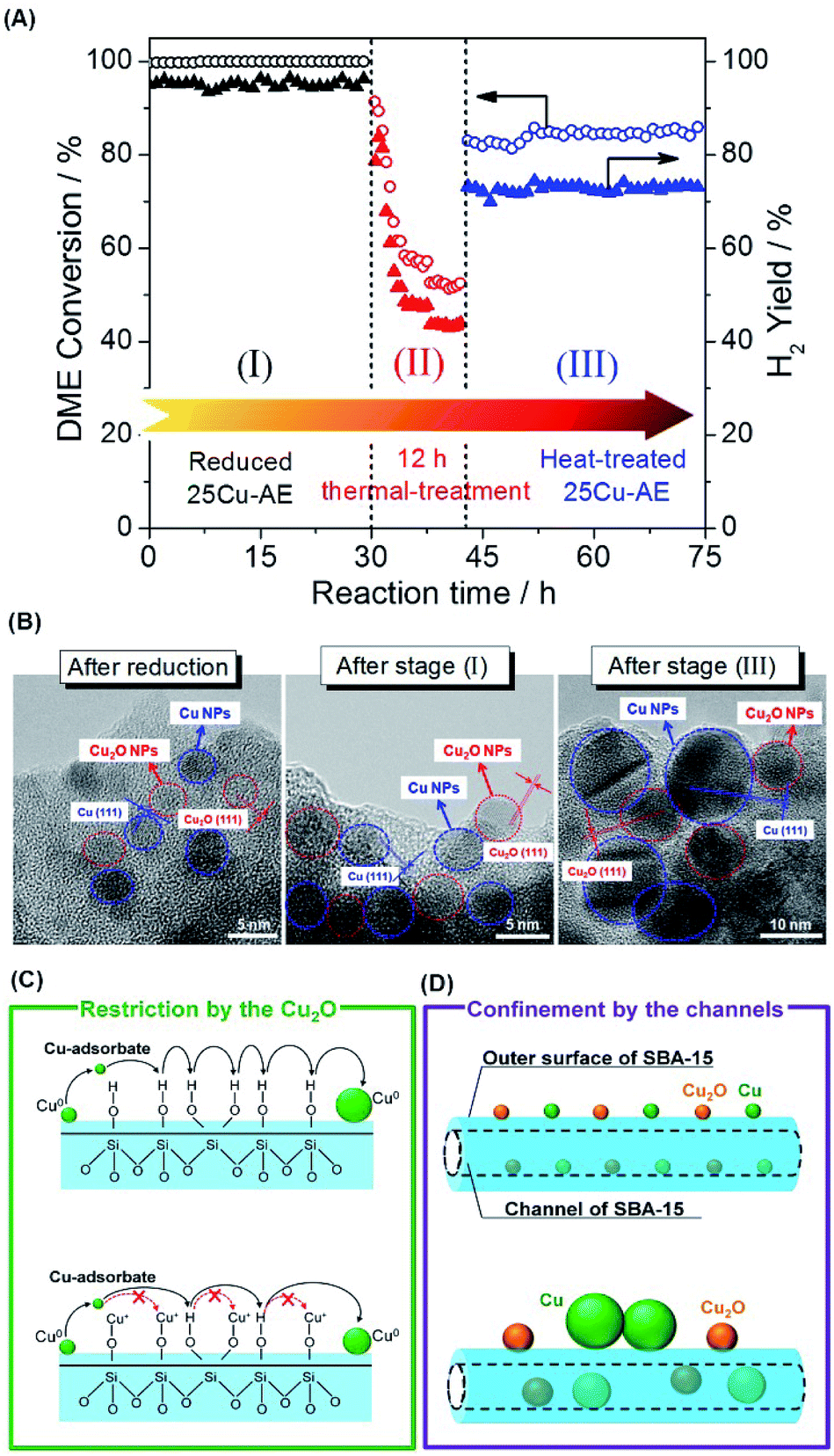 | ||
| Fig. 3 (A) Stabilities of 25Cu-AE upon thermal treatment. Reaction conditions: (I) and (III): GHSV = 18000 h−1, S/C = 2/1 (mol mol−1), T = 400 °C; (II): GHSV = 36000 h−1, S/C = 2/1 (mol mol−1), T = 450 °C; (B) TEM images of 25Cu-AE during the stability test; (C) schematic representation of the restriction by the adjacent Cu2O; (D) schematic illustration of the spatial confinement by the channels. | ||
As the reaction temperature (400 °C) is relatively lower than the Tammann temperature of copper (407 °C), the thermal migration of copper atoms in the catalysts is difficult in the absence of a reactive adsorbate. Thus, the copper particle growth in SR is probably mediated by mobile copper adsorbate species on the support (Ostwald ripening).29 The transport of Ostwald ripening Cu species depends upon the chemical nature of the support surface. As evidenced by FTIR in Fig. S1A,† the formation of copper phyllosilicates is accompanied by consuming surface hydroxyl groups, which is an efficient support functionalization to increase the distance between neighboring functional groups and consequently inhibits Cu0 diffusion, as presented in Fig. 3C. Accordingly, the structure of Cu surrounded by Cu2O in 25Cu-AE maximizes such restriction. Also, Cu2O NPs can function as a physical spacer to isolate Cu0 NPs.
After the thermal treatment (stage II), the DME conversion over 25Cu-AE drops to 84% but remains stable for another 30 h at stage III; meanwhile the TEM images of 25Cu-AE in Fig. 3B demonstrate a break of the surrounding structure of copper species. Herein, a bimodal size distribution of ∼11.2 and ∼5.8 nm is presented after stage III, which is attributed to the aggregated Cu species on the outer surface and in the channels, respectively (Fig. S13A†). For 25Cu-SiO2, the catalytic performance substantially decreases at stage III. The corresponding Cu NPs noticeably aggregate as indicated by a distribution of ∼15.0 nm after stage III (Fig. S13B†). Thus, we conclude that the spatial restriction by the channels of SBA-15 can also help inhibit the aggregation of Cu0 (Fig. 3D). Owing to such combined effects, 25Cu-AE exhibits a robust performance (DME conversion: 83.9%, H2 yield: 72.7%) even after the severe thermal treatment.
We hypothesize that the high activity of Cu-AE results from a cooperative effect of the unique dual-sites of copper species. To gain more insight into this, the respective contributions from the Cu0 and Cu+ species are required to be clarified. Thus catalysts containing pure Cu0 and Cu+ species are desired. However, the Cu0 and Cu+ always coexist in Cu-AE catalysts. For the above compared 25Cu-IM, its crystal size (29 nm) is much larger and less uniform than those of Cu-AE (<10 nm) (Table S2†). Moreover, the impregnation method inevitably brings the strong metal–support interaction to affect the chemical state and size distribution of copper. Consequently, we introduce a physical-sputtering method40 to realize a homogeneously distributed metallic Cu nano-catalyst (Cu-SP) instead of 25Cu-IM for rigorous comparison and discussion. For Cu-SP, the TEM and H2-TPR results (Fig. S14†) show that the copper NPs mainly exist as Cu0, which could be oxidized to Cu2O by N2O to obtain Cu-SP-N. Their physicochemical properties are listed in Table S5.†
We quantify the surface density of Cu+ and Cu0 sites (Tables S2 and S5†) to analyze their contributions toward the reaction. In Fig. 4A, the surface Cu0 site density is observed to scale linearly with the formation rates of H2 (rH2) in group I and II independently, suggesting that the H2 yield depends on the Cu0 density with two different reaction paths, whereas there is no clear correlation between the rH2 and the total Cu density (Fig. 4B). Moreover, the higher superficial Cu+/(Cu0 + Cu+) ratio (>0.50) over the catalysts in group I than that (<0.21) in group II is probably the main reason for their superior performance. Thus, we conclude that Cu0 is responsible for H2 desorption, and sufficient Cu+ can accelerate the reaction rate. In Fig. 4C, both the TOFDME and TOFH2 show a volcanic correlation with the increased Cu0/(Cu0 + Cu+) ratio, and reach a maximum at a Cu0/(Cu0 + Cu+) ratio equal to 0.50 (25Cu-AE). The activation energies (Ea) of the catalysts were calculated and classified into two types in Fig. 4D: the lower value at ca. 125 kJ mol−1 for 10Cu-AE, 20Cu-AE, 25Cu-AE and 30Cu-AE with sufficient Cu+ sites (denoted as Cu+-suf); and the higher value at ca. 160 kJ mol−1 for 25Cu-IM, Cu-SP and 40Cu-AE with sufficient Cu0 sites (denoted as Cu0-suf). Interestingly, Ea dropped from 164.5 to 124.5 kJ mol−1 when the Cu0 sites in Cu-SP were oxidized to Cu+ by N2O to obtain Cu-SP-N. These results demonstrate the determinant role of the sufficient Cu+ sites for reaction paths. Herein, 30Cu-AE follows the same reaction path as the other Cu+-suf catalysts, while its rH2 diverged from that of group I in Fig. 4A, which is probably ascribed to the excessive Cu0 sites on 30Cu-AE for H2 desorption. Summarily, we propose that there exist two reaction paths, and which to follow is determined by Cu+.
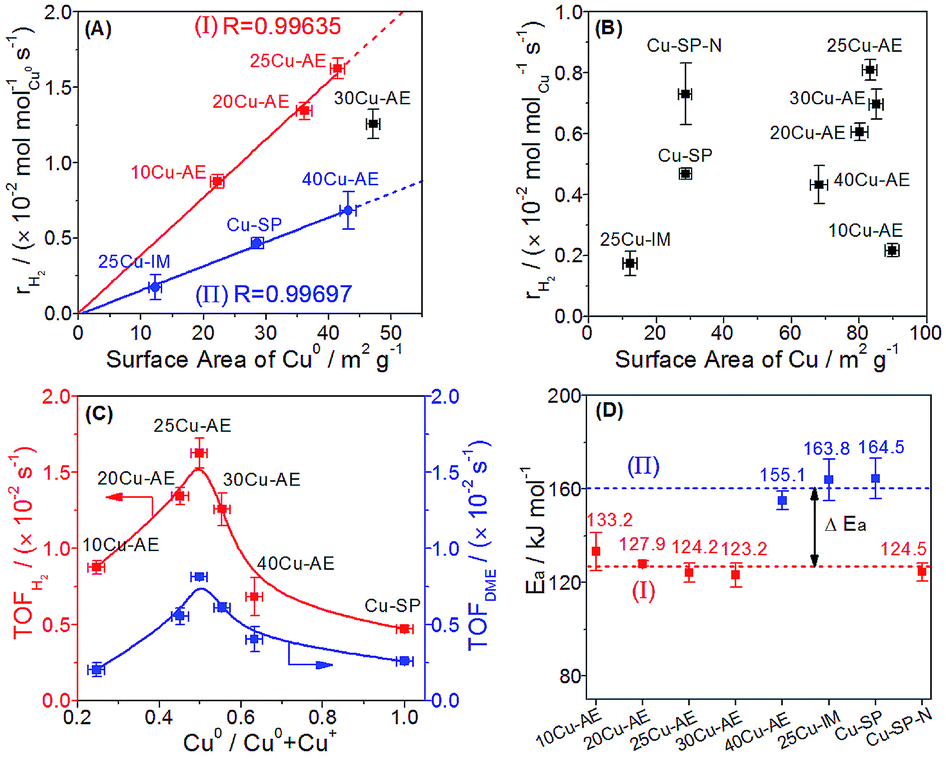 | ||
| Fig. 4 Correlation of the H2 production rate based on Cu0 sites or total Cu sites with (A) the surface area of Cu0 and (B) total surface area of Cu; (C) correlation of the TOF of H2 and DME with the Cu0/(Cu0 + Cu+) ratio; (D) apparent activation energies of the catalysts. | ||
Thereafter, we evaluated in situ DRIFTS and temperature programmed surface reaction-mass spectroscopy (TPSR-MS) for methanol SR to validate the role of the copper species in the reaction path. In Fig. 5A, we observe the bands of Cu+–OCH3 (methoxy) (1440 cm−1) over Cu+-suf and Cu0–OOCH (formate) (1345 cm−1) over Cu0-suf, respectively, which is also evidenced by the steady-state TPSR-MS (Fig. 5B and C). This suggests that the methoxy and formate groups on their corresponding catalysts are relatively stable, and their dehydrogenation affects the overall reaction rate. Importantly, dioxomethylene (CH2O2) is also detected by TPSR-MS in Fig. 5C. Along with the kinetics and Ea discrimination from Fig. 4, these findings demonstrate the different rate-determining steps: methoxy dehydrogenation for Cu+-suf and formate dehydrogenation for Cu0-suf.
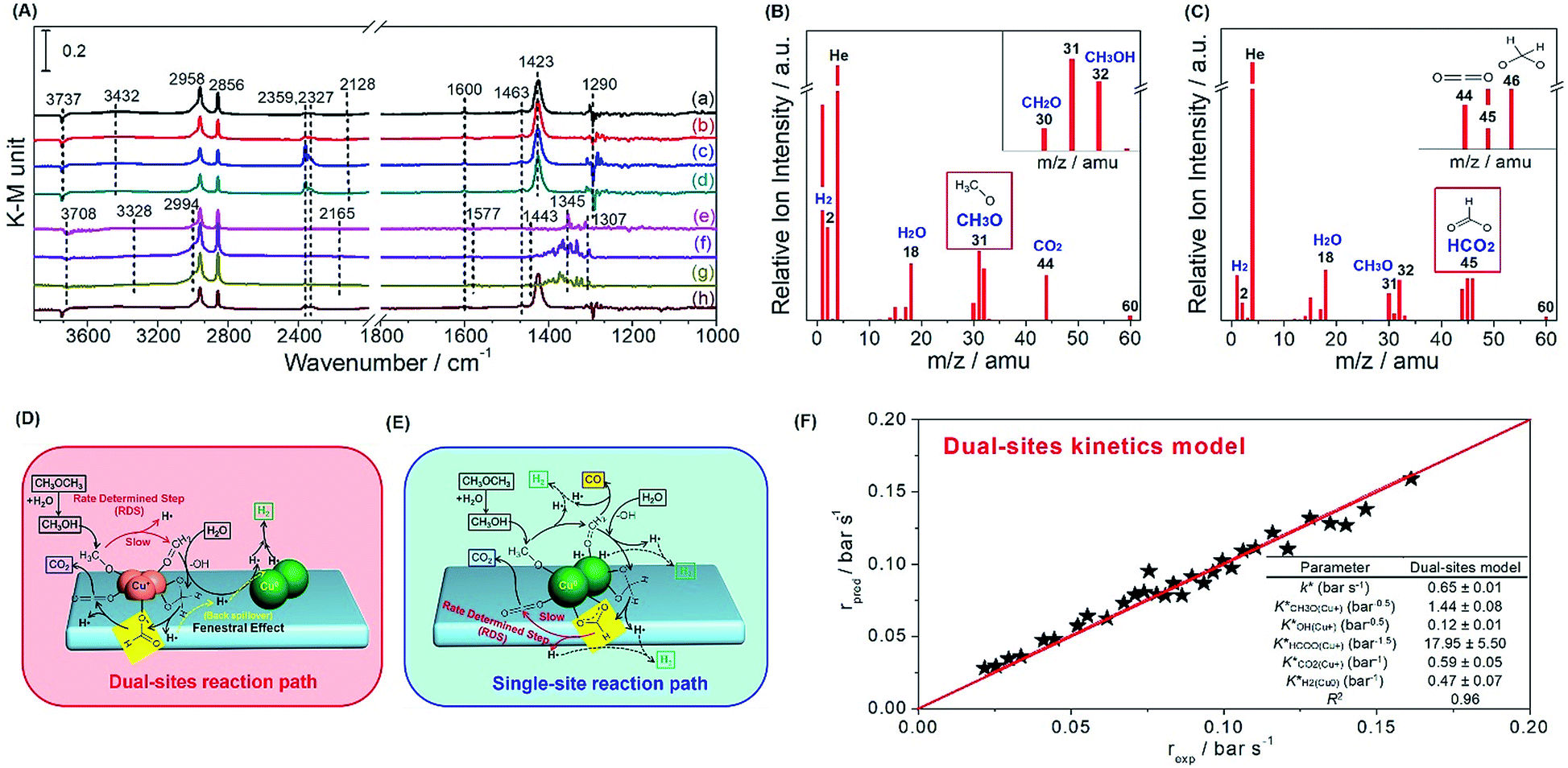 | ||
| Fig. 5 (A) Steady-state DRIFTS spectra for methanol SR on reduced (a) 10Cu-AE, (b) 20Cu-AE, (c) 25Cu-AE, (d) 30Cu-AE, (e) 40Cu-AE, (f) 25Cu-IM, and (g) Cu-SP, and (h) Cu-SP-N; TPSR-MS spectra for methanol SR over the (B) reduced 25Cu-AE (Cu+-suf) and (C) Cu-SP (Cu0-suf) at 230 °C under steady-state conditions; the hypothetical reaction paths on the (D) dual-sites of copper species; and (E) single Cu0 site; (F) parity plots of microkinetic modeling for methanol SR over 25Cu-AE, and the inset shows the fitting parameters given with the confidence interval (±5%) and stability index R2. | ||
Then we collected time-resolved in situ DRIFTS spectra of 25Cu-AE, Cu-SP and Cu-SP-N (Fig. S15–S17, Table S6†) to draw the hypothetical mechanism. Over Cu+-suf, we assume that the Cu0 sites are responsible for transferring atomic H and hydrogen desorption; and the Cu+ sites contributed to adsorbing oxygen-bonded intermediates.41 From the in situ DRIFTS results in Fig. S15,† the catalytic cycle begins with the dissociative adsorption of methanol (1008 and 1033 cm−1) on the surface. Then, dehydrogenation of the methoxy group (1440 and 1423 cm−1) to formaldehyde occurs as the first barrier. Surface hydroxyls (3737 cm−1) immediately attack the formaldehyde, resulting in the formation of dioxomethylene as a reaction intermediate, whereafter, it dehydrogenates into formate groups (1548 and 1600 cm−1) and proceeds to dehydrogenate eventually to CO2 (2359 and 2327 cm−1), adsorbing in the form of monodentate carbonate species (1463 cm−1). All H atoms produced by dehydrogenation migrate to the Cu0 sites through support due to the back spillover, and finally desorb as gaseous H2. We name it a dual-site reaction path (Fig. 5D). Over Cu0-suf in Fig. S16,† the adsorption and transition of methanol (1010 and 1030 cm−1), formaldehyde (1736 cm−1), dioxomethylene, formate (1345 and 1577 cm−1) and carbonate (1371 cm−1) occurs on the Cu0 sites alone (Fig. 5E), consistent with the reported DFT calculations on Cu(111).42 In this case, H is competitively adsorbed with oxygenated intermediates, thereby readily meeting the second barrier at formate dehydrogenation and inhibiting the overall reaction rate.
Interestingly, in Fig. 5A, monodentate formate (1600 cm−1), considered to be more active in deep dehydrogenation,43 tends to form on Cu+-suf, while both monodentate (1577 cm−1) and bidentate (1345 cm−1) species appear on Cu0-suf. Cu+ sites may function as the electrophilic sites to readily electrostatically adsorb the negative electron of O–C in formate, thereby forming more active monodentate formate to overcome the formate dehydrogenation barrier. Thus, we intrinsically reveal that the rate-determining step of the dehydrogenation of methoxy or formate relies on the valence state of Cu.
Through the evaluation on a recycle fixed-bed reactor (Fig. S18†), a microkinetic model for methanol SR over 25Cu-AE was established to verify the reliability of our proposed dual-site reaction path (see details in the Experimental section of the ESI†). In this model, the competitive adsorption of oxygenates and dissociative adsorption of hydrogen occur on Cu+ and Cu0 sites, respectively. The dehydrogenation of the methoxy group on Cu+ sites is the rate-determining step, and thus all of the other elemental reactions are in thermodynamic equilibrium. As expected, the experimental data and the dual-site microkinetic prediction are in good agreement (Fig. 5F).
Conclusions
In summary, we have successfully designed a dual Cu reforming system with a record-high and robust H2 production rate of 1145 mol kgcat−1 h−1, and provided new insights into the fundamental understanding of Cu-catalyzed SR. In fact, there exist two kinds of distinct rate-determining steps: methoxy dehydrogenation for Cu+-suf and formate dehydrogenation for Cu0-suf. Cu+ may function as the electrophilic site to adsorb the negative electron of O–C in formate, thereby more readily forming active monodentate formate. This can intrinsically accelerate the overall reaction rates. Moreover, the surrounding Cu2O and nano-channels of SBA-15 can inhibit the aggregation of metallic Cu species during SR. Our design provides a commercially achievable reforming system for fuel cells and paves a new way to related reaction mechanism investigations.Conflicts of interest
The authors declare no conflict of interest.Author contributions
X. G. L., J. L. G. and K. M. conceived the idea; K. M., J. L. G., Y. T., Z. J. Z., Q. P. C. and T. D. carried out the experiments; N. T. and T. A. prepared the sputtered catalysts; J. Z., L. R. Z. and Z. J. helped collect XAFS data; X. G. L., J. L. G. and K. M. analyzed the experimental data and wrote the manuscript. X. G. L. and J. L. G. supervised the whole project. All authors participated in discussions of the research.Acknowledgements
This work is supported by the National Key R&D Program of China (2016YFB0600901), National Natural Science Foundation of China (No. 21476159, 21476160, and 21525626) and Program of Introducing Talents of Discipline to Universities (No. B06006). We gratefully thank Prof. Ming Meng for his help with this work. We also acknowledge the staff members of the 1W1B station in Beijing Synchrotron Radiation Facility (BSRF) and BL14W1 station in Shanghai Synchrotron Radiation Facility (SSRF) for their assistance in the XAFS experiments.Notes and references
- M. Dresselhaus and I. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- U. Eberle, B. Muller and R. von Helmolt, Energy Environ. Sci., 2012, 5, 8780–8798 RSC.
- S. Krishnan and F. A. Armstrong, Chem. Sci., 2012, 3, 1015–1023 RSC.
- R. D. Cortright, R. Davda and J. A. Dumesic, Nature, 2002, 418, 964–967 CrossRef CAS PubMed.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- P. Leung, A. Tsolakis, J. Rodriguez-Fernandez and S. Golunski, Energy Environ. Sci., 2010, 3, 780–788 RSC.
- C. Dang, Y. Li, S. M. Yusuf, Y. Cao, H. Wang, H. Yu, F. Peng and F. Li, Energy Environ. Sci., 2018, 11, 660–668 RSC.
- X. Zhao, H. Zhou, V. S. Sikarwar, M. Zhao, A.-H. A. Park, P. S. Fennell, L. Shen and L.-S. Fan, Energy Environ. Sci., 2017, 10, 1885–1910 RSC.
- T. A. Semelsberger, R. L. Borup and H. L. Greene, J. Power Sources, 2006, 156, 497–511 CrossRef CAS.
- J. Sun, G. Yang, Y. Yoneyama and N. Tsubaki, ACS Catal., 2014, 4, 3346–3356 CrossRef CAS.
- R. Luque, L. Herrero-Davila, J. M. Campelo, J. H. Clark, J. M. Hidalgo, D. Luna, J. M. Marinas and A. A. Romero, Energy Environ. Sci., 2008, 1, 542–564 RSC.
- K. Faungnawakij, R. Kikuchi, N. Shimoda, T. Fukunaga and K. Eguchi, Angew. Chem., Int. Ed., 2008, 47, 9314–9317 CrossRef CAS PubMed.
- D. Li, X. Li and J. Gong, Chem. Rev., 2016, 116, 11529–11653 CrossRef CAS PubMed.
- D. R. Palo, R. A. Dagle and J. D. Holladay, Chem. Rev., 2007, 107, 3992–4021 CrossRef CAS PubMed.
- H. Oguchi, T. Nishiguchi, T. Matsumoto, H. Kanai, K. Utani, Y. Matsumura and S. Imamura, Appl. Catal., A, 2005, 281, 69–73 CrossRef CAS.
- Y. Matsumura and H. Ishibe, J. Catal., 2009, 268, 282–289 CrossRef CAS.
- Y. Matsumura and H. Ishibe, Appl. Catal., B, 2009, 86, 114–120 CrossRef CAS.
- I. Ritzkopf, S. Vukojević, C. Weidenthaler, J.-D. Grunwaldt and F. Schüth, Appl. Catal., A, 2006, 302, 215–223 CrossRef CAS.
- Y. Liu, T. Hayakawa, K. Suzuki, S. Hamakawa, T. Tsunoda, T. Ishii and M. Kumagai, Appl. Catal., A, 2002, 223, 137–145 CrossRef CAS.
- X. Wang, K. Ma, L. Guo, Y. Tian, Q. Cheng, X. Bai, J. Huang, T. Ding and X. Li, Appl. Catal., A, 2017, 540, 37–46 CrossRef CAS.
- C. Rameshan, W. Stadlmayr, S. Penner, H. Lorenz, N. Memmel, M. Hävecker, R. Blume, D. Teschner, T. Rocha, D. Zemlyanov, A. Knop-Gericke, R. Schlögl and B. Klötzer, Angew. Chem., Int. Ed., 2012, 51, 3002–3006 CrossRef CAS PubMed.
- K. M. K. Yu, W. Tong, A. West, K. Cheung, T. Li, G. Smith, Y. Guo and S. C. E. Tsang, Nat. Commun., 2012, 3, 1230 CrossRef PubMed.
- J. Huang, T. Ding, K. Ma, J. Cai, Z. Sun, Y. Tian, Z. Jiang, J. Zhang, L. Zheng and X. Li, ChemCatChem, 2018, 10, 3862–3871 CrossRef CAS.
- H. Xi, X. Hou, Y. Liu, S. Qing and Z. Gao, Angew. Chem., Int. Ed., 2014, 53, 11886–11889 CrossRef CAS PubMed.
- K. Ma, Z. Cui, Z. Zhang, J. Huang, Z. Sun, Y. Tian, T. Ding and X. Li, ChemCatChem, 2018, 10, 4010–4017 CrossRef CAS.
- L. F. Chen, P. J. Guo, M. H. Qiao, S. R. Yan, H. X. Li, W. Shen, H. L. Xu and K. N. Fan, J. Catal., 2008, 257, 172–180 CrossRef CAS.
- J. Gong, H. Yue, Y. Zhao, S. Zhao, L. Zhao, J. Lv, S. Wang and X. Ma, J. Am. Chem. Soc., 2012, 134, 13922–13925 CrossRef CAS PubMed.
- H. Yue, Y. Zhao, S. Zhao, B. Wang, X. Ma and J. Gong, Nat. Commun., 2013, 4, 2339 CrossRef PubMed.
- R. Van den Berg, T. E. Parmentier, C. F. Elkjær, C. J. Gommes, J. Sehested, S. Helveg, P. E. de Jongh and K. P. de Jong, ACS Catal., 2015, 5, 4439–4448 CrossRef CAS.
- J. Fan, X. Jiang, H. Min, D. Li, X. Ran, L. Zou, Y. Sun, W. Li, J. Yang, W. Teng, G. Li and D. Zhao, J. Mater. Chem. A, 2014, 2, 10654–10661 RSC.
- T. Tsoncheva, V. Dal Santo, A. Gallo, N. Scotti, M. Dimitrov and D. Kovacheva, Appl. Catal., A, 2011, 406, 13–21 CrossRef CAS.
- X. Wang, J. Zhuang, J. Chen, K. Zhou and Y. Li, Angew. Chem., Int. Ed., 2004, 116, 2051–2054 CrossRef.
- A. Ungureanu, B. Dragoi, A. Chirieac, C. Ciotonea, S. Royer, D. Duprez, A. S. Mamede and E. Dumitriu, ACS Appl. Mater. Interfaces, 2013, 5, 3010–3025 CrossRef CAS PubMed.
- L. F. Chen, P. J. Guo, L. J. Zhu, M. H. Qiao, W. Shen, H. L. Xu and K. N. Fan, Appl. Catal., A, 2009, 356, 129–136 CrossRef CAS.
- A. Marchi, J. Fierro, J. Santamaria and A. Monzon, Appl. Catal., A, 1996, 142, 375–386 CrossRef CAS.
- A. Yin, X. Guo, W. L. Dai and K. Fan, J. Phys. Chem. C, 2009, 113, 11003–11013 CrossRef CAS.
- X. Zheng, H. Lin, J. Zheng, X. Duan and Y. Yuan, ACS Catal., 2013, 3, 2738–2749 CrossRef CAS.
- L. S. Kau, D. J. Spira-Solomon, J. E. Penner-Hahn, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1987, 109, 6433–6442 CrossRef CAS.
- Y. Tanaka, R. Kikuchi, T. Takeguchi and K. Eguchi, Appl. Catal., B, 2005, 57, 211–222 CrossRef CAS.
- X. G. Li, C. Liu, J. Sun, H. Xian, Y. S. Tan, Z. Jiang, A. Taguchi, M. Inoue, Y. Yoneyama, T. Abe and N. Tsubaki, Sci. Rep., 2013, 3, 2813 CrossRef PubMed.
- S. Lin, D. Xie and H. Guo, J. Phys. Chem. C, 2011, 115, 20583–20589 CrossRef CAS.
- S. Lin, R. S. Johnson, G. K. Smith, D. Xie and H. Guo, Phys. Chem. Chem. Phys., 2011, 13, 9622–9631 RSC.
- A. Haghofer, D. Ferri, K. Föttinger and G. n. Rupprechter, ACS Catal., 2012, 2, 2305–2315 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00015a |
| This journal is © The Royal Society of Chemistry 2019 |