
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed oxidative C–H/N–H annulation of N-heteroaromatic compounds with alkynes†
Atsushi
Obata
,
Akane
Sasagawa
,
Ken
Yamazaki
,
Yusuke
Ano
 and
Naoto
Chatani
*
and
Naoto
Chatani
*
Department of Applied Chemistry, Faculty of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: chatani@chem.eng.osaka-u.ac.jp
First published on 4th February 2019
Abstract
The reaction of N-heteroaromatic compounds, such as 2-aryl-pyrrole, benzimidazole, imidazole, indole, and pyrazole derivatives, with alkynes in the presence of a catalytic amount of a nickel complex results in C–H/N–H oxidative annulation. The reaction shows a high functional group compatibility. While both Ni(0) and Ni(II) complexes show a high catalytic activity, Ni(0) is proposed as a key catalytic species in the main catalytic cycle. In the case of the Ni(II) system, the presence of a catalytic amount of a strong base, such as KOBut, is required for the reaction to proceed. In sharp contrast, a base is not required in the case of the Ni(0) system. The proposed mechanism is supported by DFT studies.
Introduction
Transition-metal catalyzed C–H functionalization reactions have experienced notable progress over the past two decades.1 At the early stage of the development of such reactions, noble metals, such as Pd, Rh, Ir, and Ru, were frequently used as catalysts and these metals continue to be the catalysts of choice in various C–H functionalization reactions. However, the use of first row transition metals,2 such as Fe,3 Co,4 Ni,5 and Cu,6 has attracted a great deal of interest because they are earth-abundant and therefore less expensive than noble metals. If these elements could be used in such transformations, it would permit the scope of C–H functionalization reactions to be greatly expanded. Among these metals, nickel catalysts are of particular interest. Although a pioneering example of cyclometalation with the cleavage of an ortho C–H bond was achieved by the reaction of azobenzene with a nickel complex,7 Ni-catalyzed C–H functionalization reactions have generally remained undeveloped. One of the reasons for this is the absence of a general and reliable catalytic system for Ni-catalyzed C–H functionalization. In fact, Ni-catalyzed C–H functionalization reactions were limited to specific substrates that contain an acidic C–H bond, such as pentafluorobenzene, pyridine derivatives, and azoles.5a However, since our initial report in 2013 on the use of an 8-aminoquinoline directing group8 in Ni(II)-catalyzed C–H alkylation with alkyl halides,9a a combination of Ni(II) catalysts and N,N′-bidentate directing groups has now emerged as a powerful methodology for use in Ni-catalyzed C–H functionalization reactions.5b,c Since then, the Ni(II) catalyst/8-aminoquinoline directing system has been extensively used for the development of Ni-catalyzed C–H functionalizations.5b,c,9,10In a previous study, we reported on the Ni-catalyzed oxidative annulation of C–H bonds in aromatic amides with alkynes, leading to the production of 1(2H)-isoquinolinones (Scheme 1).11 In this reaction, a specific N,N′-bidentate directing group is not required for the reaction to proceed, in contrast to our study in 2011.12 A key to the success of the reaction is the use of a catalytic amount of KOBut. We wish to report herein that this newly developed system is also applicable to the oxidative annulation of C–H bonds in various 2-aryl-N-heteroaromatic compounds (Scheme 1). A theoretical study was also conducted in connection with these experimental findings. The DFT studies indicated that the reaction proceeds via two reaction paths. One mechanism is initiated by Ni(II) and KOBut, and the other, the main catalytic cycle, is initiated by Ni(0). Although the Ni(II) system requires a base, the Ni(0) system proceeds even in the absence of KOBut. Nitrogen-containing polyaromatic scaffolds are widely found in natural products, pharmaceutical agents, and π-conjugated functional materials.13 A similar transformation was previously reported using noble metal complexes, such as [Cp*RhCl2]2 and [RuCl2(p-cymene)]2.14 The present reaction is the first example of the use of a nickel catalyst in oxidative C–H/N–H annulation reactions with alkynes.
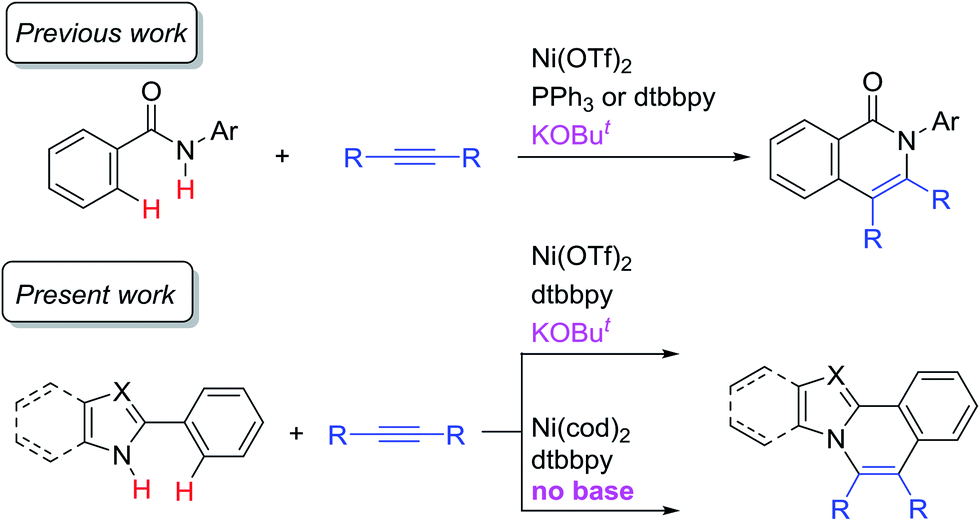 | ||
| Scheme 1 Ni-catalyzed oxidative C–H/N–H annulation of N-heteroaromatic compounds with alkynes. | ||
Results and discussion
The reaction of 2-phenylindole (1a) with diphenylacetylene was carried out under the same reaction conditions as were used in the reaction of aromatic amides.11a Unexpectedly, no reaction took place (entry 1 in Table 1). However, the use of 4,4-di-tert-butyl-2,2-dipyridyl (dtbbpy) as a ligand in place of PPh3 gave the expected product, 5,6-diphenylindolo[2,1-a]isoquinoline (2a) (entry 2). Increasing the reaction temperature to 180 °C slightly improved the product yield (entry 3). Conducting the reaction in the absence of a solvent resulted in a further improvement in the product yield (entry 4). Finally, the reaction conditions shown in entry 5 were used as standard reaction conditions. Curiously, Ni(cod)2 also showed catalytic activity (entry 6). This result will be discussed below in relation to the reaction mechanism.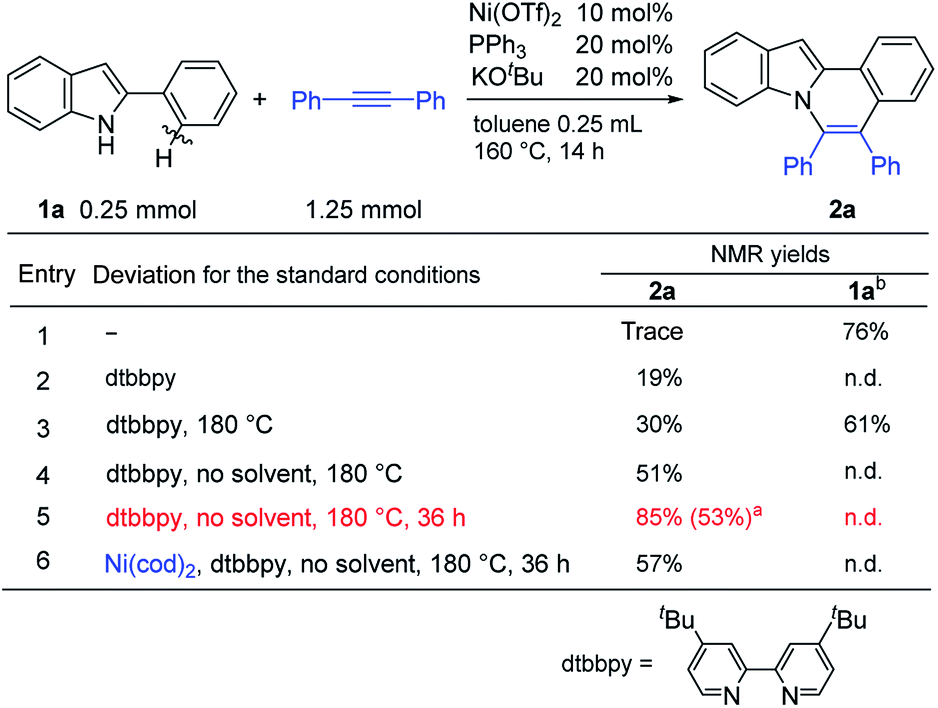
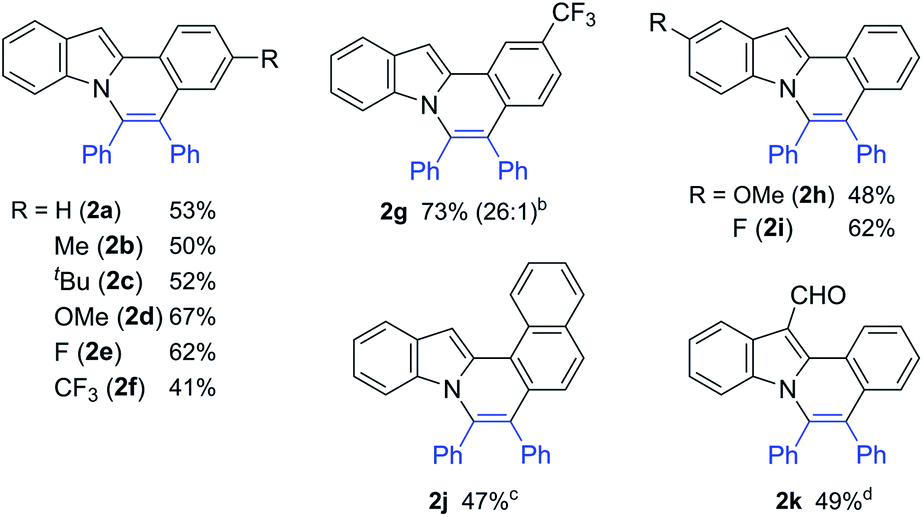
The scope of this oxidative annulation reaction with respect to 2-arylindole derivatives was also investigated (Table 2). A variety of functional groups, including methoxy, fluoride, trifluoromethyl, and even formyl groups, were tolerated in the reaction. In the reaction of 3-trifluoromethylphenylindole (2g), the less hindered C–H bond was selectively activated. In all cases, the yields of the expected product determined by 1H NMR were good to high. However, a small amount of an unidentified product was produced as a contaminant. To remove this contaminant from the expected products, the product was purified by Gel Permeation Chromatography (GPC), which reduced the product yield.
| a Reaction conditions: 2-arylindole (0.25 mmol), diphenylacetylene (1.25 mmol), Ni(OTf)2 (0.025 mmol), dtbbpy (0.05 mmol), and KOBut (0.05 mmol) at 180 °C for 36 h. Isolated by GPC. b The number in parentheses refers to the regioselectivity. c The reaction was carried out for 60 h. d Ligand (50 mol%) and toluene (0.25 mL). |
|---|
|
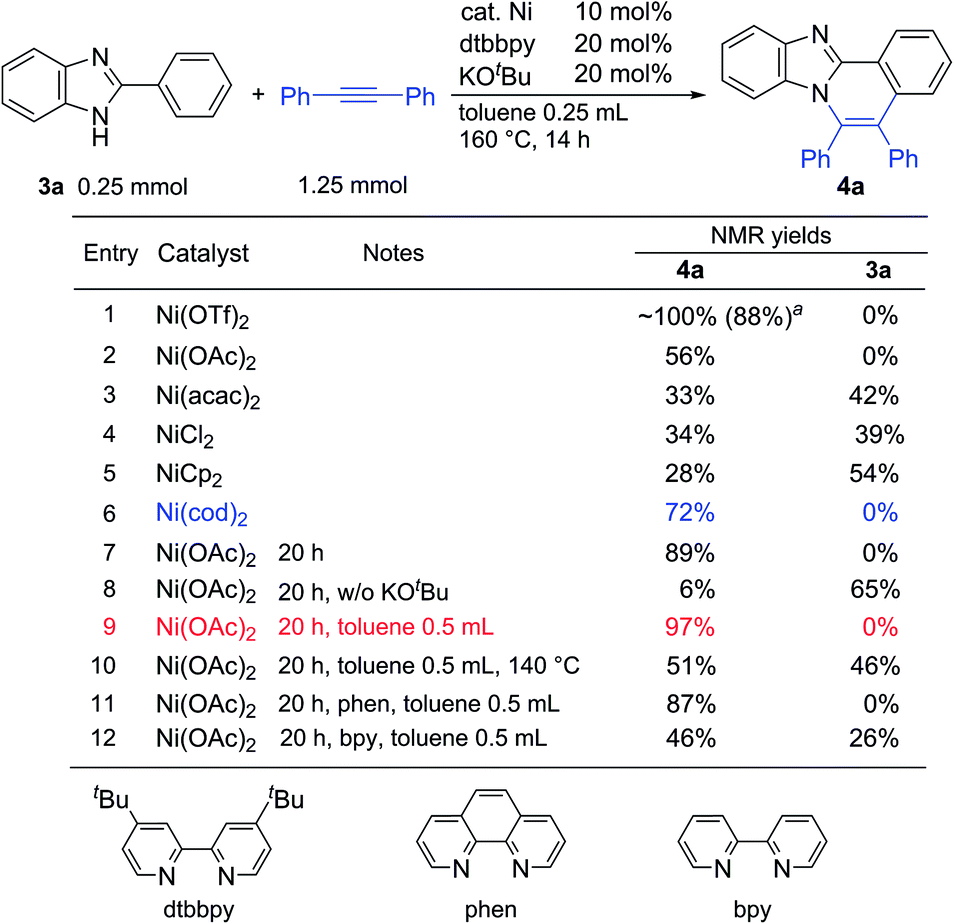
To expand the present protocol, the reaction of 2-phenylbenzimidazole (3a) with diphenylacetylene was examined under the same reaction conditions as were used in the reaction of 2-phenylindole (Table 3). As expected, the corresponding product, 5,6-diphenylbenzo[4,5]imidazo[2,1-a]isoquinoline (4a), was formed in nearly quantitative yield (entry 1). A screening of various nickel salts indicated that other Ni(II) complexes also showed catalytic activity (entries 2–5). Curiously, Ni(cod)2 again showed a high catalytic activity (entry 6). We then decided to optimize the reaction conditions using Ni(OAc)2 because Ni(OTf)2 is prepared from Ni(OAc)2 and trifluoromethanesulfonic acid in CH3CN,9a and is sensitive to moisture and air. The use of a longer reaction time resulted in an increased product yield (entry 7). In the absence of KOBut, the product yield was dramatically decreased (entry 8). The use of phenanthroline (phen) also gave 4a in high yield (entry 11), but the use of the bipyridine (bpy) ligand did not lead to optimal yields (entry 12). Finally, we determined that the reaction conditions shown in entry 9 are standard reaction conditions for the reaction of 2-phenylimidazole with alkynes.
| a Isolated yield. |
|---|
|
With the optimized reaction conditions in hand, we examined the scope of 2-arylimidazoles 3 and alkynes (Table 4). Not only diarylalkynes, but also aliphatic alkynes were applicable to the reaction. The reaction of 3 with aliphatic internal alkynes gave the corresponding benzo[4,5]imidazo[2,1-a]isoquinoline derivatives 4g and 4h in high yields. The reaction of 3 with phenylpropyne gave the corresponding product 4i in a regioselective manner, the structure of which was confirmed by X-ray crystallographic analysis.
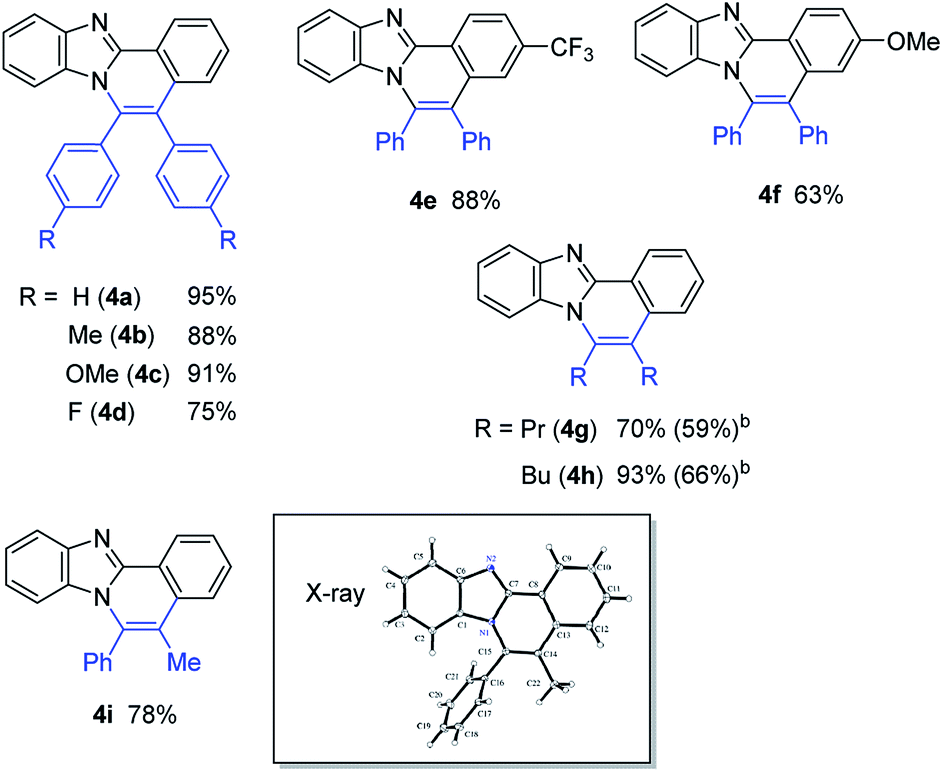
The reaction of 2-phenylimidazole (5), 3,5-diphenylpyrazole (7), and 2-phenylpyrrole (9) also gave the corresponding nitrogen-containing polyaromatic compounds, 6, 8, and 10, respectively (Table 5).
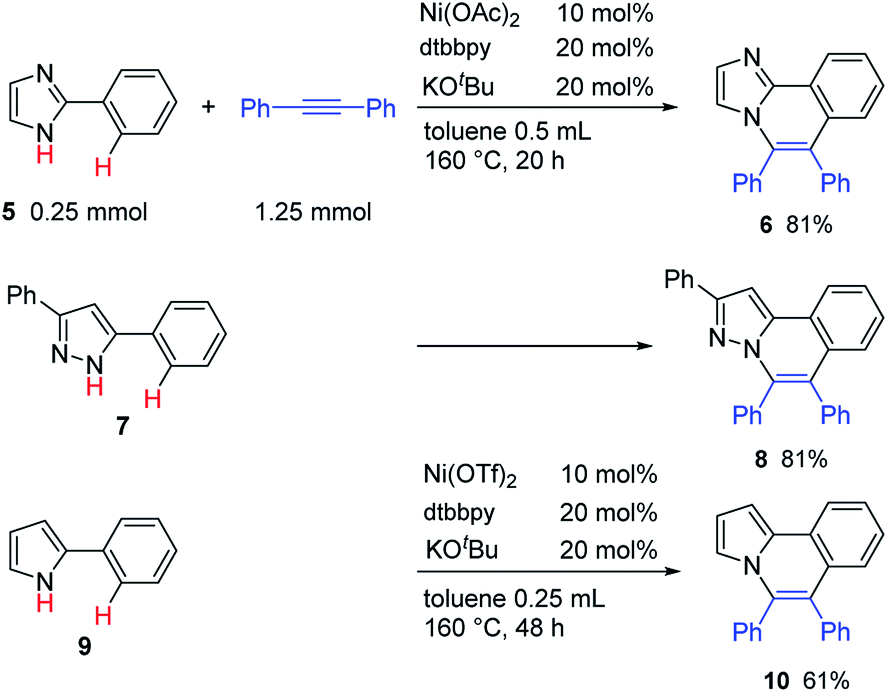
|
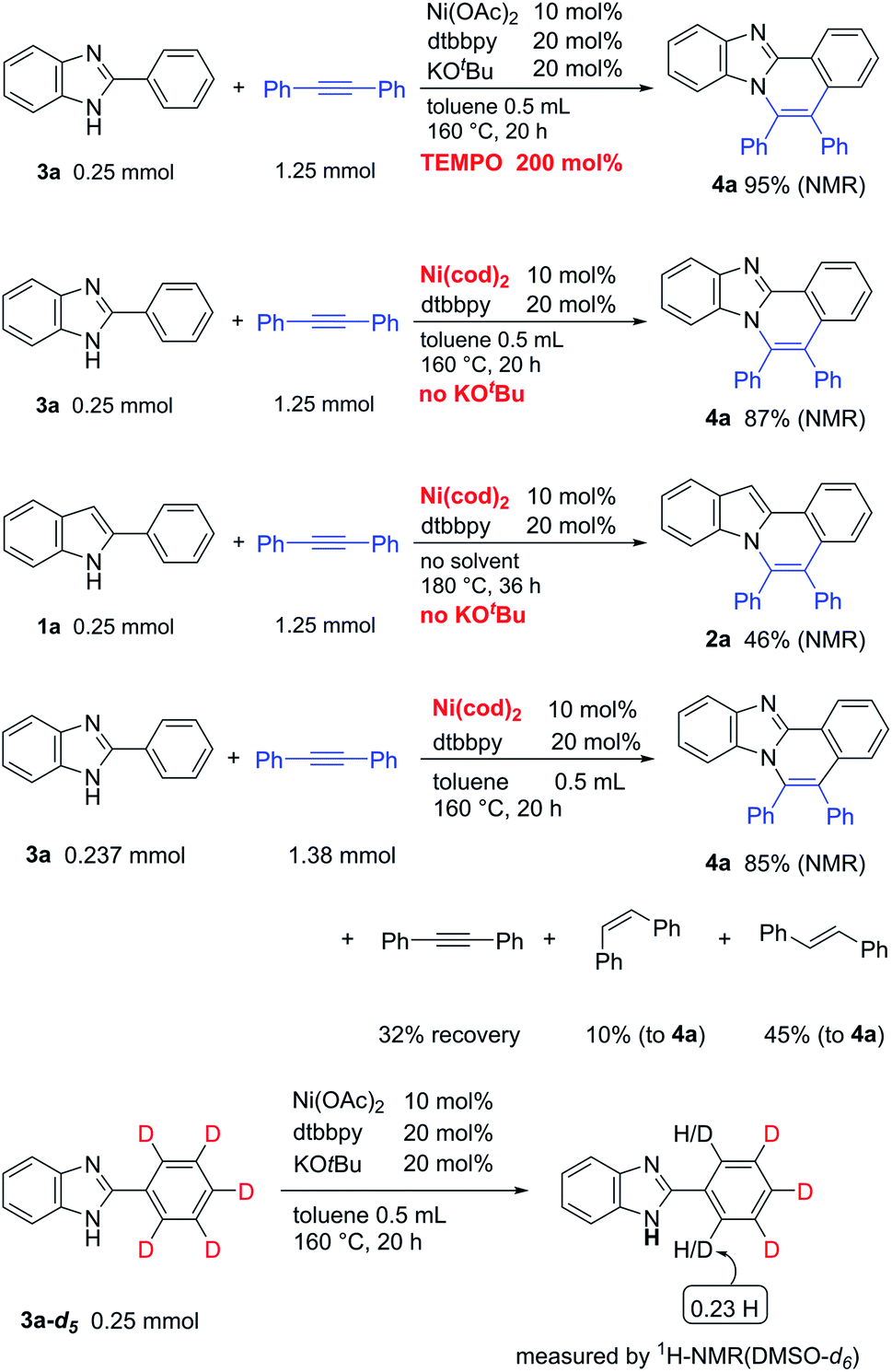
To gain insights into the reaction mechanism, the reaction was carried out in the presence of two equivalents of TEMPO. However, TEMPO did not inhibit the reaction, indicating that a radical path is not involved in the catalytic system (Scheme 2). As shown in Tables 1 and 3, the catalytic activity of Ni(cod)2 was comparable to that of Ni(II) complexes in the presence of KOBut. Curiously, it was found that the reaction proceeds in a highly efficient manner, even in the absence of KOBut. The reaction of 3a without KOBut gave 4a in a quantitative yield. Indole 1a also gave 2a. Thus, the use of KOBut is crucial for the reaction to proceed when Ni(II) complexes are used as a catalyst. In sharp contrast, KOBut is not required when Ni(0) is used as a catalyst. The reaction of 3a with diphenylacetylene in the presence of Ni(cod)2/dtbbpy gave 4a in 85% NMR yield, along with a 55% yield of stilbene relative to 4a. In the absence of 3a, no evidence was found for the formation of stilbene. These results indicate that diphenylacetylene functions as a hydrogen acceptor as well as a two-component coupling partner. A deuterium-labelling experiment in the absence of an alkyne was also carried out. H/D exchange took place only at the ortho-position.
 | ||
| Scheme 2 Mechanistic experiments. | ||
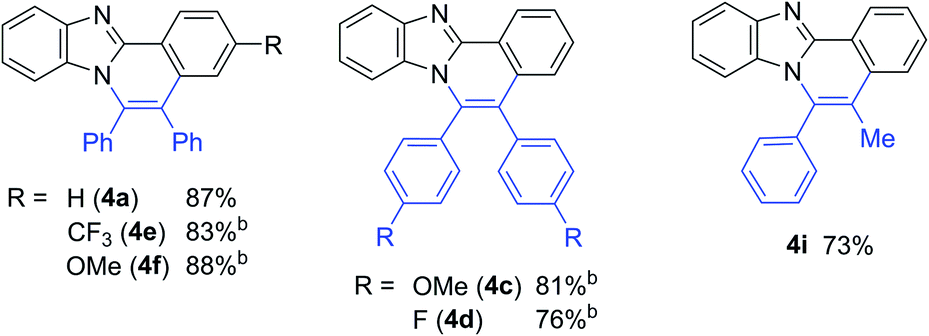
Some examples of the use of other substrates in the Ni(cod)2/no base system are summarized in Table 6.
Based on these results, a mechanism for the C–H/N–H oxidative annulation is proposed, as shown in Scheme 3. The mechanism involves two paths and is initiated by the Ni(II) complex (left scheme). An NH proton is abstracted from 3a by KOBut to generate the anion A, which reacts with Ni(II) to give the complex B. The cleavage of the ortho C–H bond followed by the insertion of an alkyne into a C–Ni bond in C gives complex D. A reductive elimination from D then gives 4a and Ni(0). However, Ni(0) cannot be re-oxidized to Ni(II) to complete a catalytic cycle under the reaction conditions employed. Instead, Ni(0) initiates another catalytic cycle (right scheme). The coordination of a N(sp2) atom in 3a to Ni(0) gives complex E, which involves a ligand-to-ligand hydrogen transfer (LLHT) mechanism,15,16 in which the ortho hydrogen atom is directly transferred to the alkyne carbon. The successive insertion of an alkyne into C–Ni bonds gives the complex G. Reductive elimination followed by deprotonation affords 4a with the regeneration of Ni(0) and the concomitant formation of an alkene. In fact, the formation of alkenes was detected by GC (Scheme 2). An alternative mechanism involves the oxidative addition of a N–H bond in 3a to Ni(0), leading to the generation of complex I. The possibility of this alternative mechanism being operative will be discussed below in conjunction with DFT studies.
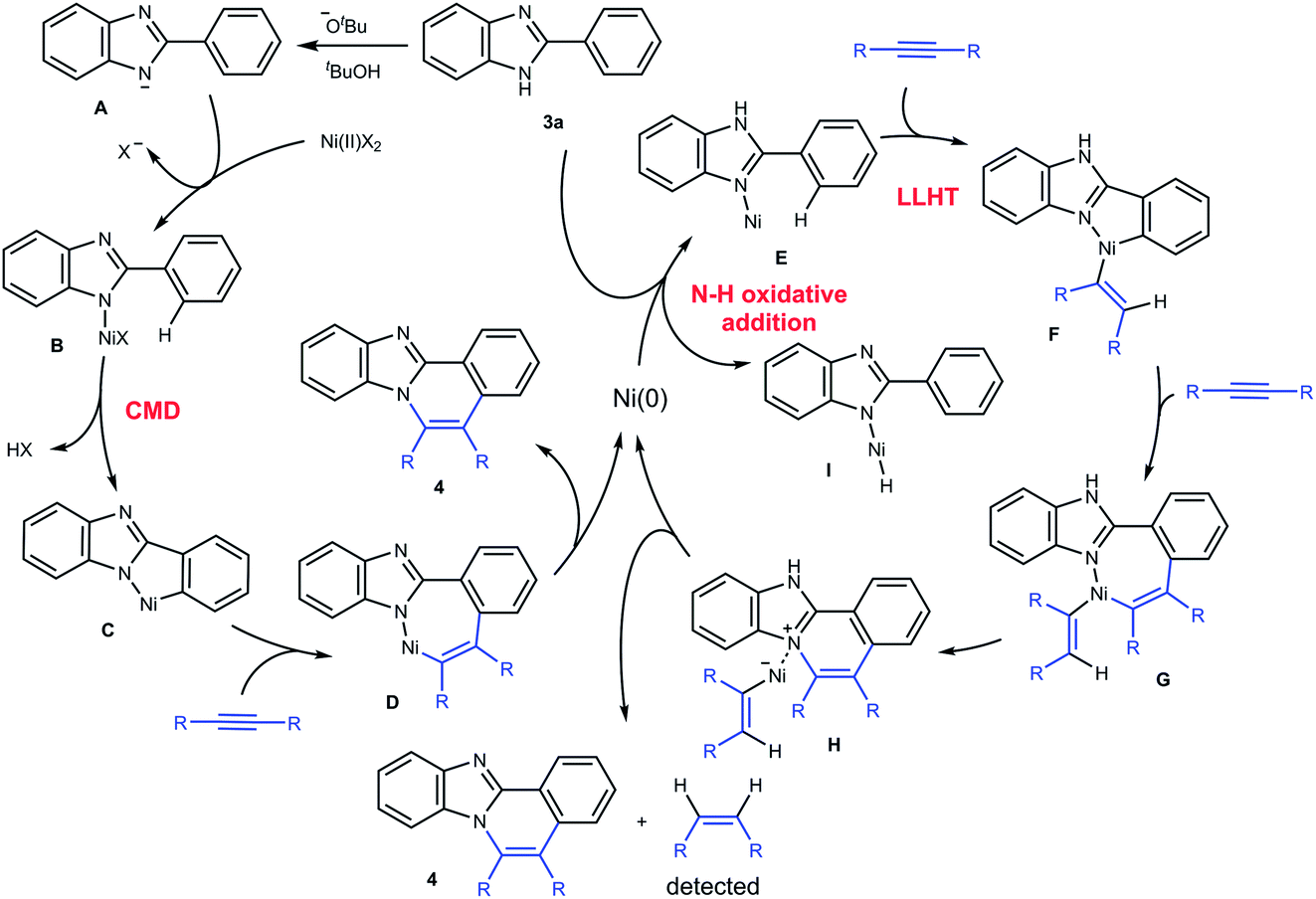 | ||
| Scheme 3 Proposed mechanism. | ||
DFT studies were conducted in an attempt to obtain a better understanding of the mechanism for this reaction. The resulting DFT studies are essentially supportive of the mechanism that is proposed above. As shown by the mechanistic details in Scheme 3, the presence of KOBut is crucial for the success of the initial reaction path (left scheme in Scheme 3), which is catalyzed by Ni(II), but it is not required for the main catalytic cycle in which Ni(0) functions as a catalyst (right scheme in Scheme 3). Therefore, we focused on the main reaction path that is initiated by Ni(0) under neutral conditions (red line in Scheme 4).17 The LLHT mechanism for the main catalytic cycle that proceeds viaTS1 has a low energy barrier (18.5 kcal mol−1). After coordination with a second alkyne molecule, the insertion of the alkyne into a Ni–C bond through TS2 proceeds smoothly, with an activation energy of 8.3 kcal mol−1. A final reductive elimination through TS3 from the relatively stable seven-membered metallacycle Int4 requires a large amount of energy to produce Int5, which is comprised of an ammonium cation and nickel anion pair. This step is followed by a proton shift to afford the product and the cis-alkene with the regeneration of Int1. While a detailed mechanism for the proton shift is not given here, the large amount of energy released during the course of the catalytic cycle is assumed to be the driving force for the reaction.
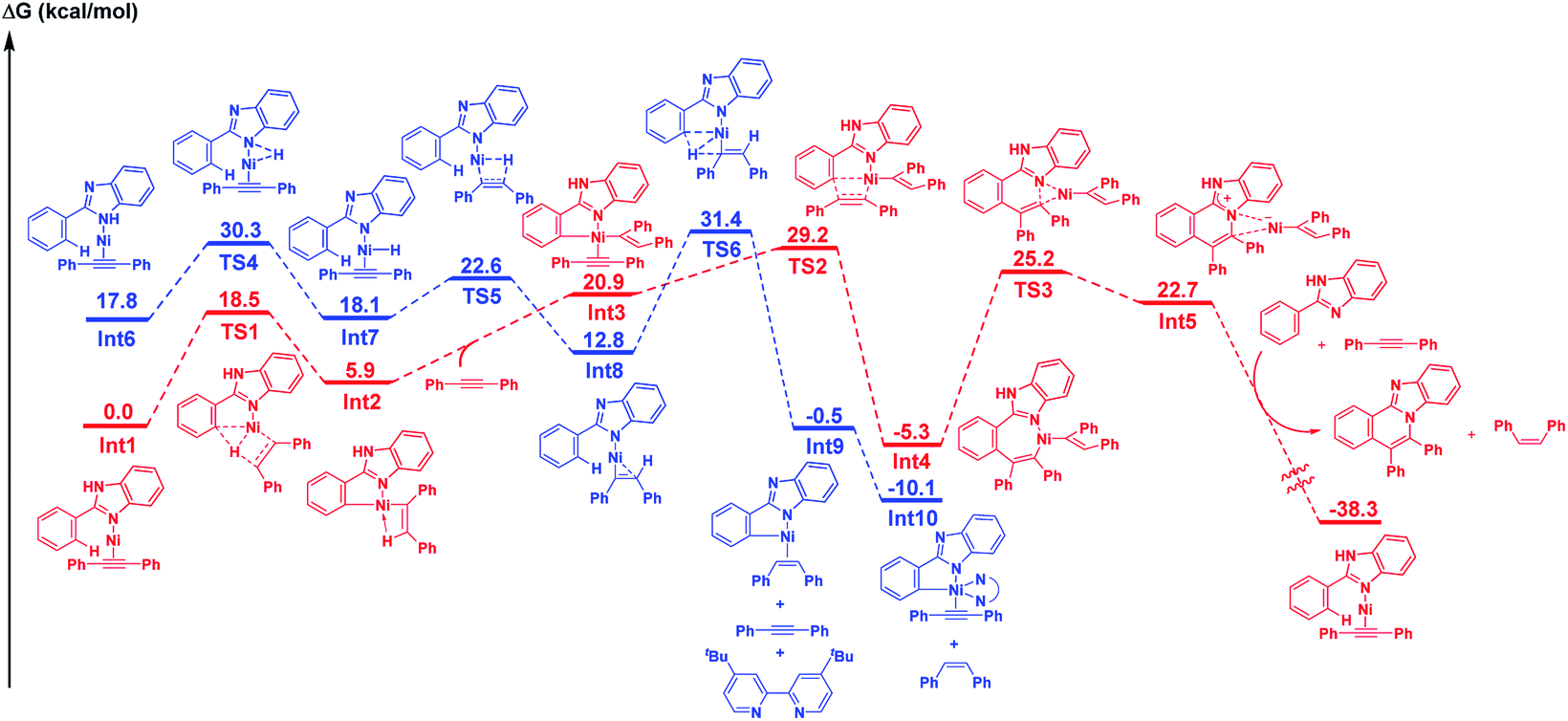 | ||
| Scheme 4 Comparison of the LLHT path (red line) and the N–H oxidative addition path (blue line) in the main catalytic cycle calculated at the B3PW91/6-311+G(2d,p)-SDD(Ni)/PCM//B3LYP/6-31+G(d)-Lanl2dz(Ni) level of theory. | ||
We also investigated the case where the main catalytic cycle is initiated by the oxidative addition of an N–H bond (complex I in Scheme 3 and the blue line in Scheme 4), because the presence of an N(sp2) atom in the substrate is not essential for the reaction to proceed, as in 2-phenylindole or pyrrole. The coordination of an N(sp2) atom to nickel, as in Int6, was found to be energetically unfavorable, compared with the coordination of an N(sp2) atom. While a LLHT path from the N–H bond of 2-phenylimidazole to an alkyne could not be located, the oxidative addition of the N–H bond in Int6 through TS4, followed by the insertion of the alkyne into a Ni–H bond is a feasible step. The σ-bond metathesis through TS6 proceeds with an activation energy of 18.6 kcal mol−1 to give a five membered metallacycle Int9. The highest Gibbs free energy for this step is the transition state of the σ-bond metathesis step (31.4 kcal mol−1), which is slightly higher than that for the LLHT mechanism (29.2 kcal mol−1). These results indicate that, while the reaction of 2-phenylimidazole proceeds through a LLHT mechanism, it is possible that the reaction of 2-phenylindole could proceed via the N–H oxidative addition-σ-bond metathesis sequence.
Conclusions
We report on the development of a new catalytic system for oxidative C–H/N–H annulation with alkynes which is promoted by a Ni(II)/KOBut or Ni(0) system. Using this system it is possible to use N-heteroaromatic compounds, such as 2-aryl-pyrrole, benzimidazole, imidazole, indole, and pyrazole, in Ni-catalyzed oxidative C–H/N–H annulation reactions with alkynes, leading to the production of highly conjugated compounds. While noble metal complexes, such as [Cp*RhCl2]2 and [RuCl2(p-cymene)]2, were used in the past in a similar transformation, the present reaction is the first example of the oxidative C–H/N–H annulation with alkynes in which a nickel catalyst is used. DFT studies indicate that the reaction involves two reaction paths. One mechanism is initiated by Ni(II) and the other by Ni(0), the latter being the main catalytic cycle. Although the Ni(II) system requires a base, the Ni(0) system proceeds, even in the absence of KOBut. C–H activation in the first step proceeds through a CMD mechanism and C–H activation in the main catalytic cycle proceeds through a LLHT mechanism.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant-in-Aid for Specially Promoted Research by The Ministry of Education, Culture, Sports, Science and Technology (17H06091).Notes and references
- For reviews on C–H functionalization, see: (a) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef PubMed; (b) J. J. Mousseau and A. B. Charette, Acc. Chem. Res., 2013, 46, 412 CrossRef CAS PubMed; (c) J. J. Topczewski and M. S. Sanford, Chem. Sci., 2015, 6, 70 RSC; (d) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC; (e) L. Yang and H. Huang, Chem. Rev., 2015, 115, 3468 CrossRef CAS PubMed; (f) J. Ye and M. Lautens, Nat. Chem., 2015, 7, 863 CrossRef CAS PubMed; (g) J. F. Hartwig and M. A. Larsen, ACS Cent. Sci., 2016, 2, 281 CrossRef CAS PubMed; (h) A. Dey, S. Agasti and D. Maiti, Org. Biomol. Chem., 2016, 14, 5440 RSC; (i) H. M. L. Davies and D. Morton, ACS Cent. Sci., 2017, 3, 936 CrossRef CAS PubMed; (j) O. Baudoin, Acc. Chem. Res., 2017, 50, 1114 CrossRef CAS; (k) L. Ping, D. S. Chung, J. Bouffard and S.-g. Lee, Chem. Soc. Rev., 2017, 46, 4299 RSC; (l) P. Nareddy, F. Jordan and M. Szostak, ACS Catal., 2017, 7, 5721 CrossRef CAS; (m) C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908 CrossRef CAS PubMed; (n) J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163 CrossRef CAS PubMed; (o) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247 CrossRef CAS PubMed; (p) K. Murakami, S. Yamada, T. Kaneda and K. Itami, Chem. Rev., 2017, 117, 9302 CrossRef CAS PubMed; (q) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333 CrossRef CAS; (r) M. P. Drapeau and L. J. Gooßen, Chem.–Eur. J., 2016, 22, 18654 CrossRef; (s) K. Hirano and M. Miura, Chem. Sci., 2018, 9, 22 RSC; (t) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62 CrossRef CAS PubMed; (u) S. Vásquez-Céspedes, X. Wang and F. Glorius, ACS Catal., 2018, 8, 242 CrossRef; (v) W.-J. Zhou, Y.-H. Zhang, Y.-Y. Gui, L. Sun and D.-G. Yu, Synthesis, 2018, 50, 3359 CrossRef CAS; (w) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603 RSC; (x) Z. Zhang, P. H. Dixneuf and J.-F. Soule, Chem. Commun., 2018, 54, 7265 RSC; (y) E. N. da Silva Júnior, G. A. M. Jardim, R. S. Gomes, Y.-F. Liang and L. Ackermann, Chem. Commun., 2018, 54, 7398 RSC; (z) S. Rej and N. Chatani, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201808159.
- For recent reviews on direct functionalization of C–H bonds by first-row transition-metal catalysis, see: (a) J. Miao and H. Ge, Eur. J. Org. Chem., 2015, 7859 CrossRef CAS; (b) G. Pototschnig, N. Maulide and M. Schnürch, Chem.–Eur. J., 2017, 23, 9206 CrossRef CAS PubMed.
- For recent reviews on Fe-catalyzed C–H functionalization reactions, see: R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086 CrossRef CAS PubMed.
- For recent reviews on Co-catalyzed C–H functionalization reactions, see: (a) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498 CrossRef CAS; (b) T. Yoshino and S. Matsunaga, Adv. Synth. Catal., 2017, 359, 1245 CrossRef CAS; (c) Y. Kommagalla and N. Chatani, Coord. Chem. Rev., 2017, 350, 117 CrossRef CAS; (d) S. Wang, S.-Y. Chen and X.-Q. Yu, Chem. Commun., 2017, 53, 3165 RSC.
- For recent reviews on Ni-catalyzed C–H functionalization reactions, see: (a) J. Yamaguchi, K. Muto and K. Itami, Eur. J. Org. Chem., 2013, 19 CrossRef CAS; (b) L. C. Misal Castro and N. Chatani, Chem. Lett., 2015, 44, 410 CrossRef; (c) N. Chatani, Top. Organomet. Chem., 2016, 56, 19 CrossRef CAS; (d) J. Yamaguchi, K. Muto and K. Itami, Top. Curr. Chem., 2016, 374, 55 CrossRef.
- For recent reviews on Cu-catalyzed C–H functionalization reactions, see: (a) K. Hirano and M. Miura, Chem. Lett., 2015, 44, 868 CrossRef CAS; (b) A. P. Jadhav, D. Ray, V. U. B. Rao and R. P. Singh, Eur. J. Org. Chem., 2016, 2369 CrossRef CAS; (c) J. Liu, G. Chen and Z. Tan, Adv. Synth. Catal., 2016, 358, 1174 CrossRef CAS; (d) W.-H. Rao and B.-F. Shi, Org. Chem. Front., 2016, 3, 1028 RSC.
- J. P. Kleiman and M. Dubeck, J. Am. Chem. Soc., 1963, 85, 1544 CrossRef CAS.
- V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed.
- (a) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2013, 135, 5308 CrossRef CAS PubMed; (b) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2014, 136, 898 CrossRef CAS PubMed; (c) Y. Aihara, M. Tobisu, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2014, 136, 15509 CrossRef CAS PubMed; (d) A. Yokota, Y. Aihara and N. Chatani, J. Org. Chem., 2014, 79, 11922 CrossRef CAS PubMed; (e) M. Iyanaga, Y. Aihara and N. Chatani, J. Org. Chem., 2014, 79, 11933 CrossRef CAS PubMed; (f) Y. Aihara, J. Wülbern and N. Chatani, Bull. Chem. Soc. Jpn., 2015, 88, 438 CrossRef CAS; (g) A. Yokota and N. Chatani, Chem. Lett., 2015, 44, 902 CrossRef CAS; (h) T. Kubo, Y. Aihara and N. Chatani, Chem. Lett., 2015, 44, 1365 CrossRef CAS; (i) L. C. Misal Castro, A. Obata, Y. Aihara and N. Chatani, Chem.–Eur. J., 2016, 22, 1362 CrossRef CAS PubMed; (j) T. Uemura, M. Yamaguchi and N. Chatani, Angew. Chem., Int. Ed., 2016, 55, 3162 CrossRef CAS PubMed; (k) T. Kubo and N. Chatani, Org. Lett., 2016, 18, 1698 CrossRef CAS PubMed; (l) Y. Aihara and N. Chatani, ACS Catal., 2016, 6, 4323 CrossRef CAS; (m) A. Sasagawa, M. Yamaguchi, Y. Ano and N. Chatani, Isr. J. Chem., 2017, 57, 964 CrossRef CAS.
- For representative papers reported by other groups, see: (a) X. Wu, Y. Zhao and H. Ge, J. Am. Chem. Soc., 2014, 136, 1789 CrossRef CAS PubMed; (b) W. Song, S. Lackner and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 2477 CrossRef CAS PubMed; (c) M. Li, J. Dong, X. Huang, K. Li, Q. Wu, F. Song and J. You, Chem. Commun., 2014, 50, 3944 RSC; (d) X. Cong, Y. Li, Y. Wei and X. Zeng, Org. Lett., 2014, 16, 3926 CrossRef CAS PubMed; (e) X. Wang, R. Qiu, C. Yan, V. P. Reddy, L. Zhu, X. Xu and S.-F. Yin, Org. Lett., 2015, 17, 1970 CrossRef CAS PubMed; (f) X. Wu, Y. Zhao and H. Ge, J. Am. Chem. Soc., 2015, 137, 4924 CrossRef CAS PubMed; (g) X. Ye, J. L. Petersen and X. Shi, Chem. Commun., 2015, 51, 7863 RSC; (h) Y.-J. Liu, Z.-Z. Zhang, S.-Y. Yan, Y.-H. Liu and B.-F. Shi, Chem. Commun., 2015, 51, 7899 RSC; (i) N. Barsu, D. Kalsi and B. Sundararaju, Chem.–Eur. J., 2015, 21, 9364 CrossRef CAS PubMed; (j) M. Sattar, Praveen, C. D. Prasad, A. Verma, S. Kumar and S. Kumar, Adv. Synth. Catal., 2016, 358, 240 CrossRef CAS; (k) C. Lin, J. Zhang, Z. Chen, Y. Liu, Z. Liu and Y. Zhang, Adv. Synth. Catal., 2016, 358, 1778 CrossRef CAS; (l) V. G. Landge, C. H. Shewale, G. Jaiswal, M. K. Sahoo, S. P. Midya and E. Balaraman, Catal. Sci. Technol., 2016, 6, 1946 RSC; (m) B. Khan, R. Kant and D. Koley, Adv. Synth. Catal., 2016, 358, 2352 CrossRef CAS; (n) Z. He and Y. Huang, ACS Catal., 2016, 6, 7814 CrossRef CAS; (o) F.-X. Luo, Z.-C. Cao, H.-W. Zhao, D. Wang, Y.-F. Zhang, X. Xu and Z.-J. Shi, Organometallics, 2017, 36, 18 CrossRef CAS; (p) M. Iwasaki, N. Miki, Y. Tsuchiya, K. Nakajima and Y. Nishihara, Org. Lett., 2017, 19, 1092 CrossRef CAS; (q) X. Wang, P. Xie, R. Qiu, L. Zhu, T. Liu, Y. Li, T. Iwasaki, C.-T. Au, X. Xu, Y. Xia, S.-F. Yin and N. Kambe, Chem. Commun., 2017, 53, 8316 RSC; (r) S. Xu, K. Takamatsu, K. Hirano and M. Miura, Angew. Chem., Int. Ed., 2018, 57, 11797 CrossRef CAS PubMed.
- (a) A. Obata, Y. Ano and N. Chatani, Chem. Sci., 2017, 8, 6650 RSC; (b) K. Yamazaki, A. Obata, A. Sasagawa, Y. Ano and N. Chatani, Organometallics, 2019, 38, 248 Search PubMed.
- H. Shiota, Y. Ano, Y. Aihara, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2011, 133, 14952 CrossRef CAS PubMed.
- (a) K. W. Bentley, Nat. Prod. Rep., 1992, 9, 365 RSC; (b) J. Zhang, S. Didierlaurent, M. Fortin, D. Lefrançois, E. Uridat and J. P. Vevert, Bioorg. Med. Chem. Lett., 2000, 10, 1351 CrossRef CAS PubMed; (c) Y. Bansal and O. Silakari, Bioorg. Med. Chem., 2012, 20, 6208 CrossRef CAS PubMed; (d) U. H. F. Bunz, J. U. Engelhart, B. D. Lindner and M. Schaffroth, Angew. Chem., Int. Ed., 2013, 52, 3810 CrossRef CAS PubMed; (e) A. Mateo-Alonso, Chem. Soc. Rev., 2014, 43, 6311 RSC; (f) Y. Deng, Y. Xie, K. Zou and X. Ji, J. Mater. Chem. A, 2016, 4, 1144 RSC.
- Ru(II): (a) L. Ackermann, L. Wang and A. V. Lygin, Chem. Sci., 2012, 3, 177 RSC; (b) N. Kavitha, G. Sukumar, V. P. Kumar, P. S. Mainkar and S. Chandrasekhar, Tetrahedron Lett., 2013, 54, 4198 CrossRef CAS; (c) R. Wang and J. R. Falck, J. Organomet. Chem., 2014, 759, 33 CrossRef CAS; (d) S. Karthik, J. Ajantha, C. M. Nagaraja, S. Easwaramoorthi and T. Gandi, Org. Biomol. Chem., 2016, 14, 10255 RSC . Rh(III): ; (e) N. Umeda, T. Tsurugi, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2008, 47, 4019 CrossRef CAS PubMed; (f) K. Morimoto, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2010, 12, 2068 CrossRef CAS PubMed; (g) X. Li and M. Zhao, J. Org. Chem., 2011, 76, 8530 CrossRef CAS; (h) H. Sun, C. Wang, Y.-F. Yang, P. Chen, Y.-D. Wu, X. Zhang and Y. Huang, J. Org. Chem., 2014, 79, 11863 CrossRef CAS PubMed; (i) J. M. Villar, J. Suárez, J. A. Varela and C. Saá, Org. Lett., 2017, 19, 1702 CrossRef CAS PubMed. Ru(II) and Rh(III): (j) A. G. Algarra, Q. B. Cross, D. L. Davies, Q. Khamker, S. A. Macgregor, C. L. Mcmullin and K. Singh, J. Org. Chem., 2014, 79, 1954 CrossRef CAS PubMed; (k) L. Zheng and R. Hua, J. Org. Chem., 2014, 79, 3930 CrossRef CAS PubMed. Co(III): (l) P. K. Dutta and S. Sen, Eur. J. Org. Chem., 2018, 5512 CrossRef CAS.
- J. Guihaumé, S. Halbert, O. Eisenstein and R. N. Perutz, Organometallics, 2012, 31, 1300 CrossRef.
- An LLHT mechanism is proposed to be involved in C–H functionalizations in which other first-row transition metal complexes, such as Ni, Mn, Fe, and Co. For recent papers in which LLHT mechanism is proposed, see: (a) A. J. Nett, W. Zhao, P. M. Zimmerman and J. Montgomery, J. Am. Chem. Soc., 2015, 137, 7636 CrossRef CAS PubMed; (b) J. S. Bair, Y. Schramm, A. G. Sergeev, E. Clot, O. Eisenstein and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 13098 CrossRef CAS PubMed; (c) L.-J. Xiao, X.-N. Fu, M.-J. Zhou, J.-H. Xie, L.-X. Wang, X.-F. Xu and Q.-L. Zhou, J. Am. Chem. Soc., 2016, 138, 2957 CrossRef CAS PubMed; (d) S. Tang, O. Eisenstein, Y. Nakao and S. Sakaki, Organometallics, 2017, 36, 2761 CrossRef CAS; (e) A. J. Nett, J. Montgomery and P. M. Zimmerman, ACS Catal., 2017, 7, 7352 CrossRef CAS; (f) S. Nakanowatari, T. Müller, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 15891 CrossRef CAS PubMed; (g) J. Diesel, A. M. Finogenova and N. Cramer, J. Am. Chem. Soc., 2018, 140, 4489 CrossRef CAS PubMed.
- Details of the energy profile for the left path are shown in the ESI (Scheme S1†).
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1868340. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05063e |
| This journal is © The Royal Society of Chemistry 2019 |