
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
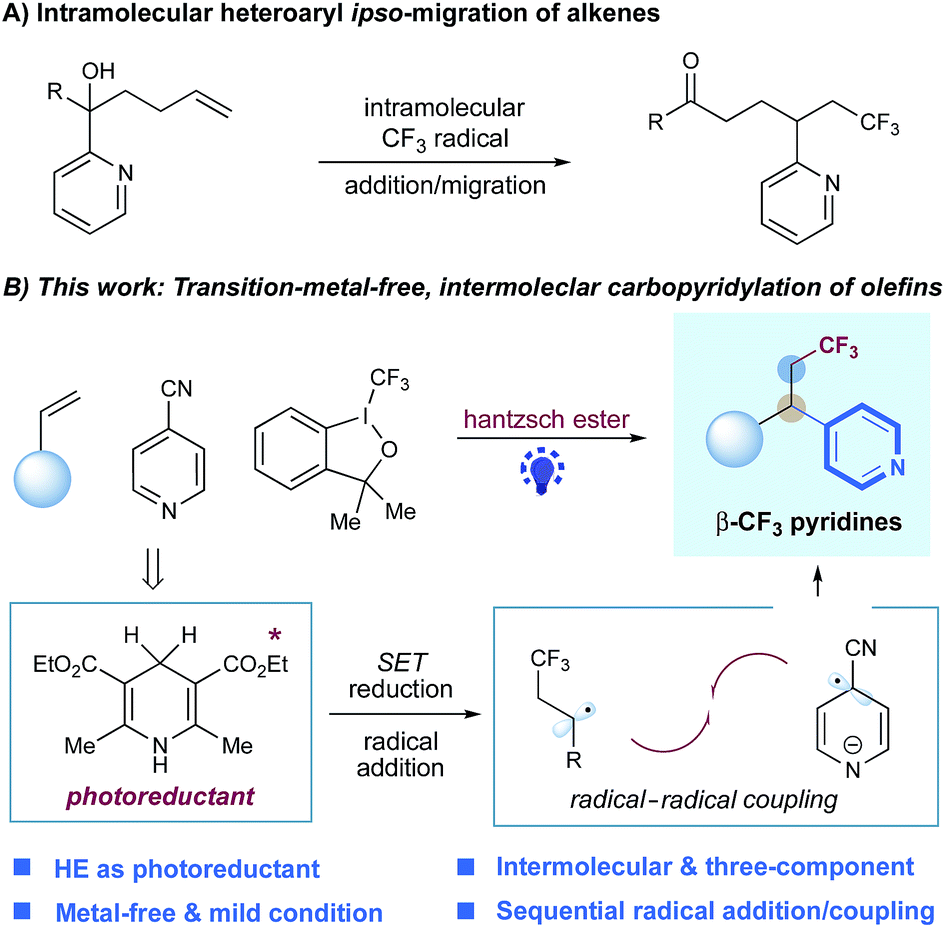
Metal-free, intermolecular carbopyridylation of alkenes via visible-light-induced reductive radical coupling†
Dan
Chen
,
Lei
Xu
,
Tianyu
Long
,
Shengqing
Zhu
,
Jun
Yang
and
Lingling
Chu
 *
*
Center for Advanced Low-dimension Materials, State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, China. E-mail: lingling.chu1@dhu.edu.cn
First published on 2nd October 2018
Abstract
An efficient, metal-free strategy for the intermolecular three-component carbopyridylation of styrenes, enabled by Hantzsch ester and visible light, has been described. This versatile protocol gives access to important β-CF3 pyridines, through the regioselective, sequential formation of two C–C bonds without the use of exogenous catalysts. The value of this benign protocol has been demonstrated through functionalizations of natural-product- and drug-based complex molecules.
Pyridines are important heterocycles widely found in bioactive natural products, pharmaceuticals, agrochemicals and functional materials.1 Top selling pharmaceuticals such as antihistamine drug loratadine and anti-depressant drug mirtazapina contain the pyridine core. Moreover, pyridines are also versatile ligand scaffolds widely employed in the areas of transition-metal catalysis.2 As a result, the development of novel and mild methodologies for the regioselective construction of complex pyridines employing simple starting materials is highly desired.
Alkene–pyridine cross-coupling represents an efficient and powerful strategy to access alkylpyridines with chemo- and regio-selectivity due to the fact that alkenes are simple and abundant building blocks in organic synthesis.3 Significant achievement has been made via transition metal catalysis, enabling the efficient intermolecular hydropyridylation of alkenes with pyridines and their derivatives (e.g. N-oxides and N-methoxy pyridinium salts).4 Recently, several elegant examples, through visible light-induced photoredox catalysis,5 of hydropyridylation of alkenes with simple pyridyl halides under mild conditions have been developed.6 Nevertheless, carbopyridylation of alkenes, which simultaneously forge two consecutive C–C bonds across double bonds and would enable rapid buildup of complex pyridines, is highly desired yet remains a challenge. To date, only a few examples of alkene carbopyridylations have been reported. The Zhu group described the visible-light-mediated fluoroalkyl-heteroarylation of alkenes via an intramolecular heteroaryl ipso-migration, mainly focusing on five-membered heteroaromatic substrates with very few examples of simple pyridines.7 Liu and co-worker also developed a Cu-catalyzed trifluoromethylarylation of alkenes, with one pyridine substrate.8 Very recently, Su and coworkers reported a visible-light induced carbo-2-pyridylation of electron-deficient alkenes with pyridinium salts via an electron donor–acceptor complex.9 Nevertheless, these elegant protocols are restricted to two-component mode.7–9 A general protocol for the intermolecular, three-component carbopyridylation of alkenes has yet to be developed.
In our continuing efforts to pursue radical functionalization of alkenes,10 we envisioned that a light-induced, sequential radical-addition/radical-coupling protocol between alkenes and pyridines could provide a generic solution to this challenging carbopyridylation of alkenes. Given the importance of trifluoromethyl groups in pharmaceuticals and agrochemicals11 as well as elegant progress in radical trifluoromethylation of alkenes,12 we focused on the development of pyridyl functionalization of alkenes with concomitant construction of C(sp3)–CF3 bonds. Herein, we reported the intermolecular, three-component carbopyridylation of olefins through visible light-induced reductive radical coupling under transition metal-free conditions (Fig. 1). Particularly, this protocol utilizes the potent redox ability of photoexcited Hantzsch ester (HE)13,14 to generate open-shell radical intermediates, thus facilitating the construction of two consecutive C–C bonds in one pot without the need for exogenous photocatalysts. Although two elegant examples of intermolecular trifluoromethylarylation of styrenes with arylboronic acids have been described recently, this Cu-catalyzed platform is inapplicable to heteroarenes.15 We expected that our new photo-chemical protocol would complement the known transition-metal protocols.
 | ||
| Fig. 1 Design of intermolecular carbopyridylation of alkenes via photoexcited Hantzsch ester-enabled reductive radical coupling. | ||
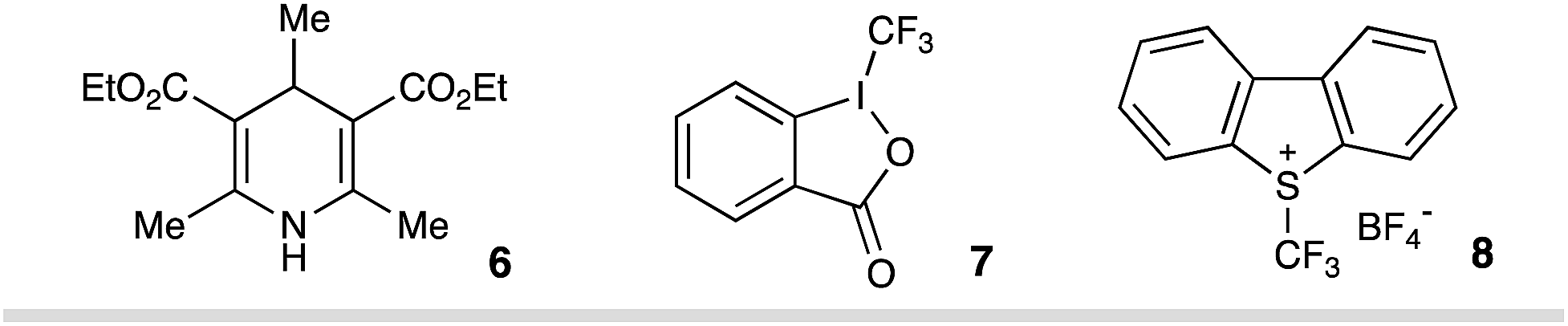
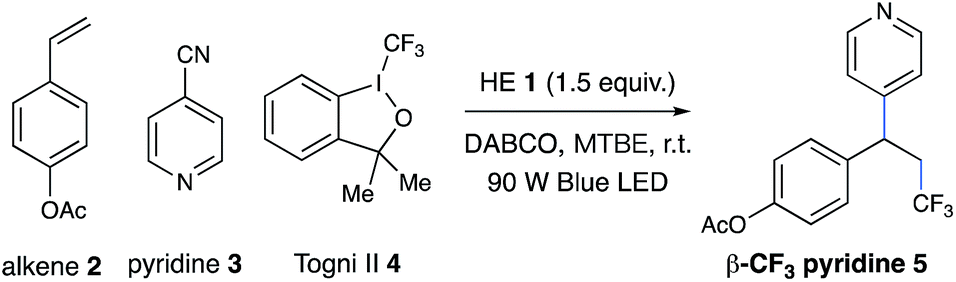
Inspired by elegant examples of radical coupling with cyanopyridines,13a,16 we chose cyanopyridines as the coupling partners and Hantzsch ester (HE) as the stoichiometric photoreductant. Irradiating a solution of styrene 2, 4-cyanopyridine 3, and 3,3-dimethyl-1-(trifluoromethyl)-1,2-benziodoxole 4 (Togni II reagent) in the presence of HE 1 and 1,4-diazabicyclo[2.2.2]octane (DABCO) with a 90 W blue LED gave the desired trifluoromethylpyridylation product 5 in 83% yield (Table 1, entry 1). Control experiments indicated that HE and visible light are required for the reductive coupling, as no products were observed in the absence of HE or under dark conditions (entries 2–4). Notably, DABCO had a dramatic influence on the reaction efficiency. Only 22% yield of product 5 was observed in the absence of DABCO (entry 5). Employing other organic or inorganic bases instead of DABCO resulted in a dramatic decrease in the reaction efficiency (entries 6–10). Additionally, replacing HE with 4-methyl Hantzsch ester 6, an analog of HE, led to the formation of product 5 with a significantly low efficiency (entry 11). Moreover, the choice of the electrophilic trifluoromethylating reagents was also found to have a dramatic effect on the reaction efficiency, with Togni reagent 4 proving to be optimal (entries 12–13).
|
|
||
|---|---|---|
| Entry | Variations from the standard conditions | Yieldb |
| a Reaction conditions: styrene 2 (0.1 mmol), 4-cyanopyridine 3 (2.0 equiv.), Togni reagent 4 (1.5 equiv.), Hantzsch ester (HE, 1.5 equiv.), DABCO (1.5 equiv.), MTBE [0.05 M], 90 W blue LED, and rt. b Yields were determined by 19F NMR using an internal standard. c Major byproducts determined were dimers of benzylic radicals; see the ESI for details. DABCO: 1,4-diazabicyclo[2.2.2]octane; MTBE: methyl tert-butyl ether. | ||
| 1 | None | 83% |
| 2 | w/o HE | 0% c |
| 3 | Dark | 0% |
| 4 | Dark, 80 °C | 0% c |
| 5 | w/o DABCO | 22% c |
| 6 | TMEDA, instead of DABCO | 43% |
| 7 | DBU, instead of DABCO | 19% |
| 8 | Et3N, instead of DABCO | 26% |
| 9 | Pyridine, instead of DABCO | 19% |
| 10 | Cs2CO3, instead of DABCO | 21% |
| 11 | 6, instead of HE | 25% |
| 12 | 7, instead of 4 | 19% |
| 13 | 8, instead of 4 | 25% |
|
||
Having identified the optimal reaction conditions for the visible light-induced reductive pyridylation of alkenes, we investigated the olefin partner using 4-cyanopyridine. As shown in Scheme 1, a variety of styrenes bearing electron-donating- and electron-withdrawing substituents are viable partners for this transformation, affording the corresponding β-CF3 pyridines in moderate to excellent yields (products 5 and 9–32, 42–86% yields). Styrenes containing reactive functional groups, including esters, amides, tosylates, alkynes, and even free amines, underwent the desired coupling with high efficiency (products 5, 16, 18–19, 22, and 29, 58–86% yields). Notably, the reaction of varied halides, from fluorides to iodides, gave the desired coupling product with halo atoms untouched (products 13–14, 21, 23–25, and 27–28, 65–85% yields). Halides are important synthetic manipulation handles via transition-metal-catalyzed cross–coupling, further indicating the complementary ability of this visible-light-induced metal-free technique. ortho-Substituents on the aryl rings have little effect on the reaction efficiency (products 23–26, 64–70% yields). Alkenes attached to electron-deficient arenes, exemplified as 1,2,3,4,5-pentafluoro-6-vinylbenzene, were found to be suitable substrates with moderate efficiency (product 31, 42% yield). Heteroarenes, in the form of indoles, were well tolerated, albeit with lower yields (product 33, 55% yield). Furthermore, 1,1-disubstituted alkenes, such as α-methyl styrene, can be successfully employed, furnishing the expected adducts with moderate efficiency (product 34, 65% yield). Notably, this three-component reductive coupling protocol can be applicable to other types of olefins. Reactions of electron-rich olefins (products 35–36) as well as un-activated alkenes (product 37) furnished the desired trifluoromethylpyridine products with moderate efficiency in the presence of 1 mol% of Ir(ppy)3 (40–52% yields). We assume that the addition of Ir(ppy)3 could facilitate the single-electron reduction of 4-cyanopyridine, thereby improving the reaction efficiency.
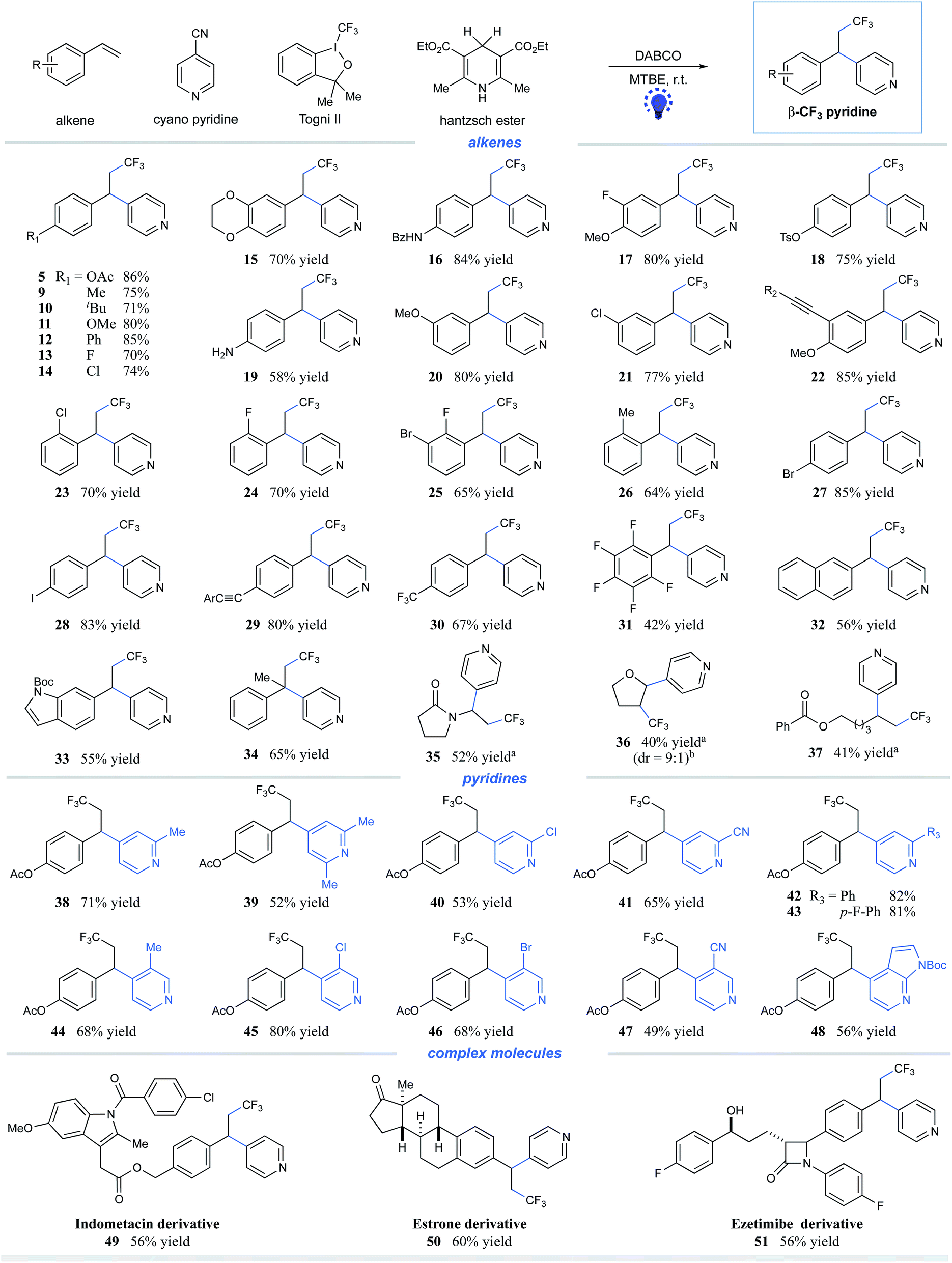 | ||
| Scheme 1 Substrate scope. Reaction conditions: alkene (0.2 mmol), cyanopyridine (2.0 equiv.), Togni II 4 (1.5 equiv.), HE 1 (1.5 equiv.), DABCO (1.5 equiv.), MTBE [0.05 M], 90W blue LED, and rt. All cited yields are isolated yields. aWith 1 mol% Ir(ppy)3. bDetermined by 19F NMR of the reaction mixture. R2 = n-C4H9; Ar = tert-Bu-phenyl. | ||
Next, we evaluated the scope of the pyridine component in this metal-free protocol. As illustrated in Scheme 1, substituted cyanopyridines reacted well under the mild conditions, furnishing the β-CF3 alkylpyridines with moderate to high efficiency. A number of substituents on the 2- or 3-position were tolerated, including alkyl, chloro, bromo, aryl, and cyano (products 38–48, 49–82% yields). Both 2,4- and 3,4-dicyanopyridines underwent selective coupling at the 4-position, affording corresponding 4-alkylated pyridines in synthetically useful yields (products 41 and 47, 65% and 49% yields, respectively). Notably, azaindole nitrile was found to readily undergo the desired three-component coupling to afford the alkylated azaindole 48 in satisfactory yield (56% yield).
To further highlight the potential application of this metal-free protocol, we have employed several natural-product- and drug-derived complex molecules in this system. As depicted in Scheme 1, derivatives of estrone, indomethacin (anti-inflammatory drug), ezetimibe (lipid-lowering drug), and nonivamide all functioned as competent coupling partners, furnishing each of the desired adducts with moderate efficiency (products 49–51, 56–60% yields; S3 in the ESI,† 55% yield, see the ESI† for details).
To probe the mechanism of this alkene carbopyridylation reaction, we have conducted some preliminary mechanistic experiments (Fig. 2). Radical trap and radical clock experiments have been conducted. The addition of TEMPO completely shut down the desired reaction, with the observation of CF3–TEMPO adduct 52 (48% yield) (Fig. 2A). Vinyl cyclopropane 53 underwent radical addition/ring opening, affording 54 as the major isolated product (40% yield, E/Z = 3.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Fig. 2B), further indicating a radical sequence involved in this novel transformation. Furthermore, light on/off experiments (Fig. 2C), as well as light control experiments (Table 1, entries 3–4), indicated that constant photoirritation is essential for this transformation. In addition, direct illumination of the reaction mixture with a commercial laser (532 nm, in which HE has no absorption) led to no product formation (see the ESI† for details), suggesting the unique role of the photoexcited HE. Importantly, Stern–Volmer fluorescence quenching analysis indicated that photoexcited HE*
:1) (Fig. 2B), further indicating a radical sequence involved in this novel transformation. Furthermore, light on/off experiments (Fig. 2C), as well as light control experiments (Table 1, entries 3–4), indicated that constant photoirritation is essential for this transformation. In addition, direct illumination of the reaction mixture with a commercial laser (532 nm, in which HE has no absorption) led to no product formation (see the ESI† for details), suggesting the unique role of the photoexcited HE. Importantly, Stern–Volmer fluorescence quenching analysis indicated that photoexcited HE* 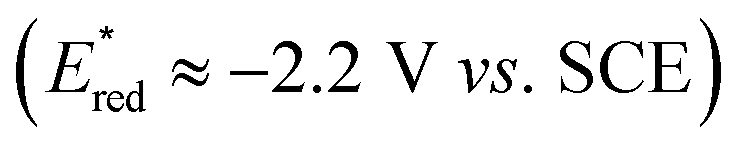 13c was quenched by Togni II 4 (Ered = −1.11 V vs. SCE in CH3CN)17 as well as 4-cyanopyridine 3 (Ered = −1.87 V vs. SCE in CH3CN),18 respectively (Fig. 2D).
13c was quenched by Togni II 4 (Ered = −1.11 V vs. SCE in CH3CN)17 as well as 4-cyanopyridine 3 (Ered = −1.87 V vs. SCE in CH3CN),18 respectively (Fig. 2D).
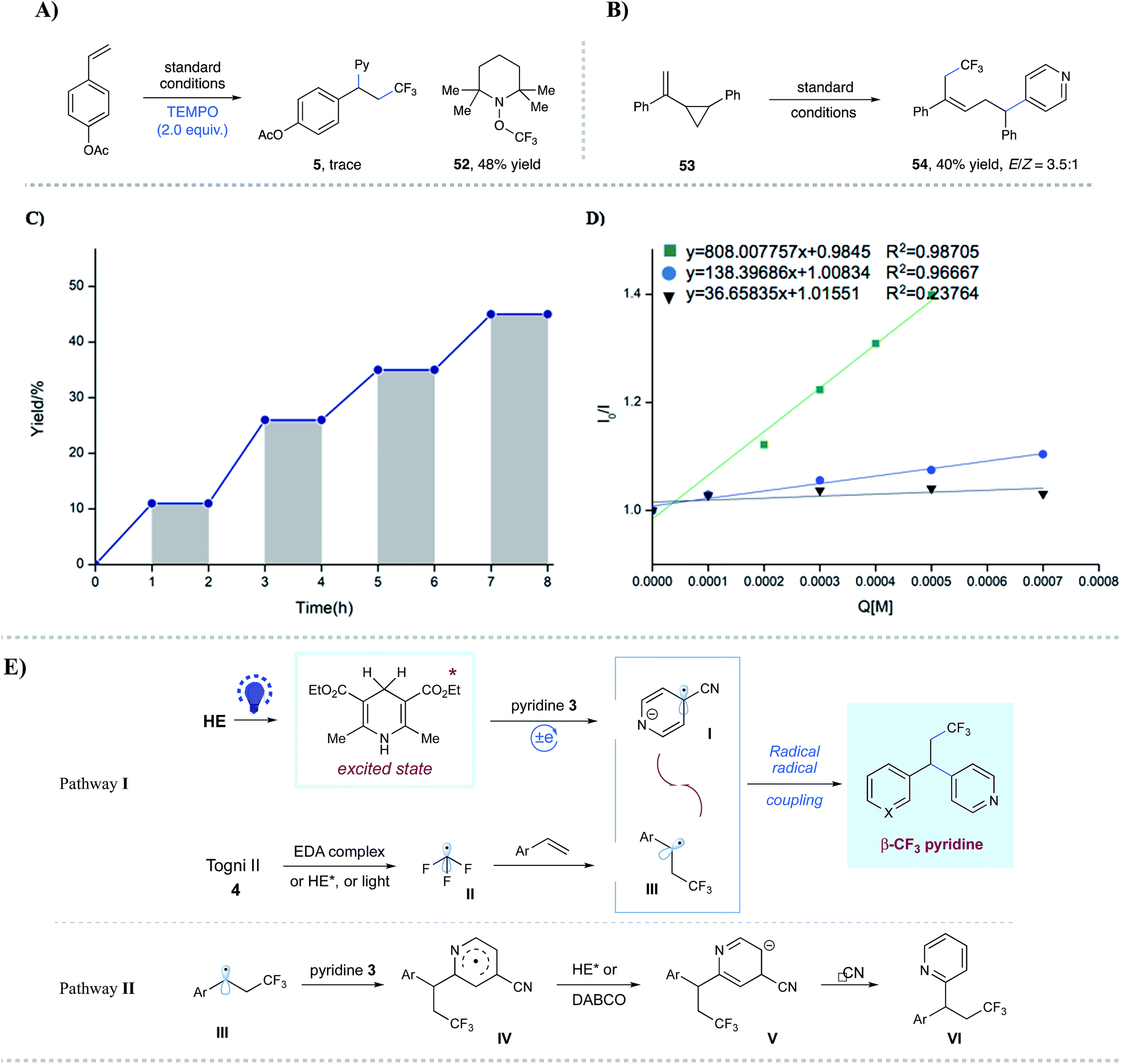 | ||
| Fig. 2 Mechanistic studies. (A) Radical inhibition reaction. (B) Radical clock reaction. (C) Light on/off experiments; (D) Stern–Volmer quenching studies. (E) Proposed mechanism. | ||
On the basis of these experimental results, a proposed mechanism has been exemplified in Fig. 2E. A thermodynamically feasible single-electron reduction between photoexcited HE* and 4-cyanopyridine 3 would produce the persistent radical anion species I. At the same time, the CF3 radical II could be generated through the SET reduction of photoexcited HE* and Togni reagent 4. Subsequent facile addition of CF3 radical II to styrene led to the formation of the nucleophilic benzylic radical III, which would undergo a selective radical–radical coupling with I to deliver the desired alkylpyridine product via the extrusion of cyanide (pathway I). At this stage, we cannot rule out the possibility that alternative pathways might be involved in this transformation. First, the generation of the CF3 radical could proceed through multiple pathways: (i) triggered by photoexcited HE, which is supported by the Stern–Volmer fluorescence quenching study; (ii) through an electron donor–acceptor (EDA) complex between the Togni reagent and DABCO, which is suggested by a bathochromic shift in UV-Vis absorption spectrometry (see Fig. S7 in the ESI† for details);19,20 (iii) triggered by visible light. Control experiments showed that the major side reactions were dimerization and hydrogen abstraction of benzyl radicals (generated via CF3 radical additions) in the absence of HE and/or DABCO, while no dimers were observed under the dark conditions (see Table S1 in the ESI† for more details). These phenomena indicated that visible light could solely promote the generation of the CF3 radical from the Togni reagent.21 Second, an alternative pathway might be involved for the coupling step between the benzylic radical and cyanopyridine: nucleophilic addition of benzylic radical III to pyridine 3 at the C2 position to give rise to aryl radical species IV;22 radical IV could be reduced by HE* or DABCO to afford aryl anion V, which undergoes elimination of cyanide to form the C2-substituted product VI (pathway II). Nevertheless, the available experimental results with no observation of VI might not support this hypothesis.
Conclusions
In conclusion, we have developed an efficient, transition metal-free strategy for the intermolecular, three-component carbopyridylation of styrenes enabled by photoexcited Hantzsch ester. This visible light-induced protocol enables a facile access to β-CF3 alkylpyridines, through the regioselective, sequential formation of two C–C bonds in one step without the need for exogenous photocatalysts. Given the importance of both pyridine and CF3 moieties in medicinal agents, we expect that the generality of this methodology and ready availability of the starting materials will allow it to enjoy extensive application in the area of organic chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21702029) and the “Thousand Plan” Youth program and the Shanghai Sailing Program (17YF1400100) for financial support.Notes and references
- (a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed; (b) J. J. Li, Heterocyclic Chemistry in Drug Discovery, John Wiley & Sons, Hoboken, NJ, 2013 Search PubMed.
- M. N. Zafar, A. H. Atif, M. F. Nazar, S. H. Sumrra, E. S. Gul and R. Paracha, Russ. J. Coord. Chem., 2016, 42, 1 CrossRef CAS.
- Y. Nakao, Synthesis, 2011, 2011, 3209 CrossRef.
- (a) G. Song, W. N. O. Wylie and Z. Hou, J. Am. Chem. Soc., 2014, 136, 12209 CrossRef CAS PubMed; (b) J. Diesel, A. M. Finogenova and N. Cramer, J. Am. Chem. Soc., 2018, 140, 4489 CrossRef CAS PubMed; (c) S. D. Friis, M. T. Pirnot and S. L. Buchwald, J. Am. Chem. Soc., 2016, 138, 8372 CrossRef CAS PubMed; (d) S. Yu, L. Sang Hui and S. Ge, Angew. Chem., Int. Ed., 2017, 56, 15896 CrossRef CAS PubMed; (e) M. W. Gribble Jr., S. Guo and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 5057 CrossRef PubMed; (f) J. C. Lewis, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2007, 129, 5332 CrossRef CAS PubMed.
- (a) C. R. J. Stephenson, T. P. Yoon and D. W. C. MacMillan, Visible Light Photocatalysis in Organic Chemistry, Wiley-VCH, 2018 CrossRef; (b) L. Marzo, S. K. Pagire, O. Reiser and B. Konig, Angew. Chem., Int. Ed., 2018, 57, 10034 CrossRef CAS PubMed; (c) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS; (d) J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563 CrossRef CAS PubMed; (e) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261 CrossRef CAS PubMed; (f) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (g) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- (a) R. A. Aycock, D. B. Vogt and N. T. Jui, Chem. Sci., 2017, 8, 7998 RSC; (b) A. J. Boyington, M.-L. Y. Riu and N. T. Jui, J. Am. Chem. Soc., 2017, 139, 6582 CrossRef CAS PubMed; (c) R. A. Aycock, H. Wang and N. T. Jui, Chem. Sci., 2017, 8, 3121 RSC; (d) I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566 CrossRef CAS PubMed; (e) A. Arora and J. D. Weaver, Acc. Chem. Res., 2016, 49, 2273 CrossRef CAS PubMed.
- (a) Z. Wu, D. Wang, Y. Liu, L. Huan and C. Zhu, J. Am. Chem. Soc., 2017, 139, 1388 CrossRef CAS PubMed; (b) J. Yu, D. Wang, Y. Xu, Z. Wu and C. Zhu, Adv. Synth. Catal., 2018, 360, 744 CrossRef CAS.
- L. Li, Q.-S. Gu, N. Wang, P. Song, Z.-L. Li, X.-H. Li, F.-L. Wang and X.-Y. Liu, Chem. Commun., 2017, 53, 4038 RSC.
- R. B. Hu, S. Sun and Y. Su, Angew. Chem., Int. Ed., 2017, 56, 10877 CrossRef CAS PubMed.
- J. Sun, P. Li, L. Guo, F. Yu, Y.-P. He and L. Chu, Chem. Commun., 2018, 54, 3162 RSC.
- (a) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (c) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed.
- (a) C. Alonso, E. Martínez de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847 CrossRef CAS PubMed; (b) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed; (c) L. L. Chu and F. L. Qing, Acc. Chem. Res., 2014, 47, 1513 CrossRef CAS PubMed; (d) T. Besset, T. Poisson and X. Pannecoucke, Chem.–Eur. J., 2014, 20, 16830 CrossRef CAS PubMed; (e) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294 CrossRef CAS PubMed; (f) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598 RSC; (g) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (h) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950 CrossRef CAS PubMed.
- (a) L. Buzzetti, A. Prieto, S. R. Roy and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 15039 CrossRef CAS PubMed; (b) M. A. Emmanuel, N. R. Greenberg, D. G. Oblinsky and T. K. Hyster, Nature, 2016, 540, 414 CrossRef CAS PubMed; (c) J. Jung, J. Kim, G. Park, Y. You and E. J. Cho, Adv. Synth. Catal., 2016, 358, 74 CrossRef CAS; (d) W. Chen, H. Tao, W. Huang, G. Wang, S. Li, X. Cheng and G. Li, Chem.–Eur. J., 2016, 22, 9546 CrossRef CAS PubMed; (e) L. I. Panferova, A. V. Tsymbal, V. V. Levin, M. I. Struchkova and A. D. Dilman, Org. Lett., 2016, 18, 996 CrossRef CAS PubMed; (f) W. Huang, W. Chen, G. Wang, J. Li, X. Cheng and G. Li, ACS Catal., 2016, 6, 7471 CrossRef CAS; (g) J. Zhang, M.-Z. Jin, W. Zhang, L. Yang and Z.-L. Liu, Tetrahedron Lett., 2002, 43, 9687 CrossRef CAS; (h) M.-Z. Jin, L. Yang, L.-M. Wu, Y.-C. Liu and Z.-L. Liu, Chem. Commun., 1998, 2451 RSC; (i) S. Fukuzumi, K. Hironaka and T. Tanaka, J. Am. Chem. Soc., 1983, 105, 4722 CrossRef CAS.
- (a) V. Balzani, P. Ceroni and A. Juris, Photochemistry and Photophysics: Concepts, Research, Applications, Wiley-VCH, 2014 Search PubMed; (b) N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, 2010 Search PubMed.
- (a) L. Wu, F. Wang, X. Wan, D. Wang, P. Chen and G. Liu, J. Am. Chem. Soc., 2017, 139, 2904 CrossRef CAS PubMed; (b) F. Wang, D. Wang, X. Mu, P. Chen and G. Liu, J. Am. Chem. Soc., 2014, 136, 10202 CrossRef CAS PubMed.
- (a) G. Wang, J. Cao, L. Gao, W. Chen, W. Huang, X. Cheng and S. Li, J. Am. Chem. Soc., 2017, 139, 3904 CrossRef CAS PubMed; (b) F. Lima, M. A. Kabeshov, D. N. Tran, C. Battilocchio, J. Sedelmeier, G. Sedelmeier, B. Schenkel and S. V. Ley, Angew. Chem., Int. Ed., 2016, 55, 14085 CrossRef CAS PubMed; (c) J. D. Cuthbertson and D. W. C. MacMillan, Nature, 2015, 519, 74 CrossRef CAS PubMed; (d) Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257 CrossRef CAS PubMed; (e) K. Qvortrup, D. A. Rankic and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 626 CrossRef CAS PubMed; (f) A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114 CrossRef CAS PubMed; (g) D. Mangion and D. R. Arnold, Acc. Chem. Res., 2002, 35, 297 CrossRef CAS PubMed; (h) R. Bernardi, T. Caronna, S. Morrocchi and M. Ursini, J. Heterocycl. Chem., 1996, 33, 1137 CrossRef CAS; (i) X. Zeng, J. Cai and Y. Gu, Tetrahedron Lett., 1995, 36, 7275 CrossRef CAS.
- Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2012, 51, 9567 CrossRef CAS PubMed.
- P. McDevitt and M. Vittimberga Bruno, J. Heterocycl. Chem., 2009, 27, 1903 CrossRef.
- For a recent review on EDA complex, see: C. G. S. Lima, T. de M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389 CrossRef CAS.
- For recent examples on EDA-enabled radical trifluoromethylation, see (a) H. Jiang, Y. He, Y. Cheng and S. Yu, Org. Lett., 2017, 19, 1240 CrossRef CAS PubMed; (b) Y. Cheng and S. Yu, Org. Lett., 2016, 18, 2962 CrossRef CAS PubMed; (c) Q. Guo, M. Wang, H. Liu, R. Wang and Z. Xu, Angew. Chem., Int. Ed., 2018, 57, 4747 CrossRef CAS PubMed; (d) M. Zhu, K. Zhou, X. Zhang and S.-L. You, Org. Lett., 2018, 20, 4379 CrossRef CAS PubMed.
- H. Wang, Q. Xu and S. Yu, Org. Chem. Front., 2018, 5, 2224 RSC.
- J. G. Traynham, Chem. Rev., 1979, 79, 323 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03493a |
| This journal is © The Royal Society of Chemistry 2018 |