
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirected evolution of mevalonate kinase in Escherichia coli by random mutagenesis for improved lycopene†
Hailin Chen ab,
Changqing Liua,
Meijie Lia,
Haibo Zhang*a,
Mo Xian*a and
Huizhou Liua
ab,
Changqing Liua,
Meijie Lia,
Haibo Zhang*a,
Mo Xian*a and
Huizhou Liua
aCAS Key Laboratory of Bio-based Materials, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, No. 189 Songling Road, Qingdao 266101, People's Republic of China. E-mail: xianmo@qibebt.ac.cn; zhanghb@qibebt.ac.cn
bSino-Danish College, University of Chinese Academy of Sciences, No. 19 (A) Yuquan Road, Beijing 100049, People's Republic of China
First published on 20th April 2018
Abstract
Lycopene is a terpenoid pigment that has diverse applications in the fields of food and medicine. Metabolic engineering in microbial hosts has shown that mevalonate kinase (MK, EC2.7.1.366) is one of the rate-limiting enzymes in the lycopene synthetic pathway. In this study, a directed evolution strategy in Escherichia coli was used to optimize the activity of Saccharomyces cerevisiae MK. Using three rounds of error-prone PCR; screening the development of a lycopene-dependent color reaction; and combinatorial site-specific saturation mutagenesis, three activity-enhancing mutations were identified: V13D, S148I, and V301E. V13D was near the MK catalytic center, in the β-sheet that forms a salt-bridge with nearby Arg-248. S148I was located in the α-helix lid and improved the stability of the α-helix. V301E may increase MK folding by influencing its secondary structure. The Km (RS)-mevalonate of purified mutant MK decreased by 74% compared with the Km (RS)-mevalonate of the wild-type MK, and the Kcat (RS)-mevalonate was improved by 26% compared with wild type. Fermentation experiments revealed that lycopene production of the mutant MK increased 2.4-fold compared with wild-type MK.
Introduction
Lycopene is a widely used carotenoid in the healthcare product market due to its potent antioxidant properties and its links to reduced risk of prostate cancer in humans.1,2 Presently, the major commercial sources of lycopene are either Blakeslea trispora or tomatoes.2,3 Despite the well-established lycopene production protocols, the long growth cycle of the tomato is disadvantageous, as is the addition of cyclase inhibitors to B. trispora which is required to inhibit the cyclase function of the bifunctional lycopene cyclase/phytoene synthase (carRA),4 which can be problematic from a food safety perspective. Alternative methods of lycopene production must be developed in order to avoid these issues. Given the recent development of metabolic engineering technologies, one promising approach is the introduction of constituent enzymes of the lycopene biosynthetic pathway into a heterologous host.1 Cellular lycopene can be synthesized by two different pathways, the 2-methyl-D-erythritol 4-phosphoric acid (MEP) pathway,5 and the mevalonate (MVA) pathway.6 For the MVA pathway, the highly regulated and well-studied 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase (EC 1.1.1.34) is the rate-limiting enzyme which metabolizes HMG-CoA.7 However, it should be noted that mevalonate kinase (MK, EC 2.7.1.366) is as important as HMG-CoA reductase in the lycopene biosynthetic pathway.8MK is the first of three successive ATP-dependent enzymes in the MVA pathway.9 The first evidence of involvement in isoprenoid biosynthesis of MK was reported in 1956 where mevalonic acid, previously identified in a Lactobaccillus strain as a growth factor,8 is a critical component of cholesterol biosynthesis.10 Anthony et al.11 optimized promoter activity to balance the expression of MK and amorphadiene synthase to alleviate two pathway bottlenecks, which yielded a 5-fold improvement in amorphadiene production. However, other studies have shown that MKs are subject to feedback inhibition by lycopene biosynthesis intermediates, such as geranyldiphosphate (GPP), farnesyldiphosphate (FPP), and geranylgeranyldiphosphate (GGPP).8 Dorsey and Porter12 first suggested that MK activity in porcine liver was subject to feedback inhibition by low GPP and FPP concentrations. Similar results were witnessed in rat and plant MK studies.13,14 Furthermore, Hinson et al.10 provided evidence to suggest that GGPP has a further increased inhibitory function compared to GPP and FPP. Recently, MK research has made significant and exciting progress. For example, high MK expression has been successfully achieved in Saccharomyces cerevisiae;15–17 however, the catalytic properties of these enzymes have been poorly characterized. Sarcophaga bullata MK has been partially characterized, showing low affinities for MVA and ATP.18 MKs with higher tolerance for long-chain isoprenoids and higher MVA and ATP affinities have previously received little attention with regards to lycopene production.
Directed evolution has emerged as a ubiquitous technique to enhance the stability, activity, and selectivity of enzymes.19 Notably, it does not rely on a priori structural or mechanistic information regarding the protein of interest,20 and thus is suitable for modifying novel enzymes. Therefore, we aimed to use directed evolution to enhance the tolerance for FPP and GGPP, to enhance the affinity of MVA and MK, and to improve lycopene production.
In this work, we integrated the bottom portion of the MVA pathway and the lycopene synthesis pathway by constructing a recombinant E. coli strain with the plasmids pET-CHL (containing the bottom MVA pathway) and pAC-LYC (containing key lycopene synthesis genes).21 An improved high-throughput MK screening method was established, which not only removed the toxicity of HMG-CoA,22 but also used FPP, GGPP and MVA23 as the screening condition for the lycopene-dependent color development reaction that was used for selection.24 Site-directed saturation mutagenesis was conducted to determine the optimal amino acid substitutions at the identified positions. Combinations of site-specific mutations were investigated to identify which combination or combinations of mutations could further enhance MK enzymatic activity.
Experimental
Chemicals and materials
Restriction enzymes, T4 DNA ligase, Taq DNA polymerase, and PCR reagents were provided by Takara Biomedical Technology (Beijing) Co., Ltd, and primers were synthesized by Jin Weizhi Biological Technology Co., Ltd. Gel Extraction Kit, PCR Purification Kit, and Plasmid Mini Kit were purchased from Omega Bio-Tek. All reagents and chemicals were of analytical grade and, unless otherwise stated, were obtained from commercial sources.Bacterial strains and plasmids
Details of the plasmids and bacterial strains used in this study are shown in Table S1.† E. coli DH5α and BL21(DE3) (Invitrogen, Carlsbad, CA, USA) were used for plasmid preparation, and protein overexpression/lycopene fermentation, respectively. Gene cloning was performed using S. cerevisiae (Invitrogen). The recombinant plasmid pCLpTrcUpper25 was built and stored by our laboratory. The recombinant plasmid pAC-LYC26 was provided by the University of Maryland (College Park, MD, USA). The vector pETDeu-1 was purchased from Invitrogen.Plasmid construction
The four key enzymes that represent the bottom portion of the MVA pathway were combined for the lycopene color development reaction screening. Through the bottom pathway, the primary phosphorylation by MK and secondary phosphorylation by phospho-mevalonate kinase (PMK, EC2.7.4.2) converts MVA to diphosphomevalonate (DPMVA). Isomerization by isopentenyl diphosphate isomerase (IDI, EC5.3.3.2) and decarboxylation by diphospho-mevalonate decarboxylase (MVD, EC4.1.1.33) produces dimethylallyl pyrophosphate (DMAPP).Four S. cerevisiae genes (PMK, MVD, MK, and IDI; ATCC201508D) were cloned into the pETDeut-1 vector. The pET-CHL1 plasmid was constructed by inserting PMK (ERG8) into the Bgl II and Aat II sites of pETDuet-1 with ERG8_F and ERG8_R primers. The plasmid pET-CHL2 was created by inserting MVD (ERG19) into the Aat II and Xho I sites of pET-CHL1 with primers ERG19_F and ERG19_R. The pET-CHL3 plasmid was established by inserting MK (ERG12) into the Sac I and Not I sites of the pET-CHL2 vector with primers ERG12_F and ERG12_R. The pET-CHL plasmid was built by the PCR amplification IDI (IDI) using primers IDI_F and IDI_R and cloning it into the Sac I and Sal I sites of the plasmid pET-CHL3.
Construction of the mutation library
Error-prone PCR was used to introduce random mutations into ERG12 within the pET-CHL vector. The error-prone PCR reaction was as follows: 10 × error-prone PCR buffer; 0.5 mmol L−1 dATP and dGTP; 2.5 mmol L−1 dCTP and dTTP; 40 pmol of each primer (ERG12_F and ERG12_R); 3 mmol L−1 MgCl2; 0.2 mmol L−1 MnCl2; 2.5 U Taq DNA polymerase, and water. The hot-start PCR amplification program annealed primers at 62 °C for 2 min, and product extension was performed at 72 °C for 2 min; this cycle was repeated 35 times.After error-prone PCR amplification, PCR products and the pET-CHL vector were purified by Sac I and Not I digestion, ligated, and transformed into competent DH5α E. coli, and positive colonies were selected and amplified to purify mutant plasmids.
Screening for mutants with high lycopene production
The pET-CHL and pAC-LYC plasmids were transformed in BL21(DE3) E. coli, and the transformation reaction was spread onto LB-agar plates, which were cultured for 15 h at 37 °C to generate the mutant screening library. Colonies were considered candidate strains and were cultured in 96-well plates in 300 μL of LB medium supplemented with MVA (3 g L−1), ampicillin (100 μg mL−1), and chloramphenicol (50 μg mL−1) before incubation for 8 h at 37 °C. Protein expression was then induced with 0.05 mmol L−1 Isopropyl β-D-1-thiogalactopyranoside (IPTG) for 48 h, and then the cells were pelleted by centrifugation at 3000 × g for 5 min. Next, the pellet was resuspended in 200 μL of acetone, and the mixture was incubated for 15 min at 55 °C, and then centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 3 min. Finally, 200 μL of the supernatant was measured at OD475 on a spectrophotometer to indirectly determine the relative lycopene content.27 Supernatant isolated from the control strain did not absorb light at 475 nm under the same conditions.
000 × g for 3 min. Finally, 200 μL of the supernatant was measured at OD475 on a spectrophotometer to indirectly determine the relative lycopene content.27 Supernatant isolated from the control strain did not absorb light at 475 nm under the same conditions.
Site-specific saturation mutagenesis
To construct MK libraries containing all possible amino acid substitutions at residues V13, S148, and V301, the target codons were replaced with NNK degenerate codons (K = G or T and N = A, T, G, or C) using the QuikChange II site-directed mutagenesis kit (Stratagene, San Diego, CA, USA). The MK-V13, MK-S148, and MK-V301 libraries were produced with the V13-F/R, S148–F/R, and V301-F/R primers, respectively (Table S1†). The constructs from all three libraries were then transformed into competent BL21(DE3) E. coli. The original pET-CHL plasmid containing wild-type ERG12 was used as the template.Site-directed mutagenesis
The double mutations V13D/S148I, V13D/V301E, S148I/V301E, and the triple mutation V13D/S148I/V301E were created using the QuikChange II site-directed mutagenesis kit (Stratagene). The S148I substitution was introduced into pET-CHL/MK-V13D using the primers S148I-F and S148I-R (Table S1†). Competent BL21(DE3) E. coli were transformed with the pET-CHL/MK-V13D/S148I plasmid by electroporation.MK activity assay
The MK activity assay was performed essentially as described previously.28 Briefly, a 0.1 M (RS)-mevalonate stock solution was prepared by dissolving mevalonic acid lactone crystals in 0.2 M KOH. Lactone was hydrolysed by incubating the solution at 37 °C for 1 h. 0.1 M HCl was added to lower the pH of the solution to 7.2. The following components were then incubated in a 1.0 mL cuvette: 5 mM MgCl2, 4 mM ATP, 0.16 mM NADH, 0.5 mM phosphoenol pyruvate, 3 mM (RS)-mevalonate, 75 units of lactate dehydrogenase and 81 units of pyruvate kinase in a 0.1 M KH2PO4/K2HPO4 buffer, pH 8.0. A spectrophotometer was used to quantify the change in absorbance as a readout of enzymatic activity. NADH oxidation was initially measured in the absence of MK for 2 min, before following the reaction for 2 min once the MK was added. One unit of MK activity was defined as the activity required to produce 1 mmol of mevalonate 5-phosphate/min. The Km for (RS)-mevalonate was quantified using a (RS)-mevalonate concentration of between 0.03 and 5 mM with 0.2 mg of pure MK in an assay buffer containing 4 mM ATP.Determining the kinetic parameters and effects of temperature and pH on MK activity
Purified MK was used to investigate the Vmax (RS)-mevalonate, Km (RS)-mevalonate, Kcat (RS)-mevalonate, pH and thermal stability of the enzyme. Km was determined by the double-reciprocal plot method.28 Phosphate buffer (0.05 mol L−1, pH 5–8), Tris–HCl buffer (0.05 mol L−1, pH 8–9) and carbonate buffer (0.05 mol L−1, pH 9–10) were prepared to determine the optimum pH for MK activity. To determine the pH-stability of MK in varying pH conditions, a 0.25 U enzyme solution was separately added to the above buffer solutions at different pH values, and then the solutions were incubated for 1 h at 25 °C to determine enzyme activity under standard conditions. To determine thermal stability and optimal temperature, the 0.25 U enzyme solution underwent heat preservation for 1 h at 25–55 °C to determine MK activity under standard conditions.Lycopene production by wild-type and mutated MK in batch fermentation
A Biostat B plus MO5L fermenter (Sartorius Stedim Biotech GmbH, Göttingen, Germany) was used to perform fed-batch fermentation reactions using 2 L of fresh M9 medium (20 g glucose, 9.8 g KH2PO4, 2.1 g citric acid, 0.3 g ammonium ferric citrate, 5 g beef extract, 25 mg MgSO4·7H2O per 1 L water, pH 7.0) at 37 °C. After the initial carbon source (20 g L−1 glucose) was almost entirely consumed, a 3 M glucose solution was added to begin the fed batch mode. A 25% ammonia solution was used to maintain a constant neutral pH. Given that oxygen is an important factor for isoprenoid synthesis, fermentation was performed under strict aerobic conditions, with the dissolved oxygen concentration maintained at 20% saturation. When the OD600 reached 20, 0.05 mM IPTG was added to induce recombinant protein expression. Fresh IPTG and antibiotics were added every 24 h, and lycopene production and cell growth were monitored every 5 h throughout the 72 h fermentation process.Simulation of 3D structure and molecular docking of substrate to mutant MK
The NCBI protein database (http://www.ncbi.nlm.nih.gov/protein/) was queried using the amino acid sequence S. cerevisiae MK. The employed protein sequence was CAA39359.1, as reported by Oulmouden et al.29 The BLAST server (http://blast.ncbi.nlm.nih.gov) was used as a search template for the chain. We applied human MK (PDB: 1KVK) as the template,30 and the homology of the amino acid sequences was determined by sequence alignment. MK homology modeling was performed with SWISS-MODEL.An in silico molecular docking analysis was executed to investigate potential binding modes between MVA, ATP, and MK using Autodock vina 1.1.2. Tertiary and quaternary MK structures were built with SWISS-MODEL. The 2D structure of MVA and ATP were drawn with ChemBioDraw Ultra 14.0 and converted to 3D structures using ChemBio3D Ultra 14.0 software. Docking input files were created with the AutoDockTools 1.5.6 package.31 The search grid of MK for ATP was determined as center_x: 43.113, center_y: 29.598, and center_z: 15.319, with dimensions size_x: 15, size_y: 15, and size_z: 15; while the search grid for MVA was determined as center_x: 42.508, center_y: 33.926, and center_z: 6.015, with dimensions size_x: 15, size_y: 15, and size_z: 15. The value of exhaustiveness was set to 20. Default Vina docking parameters were used unless otherwise described. The best-scoring model as determined by the Vina docking score was visually analyzed using PyMoL 1.7.6 (http://www.pymol.org/).
Results and discussion
Screening for recombinant strains with high lycopene production
We performed three cycles of error-prone PCR, and the capacity of the mutation library was always 10000–15000 strains. Using the lycopene color development reaction as the primary screening method, 20 objective bacteria strains were obtained in each cycle (Fig. 1A).
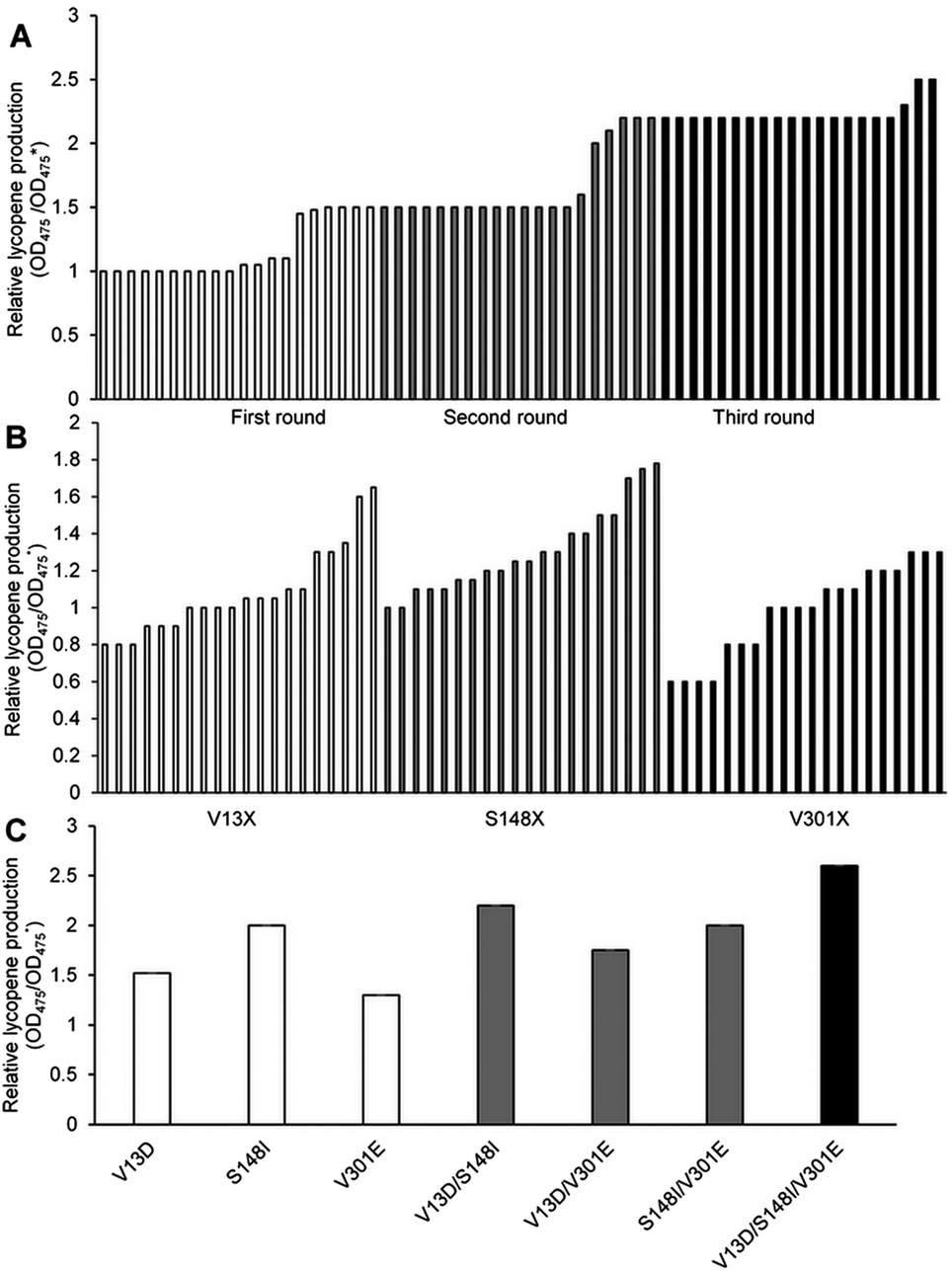 | ||
| Fig. 1 Screening results of three cycles of random mutagenesis (A); saturation mutagenesis at residues V13, S148, and V301 (B); and critical amino acid substitutions by site-directed mutagenesis (C). (A) Twenty representative strains are shown for each sequential mutagenesis step. The average relative lycopene production of the mutants increased progressively with every round of mutagenesis. Strains with the highest relative lycopene production in each round were chosen for further analysis. (B) Twenty representative strains are shown for every position to identify the most advantageous amino acid substitution at these residues. These same color columns represent different residual activities at the same position. (C) Single, double and triple mutation enzymes were constructed to determine which were sufficient and necessary for enhanced MK activity. Every mutant was necessary for MK and worked well with no interference. Wild-type MK was used as the control (OD475*). | ||
Six objective bacteria strains were identified in the first cycle of screening (Fig. 1A), the genomes of which were sequenced and used as templates to perform the second cycle of error-prone PCR. Five objective bacteria strains were identified in the second cycle (Fig. 1A), the genomes of which were sequenced and used as templates to perform the third cycle of error-prone PCR. Three objective bacteria strains emerged from the third cycle (Fig. 1A). These 14 strains were sequenced, ten of which were determined to contain base mutations, and eight to contain amino acid mutations.
Site-directed saturation mutagenesis of residues V13, S148, and V301
Substitutions V13, S148, and V301 were identified as mutations capable of enhancing lycopene production. Separate saturation mutagenesis libraries were constructed using NNK codon degeneracy (20 amino acids/32 codons), which can encode all possible amino acids at positions V13, S148, and V301 in MK. Approximately 300 colonies from each of the three libraries were screened to ensure that all possible substitutions were assessed. It has been suggested that screening 94 NNK codon degeneracy mutagenesis colonies will yield a 95% probability of evaluating all possible 32 outcomes.32Five colonies from the V13 library displayed enhanced lycopene production (Fig. 1B), Two of which contained aspartic acid substitutions (V13D), while the remaining three contained glutamic acid (V13E), which was the substitution identified in the consensus library. Amino acid analysis showed that acidic amino acids were predominate in this screening assay. The V13D variant displayed a 0.70-fold improvement in lycopene production relative to wild type, while V13E showed a 0.38-fold enhancement. Eighteen colonies from the S148 library displayed enhanced lycopene production (Fig. 1B), and all contained substitutions of residues with hydrophobic side chains: alanine, leucine, valine, isoleucine and phenylalanine. Substitutions of S148-to-Leu, -Phe, -Val, or -Ala enhanced lycopene production between 0.21- and 0.49-fold compare with the wild type, although the activity of these mutants was lower than that of S148I, which displayed a 0.80-fold increase in the validation assay. Three colonies from the V301 library, including V301E, displayed enhanced lycopene production (Fig. 1B). During the validation screen, the V301L and V301C variants with improved activity, and were also found to have a 0.25- to 0.38-fold increase in stability compared with the wild-type enzyme; however, V301E showed the highest increase in stability (0.40-fold).
Identifying critical MK mutations
The optimal amino acid substitutions for MK activity from the saturation mutagenesis screen were V13D, S148I, and V301E. Next, a combinatorial mutagenesis approach was used to identify the MK variants with the largest increase in lycopene production.V13D, S148I, and V301E showed 0.51-, 0.70- and 0.47-fold higher lycopene production compared with the wild type, respectively (Fig. 1C), indicating that these individual mutations were all beneficial and improved lycopene production.
Moreover, V13D/S148I, V13D/V301E, and S148I/V301E showed 1.43-, 1.05- and 1.25-fold higher lycopene production, respectively (Fig. 1C), indicating that double mutation combinations could further improve lycopene production. Furthermore, no double mutation combination decreased production. Finally, the triple mutant V13D/S148I/V301E showed the highest improvement (1.79-fold compared with wild type) (Fig. 1C). In summary, the enhanced catalytic activity of mutant MK resulted from additive mutations at residues 13, 148 and 301.
Determination of kinetic parameters
The kinetic parameters of wild-type and triple-mutant (V13D/S148I/V301E) MK are summarized in Table 1. The Km (RS)-mevalonate of triple-mutant MK decreased by 74% (from 31.61 ± 0.41 μM to 8.23 ± 0.12 μM) compared with wild type. Such a decrease indicated that the affinity of triple-mutant MK for substrate had increased, and that the specific activity of the enzyme had risen.33 The Kcat (RS)-mevalonate value increased from 7.36 ± 0.10 s−1 to 9.25 ± 0.12 s−1. Such an increase indicated when the enzyme was saturated with substrate, the number of substrate molecules converted per molecule of enzyme per min increased.34 The catalytic efficiency of the enzyme was therefore enhanced from 0.23 s−1 μM−1 to 1.13 s−1 μM−1.| Enzyme | Km (RS)-mevalonate (μM) | Kcat (RS)-mevalonate (s−1) | Kcat (RS)-mevalonate/Km (RS)-mevalonate (s−1 μM−1) |
|---|---|---|---|
| MK | 31.61 ± 0.41 | 7.36 ± 0.10 | 0.23 |
| MK (V13D/S148I/V301E) | 8.23 ± 0.12 | 9.25 ± 0.12 | 1.13 |
A series of mutations in the MK active-sites resulted in increased enzymatic activity. The decreased Km (RS)-mevalonate and increased Kcat (RS)-mevalonate values mirrored the enhanced performance of mutant MK in substrate catalysis. This suggested that enhanced enzymatic activity was primarily the result of two main factors: (1) improved affinity between the substrate and enzyme caused by mutations (V13E and S148I) near the active site (Arg248 and Ser148);29 and (2) increased foldability due to V301E. Our investigation of mutant MK stability and activity were in agreement with these conclusions.
Enzymatic properties of wild-type and triple-mutant MK
As shown in Fig. 2A, the enzymatic activities of wild-type and triple-mutant MK increased from 25–35 °C, reaching a maximum at 35 °C. The activity of both MK proteins were reduced at temperature above 35 °C, indicating that the optimal temperature for both MK versions was 35 °C, and that mutations did not affect the optimal temperature for MK activity.35 The thermal stabilities of triple-mutant and wild-type MK were similar, with a range of 25–55 °C (Fig. 2B). The thermal stabilities of both MK proteins continuously decreased at temperature above 35 °C, although the thermal stability of wild-type MK decreased more significantly than MK (V13D/S148I/V301E). A possible explanation for this finding may be that a salt bridge was formed by the mutation, which enhanced the rigidity of the enzyme (Fig. 5B).36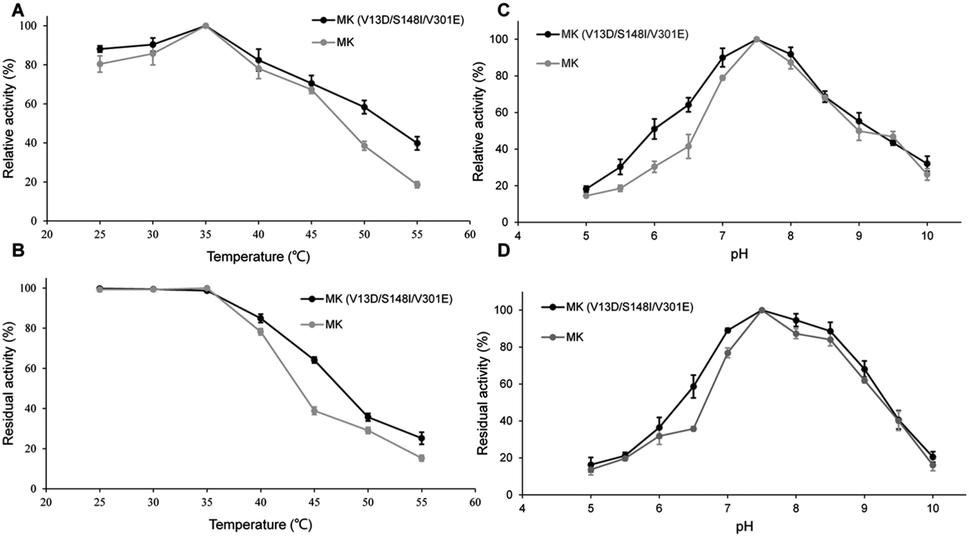 | ||
| Fig. 2 Effect of temperature on wild-type and triple-mutant MK activity (A) and stability (B); the effect of pH on wild-type and triple-mutant MK activity (C) and stability (D). (A) The optimal temperature was determined to be 35 °C. When temperature was above 35 °C, the rate of MK (V13D/S148I/V301E) activity decreased faster than wild-type MK; (B) the stability of the triple-mutant MK was improved compared with the wild type. (C) The optimal pH was 7.5 at 25 °C. Mutations did not affect parameters related to pH; (D) pH affected the stability of wild-type and V13D/S148I/V301E MK, although mutations did not change the acid-alkali tolerance of MK. | ||
As shown in Fig. 2C, at pH = 5, the activity of MK (V13D/S148I/V301E) and MK were 18% and 15% of maximum activity, respectively. The activities of both enzymes increased when pH was <7.5, and decreased when pH was >7.5, suggesting that the optimum pH for both MK proteins was 7.5, and that the mutations did not affect the optimal pH for MK activity. Fig. 2D shows that in the pH range of 5–7.5, MK (V13D/S148I/V301E) activity increased rapidly from 18% to 100%, while from pH 7.5–8.5, the activity was basically unchanged; when the pH was >8.5, activity decreased. The general trend suggested that the wild-type and mutant MKs had similar pH stability properties.37 Finally, we also showed that acidic conditions affected the pH stability of MK significantly, while alkaline conditions maintained the pH stability of MK (Fig. 2D).
Lycopene production of mutant and wild-type MK in batch fermentation assays
The performance of MK (V13D/S148I/V301E) was ultimately evaluated in terms of final lycopene yield and production through fed fermentation. For these experiments, the recombinant strain containing wild-type MK was named CHL-1, and the strain containing MK (V13D/S148I/V301E) was named CHL-2. Fed-batch fermentations were performed to test the suitability and stability of MK (V13D/S148I/V301E) for improved lycopene production.As shown in Fig. 3A and B, when IPTG was added into the bioreactor, the biomass reached OD600 = 20, while the lycopene in CHL-1 and CHL-2 were only about 0.43 and 0.37 mg g−1 dry cell weight (DCW). During the fed-batch stage, the biomass showed a steady increase, while the lycopene accumulation was dramatically increased. After 90 h of fermentation, a lycopene production of 1.43 g L−1 and 19.83 mg g−1 DCW was achieved in CHL-2 while a lycopene production of 0.72 g L−1 and 8.32 mg g−1 DCW was achieved in CHL-1. Although the fermentation process was stopped after 90 h, the lycopene accumulation in CHL-1 lasted for 70 h, while lycopene accumulation in CHL-2 was continuous throughout the whole fermentation process. The lycopene production and lycopene accumulation time suggested that the engineered triple-mutant MK efficiently improved lycopene production.
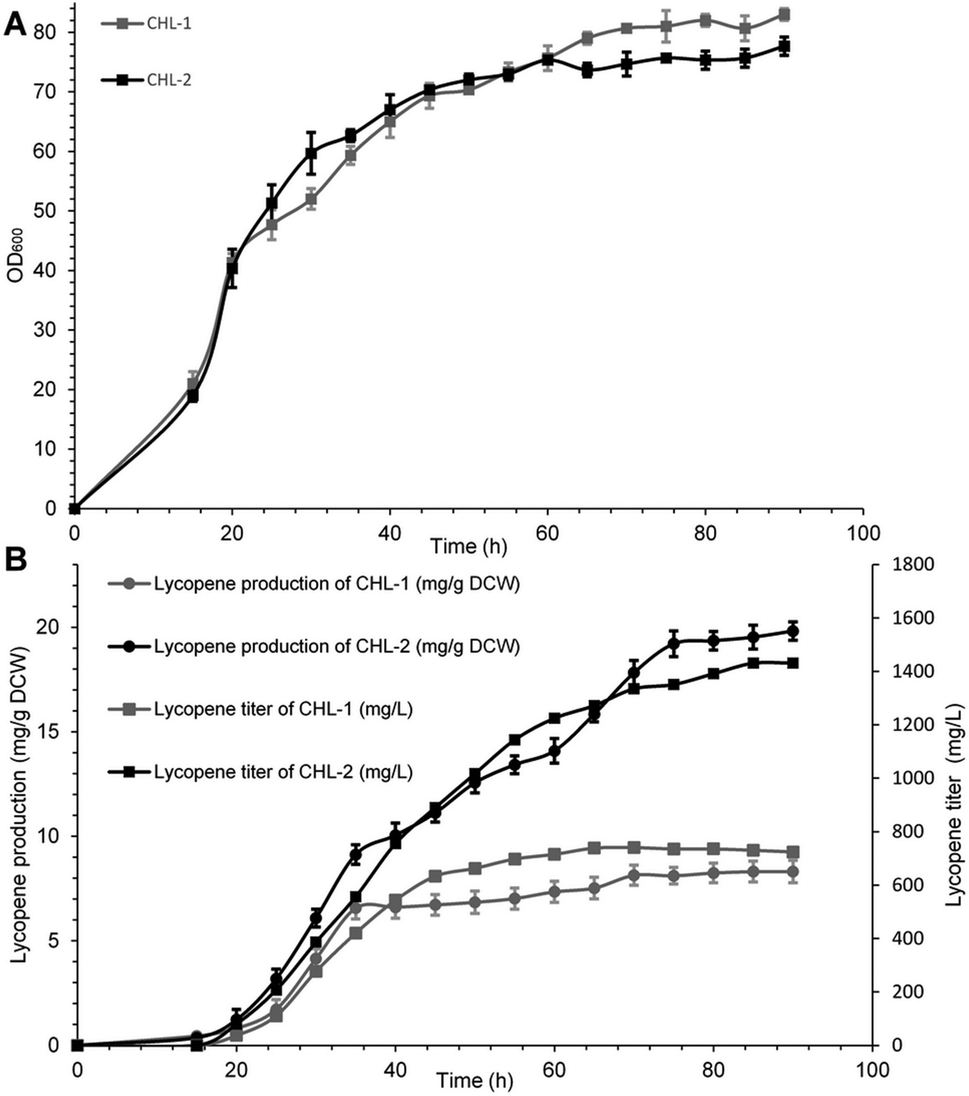 | ||
| Fig. 3 Cell growth rates (A) and lycopene production (B) during aerobic fed-batch fermentation of CHL-1 and CHL-2. (A) The cell growth rates of CHL-1 and CHL-2 were similar. (B) Lycopene production of CHL-2 was increased 2.0-fold compared with CHL-1. | ||
The lycopene content in this work (19.83 mg g−1 DCW) was much higher than previous reports of lycopene production from engineered S. cerevisiae strains,38,39 or natural producers such as tomato40 and B. trispora.41 Although it was still lower than the maximum E. coli production system (32 mg g−1 DCW),42 we believe that optimizing media and fermentation conditions in subsequent studies will further enhance lycopene production.43
Structural simulation and molecular docking of MK
The results of in silico structural modelling and molecular docking of MK are shown in Fig. 4A. The theoretical binding mode of ATP and MVA to the binding MK sites are illustrated in Fig. 4B. ATP assumes a compact conformation to bind inside the MK binding pocket.34 The amino group of the adenine ring is hydrogen-bonded to the hydroxyl group of Ser-138 and to the carbonyl group of Asp-56. The magnesium ion within the magnesium triphosphate moiety is coordinated to both β- and γ-phosphates and is stabilized by a surrounding glycine-rich loop. It is also coordinated to the hydroxyl group of Ser-149 and the carboxyl group of Glu-191, both of which share a hydrogen-bond. A hydrogen-bond is formed between the hydroxyl group of Ser-149 and β-PO4. The interaction between Lys-12 and Asp-202 was of particular interest, as there is a salt bridge linking these two residues. Lys-12 forms a close interaction with the ATP γ-PO4. The carboxyl group of MVA binds nearby residues Arg-248, Thr-350, and Ala-352. Binding of MVA is stabilized through the formation of a salt bridge between the substrate carboxyl group and Arg-248. Additionally, the substrate carboxyl group is hydrogen bonded with the main-chain amide nitrogen of Ala-352. These interactions facilitated MVA anchorage within the MK binding site. Our 3D homology model of MK was built to advance our appreciation of the consequence of mutagenesis on MK activity and stability. The locations of the three mutation sites in the MK molecule are shown in Fig. 4A.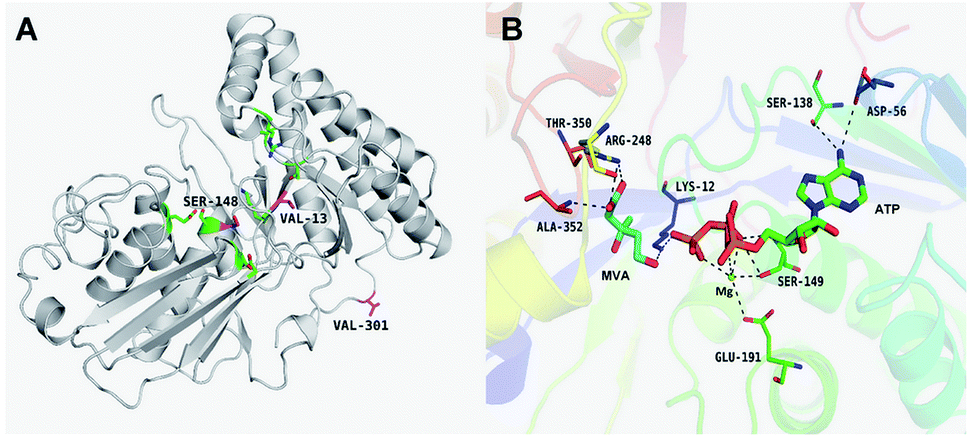 | ||
| Fig. 4 Structural (A) and docking (B) models of wild-type MK. (A) The location of the beneficial mutations are marked in magenta, and the active site catalytic triad residues (Arg-248, Thr-350 and Ala-352) are marked in green. (B) The active site catalytic triad residues and the ATP-binding residues (Lys-12, Asp56, Ser138, Ser-149 and Glu-191) are respectively marked. | ||
Glu-301 is situated in the loop on the periphery of MK. Using 3D structural simulation and comparison analysis with wild-type MK, it was considered that the site mutation was far away from the catalytic center (Fig. 4A). For directed evolution, enzyme activity is only one mutagenic effect, and foldability, or the role of amino acid residues in protein folding, should also be considered. Protein secretion have a strict quality assurance system, misfolded or folded partially proteins are degraded by the proteasome. Glu-301 may increase the foldability of MK, allowing more effective MK to be produced.29,44
Asp-13 is located in the β-sheet inside the tertiary structure of MK. The molecular docking results suggested that the loop is located close to the MVA binding site (Fig. 5A and B). Asp-13 is very close to the MK active site Arg-248 in the 3D structure. Furthermore, Asp 13 is an acidic amino acid, therefore it can bond with Arg-248 (positively charged) in the hatchway loop that leads to the active center to form a salt bridge.45 The electrostatic attraction draws the loop in the opposite direction of the hatchway, thus reducing the hindrance of the loop for substrate molecules to enter the active site. The positively charged amidogen in Arg-248 forms mutual affinity with the negatively charged carboxyl group in Asp-13, thus forming a salt bridge (Fig. 5A and B). Additionally, this affinity narrows the distance between the loop and the backbone of the β-sheet from the original 6.50 Å to 6.23 Å. Thus, it could be inferred that the hatchway for substrate molecules to enter the enzyme active site will become larger, reducing the hindrance accordingly and enhancing enzyme activity. The molecular docking results revealed that the binding energy between the substrate and the mutant enzyme was reduced to 3.21 kcal mol−1 from the original 3.87 kcal mol−1,46 which further indicated that the hindrance for substrate to enter the enzyme active site an bind to the enzyme was reduced. Thus, binding was more stable, enhancing enzymatic activity.
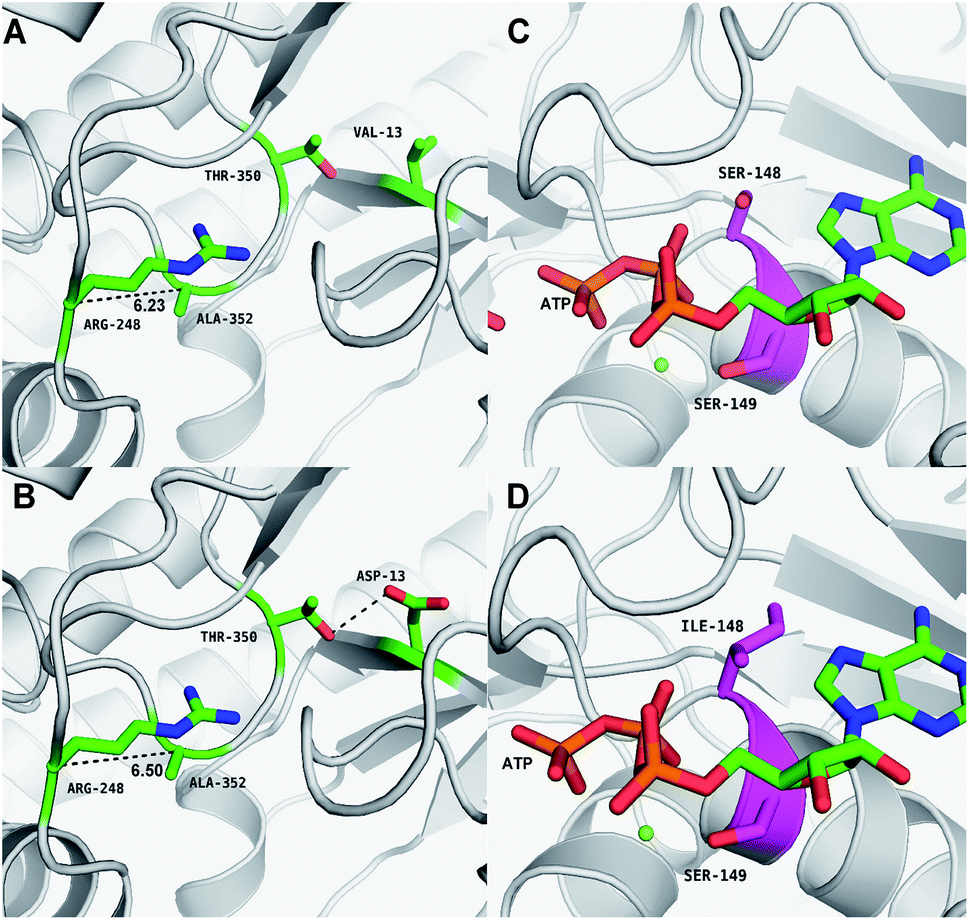 | ||
| Fig. 5 Structural models of the mutations: (A) wild-type V13; (B) V13D; (C) wild-type S148; and (D) S148I. (A and B) The V13D substitution shows Asn-13 forming a salt-bridge with Arg-248, which enlarged the MK catalytic domain. (C and D) The S148I substitution is located in the α-helix lid and improved the stability of the α-helix. | ||
Ile-148 is located in the α-helix lid, in close proximity to the key site Ser149 of MK (Fig. 5C and D). The α-helix lid is a key factor in maintaining MK stability and activity,47,48 as it covers the active site, keeping out water soluble factors.46 Ile-148 did not affect Ser149 and made the structure of the α-helix lid more stable, slightly enhancing enzymatic activity.
Conclusions
In conclusion, we have successfully engineered MK to improve its catalytic activity and lycopene production via a random mutagenesis approach. In this study, we used error-prone PCR to obtain a mutant MK library and developed a lycopene-dependent color development reaction to screen for activity-enhancing mutations. The best mutants produced by this approach maintained catalytic activity while also displaying improved thermo- and pH stability. These properties could make it a potent and attractive biocatalyst for the production of biochemicals. Fermentation experiments revealed that the lycopene production of triple-mutant MK was 2.4-fold higher than wild-type MK; however, due to the inherent deficiency of error-prone PCR, it was difficult to substantially increase MK activity. The MK performance in lycopene production could be further optimized by using additional or alternative protein engineering techniques, such as DNA shuffling,49 rational design,50 B-FIT, or ISM approaches.51 The X-ray crystal structures of these engineered enzymes will also be further investigated to further elucidate the molecular mechanisms that govern catalytic activity.Conflicts of interest
The authors declare no financial or commercial conflict of interest.Acknowledgements
This research was financially supported by National Natural Science Foundation of China (No. 21572242), Taishan Scholars Climbing Program of Shandong (No. tspd20150210), National Natural Science Foundation of China (NSF No. 31400084) and Youth Innovation Promotion Association CAS No. 2017252.References
- A. V. Rao and S. Agarwal, Nutr. Res., 1999, 19, 305–323 CrossRef CAS.
- L. Alexander and D. Grierson, J. Exp. Bot., 2002, 53, 2039–2055 CrossRef CAS PubMed.
- S. M. Choudhari, L. Ananthanarayan and R. S. Singhal, Bioresour. Technol., 2008, 99, 3166–3173 CrossRef CAS PubMed.
- N. Arrach, R. Fernández-Martín, E. Cerdá-Olmedo and J. Avalos, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1687–1692 CrossRef CAS.
- M. A. Phillips, P. León, A. Boronat and M. Rodríguez-Concepción, Trends Plant Sci., 2008, 13, 619–623 CrossRef CAS PubMed.
- J. Yang, G. Zhao, Y. Sun, Y. Zheng, X. Jiang, W. Liu and M. Xian, Bioresour. Technol., 2012, 104, 642–647 CrossRef CAS PubMed.
- J. A. Tobert, Nat. Rev. Drug Discovery, 2003, 2, 517 CrossRef CAS PubMed.
- S. M. Houten, R. J. A. Wanders and H. R. Waterham, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2000, 1529, 19–32 CrossRef CAS.
- H. M. Miziorko, Arch. Biochem. Biophys., 2011, 505, 131–143 CrossRef CAS PubMed.
- D. D. Hinson, K. L. Chambliss, M. J. Toth, R. D. Tanaka and K. M. Gibson, J. Lipid Res., 1997, 38, 2216–2223 CAS.
- J. R. Anthony, L. C. Anthony, F. Nowroozi, G. Kwon, J. D. Newman and J. D. Keasling, Metab. Eng., 2009, 11, 13–19 CrossRef CAS PubMed.
- J. K. Dorsey and J. W. Porter, J. Biol. Chem., 1968, 243, 4667–4670 CAS.
- R. D. Tanaka, B. L. Schafer, L. Y. Lee, J. S. Freudenberger and S. T. Mosley, J. Biol. Chem., 1990, 265, 2391–2398 CAS.
- J. C. Gray and R. G. O. Kekwick, Biochim. Biophys. Acta, Gen. Subj., 1972, 279, 290–296 CrossRef CAS.
- V. J. J. Martin, D. J. Pitera, S. T. Withers, J. D. Newman and J. D. Keasling, Nat. Biotechnol., 2003, 21, 796 CrossRef CAS PubMed.
- M. A. Lluch, A. Masferrer, M. Arró, A. Boronat and A. Ferrer, Plant Mol. Biol., 2000, 42, 365–376 CrossRef CAS PubMed.
- S. Hogenboom, J. J. M. Tuyp, M. Espeel, J. Koster, R. J. A. Wanders and H. R. Waterham, J. Cell Sci., 2004, 117, 631–639 CrossRef CAS PubMed.
- R. D. Goodfellow and F. J. Barnes, Insect Biochem., 1971, 1, 271–282 CrossRef CAS.
- F. H. Arnold and A. A. Volkov, Curr. Opin. Chem. Biol., 1999, 3, 54–59 CrossRef CAS PubMed.
- N. J. Turner, Nat. Chem. Biol., 2009, 5, 567 CrossRef CAS PubMed.
- L. Ye, C. Zhang, C. Bi, Q. Li and X. Zhang, Microb. Cell Fact., 2016, 15, 202 CrossRef PubMed.
- C. Denoyelle, M. Vasse, M. Körner, Z. Mishal, F. Ganné, J.-P. Vannier, J. Soria and C. Soria, Carcinogenesis, 2001, 22, 1139–1148 CrossRef CAS PubMed.
- S. H. Yoon, Y. M. Lee, J. E. Kim, S. H. Lee, J. H. Lee, J. Y. Kim, K. H. Jung, Y. C. Shin, J. D. Keasling and S. W. Kim, Biotechnol. Bioeng., 2006, 94, 1025–1032 CrossRef CAS PubMed.
- H. Yokoyama, C. DeBenedict, C. W. Coggins and G. L. Henning, Phytochemistry, 1972, 11, 1721–1724 CrossRef CAS.
- M. A. Cervin, G. K. Chotani, F. J. Feher, R. La Duca, J. C. McAuliffe, A. Miasnikov, C. M. Peres, A. S. Puhala, K. J. Sanford and F. Valle, US pat. 9260727, 2016.
- R. V. Vadali, Y. Fu, G. N. Bennett and K. Y. San, Biotechnol. Prog., 2005, 21, 1558–1561 CrossRef CAS PubMed.
- H. Alper, K. Miyaoku and G. Stephanopoulos, Nat. Biotechnol., 2005, 23, 612 CrossRef CAS PubMed.
- K.-x. Huang, A. Scott and G. N. Bennett, Protein Expression Purif., 1999, 17, 33–40 CrossRef CAS PubMed.
- A. Oulmouden and F. Karst, Curr. Genet., 1991, 19, 9–14 CrossRef CAS PubMed.
- B. Schafer, R. Bishop, V. Kratunis, S. Kalinowski, S. Mosley, K. Gibson and R. Tanaka, J. Biol. Chem., 1992, 267, 13229–13238 CAS.
- A. Munir, S. Azam, S. Fazal, Z. Khan and A. Mehmood, Int. J. BIOautom., 2016, 20 CAS.
- M. T. Reetz, D. Kahakeaw and R. Lohmer, ChemBioChem, 2008, 9, 1797–1804 CrossRef CAS PubMed.
- J. Gray and R. Kekwick, Biochem. J., 1973, 133, 335–347 CrossRef CAS PubMed.
- D. Potter and H. M. Miziorko, J. Biol. Chem., 1997, 272, 25449–25454 CrossRef CAS PubMed.
- A. Simon, L. Cuisset, M.-F. Vincent, S. D. van der Velde-Visser, M. Delpech, J. W. van der Meer and J. P. Drenth, Ann. Intern. Med., 2001, 135, 338–343 CrossRef CAS PubMed.
- N. E. Voynova, S. E. Rios and H. M. Miziorko, J. Bacteriol., 2004, 186, 61–67 CrossRef CAS PubMed.
- I. Williamson and R. Kekwick, Biochem. J., 1965, 96, 862 CrossRef CAS PubMed.
- A. Bahieldin, N. O. Gadalla, S. M. Al-Garni, H. Almehdar, S. Noor, S. M. Hassan, A. M. Shokry, J. S. Sabir and N. Murata, Plasmid, 2014, 72, 18–28 CrossRef CAS PubMed.
- S. Yamano, T. Ishii, M. Nakagawa, H. Ikenaga and N. Misawa, Biosci., Biotechnol., Biochem., 1994, 58, 1112–1114 CrossRef CAS PubMed.
- A. Agarwal, H. Shen, S. Agarwal and A. Rao, J. Med. Food, 2001, 4, 9–15 CrossRef CAS PubMed.
- F. Mantzouridou and M. Z. Tsimidou, Trends Food Sci. Technol., 2008, 19, 363–371 CrossRef CAS.
- S.-H. Yoon, J.-E. Kim, S.-H. Lee, H.-M. Park, M.-S. Choi, J.-Y. Kim, S.-H. Lee, Y.-C. Shin, J. D. Keasling and S.-W. Kim, Appl. Microbiol. Biotechnol., 2007, 74, 131–139 CrossRef CAS PubMed.
- Y.-S. Kim, J.-H. Lee, N.-H. Kim, S.-J. Yeom, S.-W. Kim and D.-K. Oh, Appl. Microbiol. Biotechnol., 2011, 90, 489–497 CrossRef CAS PubMed.
- Z. Fu, N. E. Voynova, T. J. Herdendorf, H. M. Miziorko and J.-J. P. Kim, Biochemistry, 2008, 47, 3715–3724 CrossRef CAS PubMed.
- Z. Fu, M. Wang, D. Potter, H. M. Miziorko and J.-J. P. Kim, J. Biol. Chem., 2002, 277, 18134–18142 CrossRef CAS PubMed.
- D. Yang, L. W. Shipman, C. A. Roessner, A. I. Scott and J. C. Sacchettini, J. Biol. Chem., 2002, 277, 9462–9467 CrossRef CAS PubMed.
- J. L. Andreassi, P. W. Bilder, M. W. Vetting, S. L. Roderick and T. S. Leyh, Protein Sci., 2007, 16, 983–989 CrossRef CAS PubMed.
- T. Sgraja, T. K. Smith and W. N. Hunter, BMC Struct. Biol., 2007, 7, 20 CrossRef PubMed.
- W. P. Stemmer, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 10747–10751 CrossRef CAS.
- M. A. Wambaugh, V. P. Shakya, A. J. Lewis, M. A. Mulvey and J. C. Brown, PLoS Biol., 2017, 15, e2001644 Search PubMed.
- I. V. Korendovych in Protein Engineering, Springer, 2018, pp. 15–23 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01783b |
| This journal is © The Royal Society of Chemistry 2018 |