
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of 2-arylbenzo[h]quinolone analogs as selective CYP1B1 inhibitors†
Jinyun Dong ,
Zengtao Wang,
Qingqing Meng,
Qijing Zhang,
Guang Huang,
Jiahua Cui* and
Shaoshun Li*
,
Zengtao Wang,
Qingqing Meng,
Qijing Zhang,
Guang Huang,
Jiahua Cui* and
Shaoshun Li*
School of Pharmacy, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai, China. E-mail: cpucjh@sjtu.edu.cn; ssli@sjtu.edu.cn; Fax: +86 21 34204775; Tel: +86 21 34204775
First published on 20th April 2018
Abstract
The CYP1B1 enzyme is regarded as a potential target for cancer prevention and therapy. Based on the structure of α-naphthoflavone (ANF), diverse 2-arylbenzo[h]quinolone derivatives were designed, synthesized and evaluated as selective CYP1B1 inhibitors. Compared with ANF, although few of the title compounds possessed comparable or slightly higher CYP1B1 inhibitory activity, these compounds displayed a significantly increased selectivity toward CYP1B1 over CYP1A2. Among them compounds 5e, 5g and 5h potently inhibited the activity of CYP1B1 with IC50 values of 3.6, 3.9 and 4.1 nM respectively, paralleled by an excellent selectivity profile. On the basis of predicted clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values, these target compounds may exhibit improved water-solubility compared to ANF. In particular, 5h showed a great superiority in the reversal of CYP1B1-mediated docetaxel resistance in vitro. The current study may serve as a good starting point for the further development of more potent as well as specific CYP1B1 inhibitors capable of reversing CYP1B1-mediated anticancer-drug resistance.
P values, these target compounds may exhibit improved water-solubility compared to ANF. In particular, 5h showed a great superiority in the reversal of CYP1B1-mediated docetaxel resistance in vitro. The current study may serve as a good starting point for the further development of more potent as well as specific CYP1B1 inhibitors capable of reversing CYP1B1-mediated anticancer-drug resistance.
1 Introduction
Human cytochrome P450 enzymes (CYPs) are composed of different families of hemeproteins that play a predominant role in the oxidative metabolism of both exogenous and endogenous substances. The CYP1 subfamily of enzymes (CYP1s) contains three extensively studied members, CYP1A1, CYP1A2, and CYP1B1, due to their critical roles in pro-carcinogen-elicited tumorigenesis and inducing drug resistance.1 They are expressed in a tissue-specific manner, and are induced via the aryl hydrocarbon receptor (AhR), which is a ligand-activated transcription factor.2 Planar aromatic molecules such as 2,3,7,8-tetra-chlorodibenzo-p-dioxin (TCDD) and 7,12-dimethylbenz[a]anthracene (DMBA) have been identified as potent AhR ligands that induce the overexpression of CYP1 enzymes.3–5 A large number of pro-carcinogens such as polycyclic aromatic hydrocarbons (PAHs), polyhalogenated aromatic hydrocarbons (PHAHs), aryl amines, heterocyclic aryl amines and estrogens can be converted into cytotoxic, mutagenic and carcinogenic chemicals in the presence of overexpressed CYP1 enzymes.1Among the three isoforms mentioned above, CYP1B1 is the most promising target. On one hand, it has been regarded as an important enzyme in the carcinogenic action of 17β-estradiol (E2) by catalyzing the hydroxylation of E2 at C-4 regiospecifically to generate the carcinogenic form 4-hydroxy estradiol (4-OHE2). Subsequent oxidation of 4-OHE2 to E2-3,4-quinone promotes the formation of a covalent quinone-DNA adduct, which is responsible for estrogen-related carcinogenesis. Conversely, the other two members of the CYP1 family are prone to generate 2-hydroxy estradiol (2-OHE2); its further-oxidized product E2-2,3-quinone has been proven to be non-mutagenic.6,7 On the other hand, the overexpressed CYP1B1 isoform can speed up the metabolic inactivation of a structurally diverse range of anticancer drugs, such as docetaxel, doxorubicin, paclitaxel, and mitoxantrone.8 As a result, the cellular efficacy of cytotoxic drugs is decreased and cancer cells will eventually develop resistance to these chemotherapeutic agents. Many studies have confirmed that the CYP1B1 isoform is significantly and consistently overexpressed in a wide variety of tumor tissues such as mammary, prostate, ovary, uterus, pituitary, and also in skin and lung cancers, while it is expressed at very low levels or not at all in normal tissues.9–12 Hence, selective inhibition of this enzyme will have a great influence on cancer prevention and the reversal of drug resistance.
α-Naphthoflavone (ANF), a synthetic flavonoid, bears a phenyl ring annulated at C-7,8 of the A-ring of flavone. It has been recognized as a potent CYP1 inhibitor which exhibits strong inhibitory effects on recombinant human CYP1B1 and CYP1A2 with IC50 values of 5 and 6 nM respectively, but to a much lesser extent on CYP1A1 (with an IC50 value of 60 nM).13 Evidence has accumulated during the past several years that exposure to ANF can reduce drug resistance and enhance the sensitivity of CYP1B1-expressing cell lines to anticancer drugs.14,15 However, its further development is still restricted due to its poor water solubility and very limited selectivity toward CYP1B1 over CYP1A2.8 From the X-ray crystallographic structures of CYP1 enzymes and ANF, it can be observed that the amino acid sequences and the orientations of ANF in the binding cavity are different among CYP1 members.16–18 Asp333 on the I helix of CYP1B1 is adjacent to the oxygen atom in the C-ring of ANF, with a short distance of 4.6 Å, while Asp320 on the I helix of CYP1A1 or CYP1A2 is close to the carbonyl oxygen atom of ANF. Inspired by a hypothesis that an alkaline group can be introduced at a suitable location to enhance interactions with the acidic amino acid Asp333, replacement of the oxygen atom on the C-ring of ANF with a bioisosteric hydrophilic imino group was carried out with the aim to improve its selectivity towards CYP1B1 and water solubility. In continuation of our previous work in this area,8 we synthesized and biologically evaluated different classes of 2-arylbenzo[h]quinolones as novel CYP1 inhibitors (Fig. 1).
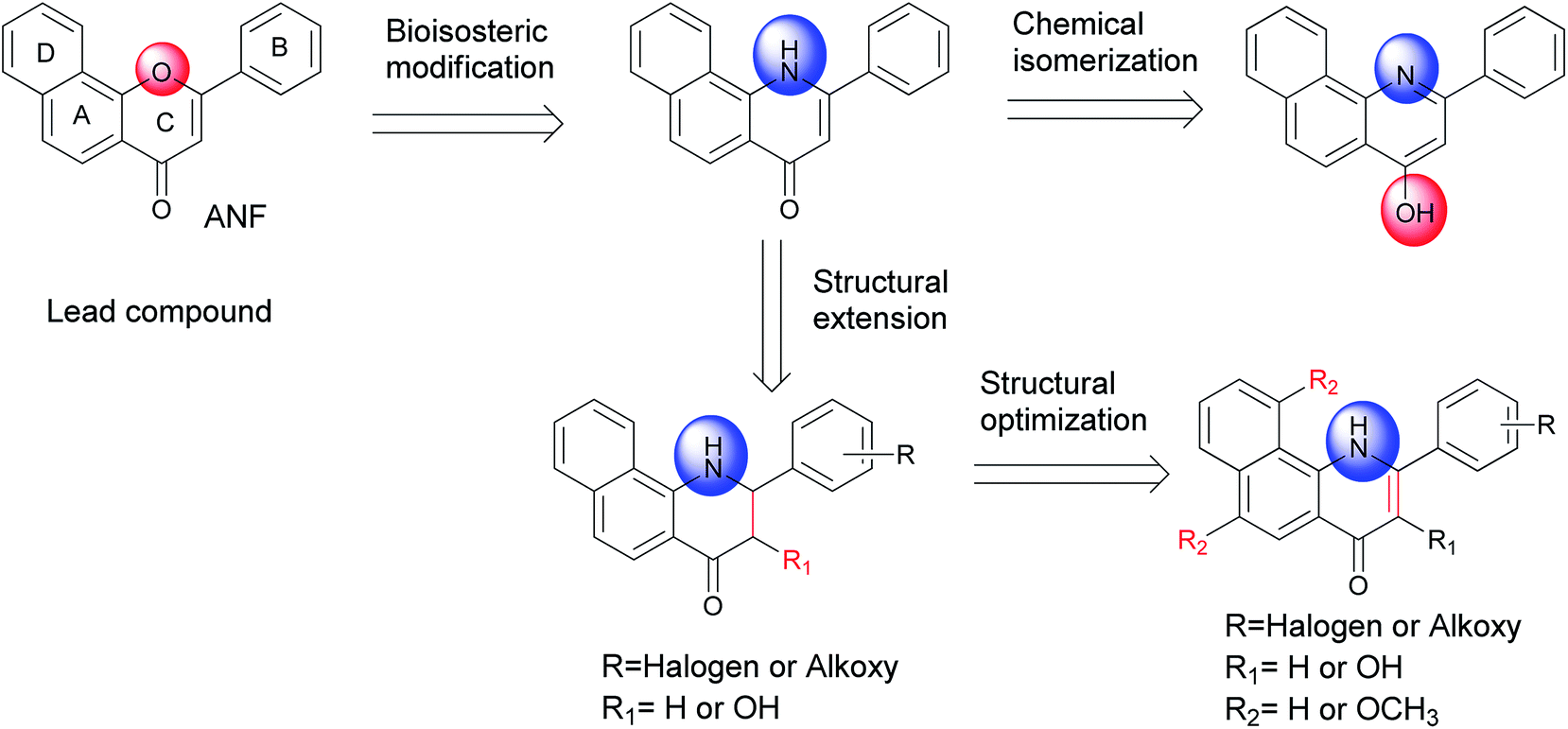 | ||
| Fig. 1 Design of a series of 2-arylbenzo[h]quinolones as CYP1 inhibitors. | ||
2 Results and discussion
2.1 Synthesis
The 2-aryl-2,3-dihydrobenzo[h]quinolin-4(1H)-ones 1a–d and their 3-hydroxy derivatives 2a,b were prepared from 1-nitronaphthalene (6) (Scheme 1). Treatment of 6 with ethyl cyanoacetate and KCN in the presence of KOH in DMF at 55 °C for 36 h formed an intermediate, which was followed by hydrolysis to furnish 1-amino-2-naphthonitril (7) in 40% yield.19 Several attempts to improve the yield of compound 7 including variation of time, temperature and replacement of ethyl cyanoacetate with dicyanomethane were made by our research group–unfortunately the yield was not obviously improved. Subsequently, intermediate 7 was reacted with methylmagnesium iodide, and this was followed by a hydrolysis reaction to give 1-amino-2-acetylnaphthalene (8).20 The 2-aminochalcone derivatives 10a–d were obtained by the condensation of 8 with benzaldehydes 9a–d under alkaline conditions in THF. On one hand, the cyclizations of 10a–d to the corresponding 2-aryl-2,3-dihydrobenzo[h]quinolin-4(1H)-ones 1a–d were accomplished by the addition of antimony trichloride (SbCl3) in CH3CN.21 Considering its toxicity, we also made efforts in exploring safe catalysts such as ZnCl2 and AlCl3, nevertheless, none of these Lewis acids were identified as an efficient catalyst for this intramolecular aza-Michael reaction. On the other hand, oxidation of 10a–d by alkaline hydrogen peroxide afforded the stable 2-aminochalcone epoxide derivatives 11a,b in very good yields,22 which were cyclized to 3-hydroxy-2-aryl-2,3-dihydrobenzo[h]quinolin-4(1H)-ones 2a,b via an intramolecular nucleophilic reaction by addition of a catalytic amount of AlCl3. It is of note that dilute HCl and HOAc as catalysts were also used in this reaction, but we failed to obtain any title compound.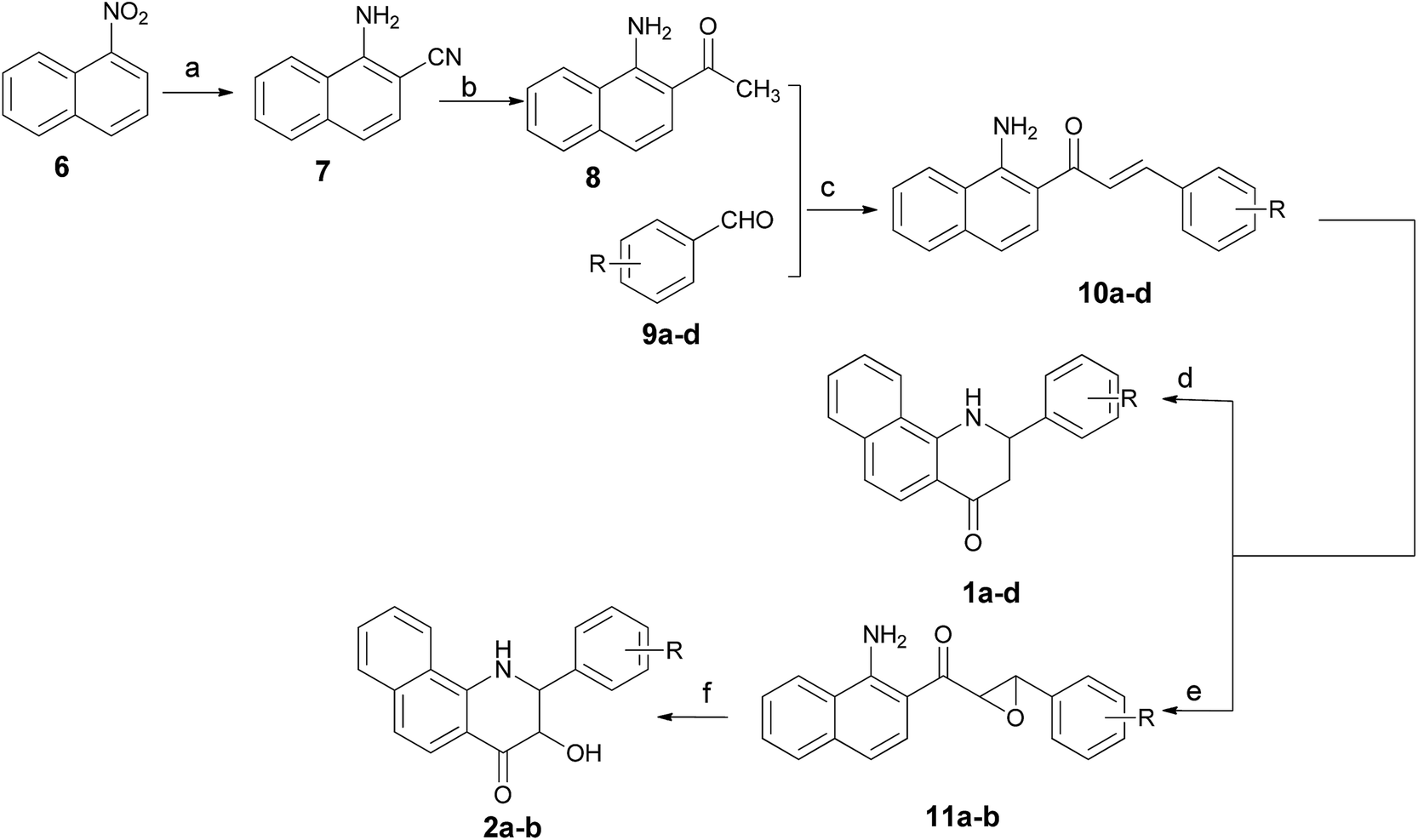 | ||
| Scheme 1 Synthetic routes for compounds 1a–d and 2a,b. Reagents and conditions: (a) KCN, NCCH2COOEt, KOH, DMF, 60 °C for 24 h and then reflux in NaOH–H2O (5%); (b) Mg, CH3I, Et2O, N2, HCl (2 N); (c) CH3ONa, THF; (d) SbCl3, CH3CN, 80 °C; (e) H2O2, NaOH, MeOH; (f) AlCl3, DCM. | ||
For the preparation of 3-hydroxy-2-arylbenzo[h]quinolin-4(1H)-ones 3a–d, we used the synthetic strategy described in Scheme 2. 1-Amino-2-naphthonitril (7) was hydrolyzed with 20% NaOH to provide the corresponding L-amino-2-naphthoic acid (12) in 62% yield. Treatment of 12 with 2-bromo-1-arylethan-1-ones 13a–d in the presence of K2CO3 in DMF gave intermediates 14a–d in excellent yields, which was followed by a cyclization under acidic conditions using polyphosphoric acid (PPA) at 130 °C to yield title compounds 3a–d.
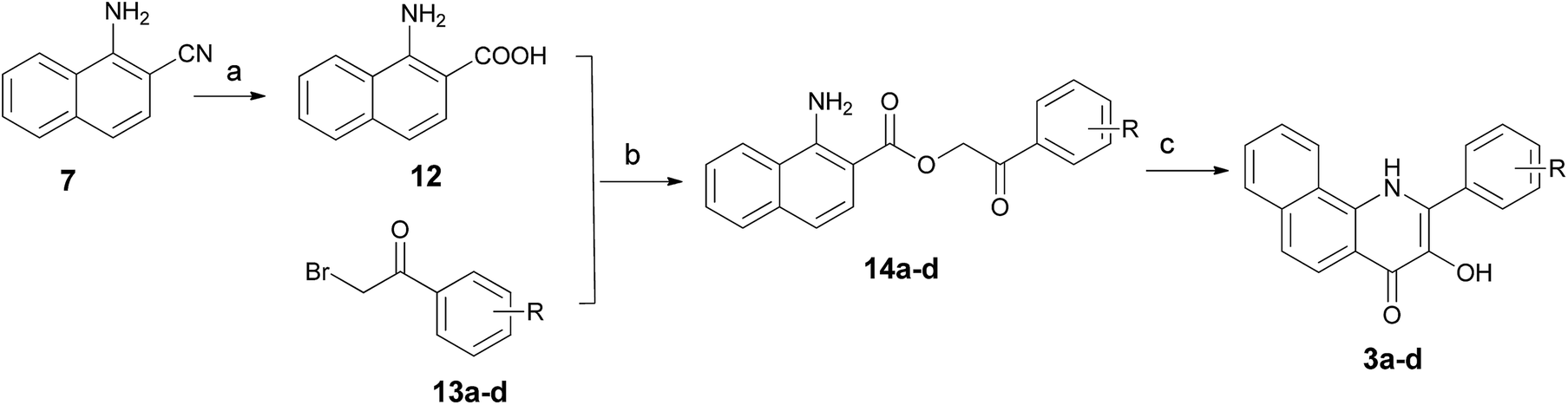 | ||
| Scheme 2 Synthetic routes for compounds 3a–d. Reagents and conditions: (a) NaOH solution (20%), EtOH, reflux, conc. HCl; (b) K2CO3, DMF, 90 °C; (c) PPA, 120 °C. | ||
The 2-arylbenzo[h]quinolin-4(1H)-one series 3e and 5a–j was prepared using the synthetic method described in Scheme 3. Ethyl benzoylacetate derivatives 17a–j as key intermediates were obtained by the reaction of potassium ethyl malonate with different benzoic acids 16a–j in the presence of 1,1′-carbonyl-bis-1H-imidazole (CDI) in an acceptable yield.23 1-Naphthylamine (18) was commercially available and its derivative 20 was prepared effectively from the starting material 19 using our previously reported method.24 The condensation reaction of these naphthylamine derivatives with 17a–j in the presence of PPA provided title compounds in low yields. It is interesting to note that 4a, the isomer of 3e, also could be isolated in high yield during the process for the preparation of 3e. Lastly, methylation of 4a was carried out using methyl iodide under alkaline conditions to give 4b in high yield.
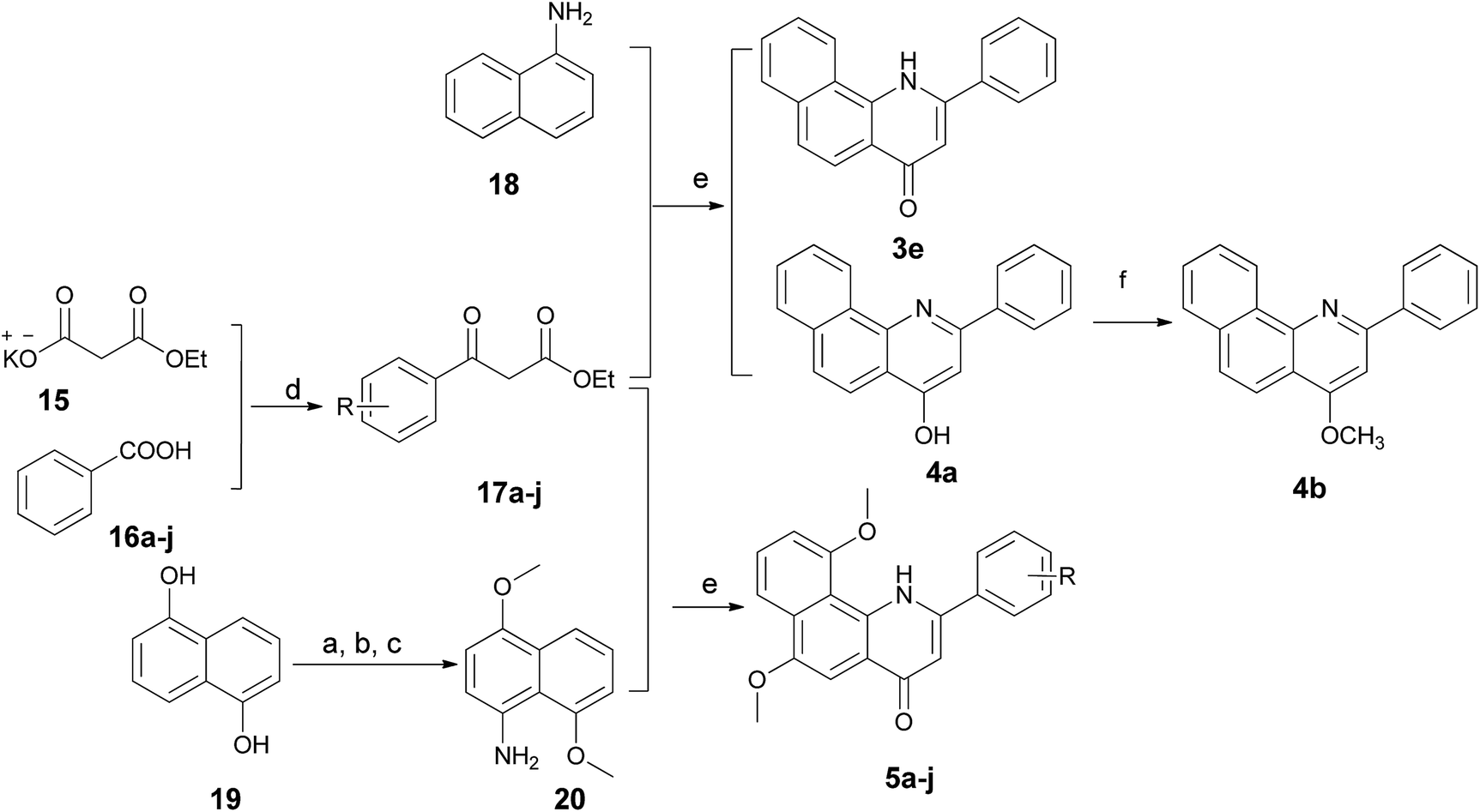 | ||
| Scheme 3 Synthetic routes for compounds 3e, 4a, 4b and 5a–j. Reagents and conditions: (a) (CH3)2SO4, NaOH, THF; (b) HNO3, HOAc; (c) NH2NH2–H2O, Zn; (d) CDI, MgCl2, TEA, THF; (e) PPA, 130 °C; (f) NaH, CH3I, DMF. | ||
2.2 CYP1 enzyme inhibitory activities
The ethoxyresorufin-O-deethylase (EROD) assay, a widely used method for the evaluation of CYP1 activities, was used to determine the inhibitory abilities of these synthesized compounds toward recombinant human CYP1B1, CYP1A1, and CYP1A2 enzymes. Initially, we synthesized several 2-aryl-2,3-dihydrobenzo[h]quinolin-4(1H)-ones and their 3-hydroxy derivatives that were lacking the C2–C3 double bond and/or a hydroxyl group at the C3 position on the C-ring (1a–d and 2a,b), with the view that this would possibly improve the poor water solubility of ANF by reducing its molecular planarity. The results are presented in Table 1. Generally, it was observed that all compounds displayed selective inhibition of CYP1B1 over CYP1A1 and CYP1A2, which was consistent with our expectation. However, all of the tested compounds showed moderate to poor inhibitory activities toward CYP1s, and their inhibitory activities decreased dramatically compared to that of ANF. Among them, 1b was identified as the most potent compound in inhibiting CYP1B1 and CYP1A1, with IC50 values of 20.2 and 44.6 nM respectively, but was much less effective in inhibiting CYP1A2. However, addition of a hydroxyl group at the C3 position (2b) dramatically reduced the inhibitory effects on CYP1B1 and CYP1A1. On the basis of the results above, we presume that the double bond at the C2–C3 position may be an indispensable structural feature for inhibitory action, while the hydroxyl group at the C3 position is an unfavorable structural characteristic for exerting an inhibitory effect on CYP1s.
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R | IC50 values (nM) | IC50 ratio | |||
| 1B1 | 1A1 | 1A2 | 1A1/1B1 | 1A2/1B1 | |||
| 1a | H | H | 131.4 | 404.6 | 952.8 | 3.1 | 7.3 |
| 1b | H | 3,4,5-TriOCH3 | 20.2 | 44.6 | >5000 | 2.2 | >247.5 |
| 1c | H | o-F | 139.3 | 577.5 | 1097 | 4.1 | 7.9 |
| 1d | H | m-F | 98.6 | 493.2 | 1045 | 5.0 | 10.6 |
| 2a | OH | H | 259.6 | 587.9 | 1411 | 2.3 | 5.4 |
| 2b | OH | 3,4,5-TriOCH3 | 302.5 | 1161 | >5000 | 3.8 | >16.5 |
| ANF | — | — | 5.6 | 62.8 | 14.1 | 11.2 | 2.5 |
Next we prepared and evaluated a series of 3-hydroxy-2-arylbenzo[h]quinolin-4(1H)-ones 3a–d and a 2-arylbenzo[h]quinolin-4(1H)-one (3e) with a C2–C3 double bond. As can be appreciated from Table 2, the inhibitory activities of compounds 3a–d against CYP1B1 were significantly higher than those against CYP1A2 (IC50 > 5000 nM). Also, they poorly inhibited the catalytic activity of CYP1A1. It seems that substituents on the B-ring exert no obvious effect on CYP1 inhibition, since compounds 3b–d displayed similar potencies against the CYP1s to that of 3a. It was noteworthy that 3a, with a double bond introduced at the C2–C3 position, was 3 times more potent than 2a in the CYP1B1 enzymatic inhibition assay. Compound 3e, a 3-dehydroxylated compound of 3a, exhibited an increased ability to inhibit CYP1B1, with an IC50 value of 17.3 nM, and is about 5–8 times more potent than 3a and 1a. However, comparison of the activities of 3e and ANF against the CYP1s showed that replacement of the oxygen atom in ANF with a nitrogen atom in 3e led to a loss of inhibitory potency. This result may be explained by the fact that introduction of a nitrogen atom resulted in a reduced planarity of the backbone of ANF. Interestingly, compared with ANF this compound had an excellent selectivity profile, as it was 27.9-fold selective for CYP1B1 over CYP1A2, suggesting that 3e is a promising lead compound for the further development of selective and potent CYP1B1 inhibitors. Together with the data presented in Tables 1 and 2, it can be concluded that the C2–C3 double bond contributes to improving the inhibitory efficiency toward CYP1 enzymes, while the C3-hydroxyl group results in a loss in potency. To our surprise, compound 4a as a chemical isomer of 3e could strongly inhibit the activities of CYP1B1 and CYP1A1 with IC50 values of 7.6 and 18.5 nM respectively, and was 2 and 5 times more potent than 3e. Additionally, it showed 19.3-fold selectivity for CYP1B1 over CYP1A2 (with an IC50 value of 146.9 nM for CYP1A2). However, when this hydroxyl group was methylated, the obtained compound 4b was no longer a CYP1 inhibitor since its concentration-dependent activity was not observed. It can be speculated that the hydroxyl group may form an essential hydrogen bond with other residue(s).
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R | IC50 values (nM) | IC50 ratio | |||
| 1B1 | 1A1 | 1A2 | 1A1/1B1 | 1A2/1B1 | |||
| a NI = no inhibition. | |||||||
| 3a | OH | H | 86.7 | 897.7 | >5000 | 10.4 | >57.7 |
| 3b | OH | o-F | 85.9 | 1783 | >5000 | 20.8 | >58.2 |
| 3c | OH | p-OH | 141.7 | 525.1 | >5000 | 3.7 | >35.3 |
| 3d | OH | p-F | 93.3 | 333.5 | >5000 | 3.6 | >53.6 |
| 3e | H | H | 17.3 | 103.2 | 483.5 | 6.0 | 27.9 |
| 4a | OH | — | 7.6 | 18.5 | 146.9 | 2.4 | 19.3 |
| 4b | OCH3 | — | NIa | NI | NI | — | — |
The crystal structure of human CYP1B1 with ANF disclosed that π–π stacking between the naphthalene part of ANF and Phe231 plays an influential role in maintaining the high binding affinity of ANF with human CYP1B1.16 Considering that enhancing the electron density of the naphthalene part will intensify the π–π stacking interaction with Phe231, a series of 6,10-dimethoxy-2-arylbenzo[h]quinolin-4(1H)-ones were synthesized. The results are summarized in Table 3. In general, it was observed that the potency loss caused by replacing the oxygen atom in ANF with a nitrogen atom was able to be partially or completely restored by introducing two methoxy groups on the naphthalene moiety. The resulting inhibitors (5a–j) suppressed the CYP1 activities in a substituent-dependent manner. Consistent with our previous results, all of the inhibitors except 5a, 5b and 5d showed high selectivity toward CYP1B1 over CYP1A1 and CYP1A2. Among them, compounds 5e and 5g exhibited the highest potency for inhibition of CYP1B1 with IC50 values of 3.6 and 3.9 nM respectively, and were more potent than ANF. Furthermore, these two compounds also displayed very high selectivity toward CYP1B1 over CYP1A2 (more than 163 and 540 times higher than those observed for CYP1A2, respectively), more than 65 and 216 times higher than that of ANF. Besides, compounds 5c, 5f and 5h displayed similar potencies and notable selectivity against CYP1B1 to those of 5e and 5g with IC50 values of 7.8, 7.1 and 4.1 nM, respectively. Conversely, when three methoxy groups were introduced into the B-ring, the resulting compound 5j suffered dramatic potency losses in CYP1 inhibitory activities. This might be due to the fact that the substituted groups can form steric clashes which affect 5j binding to the active site cavities of CYP1 enzymes with a suitable conformation.
|
||||||
|---|---|---|---|---|---|---|
| Compound | R | IC50 values (nM) | IC50 ratio | |||
| 1B1 | 1A1 | 1A2 | 1A1/1B1 | 1A2/1B1 | ||
| 5a | H | 16.6 | 12.6 | 147.6 | 0.8 | 8.9 |
| 5b | o-F | 27.9 | 26.4 | 174 | 0.9 | 6.2 |
| 5c | m-F | 7.8 | 39.3 | 330.9 | 5.0 | 42.4 |
| 5d | m-Cl | 27.6 | 29.4 | 84.5 | 1.1 | 3.1 |
| 5e | m-Br | 3.6 | 19.8 | 587.6 | 5.5 | 163.2 |
| 5f | m-OCH3 | 7.1 | 35.8 | 190.1 | 5.0 | 26.8 |
| 5g | p-F | 3.9 | 10.5 | 2107 | 2.7 | 540.3 |
| 5h | p-OCH3 | 4.1 | 64.7 | >5000 | 15.8 | >1219.5 |
| 5i | p-CF3 | 34.4 | 300.4 | 1337 | 8.7 | 38.9 |
| 5j | 3,4,5-TriOCH3 | 163.9 | 809 | >5000 | 4.9 | >30.5 |
As shown in Fig. 2, on the basis of the inhibitory potency and selectivity of the aforementioned 2-arylbenzo[h]quinolones as CYP1B1 inhibitors, we could draw the following conclusions related to their structure–activity relationships (SARs): (1) introduction of methoxy groups at C-6 and C-10 on the naphthalene ring greatly increased the inhibitory potency towards CYP1B1; (2) the double bond between C-2 and C-3 contributed to the CYP1B1 inhibitory efficiency; (3) the nitrogen atom in the C-ring caused detrimental effects to the CYP1B1 inhibitory efficiency, but the presence of this atom had somewhat positive effects towards water solubility; (4) introduction of the C-3 hydroxyl group led to a great loss in the potency of CYP1 inhibition; (5) the nature of the substituents on the B-ring also has a strong influence on both CYP1B1 potency and selectivity. Balancing all of the above factors, we have chosen 4a and 5e for further molecular docking investigations.
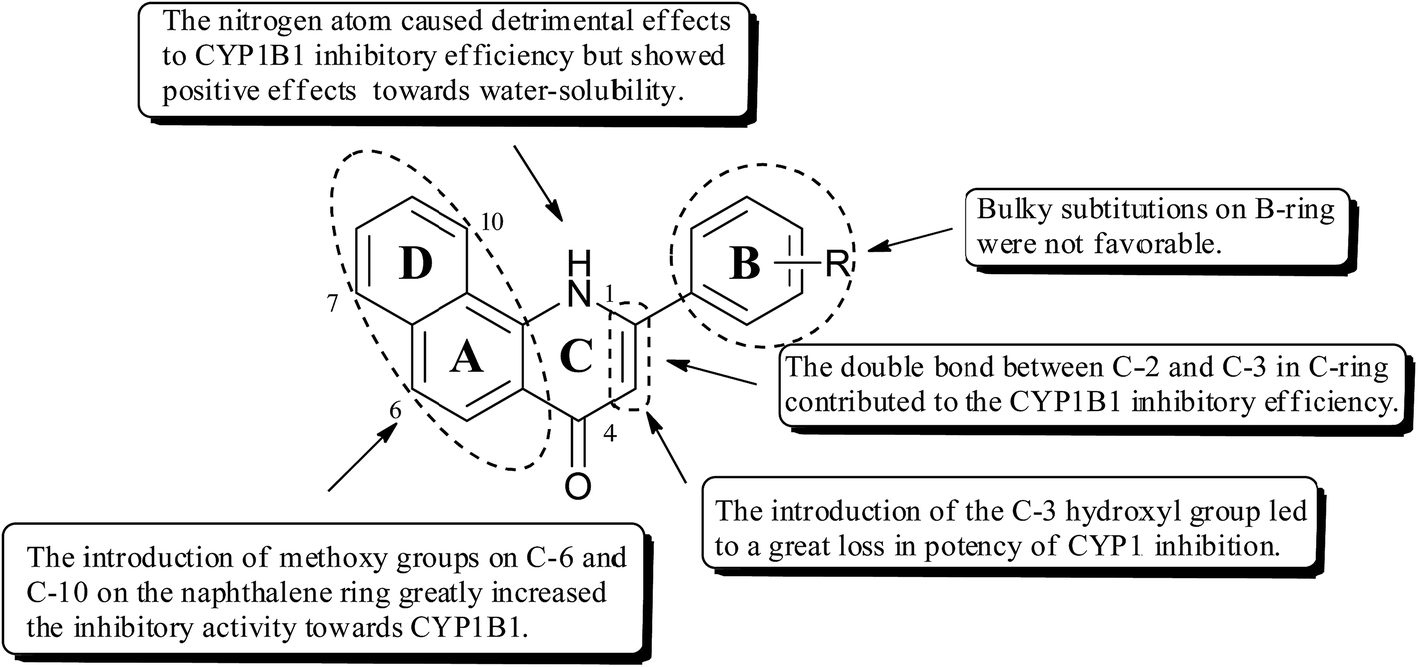 | ||
| Fig. 2 The SARs for the 2-arylbenzo[h]quinolones. | ||
2.3 Molecular docking
To investigate the binding modes and rationalize the observed efficiency and selectivity of CYP1 inhibition by compound 4a and the most potent inhibitor 5e, molecular docking studies were carried out based on the crystal structures 3PM0 and 2HI4, which are from CYP1B1 and CYP1A2 complexed with α-naphthoflavone. These two crystal structures were obtained from the Protein Data Bank (RCSB PDB). As shown in Fig. 3(A and B), compound 4a tightly fitted the active site of CYP1B1, and the naphthalene part of this molecule was covered by the phenyl ring of Phe231. In addition to this π–π stacking interaction, two hydrogen bonds formed by the hydroxyl group of 4a and Asp326 (with lengths of 1.7 and 2.4 Å, respectively) provided an enhanced binding interaction. Also, it offered a reasonable explanation for the phenomenon that methylation of this hydroxyl group led to a dramatically decreased binding affinity of compound 4b with CYP1B1. However, when 4a was bound to CYP1A2, no hydrogen bond was formed and its binding affinity arose mainly from the π–π stacking interaction with Phe226.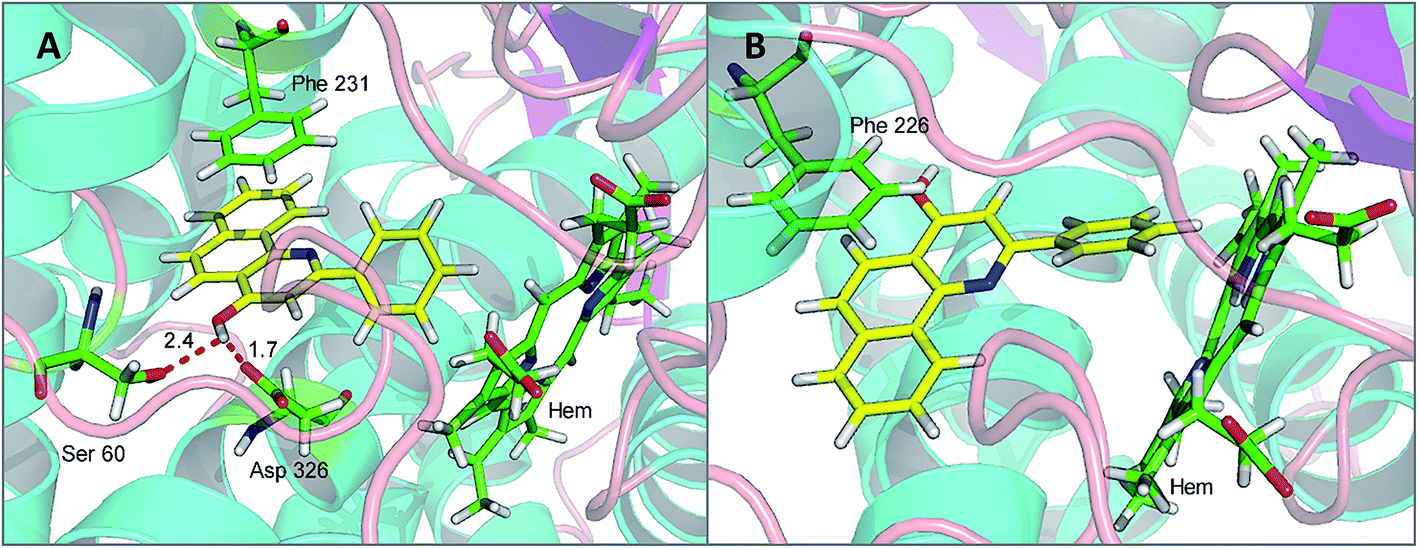 | ||
| Fig. 3 The binding models: compound 4a with CYP1B1 (A) and CYP1A2 (B). | ||
Compound 5e, a potent CYP1B1 inhibitor, exhibited excellent selectivity over CYP1A2. Molecular docking provided a plausible explanation for this observation. The results suggested that the binding affinity of this compound to CYP1B1 is much higher than that to CYP1A2 (Fig. 4(A and B)). A hydrogen bond was formed between the carbonyl group of 5e and Asp326 of CYP1B1 with a distance of 3.0 Å. Additionally, the alkaline nitrogen atom at the 1-position was adjacent to the acidic amino acid Asp333 with a short distance of 3.6 Å, which may play an essential role in improving its binding affinity to CYP1B1. Besides, the π–π stacking interaction between 5e and Phe231 also contributed to this binding affinity. However, in the case of CYP1A2, the π–π stacking interaction between 5e and Phe226 appeared to be the only direct interaction.
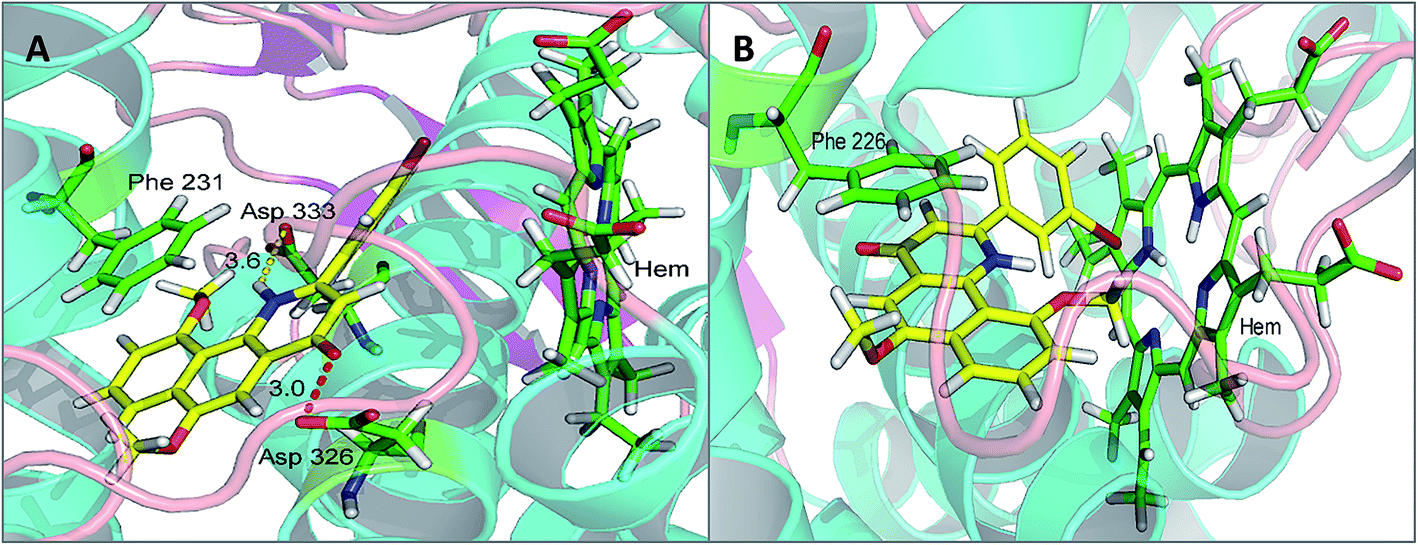 | ||
| Fig. 4 The binding models: compound 5e with CYP1B1 (A) and CYP1A2 (B) (hydrogen bonding is depicted by red dashed lines, whereas yellow dashed lines represent distance only). | ||
2.4 Reversal of drug-resistance in MCF-7 cells
Docetaxel as an effective chemotherapeutic agent has been extensively applied for the treatment of solid tumors in clinics. Nevertheless, after a positive initial response to treatment with these chemotherapeutic drugs, a large proportion of patients relapse. This event is mainly attributed to the intrinsic or acquired drug resistance that greatly compromises the efficacy of anticancer drugs. Previous studies demonstrated that acquired resistance to docetaxel is due in particular to the overexpression of the CYP1B1 enzyme in cancer cells.25 Taking into account the fact that the level of CYP1B1 expression is higher in hormone-dependent tissues such as those of the uterus, ovary and breast,26 MCF-7 cells were chosen as a model in which CYP1B1 expression was studied. In addition, early research clearly indicated its cytotoxicity can be restored by co-incubation of the CYP1B1-overexpressing cells with the CYP1B1 inhibitor ANF.14 Stimulated by this finding, we selected six potent inhibitors 4a, 5e, 5g, 5c, 5f and 5h to evaluate their potential to reverse docetaxel resistance in MCF-7 cells.It is well known that CYP1B1 expression is substantially induced by exposure of the MCF-7 cell line to TCDD.27,28 According to the reported methods and our previously presented study, CYP1B1-expressing MCF-7 cells (defined as MCF-7/1B1) could be generated by treatment of parental MCF-7 cells with a low concentration of TCDD for several days. As a result, an enhanced expression of CYP1B1 protein is readily measurable in MCF-7/1B1 cells by western-blot techniques as compared to parental cells.8,27 In this study, MCF-7/1B1 cells were obtained using the same method. Subsequently, the anti-proliferation assay was carried out to confirm whether the cells were resistant to docetaxel. Not surprisingly, the results indicated that MCF-7/1B1 cells were 5-fold more resistant to docetaxel than parental MCF-7 cells (the IC50 values for docetaxel in parental MCF-7 cells and MCF-7/1B1 cells were 29.5 and 158.6 μM, respectively).
Initially, MCF-7/1B1 cells were treated with 20 μM docetaxel alone or in combination with 10 μM CYP1B1 inhibitors to preliminarily evaluate the chemosensitization effects of these selected compounds. A percentage inhibition value for each combination was obtained after testing twice, and the results are summarized in Table 4. Generally, the percentage inhibition values for combinations of docetaxel with a CYP1B1 inhibitor were higher than that for treatment with docetaxel alone. To our surprise, among these tested compounds it was 5h–but not the most potent CYP1B1 inhibitor 5e–that was found to be the most promising compound, since it exhibited the highest ability to restore the cytotoxicity of docetaxel. Next, to further confirm whether this effect was caused by the cytotoxicity of 5e, MCF-7/1B1 cells were exposed to varying concentrations of 5e alone, but a significant antiproliferative activity was not observed. A plausible explanation was that 5h may possess a better water-solubility than 5e, as the predicted clogP value of the former is lower than that of the latter. Finally, the IC50 values of docetaxel in combination with 10 μM 5h in MCF-7/1B1 cells were determined. As described in Fig. 5, compound 5h was shown to be more active than ANF in the reversal of CYP1B1-mediated docetaxel resistance. This may be ascribed to a potent CYP1B1 inhibitory activity and an improved water-solubility. Overall, the co-incubation of MCF-7/1B1 cells with 10 μM 5h caused a complete reversal of docetaxel-resistance, since the IC50 value was decreased to 33.9 ± 5.1 μM, which was comparable to that in the parental MCF-7 cells.
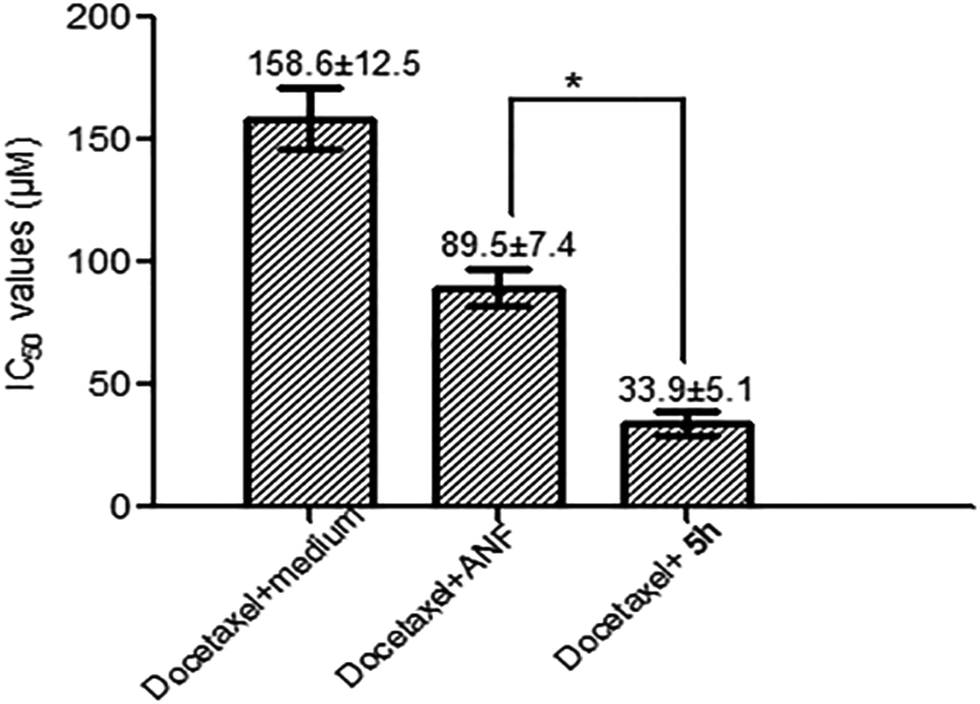 | ||
| Fig. 5 Reversal of docetaxel resistance in combination with 10 μM 5h in MCF-7/1B1 cells. IC50 values of docetaxel are shown as the mean (± SD) of three independent measurements. Significant differences from IC50 values are marked with an asterisk (P < 0.01). | ||
3 Experimental
3.1 Chemistry: general procedure
Chemicals and solvents were used as purchased from commercial suppliers. All anhydrous solvents were dried according to standard methods. Column chromatography was conducted on silica gel (200–300 mesh) from Qingdao Ocean Chemical Factory. The 1H and 13C NMR spectra were obtained on a Varian Mercury-300 (400 MHz) spectrometer using DMSO-d6, CDCl3 and CD3OD as solvents and TMS as an internal standard. All chemical shifts are given in ppm and coupling constants are given in Hz. Mass spectra were recorded with HRMS (ESI, Agilent). Reaction progress was monitored by thin-layer chromatography TLC (silica gel GF254) and visualized with UV light (254 or 365 nm). The purity of compounds was determined with an Agilent 1260 HPLC system (column: Eclipse plus C18, 3.5 μm, 4.6 × 100 mm (Agilent); flow rate: 0.8 ml min−1; mobile phase: A: MeOH, B: H2O).3.2 Enzyme assays
The recombinant human CYP1B1, CYP1A1, and CYP1A2 enzymes, each equipped with P450 reductase (Supersomes), were purchased from BD Genetest. 7-Ethoxyresorufin (7-ER) was obtained from Sigma-Aldrich. NADP+, D-glucose-6-phosphate (G-6-P) and glucose-6-phosphate dehydrogenase (G-6-PD) were purchased from Biosharp. Other solvents and reagents used in the biological evaluation were of the highest quality and commercial availability. In the enzyme assay, FlexStation 3 apparatus was used for recording the fluorescence intensity with excitation and emission filters at 544 and 590 nm, respectively.The inhibitory activities of these prepared compounds against CYP1B1, CYP1A1, and CYP1A2 enzymes were determined using the EROD assay as reported earlier.29 All tested compounds were dissolved as stock solutions (10 mM) in DMSO and diluted to working solutions with a Tris–HCl buffer at pH 7.4, and the final concentration of organic solvent was <1% (v/v) in all cases. Next, a mixture with a final volume of 200 μl containing different concentrations of tested compounds (except positive and negative control wells), an enzyme source (20 fmol CYP1B1, 10 fmol CYP1A1 and 60 fmol CYP1A2), 150 nM 7-ER, 1.3 mM NADP+, 3.3 mM G-6-P, 0.5 U ml−1 G-6-PD and 3.3 mM MgCl2 was incubated in a black 96-well flat-bottomed microplate at 37 °C for different times (incubation time for CYP1B1, CYP1A1, and CYP1A2 was 35, 15 and 50 min, respectively). Finally, the reaction was stopped by the addition of 100 μl of methanol to all wells. The IC50 value for each compound was calculated with the GraphPad Prism Software (Version 5.0) using the non-linear regression formula: log (inhibitor) vs. normalized response-variable slope.
3.3 Cancer cell growth inhibition assays
RPMI-1640 culture medium, fetal bovine serum (FBS), and PBS (Hyclone) were purchased from Thermo Fisher Scientific. TCDD was obtained from AccuStandard. The MCF-7 cell line was cultured in RPMI 1640 medium with 10% FBS at 37 °C in a humidified atmosphere with 5% CO2. MCF-7/1B1 cells were obtained by coincubation of parental MCF-7 cells with TCDD (10 nM) for five days. The in vitro cytotoxicity was determined by combination of docetaxel with CYP1B1 inhibitors using the standard MTT assay.30,31 Approximately 3000 cells, suspended in RPMI 1640 medium, were seeded into each well of a 96-well plate and incubated at 37 °C in a humidified atmosphere with 5% CO2 for 24 h. Then, six different concentrations of docetaxel with or without 10 μM CYP1B1 inhibitors were added. Absorbance values were determined by a microplate reader at 490 nm, and the IC50 values were calculated with the GraphPad Prism Software (Version 5.0) using the non-linear regression formula: log (inhibitor) vs. normalized response-variable slope. Student’s t tests were performed to compare their IC50 values using the PASW Statistics 18 software.3.4 Molecular docking
In order to understand the binding mode and selectivity of the title compounds with both CYP1B1 and CYP1A2, we performed a molecule docking experiment using the docking program in MOE 2008 based on the crystal structures of CYP1B1 (PDB ID: 3PM0) and CYP1A2 (PDB ID: 2HI4). The initial 3D conformation of compounds 4a and 5e were optimized in ChemBio3D Ultra using the MM2 energy minimization method. All molecules were removed from the crystal structures before the experiment. Hydrogens and partial charges were added with the protonate 3D application. The residues within a radius of 8.0 Å around the ligands were selected as docking sites. Docking parameters were kept at default, except for the first scoring function, where ASE Scoring was used instead of the default London dG. The best pose was characterized by the scoring results. To test whether the docking program is suitable for the ligands binding to human CYP1B1 and CYP1A2, the CYP1B1-ANF and CYP1A2-ANF complexes (PDB ID: 3PM0 and PDB ID: 2HI4, respectively) were initially chosen as models. The results revealed that the docking compounds with ANF were comparable to their corresponding crystallographic structures. It was implied that the MOE docking program used is suitable for the current study.4 Conclusions
The structural modification of α-naphthoflavone gave new 2-arylbenzo[h]quinolone derivatives, which exhibited varying potency and selectivity for CYP1B1 inhibition. Compared with ANF, replacement of the oxygen atom on the C-ring by a nitrogen atom caused a mild potency loss. However, this decreased inhibitory activity could be restored by enhancing the electron-density of the naphthalene part to intensify the π–π stacking interaction. It is noteworthy that 2-arylbenzo[h]quinolones have shown a significantly increased selectivity toward CYP1B1 over CYP1A2. SAR analysis indicated that the double bond at the C2–C3 position is an indispensable structural feature for inhibitory action, while the hydroxyl group at the C3 position is an unfavorable structural characteristic. A molecular docking study disclosed that the most potent compound 5e tightly fitted the binding cavity of CYP1B1; hydrogen bonds, hydrophobic interactions and π–π stacking interaction contribute to this high binding affinity, while these interactions were markedly reduced when it bound to CYP1A2. On the basis of the predicted clogP values, these target compounds may exhibit an improved water-solubility compared to ANF. Among them, 5h was found to be a potential compound to reverse CYP1B1-mediated drug resistance, since the cytotoxicity of docetaxel could be restored by pretreating MCF-7/1B1 cells with 5h at 10 μM. All of these results indicated that further development of these 2-arylbenzo[h]quinolones as more potent and specific CYP1B1 inhibitors may result in the discovery of promising candidates to reverse CYP1B1-mediated anticancer-drug resistance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No. 21602132), the Shanghai Science and Technology Innovation Program (No. 15431900700) and the Shanghai Natural Science Fund (No. 16ZR1418100). Jinyun Dong also gratefully acknowledges the help and encouragement of Haixia Zhang.Notes and references
- J. Dong, Q. Zhang, Q. Cui, G. Huang, X. Pan and S. Li, ChemMedChem, 2016, 11, 2102–2118 CrossRef CAS PubMed.
- H. Doostdar, M. D. Burke and R. T. Mayer, Toxicology, 2000, 144, 31–38 CrossRef CAS PubMed.
- J. Mimura and Y. Fujii-Kuriyama, Biochim. Biophys. Acta, 2003, 1619, 263–268 CrossRef CAS.
- T. Muto, T. Watanabe, M. Moto, M. Okamura, Y. Kashida, Y. Kanai, K. Mitsumori and H. Endou, J. Toxicol. Pathol., 2003, 16, 287–290 CrossRef CAS.
- B. N. Zordoky and A. O. El-Kadi, Toxicol. In Vitro, 2010, 24, 863–871 CrossRef CAS PubMed.
- A. J. Lee, M. X. Cai, P. E. Thomas, A. H. Conney and B. T. Zhu, Endocrinology, 2003, 144, 3382–3398 CrossRef CAS PubMed.
- J. G. Liehr and M. J. Ricci, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 3294–3296 CrossRef CAS.
- J. Cui, Q. Meng, X. Zhang, Q. Cui, W. Zhou and S. Li, J. Med. Chem., 2015, 58, 3534–3547 CrossRef CAS PubMed.
- G. I. Murray, M. C. Taylor, M. C. McFadyen, J. A. McKay, W. F. Greenlee, M. D. Burke and W. T. Melvin, Cancer Res., 1997, 57, 3026–3031 CAS.
- R. D. Bruno and V. C. Njar, Bioorg. Med. Chem., 2007, 15, 5047–5060 CrossRef CAS PubMed.
- M. C. E. McFadyen, M. E. Cruickshank, I. D. Miller, H. McLeod, W. T. Melvin, N. E. Haites, D. Parkin and G. I. Murray, Br. J. Cancer, 2001, 85, 242–246 CrossRef CAS PubMed.
- L. H. Patterson and G. I. Murray, Curr. Pharm. Des., 2002, 8, 1335–1347 CrossRef CAS PubMed.
- T. Shimada, H. Yamazaki, M. Foroozesh, N. E. Hopkins, W. L. Alworth and F. P. Guengerich, Chem. Res. Toxicol., 1998, 11, 1048–1056 CrossRef CAS PubMed.
- M. C. McFadyen, H. L. McLeod, F. C. Jackson, W. T. Melvin, J. Doehmer and G. I. Murray, Biochem. Pharmacol., 2001, 62, 207–212 CrossRef CAS PubMed.
- A. Datta, N. Bhasin, H. Kim, M. Ranjan, B. Rider, Z. Y. A. Elmageed, D. Mondal, K. C. Agrawal and A. B. Abdel-Mageed, Cancer Lett., 2015, 362, 25–35 CrossRef CAS PubMed.
- A. Wang, U. Savas, C. D. Stout and E. F. Johnson, J. Biol. Chem., 2011, 286, 5736–5743 CrossRef CAS PubMed.
- S. Sansen, J. K. Yano, R. L. Reynald, G. A. Schoch, K. J. Griffin, C. D. Stout and E. F. Johnson, J. Biol. Chem., 2007, 282, 14348–14355 CrossRef CAS PubMed.
- A. A. Walsh, G. D. Szklarz and E. E. Scott, J. Biol. Chem., 2013, 288, 12932–12943 CrossRef CAS PubMed.
- Y. Tomioka, K. Ohkubo and M. Yamazaki, Chem. Pharm. Bull., 1985, 33, 1360–1366 CrossRef CAS.
- W. Zhang, K. F. Koehler, B. Harris, P. Skolnick and J. M. Cook, J. Med. Chem., 1994, 37, 745–757 CrossRef CAS PubMed.
- R. Bhattacharya, P. Kundu and G. Maiti, Synth. Commun., 2010, 40, 476–481 CrossRef CAS.
- J. A. Donnelly and D. F. Farrell, J. Org. Chem., 1990, 55, 1757–1761 CrossRef CAS.
- K. Machida, Y. Hirose, S. Fuse, T. Sugawara and T. Takahashi, Chem. Pharm. Bull., 2010, 58, 87–93 CrossRef CAS PubMed.
- J. Dong, Q. Zhang, G. Huang, Q. Meng and S. Li, Russ. J. Gen. Chem., 2017, 87, 837–841 CrossRef CAS.
- I. Chang, Y. Mitsui, S. Fukuhara, A. Gill, D. K. Wong, S. Yamamura, V. Shahryari, Z. L. Tabatabai, R. Dahiya and D. M. Shin, Oncotarget, 2015, 6, 7774–7787 Search PubMed.
- D. C. Spink, C. L. Hayes, N. R. Young, M. Christou, T. R. Sutter, C. R. Jefcoate and J. F. Gierthy, J. Steroid Biochem. Mol. Biol., 1994, 51, 251–258 CrossRef CAS PubMed.
- D. C. Spink, B. C. Spink, J. Q. Cao, J. A. DePasquale, B. T. Pentecost, M. J. Fasco, Y. Li and T. R. Sutter, Carcinogenesis, 1998, 19, 291–298 CrossRef CAS PubMed.
- V. Martinez, R. O’connor, Y. Liang and M. Clynes, Br. J. Cancer, 2008, 98, 564–570 CrossRef CAS PubMed.
- S. Yamaori, M. Kushihara, I. Yamamoto and K. Watanabe, Biochem. Pharmacol., 2010, 79, 1691–1698 CrossRef CAS PubMed.
- W. Zhou, Y. Peng and S. Li, Eur. J. Med. Chem., 2010, 45, 6005–6011 CrossRef CAS PubMed.
- A. Regielfutyra, M. Kusliśkiewicz, V. Sebastian, S. Irusta, M. Arruebo, A. Kyzioł and G. Stochel, RSC Adv., 2017, 7, 52398–52413 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00465j |
| This journal is © The Royal Society of Chemistry 2018 |