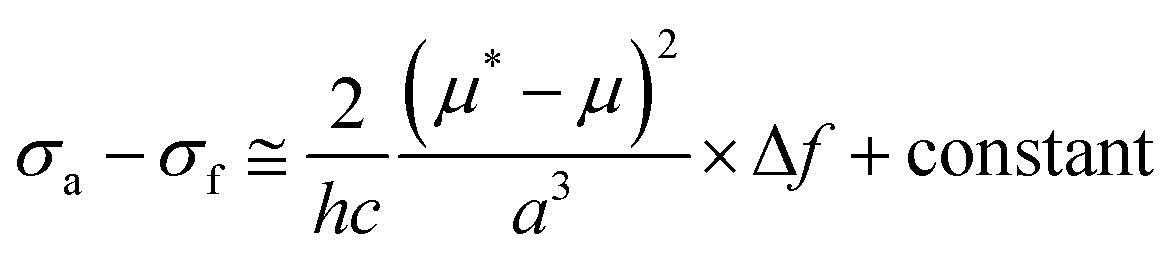DOI:
10.1039/C8QM00184G
(Research Article)
Mater. Chem. Front., 2018,
2, 1498-1507
The unusual aggregation-induced emission of coplanar organoboron isomers and their lipid droplet-specific applications†
Received
24th April 2018
, Accepted 28th May 2018
First published on 30th May 2018
Abstract
Luminogens with aggregation-induced emission (AIE) characteristics generally possess twisted structures to prevent emission quenching in the solid state by π–π stacking interactions. In this work, new organoboron derivatives with coplanar structures were synthesized and found to be AIE-active. Analysis by photoluminescence spectroscopy, single crystal X-ray diffraction and theoretical calculations depicted that the distinct luminescence behaviors of the molecules stemmed from the different extent of excited-state double-bond reorganization (ESDBR), which consumed the energy of the excitons through non-radiative pathways. This process was restricted in a highly viscous medium or in the solid state to enable the molecules to emit efficiently. The organoboron derivatives based on the ESDBR mechanism not only provide a new strategy to design coplanar AIE luminogens but also act as biological probes excellently and specifically targeting lipid droplets.
Introduction
Luminescent materials have attracted much attention because of their potential applications in optics, electronics and biomedicine science and engineering. Traditional organic luminophores often show efficient light emission in the solution state. However, in the solid state, they tend to suffer strong intermolecular π–π stacking interactions due to their planar aromatic structures to cause emission annihilation.1 Such a phenomenon is known as aggregation-caused quenching (ACQ) and has limited their applications in fields such as optoelectronic devices, bio/chemo sensors and biological imaging.2 Luminogens with aggregation-induced emission (AIE) characteristics show dim or no emission in dilute solutions but strong fluorescence in the solid state.3,4 Thus, AIE is the exact opposite of ACQ. Unlike ACQ luminogens, AIE luminogens (AIEgens) normally possess twisted structures to prevent detrimental π–π stacking interactions in the solid state.5–10 The mechanism of restriction of intramolecular motions has been proposed to explain the AIE effect11 but other causes such as twisted intramolecular charge transfer (TICT),12–14E/Z isomerization (EZI),15,16 excited-state intramolecular proton transfer,17 vibration-induced emission,18 and block of nonradiative decay channels19 have been used to explain the AIE phenomenon of other systems. Thanks to the enthusiastic efforts of scientists, different types of AIEgens have been developed and some of them even carry a planar structure (Scheme S1, in the ESI†).20–27 However, why these molecules are AIE-active is rarely investigated. On the other hand, compared with twisted molecules, those with coplanar structures show tight molecular packing to impart higher charge mobility. If they are AIE-active, they may provide competitive applications in fluorescence imaging and light emitting diodes. Deciphering their working mechanism will further assist molecular design of new AIEgens for high technology applications.
Lipid droplets (LDs) are well known as the place of energy storage for neutral lipids and are found in the cytoplasm of normal cells like adipocytes, hepatocytes and myocytes. They interact with a variety of cell organelles, including the endoplasmic reticulum, mitochondria and peroxisomes,28 and involve some significantly important bio-functions, such as dynamic regulation of the storage and metabolism of neutral lipids, membrane maintenance, and protein trafficking, maturation and degradation.29 Aberrations in neutral lipid storage of LDs will lead to several common metabolic diseases, including atherosclerosis, fatty liver, obesity and type II diabetes,30 and/or the induction of cancer cells and tissues, especially in cancer stem cells.29 Though biological fluorometric technology has been utilized extensively in detecting LDs, the commercially-available LD probes still have more disadvantages to be overcome, for example poor photostability and specificity for the Nile red probe.31 Thus, the development of highly sensitive and selective fluorescent probes for LDs is quite significant. In some cases, luminescent organoboron materials like BODIPY without special substituents can specifically target LDs directly.32–34
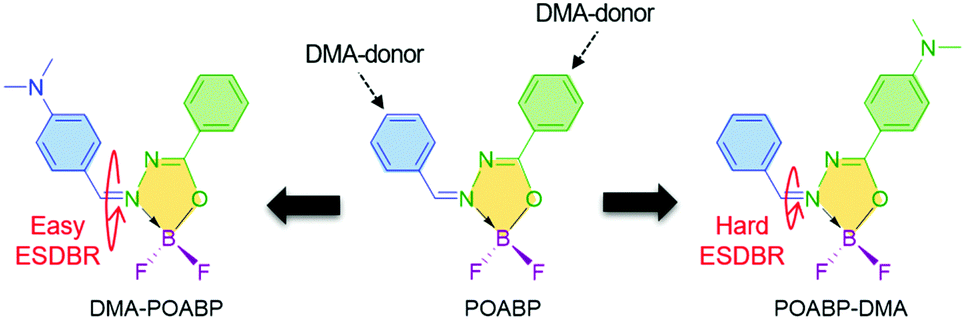
Organoboron materials often possess a coordinated boron center to rigidify the ligand backbone and impart molecular planarity. This decreases the nonradiative relaxation process to enable the molecule to show strong emission with high quantum yield.35–37 Compared with the previous non-planar organoboron AIEgens,5,8,38 we are aware of several interesting features of a heterocyclic organoboron ring named 2,2-difluoro-1,3,4,2-oxadiazaborol-3-ium-2-uide (OAB). OAB (i) is more flexible than an aromatic ring, (ii) adopts a low steric and relatively coplanar structure to favor electronic communication with the nearby groups, and (iii) shows strong electron-withdrawing ability to benefit the construction of a donor (D)–acceptor (A) architecture to regulate its optical properties. By introducing phenyl rings and a dimethylamino (DMA) group to its periphery, coplanar organoboron isomers, abbreviated as DMA-POABP and POABP-DMA, were developed (Scheme 1). The compound called POABP was used for comparison purposes.39
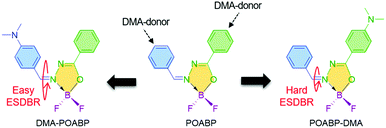 |
| | Scheme 1 Structures of coplanar organoboron luminogens. | |
In this investigation, we showed that the restriction of intramolecular double-bond rotation in the excited state served as the main reason for the AIE phenomenon of these planar AIEgens. Such phenomenon was defined as excited-state double-bond reorganization (ESDBR), which chiefly involved the vibration relaxation of the double bond and EZI in the excited state. It is worth noting that ESDBR is seldom associated with the emission of luminophores in solution. However, we indeed detected and demonstrated its important role for the weak emission of DMA-POABP and strong emission of POABP-DMA in THF solution by NMR analysis, viscosity dependent fluorescence measurement and potential surface energy calculation. The ESDBR was restricted in the aggregated state to enable the coplanar AIEgens to emit intensely. Significantly, ESDBR integrated the motion of the double bond in the excited state and explained the AIE characteristics of rare coplanar AIEgens.
Results and discussion
Synthesis and thermal properties
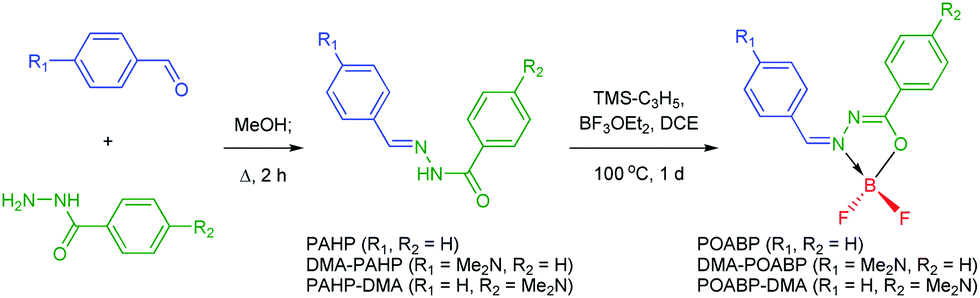
The synthetic protocols for POABP, DMA-POABP and POABP-DMA are illustrated in Scheme 2, and experimental details are provided in the experimental section. The intermediates, PAHP, DMA-PAHP and PAHP-DMA, were synthesized via the condensation between arylaldehyde and aryloyl hydrazine and used without further purification.40,41 After boronation with allyltrimethylsilane and boron trifluoride etherate, POABP, DMA-POABP and POABP-DMA were obtained in 62–80% yield via a two-step organic reaction. Associated structural data of Nuclear Magnetic Resonance (NMR) spectroscopy and high-resolution mass spectrometry (HRMS) are given in the ESI† and Fig. S1–S9.
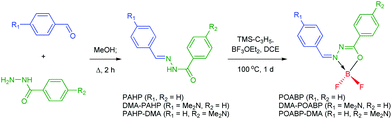 |
| | Scheme 2 Synthetic routes of the organoboron luminogens. | |
Optical properties
The photophysical properties were investigated and the results are given in Fig. 1A, Fig. S10 and S11 (ESI†) and Table 1. The UV spectrum progressively red-shifted from POABP to DMA-POABP and then to POABP-DMA possibly due to the enhancement of the intramolecular charge transfer (ICT) effect. This was further proved by the theoretical calculation data provided in Table S1 (ESI†). Similarly, POABP-DMA showed a red-shifted emission spectrum and larger Stokes shift than POABP and DMA-POABP. The Stokes shift versus the solvent orientation polarizability (Δf) in POABP-DMA was about three times larger than that in DMA-POABP (Fig. 1B and Table S2, ESI†), implying that its optical property was more sensitive to the solvent polarity change. According to the Lippert–Mataga relationship described by the following equation,
where σa − σf is the energy difference between the absorption and emission maxima, μ and μ* are the dipole moments of the molecule in the ground and excited states, respectively, a is the cavity radius in which the fluorophore resides, h is Planck's constant and c is the speed of light. The Stokes shift is ascribed mainly to an ICT process and is consistent with the difference of dipole moment (Δμ) between the ground and excited states (Fig. S12 and Table S3, ESI†). The Δμ value of POABP-DMA (13.6 D) is calculated to be 2.5 times larger than that of DMA-POABP (5.5 D). This indicated that the electron-donating effect was more effective when the aromatic ring near the organoboron was substituted with a DMA group (Fig. S13, ESI†).42 Thus, POABP-DMA showed a more distinct solvatochromism effect (Fig. 1C, D and Fig. S14, ESI†), in which its emission red-shifted from non-polar (465 nm in hexane) to polar solvent (606 nm in DMF), arising from a strong ICT effect. In contrast, DMA-POABP showed only 28 nm of red-shifted emission (455 nm in hexane and 483 nm in DMF).
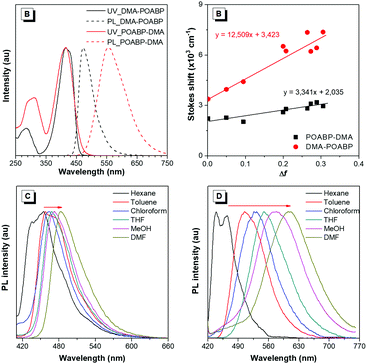 |
| | Fig. 1 (A) Normalized UV-vis absorption and PL spectra of the organoboron compounds in THF (10 μM). (B) Lippert–Mataga plots of Stokes shifts versus the solvent orientation polarizability of DMA-POABP and POABP-DMA. (C and D) Normalized PL spectra of (C) DMA-POABP and (D) POABP-DMA in different solvents. | |
Table 1 Photophysical properties of POABP, DMA-POABP and POABP-DMA
| Luminogen |
THF solutiona |
Solid powder |
α
AIE
|
|
λ
abs (nm) |
λ
em (Φ)b (nm, %) |
τ (kr, knr)c (ns, ns−1, ns−1) |
Δ
THF
(nm) |
E
g
(eV) |
λ
em (Φ) (nm, %) |
τ (kr, knr) (ns, ns−1, ns−1) |
|
Recorded at 298 K (10 μM).
Quantum yield (Φ) measured by a calibrated integrating sphere.
Rate constants for radiative (kr) and nonradiative decay (knr) calculated from Φ and lifetime (τ) values according to kr = Φ/τ and knr = (1 − Φ)/τ.
Δ
THF = Stokes shift = λem − λabs.
Band gap (Eg) calculated from the onset wavelength (λonset) of the absorption spectra, Eg = 1240/λonset.
α
AIE = Φsolid/ΦTHF.
|
| POABP |
338 |
402 (0.7) |
0.70 (0.010, 1.419) |
64 |
3.25 |
435 (1.1) |
0.76 (0.014, 1.301) |
1.6 |
| DMA-POABP |
415 |
473 (0.6) |
0.42 (0.014, 2.367) |
58 |
2.76 |
540 (25.0) |
1.94 (0.129, 0.386) |
41.7 |
| POABP-DMA |
415 |
555 (16.3) |
1.14 (0.143, 0.734) |
140 |
2.55 |
590 (34.9) |
27.4 (0.013, 0.024) |
2.1 |
AIE properties
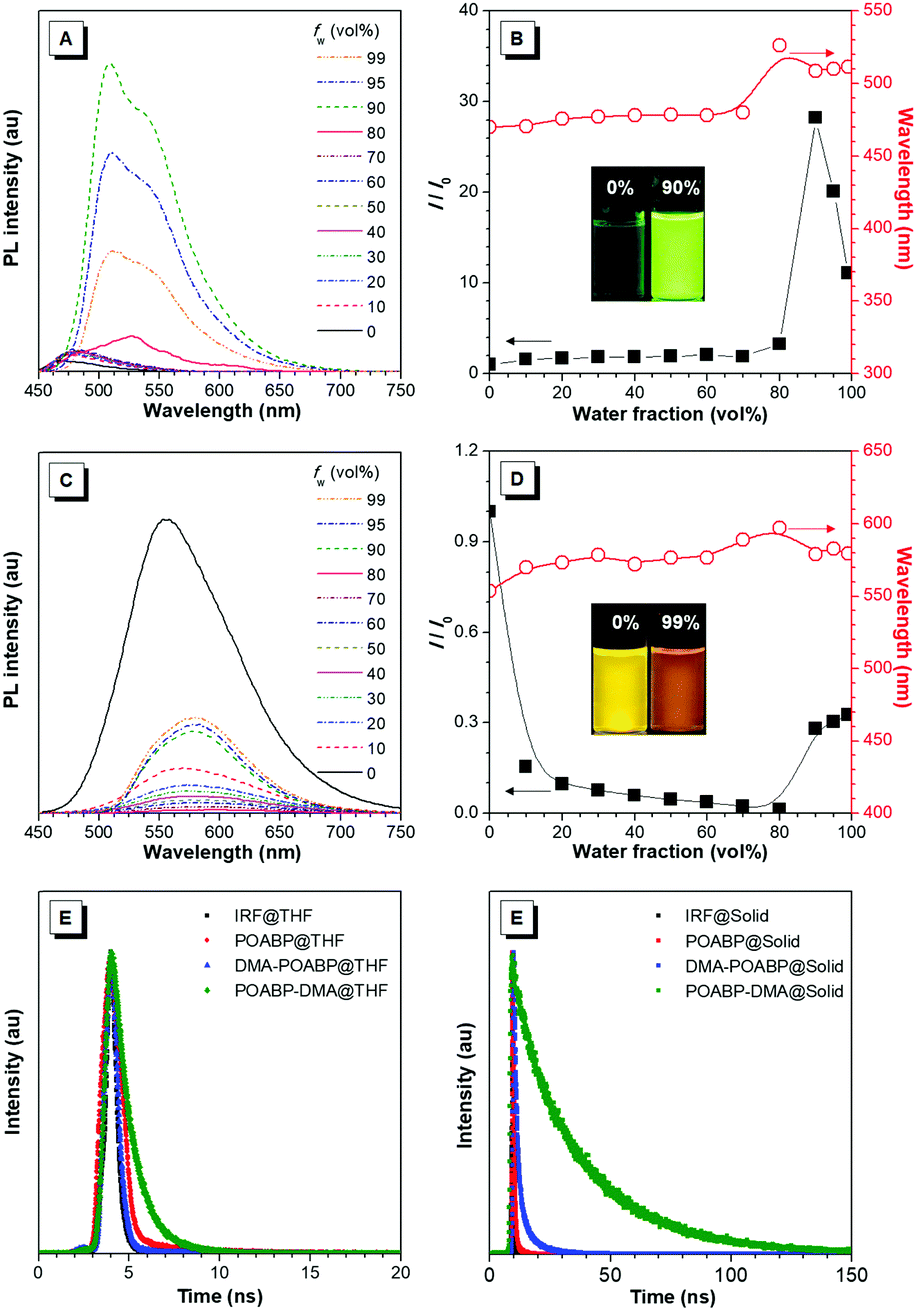
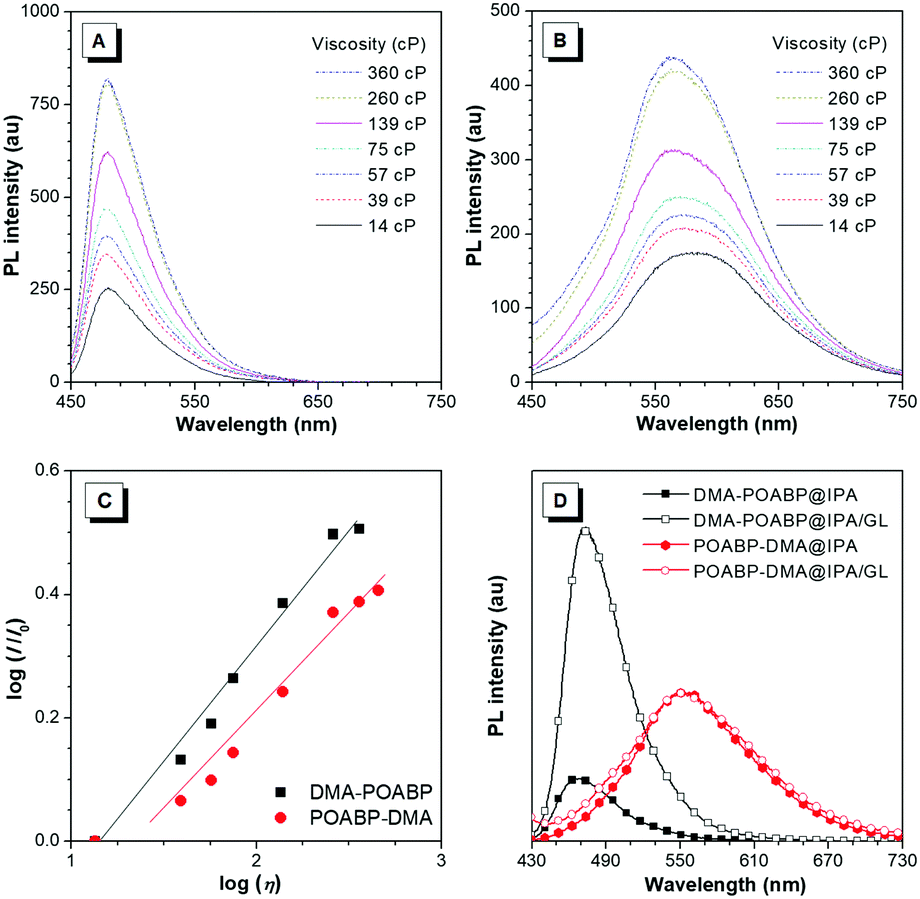
The fluorescence properties of the molecules were further investigated in THF and THF/water mixtures with different water fractions. As shown in Fig. 2A–D, Fig. S15 and S16 (ESI†), DMA-POABP emitted weak blue-green fluorescence in THF at 473 nm, whose intensity gradually increased by ca. 28-fold by gradually increasing the water fraction (fw) from 0 to 90 vol%. Exceeding 90 vol% of fw, the relative intensity (I/I0) declined due to the formation of large particles. This demonstrated a typical AIE behavior. The emission of POABP-DMA became weaker upon addition of water into its THF solution due to a stronger ICT effect but increased slightly at high water fraction owing to the AIE effect. The AIE effect (αAIE) was calculated by dividing the quantum yield of the solid powder (Φsolid) by that of the THF solution (ΦTHF), and was equal to 41.7 and 2.1 for DMA-POABP and POABP-DMA, respectively. (i) Why did DMA-POABP show weak emission but POABP-DMA strong emission in THF and (ii) why did they show stronger emission in the solid state? These will be explained through time-resolved fluorescence spectroscopy, single X-ray diffraction and theoretical calculation.
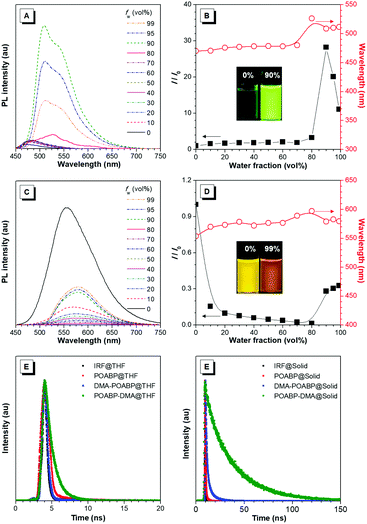 |
| | Fig. 2 (A and C) PL spectra in THF and THF/water mixtures with different water fractions (fw) of (A) DMA-POABP and (C) POABP-DMA. (B and D) Change in relative emission intensity (I/I0) and emission maximum versus the water fraction of the THF/water mixtures of (C) DMA-POABP and (D) POABP-DMA. I0 is the emission intensity in pure THF. (E and F) Fluorescence decay curves of POABP, DMA-POABP and POABP-DMA in (E) THF and (F) the solid state. | |
De-excited route of excited molecules
Time-resolved fluorescence spectra (Fig. 2E and F) revealed that the fluorescence lifetime of DMA-POABP in THF was 0.42 ns, which is shorter than that of POABP-DMA (1.14 ns). In a solid, both of the lifetimes increased to 1.94 and 27.4 ns, respectively. Their radiative and nonradiative decay rates were estimated and listed in Table 1. In THF, the nonradiative decay rate (2.4 × 109 s−1) of DMA-POABP was remarkably larger than the radiative decay rate (1.4 × 107 s−1) by two orders of magnitude, whereas those difference values of POABP-DMA were the same order of magnitude (1.43 × 108 s−1 of krvs. 7.34 × 108 s−1 of knr). However, in a solid, the nonradiative decay rate of DMA-POABP decreased drastically to 3.9 × 108 s−1, which is close to the radiative rate (1.3 × 108 s−1). These results suggest that nonradiative decay is dominant for DMA-POABP in solution, but is weakened to the same level as radiative decay in a solid, i.e., the ratio of nonradiative decay rate to radiative rate in THF was dramatically decreased in comparison with that in the solid state (knr/kr; 169.1 vs. 3.0). Moreover, the ratio for POABP-DMA was also decreased in THF compared to that in the solid state (5.1 vs. 1.8). This implies that the radiative rate becomes a major de-excited pathway in the solid, resulting in more solid-luminescence.
Single crystal analysis
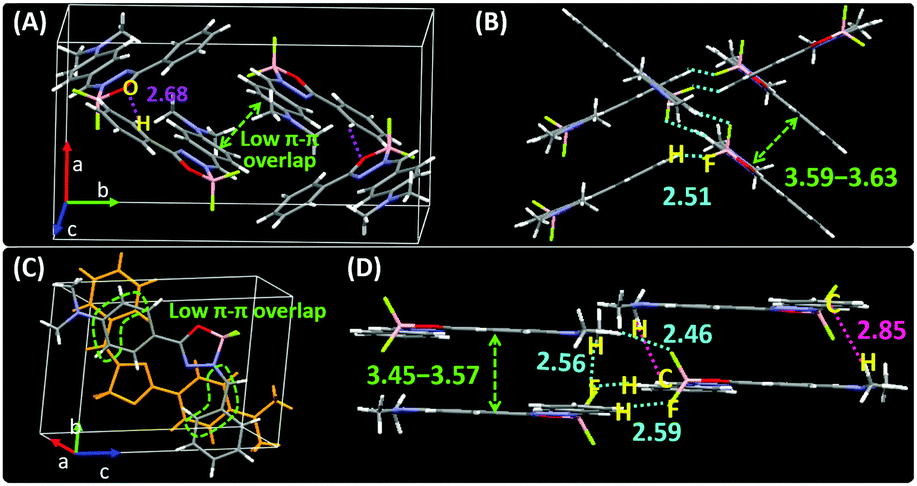
To gain insight into the solid-state emission of the isomers, their crystal structures and data were carefully analyzed (Fig. 3, Fig. S17, S18, Tables S4 and S5, ESI†). The results showed that (i) all the molecules adopted a Z-structure, presumably due to the existence of an intramolecular hydrogen bond (H7⋯N2) with distances in the range of 2.33–2.36 Å, (ii) their overall architectures showed high coplanarity with smaller dihedral angles (only 0.43–6.75°) than any known organoboron materials (Fig. S19, ESI†), (iii) weak intermolecular π–π interactions were observed and (iv) the molecular motions of the molecules were hampered by multiple intermolecular interactions, including short contacts such as C–H⋯F (2.46–2.59 Å), C–H⋯O (2.68 Å) and C–H⋯π (2.85 Å). The collective force of these weak intermolecular interactions could rigidify the molecular conformation in the solid and decrease the nonradiative rate (knr) of energy loss to trigger strong solid-state emission.
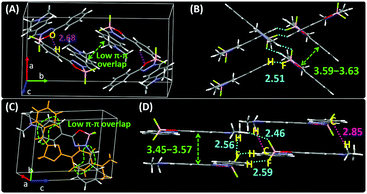 |
| | Fig. 3 XRD crystallographic packing unit and structure viewed laterally of (A and B) DMA-POABP and (C and D) POABP-DMA. | |
Theoretical calculation study
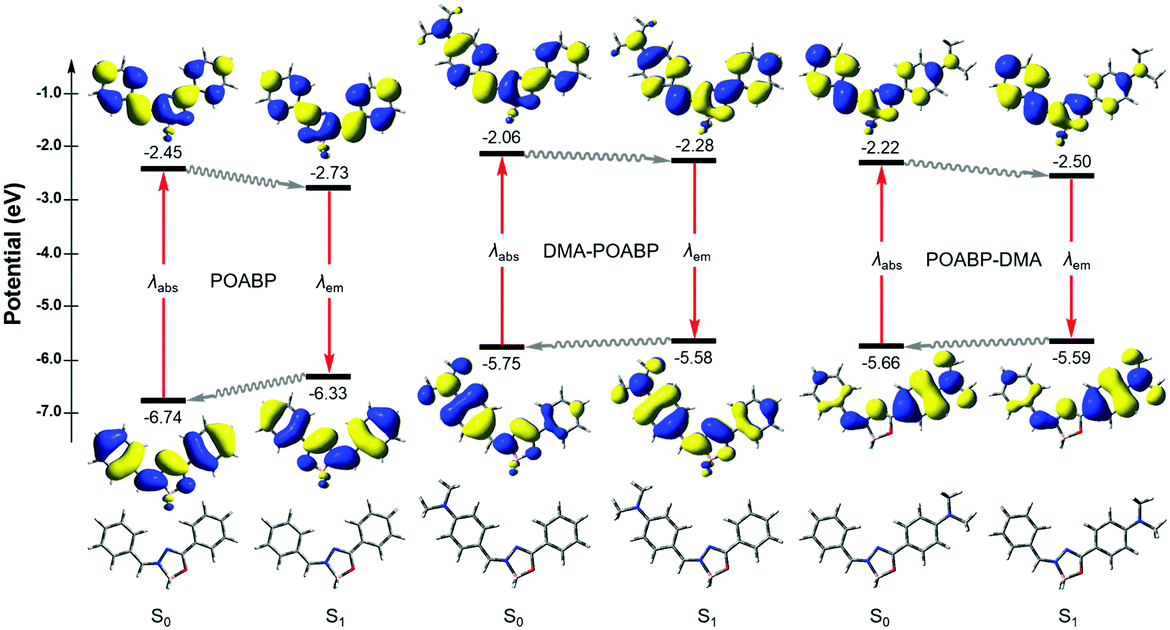
Theoretical calculations can provide detailed mechanistic and dynamical information of the molecules in the ground and excited states. Thus, they are useful in investigating the fluorescence quenching and aggregation-induced emission. On the basis of the theoretical calculations of their optimized molecular geometries, frontier orbitals and electrostatic potential data (Fig. 4, Fig. S20–S22, Tables S6–S9, ESI†), we found that the electron cloud of the HOMO was mainly delocalized from the DMA unit to the whole molecule, whereas that of the LUMO was contributed by the orbitals of the organoboron ring and the neighboring aromatic rings. By contrast, the orbitals of HOMO and LUMO of POABP without an electron donating group were delocalized throughout the whole molecule. The frontier orbitals showed that charge transfer character is gradually obvious from POABP and DMA-POABP to POABP-DMA. After further calculating the excited (S1) state by the standard linear response or state-specific polarizable continuum model method based on solvation of THF,42 high values of the oscillator strengths (fos) were obtained for both DMA-POABP and POABP-DMA (Tables S6 and S7, ESI†) and all the dihedral angles of the fluorogens were close to zero degree in the S1 state. This suggested that the main configurations of the S1 states were evidently mixed and implied strong interaction between the ground and the excited states, i.e., the greater overlapping of the molecular orbitals between the HOMO and LUMO should result in strong emission in THF solution. However, in fact, only POABP-DMA showed obvious emission in THF, whereas DMA-POABP emitted no light at all.
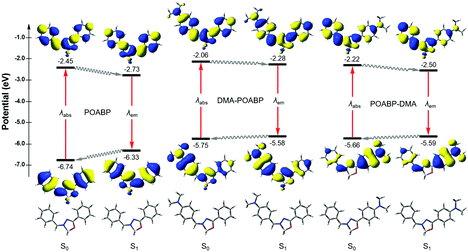 |
| | Fig. 4 Molecular geometries, orbitals and energy levels of POABP, DMA-POABP and POABP-DMA in the ground (S0) and excited (S1) states calculated with TD-DFT at the level of PBE/6-31G* based on solvation of THF. | |
To decipher the emission mechanism in THF, the potential energy surface (PES) of their S1 states as a function of the dihedral angles between (i) the DMA group and the phenyl-ring (θAP), (ii) the phenyl ring and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond (θPD), and (iii) the phenyl ring and the organoboron ring (θPB) and the benzyl group and the organoboron ring (θZE) were further calculated by scanning the torsional angle from 0° to 180° at 10° intervals. (Fig. 5A–C and Fig. S23, ESI†).43 The θAP, θPD, and θPB were associated with the rotations of the single bonds while θZE was related to double bond rotation. A high rotational energy barrier existed between the non-substituted phenyl ring and the neighboring group. According to the thermodynamics, rotation with an energy barrier of below 0.026 eV (2.5 kJ mol−1; room temperature) was free and that above 0.88 eV (83.7 kJ mol−1) was hindered. Thus, owing to the low energy barriers of S1, all rotations were allowed in the excited state to bring about nonradiative decay. In contrast, the diminished energy gaps between S1 and its vertical projected S0 state (S0//S1) could cause an increased rate of internal conversion (IC) and fast non-radiative decay according to the energy-gap law.44
N double bond (θPD), and (iii) the phenyl ring and the organoboron ring (θPB) and the benzyl group and the organoboron ring (θZE) were further calculated by scanning the torsional angle from 0° to 180° at 10° intervals. (Fig. 5A–C and Fig. S23, ESI†).43 The θAP, θPD, and θPB were associated with the rotations of the single bonds while θZE was related to double bond rotation. A high rotational energy barrier existed between the non-substituted phenyl ring and the neighboring group. According to the thermodynamics, rotation with an energy barrier of below 0.026 eV (2.5 kJ mol−1; room temperature) was free and that above 0.88 eV (83.7 kJ mol−1) was hindered. Thus, owing to the low energy barriers of S1, all rotations were allowed in the excited state to bring about nonradiative decay. In contrast, the diminished energy gaps between S1 and its vertical projected S0 state (S0//S1) could cause an increased rate of internal conversion (IC) and fast non-radiative decay according to the energy-gap law.44
However, the rotation of a single-bond still did not explain the unusual emission behaviors of the organoboron derivatives. Apparently, the low emissions of POABP and DMA-POABP in THF were mainly due to the ease of rotation of the imino-double bond (CN) in the S1 state. According to the PES of the rotational double-bond (Fig. 5A–C), the low energy barrier (ΔE*) of Z to E isomerization of POABP (0.14 eV) and DMA-POABP (0.29 eV) favored its occurrence, while the relatively high value (0.56 eV) of POABP-DMA suggested that this process was less likely to occur. Briefly, when configurational change between Z and E forms occurred, the conical intersection would be clearly found in the plot of PES versus the dihedral angles of θZE.15 This revealed that the excited state would be efficiently deactivated to the S0//S1 state through the conical intersection of the nonradiative IC pathway, followed by the vibrational relaxation to its original ground state or isomer. This explained why the emissions of POABP and DMA-POABP were much weaker in THF.
EZI study by NMR monitoring
Then, we used 1H NMR spectroscopy to monitor the EZI of DMA-POABP and POABP-DMA in THF-d8 under the irradiation of blue LED light. After irradiation, the proton peaks at 3.16 and 3.09 ppm became weaker, while new peaks at 3.14 and 3.07 ppm emerged due to the transformation of the Z-form to E-form (Fig. S24 and S25, ESI†). The fraction of E-form was calculated from their integrals and plotted against the irradiation time (Fig. 5D). The E-fraction of DMA-POABP increased rapidly at the beginning, implying its easy EZI due to its more stable diradical structure (Fig. S26, ESI†). This suggests that the double-bond rotation of DMA-POABP in the excited state is easier than that of POABP-DMA, resulting from its lower ΔE* value (vide supra calculation). Thus, the torsion of the double-bond in the excited state consumed the energy of the excited state nonradiatively. Such excited-state double-bond reorganization (ESDBR) will be prohibited or weakened in a highly viscous solution and aggregated or solid state to enable the molecules to show strong light emission.
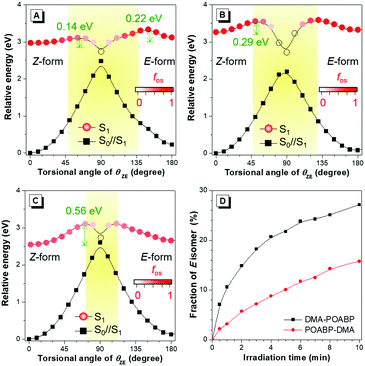 |
| | Fig. 5 (A–C) Potential energy surfaces of E/Z-isomerization of (A) POABP, (B) DMA-POABP and (C) POABP-DMA. (D) Isomerization of pure Z-isomers of DMA-POABP and POABP-DMA with time by irradiating their solutions in THF-d8 (40 mM) with a blue LED lamp. S1 was the energy of the first excited transition in the S1 state, S0//S1 was the vertical projected energy of the S0 state based on the S1 geometries, and fos was the oscillator strength of an electronic dipole-allowed transition between S0//S1 and S1. | |
Viscosity study
In the viscosity-dependent photoluminescence (PL) measurement based on the different fractions of ethylene glycol and glycerol (GL) at a concentration of 10 μM, according to the Förster–Hoffmann equation, the logarithmic relative PL intensity, log(I/I0), follows a power-law relationship with the logarithmic viscosity, log(η). As shown in Fig. 6A–C, the dye-dependence constants were calculated to be 0.38 for DMA-POABP and 0.28 for POABP-DMA, respectively, indicating that the motions of the former were restricted by ca. 10 times higher than those of the latter one in a highly viscous environment. In addition, in another PL measurement based on isopropanol (IPA) with or without GL at a concentration of 10 μM, the intensity of DMA-POABP increased by ca. five times from IPA to IPA/GL mixtures (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, v/v), whereas that of POABP-DMA remained almost unchanged (Fig. 6D). These clearly proved that the radiative decay of DMA-POABP was largely promoted by the restriction of ESDBR (Table 1), as proved by the large decrease of the knr/kr ratio (169.1 to 3.0) from THF solution to the solid state.
:2, v/v), whereas that of POABP-DMA remained almost unchanged (Fig. 6D). These clearly proved that the radiative decay of DMA-POABP was largely promoted by the restriction of ESDBR (Table 1), as proved by the large decrease of the knr/kr ratio (169.1 to 3.0) from THF solution to the solid state.
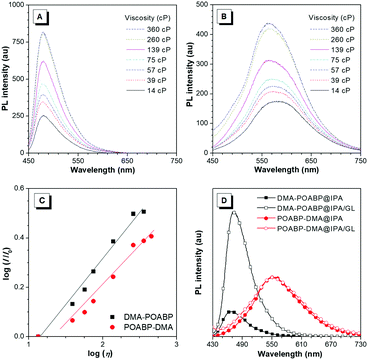 |
| | Fig. 6 (A and B) PL spectra of (A) DMA-POABP and (B) POABP-DMA in glycerol/ethylene glycol mixtures with different viscosities (η). (C) Plot of log(I/I0) versus log(η) of glycerol/ethylene glycerol mixtures of DMA-POABP and POABP-DMA, where I and IE are the PL intensity in a glycerol/ethylene glycol mixture and ethylene glycol, respectively. Inset: Linear fitting of DMA-POABP (χ = 0.38, R2 = 0.98) and POABP-DMA (χ = 0.28, R2 = 0.97). (D) PL spectra of DMA-POABP and POABP-DMA in i-propanol (IPA) and IPA/glycerol (GL) mixtures. | |
ESDBR mechanism
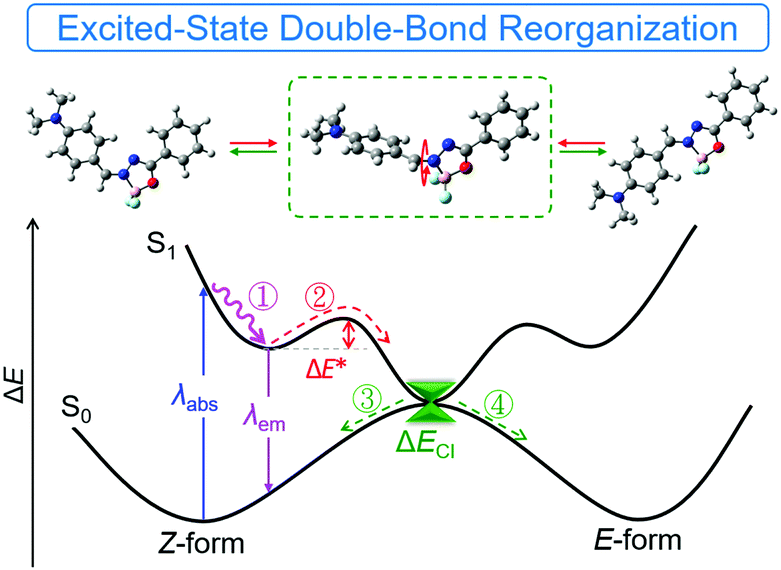
Based on the previous experimental data and discussion, we proposed a simple mechanistic model as shown in Fig. 7 and Fig. S27 (ESI†). After excitation to the S1 state, reorganization of the double bond via bond stretching, bending and rotation occurred to relax the exciton to the minimum energy level (route ①). Further reorganization to another locally-excited state (route ②) will occur if the energy barrier (ΔE*) of EZI is overcome by the environmental energy. The total reorganization processes were designated as ESDBR. The relaxation of the exciton through the conical intersection between the S0 and S1 states regenerated the ground-state molecules with either Z or E-form (route ③ and ④). The energy will be lost nonradiatively to contribute weak emission in solution. In contrast, the high ΔE* limits the ESDBR to allow it to show strong fluorescence with high quantum yield in THF (Fig. S27B, ESI†). As the ESDBR will be hampered in a highly viscous environment and aggregated or solid state, the radiative process and quantum yield will be greatly enhanced compared to those in solution.
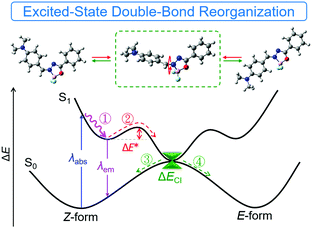 |
| | Fig. 7 A proposed model for the excited-state behavior of E/Z photoisomerization of DMA-POABP. ΔE* denotes the energy barrier from Z to E-form, and ΔECI denotes the energy gap of the de-excitation pathway through the conical intersection between the S0 and S1 states. Route ① and ② were the reorganization process in the excited state, and route ③ and ④ and λem were the de-excited process to the S0 state. | |
Lipid droplet markers for living cell imaging
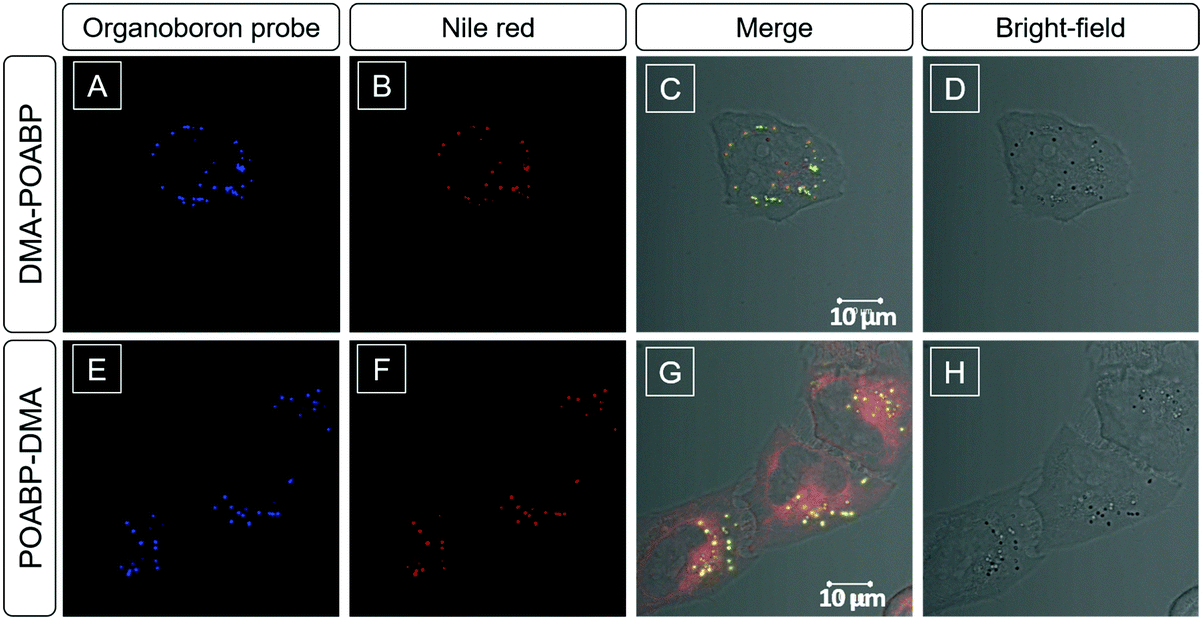
Considering the lipophilic structure of organoboron isomers (Scheme S2, ESI†), it was envisioned that they could be used as selective probes for hydrophobic organelles, such as lipid droplets. The fluorescent bioimaging potential of DMA-POABP and POABP-DMA was evaluated by co-staining experiments using the commercially-available LD probe, Nile red (Fig. 8), where human cervical (HeLa) cancer cells were used. After 30 min incubation with the probes (5 μM for DMA-POABP and POABP-DMA; 100 nM for Nile red), DMA-POABP and POABP-DMA specifically targeted to the LDs with bright-blue fluorescence (Fig. 8A and E) and exhibited better resolution and higher contrast in comparison with Nile red. From the merged images (Fig. 8C and G), we observed that Nile red simultaneously stained the LDs (yellow spots) and other intracellular organelles like mitochondria (red emission). The poor specificity of Nile red for the LDs made it difficult to distinguish them from other organelles and limits some in depth research such as that on the dynamic motions of LDs. On the other hand, DMA-POABP and POABP-DMA as lipophilic probes not only were prone to accumulate in the hydrophobic LDs but also enhanced the fluorescence intensity in the LDs with the increase of their concentration in the staining process, arising from their good AIE property.31
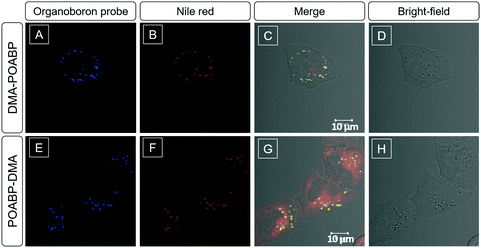 |
| | Fig. 8 Co-localization imaging of HeLa cells stained with DMA-POABP, POABP-DMA and Nile red. (A–C and E–G) Confocal images and (D and H) bright-field images of HeLa cells co-stained with (A–D) DMA-POABP and Nile red and (E–H) POABP-DMA and Nile red. (C and G) Merged images of the corresponding bright-field and fluorescence images. Excitation wavelength: 405 nm for DMA-POABP and POABP-DMA and 560 nm for Nile red (1% laser power). Concentrations: 5 μM for DMA-POABP and POABP-DMA and 100 nM for Nile red. Scale bar = 10 μm. | |
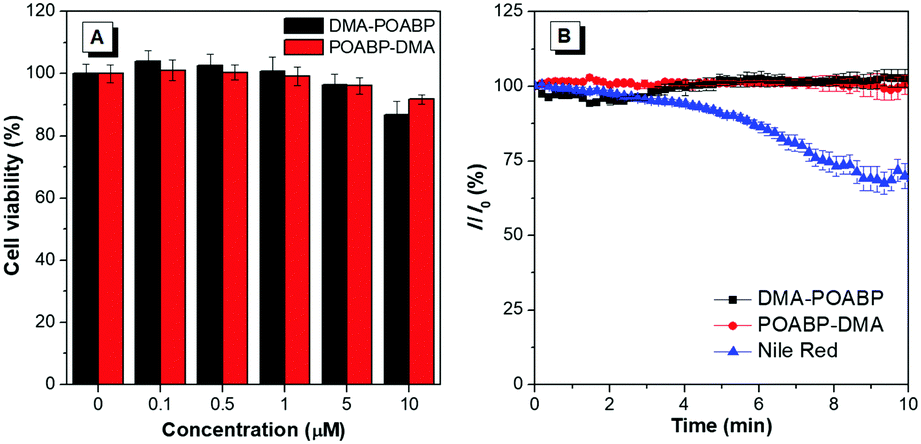
The cytotoxicity of DMA-POABP and POABP-DMA was further evaluated by the method of a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) assay (Fig. 9A).45 Although the MTT assay had some experimental errors, resulting from factors such as the cellular stress effect and interference of probes taken up by the cells,46,47 the overall results showed that no demonstrable inhibitory effect was observed on the growth of cervical cancer HeLa cells as DMA-POABP and POABP-DMA were respectively added to the culture medium at a high concentration of up to 10 μM. The excellent biocompatibility of DMA-POABP and POABP-DMA helped to gain a better understanding for their potential biological applications, such as dynamic monitoring of LD formation, metabolism and death in living cells.
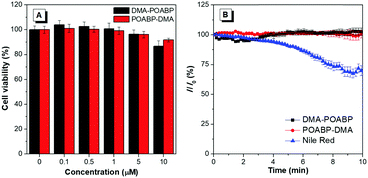 |
| | Fig. 9 (A) Cytotoxicity of DMA-POABP and POABP-DMA in HeLa cells determined by MTT assay. (B) The relative signal of fluorescence intensity of DMA-POABP, POABP-DMA and Nile red in HeLa cells with the scanning time. | |
Photostability was a key parameter for evaluating the fluorescent probes, so the oleic acid-treated HeLa cells stained with DMA-POABP and POABP-DMA were examined by scanning with a confocal microscope (Fig. 9B). Compared with the initial fluorescence intensity, they had no signal loss after 66 scans with a total irradiation time of over 10 min, whereas Nile red showed a more apparent signal loss of fluorescence intensity of about 30%. Accordingly, DMA-POABP and POABP-DMA had better photostability as well as specificity targeting to LDs than the commercially-available probe, Nile red.
Green algae imaging
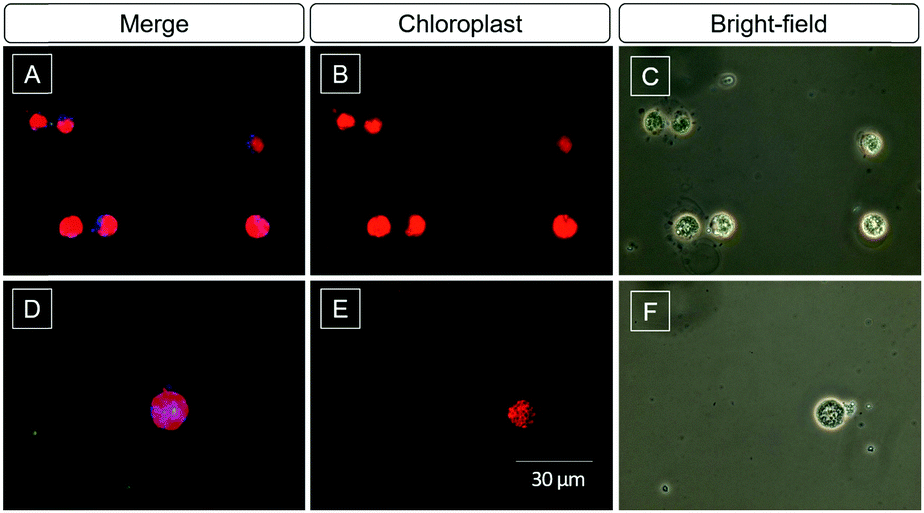
Algae biofuel for sustained energy research has been paid attention in the last decade because of its potential to be an environment-friendly alternative to fossil fuels.48 The content of lipids is an important parameter to evaluate the value of the algae. The high selectivity and light-up property of DMA-POABP and POABP-DMA motivated us to further investigate their imaging of LDs in algae. After being incubated with DMA-POABP and POABP-DMA (10 μM) at room temperature for 30 min, the lipid content of green algae Chlamydomonas reinhardtii was identified and acquired by the blue emission of the probe (Fig. 10). Here, the red emission color originates from the chloroplasts of the algae itself (Fig. 10B and E), and the little greenish blue color is the aggregate of the probe without staining the algae (Fig. 10D), which could be differentiated from the LDs under a fluorescence microscope. Thus, the dimension of blue-emission imaging mainly expressed the content of LDs, which validates the practicability of using DMA-POABP and POABP-DMA for high-throughput screening of algae species with high biofuel contents.
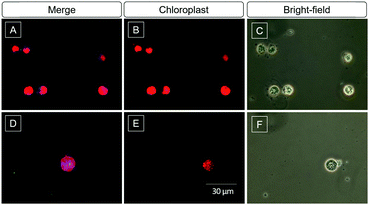 |
| | Fig. 10 (A, B, D and E) Fluorescent images and (C and F) bright-field images of green algae stained with (A) DMA-POABP and (D) POABP-DMA (10 μM). Excitation wavelength: 330–385 nm. The blue and red emissions are from the LDs and chloroplasts, respectively. | |
Conclusions
In this work, we investigated the optical properties of new coplanar organoboron isomers and observed distinctly different behaviours. DMA-POABP showed bluer but weaker emission than POABP-DMA in solution due to its stronger ESDBR. All the molecules showed strong emission in a viscous medium and aggregated state owing to the restriction of ESDBR to display a phenomenon of AIE. The organoboron isomers were not only rare AIEgens for their coplanar structures but also their AIE phenomenon was stemmed from the ESDBR mechanism. The present results suggested that the AIEgens were not necessarily derived from specific substituents and provided a new approach to design new AIEgens by controlling the ESDBR property. As a biological fluorescence probe, the organoboron isomers exhibited better photostability and specificity targeting lipid droplets in comparison with commercial Nile red. This mechanistic elucidation eliminated the structural limit for AIEgen design and helped to develop new coplanar AIEgens with novel material properties for high-technology applications.
Experimental section
Materials and methods
Chemicals were purchased from J&K, Sigma-Aldrich and TCI and used directly without further purification. Tetrahydrofuran (THF) was distilled from sodium benzophenone ketyl under dry N2 immediately before use. Other solvents were directly used without further purification. 1H and 13C NMR spectra were recorded on a Bruker ARX 400 spectrometer and Varian VNMRS 400 using CDCl3 or THF-d8 as solvent and tetramethylsilane (TMS) as an internal standard. High-resolution mass spectra (HRMS) were recorded on a GCT premier CAB048 mass spectrometer operated in MALDI-TOF mode. FT-IR spectra and UV-vis absorption spectra were recorded on a Bruker Tensor II and a Shimadzu UV-2600 spectrometer, respectively. Photoluminescence (PL) spectra were recorded on the Perkin-Elmer LS 55 fluorescence spectrometer. The absolute quantum yields of the solid powder and solution were determined using a Hamamatsu C11347-11 Quantaurus-QY Analyzer. The lifetime was recorded on an Edinburgh FLS 980 instrument and measured using a time-correlated single-photon counting method.
Synthesis of organoboron derivatives
Benzohydrazide (1 eq.) and benzaldehyde (1 eq.) were dissolved in methanol (0.5 M), and the resulting mixture was then refluxed overnight. The mixture was cooled to room temperature, and the precipitate was filtered, washed with cool ethanol, dried under vacuum, and used without purification. The above hydrazone intermediate was suspended in 1,2-dichloroethane (0.2 M), and allyltrimethylsilane (1.3 eq.) and boron trifluoride etherate (BF3·OEt2; 1.5 eq.) were successively added. The reaction flask was immersed in a boiling oil bath of ca. 120 °C and stirred overnight. The mixture was slowly cooled to room temperature, quenched with salt water, and extracted with dichloromethane. After the removal of solvent, the crude product was purified by silica-gel column chromatography using hexane/petroleum ether as the eluent. Single crystals for single crystal X-ray analysis were obtained in tetrahydrofuran/hexane with slow layer-to-layer diffusion.
Characterization data of POABP.
White solid, 80% yield. 1H NMR (400 MHz, CDCl3), δ (ppm): 8.46 (d, 2H, J = 7.6 Hz), 8.22 (d, 2H, J = 7.2 Hz), 7.90 (s, 1H), 7.70–7.59 (m, 4H), 7.53–7.49 (t, 2H, J = 7.6 Hz). 13C NMR (100 MHz, CDCl3), δ (ppm): 173.07, 150.90, 134.84, 134.44, 133.75, 129.37, 129.09, 128.96, 128.78, 126.90. HRMS (MALDI-TOF): calcd for C14H11BF2N2O [M]+: 273.0932, found: 273.0999.
Characterization data of DMA-POABP.
Yellow solid, 77% yield. 1H NMR (400 MHz, THF-d8), δ (ppm): 8.45–8.43 (m, 2H), 8.16 (d, 2H, J = 7.2 Hz), 7.86 (s, 1H), 7.59–7.55 (m, 1H), 7.51–7.49 (m, 2H), 6.89–6.87 (m, 2H), 3.16 (s, 6H). 13C NMR (100 MHz, THF-d8), δ (ppm): 170.65, 155.63, 151.25, 138.14, 133.58, 129.66, 129.41, 129.27, 128.47, 117.98, 112.88, 112.53, 40.54, 40.22. HRMS (MALDI-TOF): calcd for C16H16BF2N3O [M]+: 316.1354, found, 316.1354.
Characterization data of POABP-DMA.
Orange solid, 62% yield. 1H NMR (400 MHz, THF-d8), δ (ppm): 8.06 (d, 2H, J = 7.6 Hz), 8.05–8.02 (m, 3H), 7.64–7.58 (m, 3H), 6.80–6.78 (m, 2H), 3.09 (s, 6H). 13C NMR (100 MHz, THF-d8), δ (ppm): 174.34, 155.35, 148.68, 135.04, 134.68, 131.76, 131.43, 130.05, 114.29, 112.20, 40.27. HRMS (MALDI-TOF): calcd for C16H16BF2N3O [M]+: 315.1354, found, 315.1364.
Theoretical calculations
The optimized geometries of the ground-state (S0) and excited-state (S1) minima were calculated by Gaussian 13 with time-dependent density functional theory (TD-DFT) at the level of PBE0/6-31G* based on solvation of THF, in which the polarizable continuum model (PCM) with computationally efficient vertical excitation as linear response solvation were used. Furthermore, we used the state-specific PCM method to correct POABP-DMA with a stronger charge transfer. Its first excited transition was close to the experimental emission peak (535 vs. 555 nm) after correction. A QM/MM model at the ONIOM (TDDFT:UFF) level for a cluster of organoboron molecules cut from the crystal structure with the central one as a QM region and the surrounding 37 molecules for DMA-POABP and 41 molecules for POABP-DMA as an MM region was used to explore the solid-state luminophore.
Monitoring the E/Z photoisomerization
DMA-POABP or POABP-DMA solutions of THF-d8 (0.5 mL) at a concentration of 40 mM were prepared and added into NMR tubes. After capping, the NMR tubes containing the solutions were irradiated with light from a blue LED lamp at the same position. The process was monitored by 1H NMR spectroscopy.
Viscosity effects on PL intensity
30 μL of DMA-POABP or POABP-DMA in THF (1 mM) was added into 2.97 mL of the mixture solvent of glycerol and ethylene glycol and mixed thoroughly. The PL intensity (I) of each mixture was measured at 480 and 575 nm for DMA-POABP and POABP-DMA, respectively. Compared with the intensity of the pure ethylene glycol solution (I0), log(I/I0) was plotted against log(η) and the slope (χ) was obtained by fitting a linear relationship or the Förster–Hoffmann power-law relationship.
Cell imaging and confocal colocalization
HeLa cells were cultured in MEM containing 10% FBS and antibiotics (100 units mL−1 penicillin and 100 mg mL−1 streptomycin) in a 5% CO2 humidity incubator at 37 °C. For co-staining with Nile red, HeLa cells were first incubated with AIEgen (DMA-POABP or POABP-DMA, 5 μM) and Nile red (100 nM) at 37 °C for 30 min. The medium was then removed and the cells were rinsed with PBS three times and then imaged under a confocal microscope (LSM710). The emission filter was 448–548 nm for DMA-POABP and POABP-DMA and 598–753 nm for Nile red. The excitation was 405 nm for DMA-POABP and 560 nm for Nile red.
Cytotoxicity study
MTT assays were used to evaluate the cytotoxicity of the presented AIEgens. Cells were seeded in 96-well plates (Costar, IL, USA) at a density of 5000 cells per well. After overnight culturing, the medium in each well was replaced by 100 μL of fresh medium containing different concentrations of the presented AIEgens. The same volume fraction of DMSO (0.1%) is used for each concentration of AIEgens. 24 hours later, 10 μL MTT solution (5 mg mL−1 in PBS) was added into each well. After 4 hours of incubation, 100 μL DMSO was added to each well and then vibrated for 15 min. The absorption of each well at 595 nm was recorded via a plate reader (Perkin-Elmer Victor3™). Each trial was performed with 6 wells parallel.
Photostability
The dye-labelled HeLa cells were imaged by using a laser scanning confocal microscope (LSM710) with the rate of 11 s per scan and analysed with ZEN 2009 software (Carl Zeiss). Conditions: excitation wavelength: 405 nm for DMA-POABP and POABP-DMA and 560 nm for Nile red with 1% laser power.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for financial support from the National Science Foundation of China (21788102, 21490570 and 21490574), the Research Grants of Council of Hong Kong (16308016, C2014-15G and A-HKUST665/16), the Innovation of Technology Commission (ITC-CNERC14SC01), the Science and Technology Plan of Shenzhen (JCYJ20170307173739739 and JCYJ20160229205601482) and China Postdoctoral Science Foundation (2016M602538).
Notes and references
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef PubMed.
- Y. Yuan, S. Xu, X. Cheng, X. Cai and B. Liu, Angew. Chem., Int. Ed. Engl., 2016, 55, 6457 CrossRef PubMed.
- K. Keshav, M. K. Kumawat, R. Srivastava and M. Ravikanth, Mater. Chem. Front., 2017, 1, 1207 RSC.
- J. Yang, Y. Gao, T. Jiang, W. Liu, C. Liu, N. Lu, B. Li, J. Mei, Q. Peng and J. Hua, Mater. Chem. Front., 2017, 1, 1396 RSC.
- R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 18131 CrossRef PubMed.
- H. Nie, K. Hu, Y. Cai, Q. Peng, Z. Zhao, R. Hu, J. Chen, S.-J. Su, A. Qin and B. Z. Tang, Mater. Chem. Front., 2017, 1, 1125 RSC.
- F. Wang, C. A. DeRosa, M. L. Daly, D. Song, M. Sabat and C. L. Fraser, Mater. Chem. Front., 2017, 1, 1866 RSC.
- M. Yamaguchi, S. Ito, A. Hirose, K. Tanaka and Y. Chujo, Mater. Chem. Front., 2017, 1, 1573 RSC.
- W. Qin, P. Zhang, H. Li, J. W. Y. Lam, Y. Cai, R. T. K. Kwok, J. Qian, W. Zheng and B. Z. Tang, Chem. Sci., 2018, 9, 2705 RSC.
- X. Zhang, Z. Chi, H. Li, B. Xu, X. Li, W. Zhou, S. Liu, Y. Zhang and J. Xu, Chem. – Asian J., 2011, 6, 808 CrossRef PubMed.
- F. Bu, R. Duan, Y. Xie, Y. Yi, Q. Peng, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2015, 54, 14492 CrossRef PubMed.
- S. Sasaki, S. Suzuki, W. M. Sameera, K. Igawa, K. Morokuma and G. Konishi, J. Am. Chem. Soc., 2016, 138, 8194 CrossRef PubMed.
- S. Wiedbrauk, B. Maerz, E. Samoylova, A. Reiner, F. Trommer, P. Mayer, W. Zinth and H. Dube, J. Am. Chem. Soc., 2016, 138, 12219 CrossRef PubMed.
- K. Li, Y. Liu, Y. Li, Q. Feng, H. Hou and B. Z. Tang, Chem. Sci., 2017, 8, 7258 RSC.
- M. Jiang, Z. He, Y. Zhang, H. H. Y. Sung, J. W. Y. Lam, Q. Peng, Y. Yan, K. S. Wong, I. D. Williams, Y. Zhao and B. Z. Tang, J. Mater. Chem. C, 2017, 5, 7191 RSC.
- J. B. Xiong, Y. X. Yuan, L. Wang, J. P. Sun, W. G. Qiao, H. C. Zhang, M. Duan, H. Han, S. Zhang and Y. S. Zheng, Org. Lett., 2018, 20, 373 CrossRef PubMed.
- P. Kaewmati, Y. Yakiyama, H. Ohtsu, M. Kawano, S. Haesuwannakij, S. Higashibayashi and H. Sakurai, Mater. Chem. Front., 2018, 2, 514 RSC.
- W. Chen, C.-L. Chen, Z. Zhang, Y.-A. Chen, W.-C. Chao, J. Su, H. Tian and P.-T. Chou, J. Am. Chem. Soc., 2017, 139, 1636 CrossRef PubMed.
- Q. Peng, Y. Yi, Z. Shuai and J. Shao, J. Am. Chem. Soc., 2007, 129, 9333 CrossRef PubMed.
- R. Katoh, K. Suzuki, A. Furube, M. Kotani and K. Tokumaru, J. Phys. Chem. C, 2009, 113, 2961 Search PubMed.
- G. Han, D. Kim, Y. Park, J. Bouffard and Y. Kim, Angew. Chem., 2015, 127, 3984 CrossRef.
- Y.-X. Li, X.-F. Yang, J.-L. Miao and G.-X. Sun, J. Phys. Chem. C, 2016, 120, 21722 Search PubMed.
- Y. Zhou, Y. Liu, Y. Guo, M. Liu, J. Chen, X. Hung, W. Gao, J. Ding, Y. Cheng and H. Wu, Dye Pigm., 2017, 141, 428 CrossRef.
- S. Sasaki, S. Suzuki, K. Igawa, K. Morokuma and G.-I. Konishi, J. Org. Chem., 2017, 82, 6865 CrossRef PubMed.
- J. Zhang, S. Ma, H. Fang, B. Xu, H. Sun, I. Chan and W. Tian, Mater. Chem. Front., 2017, 1, 1422 RSC.
- J. N. Zhang, H. Kang, N. Li, S. M. Zhou, H. M. Sun, S. W. Yin, N. Zhao and B. Z. Tang, Chem. Sci., 2017, 8, 577 RSC.
- X. Zhang, Z. Chi, J. Zhang, H. Li, B. Xu, X. Li, S. Liu, Y. Zhang and J. Xu, J. Phys. Chem. B, 2011, 115, 7606 CrossRef PubMed.
- M. Beller, K. Thiel, P. J. Thul and H. Jackle, FEBS Lett., 2010, 584, 2176 CrossRef PubMed.
- L. Tirinato, F. Pagliari, T. Limongi, M. Marini, A. Falqui, J. Seco, P. Candeloro, C. Liberale and E. Di Fabrizio, Stem Cells Int., 2017, 2017, 1656053 Search PubMed.
- T. C. Walther and R. V. Farese, Jr., Annu. Rev. Biochem., 2012, 81, 687 CrossRef PubMed.
- E. Wang, E. Zhao, Y. Hong, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem. B, 2014, 2, 2013 RSC.
- F. M. Santos, J. N. Rosa, N. R. Candeias, C. P. Carvalho, A. I. Matos, A. E. Ventura, H. F. Florindo, L. C. Silva, U. Pischel and P. M. Gois, Chem. – Eur. J., 2016, 22, 1631 CrossRef PubMed.
- B. Lee, B. G. Park, W. Cho, H. Y. Lee, A. Olasz, C. H. Chen, S. B. Park and D. Lee, Chem. – Eur. J., 2016, 22, 17321 CrossRef PubMed.
- B. Zhang, G. Feng, S. Wang and X. Zhang, Dyes Pigm., 2018, 149, 356 CrossRef.
- S. Mukherjee and P. Thilagar, J. Mater. Chem. C, 2016, 4, 2647 RSC.
- P. Galer, R. C. Korosec, M. Vidmar and B. Sket, J. Am. Chem. Soc., 2014, 136, 7383 CrossRef PubMed.
- J. Roose, A. C. S. Leung, J. Wang, Q. Peng, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Chem. Sci., 2016, 7, 6106 RSC.
- J. Sun, J. Sun, W. Mi, P. Xue, J. Zhao, L. Zhai and R. Lu, New J. Chem., 2017, 41, 763 RSC.
- A. D. Dilman, D. E. Arkhipov, V. V. Levin, P. A. Belyakov, A. A. Korlyukov, M. I. Struchkova and V. A. Tartakovsky, J. Org. Chem., 2008, 73, 5643 CrossRef PubMed.
- M. Kozaki, A. Isoyama, K. Akita and K. Okada, Org. Lett., 2005, 7, 115 CrossRef PubMed.
- Y.-S. Yen, J.-S. Ni, T.-Y. Lin, W.-I. Hung, J. T. Lin and M.-C. P. Yeh, Eur. J. Org. Chem., 2015, 7367 CrossRef.
- D. Fan, Y. Yi, Z. Li, W. Liu, Q. Peng and Z. Shuai, J. Phys. Chem. A, 2015, 119, 5233 CrossRef PubMed.
- T. Kobayashi, T. Saito and H. Ohtani, Nature, 2001, 414, 531 CrossRef PubMed.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145 CrossRef.
- R. T. Kwok, C. W. Leung, J. W. Lam and B. Z. Tang, Chem. Soc. Rev., 2015, 44, 4228 RSC.
- X. Zhang, X. Zhang, S. Wang, M. Liu, L. Tao and Y. Wei, Nanoscale, 2013, 5, 147 RSC.
- X. Zhang, K. Wang, M. Liu, X. Zhang, L. Tao, Y. Chen and Y. Wei, Nanoscale, 2015, 7, 11486 RSC.
- Y. Ding, S. Zhang, L. Yang, H. Na, P. Zhang, H. Zhang, Y. Wang, Y. Chen, J. Yu, C. Huo, S. Xu, M. Garaiova, Y. Cong and P. Liu, Nat. Protoc., 2013, 8, 43 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Compound characterization, UV-vis and PL spectra of POABP, PL spectra of DMA-POABP and POABP-DMA in different solvents, monitoring the E/Z isomerization and detailed calculation data. CCDC 1832353 and 1832354. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qm00184g |
|
| This journal is © the Partner Organisations 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Haixiang
Liu
ab,
Junkai
Liu
cd,
Meijuan
Jiang
a,
Haixiang
Liu
ab,
Junkai
Liu
cd,
Meijuan
Jiang