
The unusual aggregation-induced emission of coplanar organoboron isomers and their lipid droplet-specific applications†

Abstract
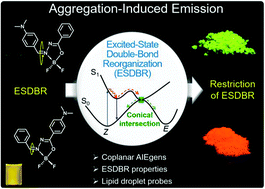
Luminogens with aggregation-induced emission (AIE) characteristics generally possess twisted structures to prevent emission quenching in the solid state by π–π stacking interactions. In this work, new organoboron derivatives with coplanar structures were synthesized and found to be AIE-active. Analysis by photoluminescence spectroscopy, single crystal X-ray diffraction and theoretical calculations depicted that the distinct luminescence behaviors of the molecules stemmed from the different extent of excited-state double-bond reorganization (ESDBR), which consumed the energy of the excitons through non-radiative pathways. This process was restricted in a highly viscous medium or in the solid state to enable the molecules to emit efficiently. The organoboron derivatives based on the ESDBR mechanism not only provide a new strategy to design coplanar AIE luminogens but also act as biological probes excellently and specifically targeting lipid droplets.
- This article is part of the themed collection: Recent Progress on Aggregation-Induced Emission
 Please wait while we load your content...
Please wait while we load your content...

