
Metal coordination and metal activation abilities of commonly unreactive chloromethanes toward metal–organic frameworks
Jinhee
Bae
 ,
Eun Ji
Lee
and
Nak Cheon
Jeong
*
,
Eun Ji
Lee
and
Nak Cheon
Jeong
*
Department of Emerging Materials Science, Daegu Gyeongbuk Institute of Science & Technology (DGIST), Daegu 42988, Korea. E-mail: nc@dgist.ac.kr
First published on 1st May 2018
Abstract
Over the last two decades, metal–organic frameworks (MOFs) have received particular attention because of their attractive properties such as permanent nanoporosity and the extraordinary functionality of open coordination sites (OCSs) at metal nodes. In particular, MOFs with open-state OCSs have shown potential in applications such as chemical separation, molecular sorption, catalysis, ionic conduction, and sensing. Thus, the activation of OCSs, i.e., the removal of coordinated solvent to produce open-state OCSs, has been viewed as an essential step that must be performed prior to the use of the MOFs in the aforementioned applications. This Feature Article focuses on the chemical functions of the commonly unreactive chloromethanes, i.e., dichloromethane (DCM) and trichloromethane (TCM), including their coordination to OCSs and activation of OCSs. Treatment with a chloromethane is a chemical route to activate OCSs that does not require an additional supply of external thermal energy. Importantly, a plausible mechanism for the chemical process, in which DCM and TCM weakly coordinate to the OCSs and then spontaneously dissociate in an intermediate step, which is proposed based on the results obtained from Raman studies will be discussed. Possible applications of chloromethane treatment to activate large-area MOF films and MOF–polymer mixed matrices, which can be propagated in molecular capture, will also be described.
 Jinhee Bae | Jinhee Bae joined the Jeong group at DGIST after receiving her BSc in 2015 from Andong National University and is currently working on her PhD. Her current research is focused on the coordination chemistry of MOFs. |
 Eun Ji Lee | Eun Ji Lee received her BSc in 2016 from Gyeongsang National University. She joined the Jeong group in 2017 and is currently working on her MSc. She conducts research on the photochemistry and electrochemistry of MOFs. |
 Nak Cheon Jeong | Nak Cheon Jeong is currently an Associate Professor of chemistry at DGIST. After receiving his MSc and PhD from the Sogang University, Korea, in 2004 and 2009, respectively, under the supervision of Prof. Kyung Byung Yoon, he spent three years as a postdoctoral researcher in the group of Joseph T. Hupp at Northwestern University, USA. In 2012, he moved to Daegu and took up his current independent position. His research is focused on the photochemistry, electrochemistry and solid-state coordination chemistry of MOFs. |
1. Introduction
Extensive study over the past few decades has been intensively focused on the synthesis, characterization, and application of microporous materials.1,2 Metal–organic frameworks (MOFs) are a highly crystalline subset of these materials that are assembled by the formation of multiple coordination bonds between inorganic metal nodes (either metal ions or metal oxide clusters) and multidentate organic ligands. The components of MOFs are largely classified into (i) metal nodes, (ii) organic ligands, and (iii) nanosized internal open spaces that are spontaneously formed by self-assembly of the metal and ligand components. Metal nodes are considered an important part of MOFs because many MOFs possess open coordination sites (OCSs) at the metal nodes, typically where Lewis base molecules can ligate through coordination bonding, and in many instances, the OCSs play important roles in a wide variety of applications of MOFs, such as chemical separation,3–10 gas storage,8–27 heterogeneous catalysis,8,28–44 sensing,8,45–51 and ionic conduction.52–55Two types of solvents reside in as-synthesized MOFs with OCSs: (i) pore-filling solvent that occupies the nanospaces via physical interaction with the walls lining pores; and (ii) coordinating solvent that binds to the OCSs via chemical bonding (see Fig. 1). To use the OCSs in MOFs for the aforementioned applications, an activation process to remove pore-filling solvent and coordinating solvent from the pore and OCSs, respectively, is required.56 To date, several strategies57,58 to activate MOFs have been developed, including (i) thermal activation (TA), which is typically performed by applying heat energy and vacuum;58–63 (ii) solvent exchange, which is typically conducted in solvents with a low boiling point;64,65 (iii) supercritical carbon dioxide (CO2) exchange;58,65 (iv) freeze-drying;66 and (v) acid treatment, which is typically performed with HCl only in the cases where the corresponding MOF is exceptionally stable in a strongly acidic environment.30,44,67 However, all of these methods except for TA have been restricted to the removal of pore-filling solvent. TA has been considered as a unique method to remove both pore-filling solvent and coordinating solvent because it easily supplies the energy required to dissociate coordinating solvent and evaporate pore-filling solvent. Thus, researchers have used TA to remove both pore-filling solvent and coordinating solvent despite the negative influence of TA on the structural integrity of MOFs.
 | ||
| Fig. 1 Schematic illustrations of a typical unit cell of an metal–organic framework (MOF) (a) before and (b) after eliminating coordinated solvent and pore-filling solvent by chloromethane treatment. | ||
Chloromethanes with a low boiling point like dichloromethane (DCM) and trichloromethane (TCM) can be used to replace other pore-filling solvent to lower the activation temperature needed in the TA of MOFs. However, another function of DCM and TCM, i.e., dissociation of solvent coordination bonds, was recently communicated.68–70 It was found that soaking MOFs in fresh DCM or TCM for several minutes at room temperature and subsequent repetition of this process several times led to not only the replacement of pore-filling solvent but also the dissociation of coordinating solvent bound at the OCSs (see Fig. 1). The following sequential two-step reaction was proposed as a mechanism for this process: (i) coordination exchange of precoordinated solvent molecules with DCM or TCM and (ii) spontaneous dissociation of the associated DCM or TCM at room temperature. Although both DCM and TCM have been frequently used in pretreatment steps prior to TA to replace pore-filling solvent (see above) and thereby lower the temperature required to remove pore-filling solvent during TA, the recent communications68–70 noted above are the first to consider the function of chloromethanes in the dissociation of coordinating solvent. These communications called chloromethane treatment a chemical activation process because it offers a chemical route to activate OCSs by removing coordinating solvent.
This Feature Article examines attempts to address the chemical function of chloromethanes to remove coordinating solvent from the OCSs in MOFs. A particular emphasis of this article is how the chloromethanes can chemically activate OCSs at a fundamental level and how this behavior can be exploited to engender the activation of large-area MOF films and MOF–polymer mixed matrix membranes (MMMs) at an applied level. In terms of the mechanism of their chemical function, the possible coordination of chloromethanes and their coordination exchanges are considered based on Raman and 1H nuclear magnetic resonance (NMR) spectroscopic results.
2. Coordination of chloromethanes to metal centers
DCM and TCM are frequently used as solvents in various organic and inorganic chemical reactions. In general, solvents should satisfy the following two prerequisite conditions: (i) miscibility with solutes that must be dissolved for homogeneous chemical reaction and (ii) more importantly, non-reactivity (or inertness) with solutes that participate in a chemical reaction. In light of the second factor, chloromethanes must be inert if a chloromethane is used in a chemical reaction. This implies that the neutral chlorine (Cl) atoms in chloromethanes should be unreactive even though the Cl atom possesses a lone pair of electrons. However, considering the primitive assumption that chloromethanes are also a weak Lewis base with a lone pair of electrons even though the polarity of chloromethanes is low, one might question whether chloromethanes can bind to transition metal ions as a Lewis base. In the late 1980s, Strauss and colleagues demonstrated the possible coordination of neutral Cl atoms in DCM and dichloroethane to soft metal ions such as Ru(I), Ag(I), and Re(I) by examining several molecular complexes.71–74 However, during the last three decades, no researchers have demonstrated the formation of coordination bonds between chloromethanes and hard (or intermediate) metal ions such as Cu(II). In the aforementioned communications,68–70 Cu(II)-based HKUST-1 was used as a model compound to demonstrate whether or not chloromethanes can coordinate to hard metal ions because (i) the Cu(II) ion in HKUST-1 is definitely a hard transition metal ion; (ii) HKUST-1 is sufficiently stable to maintain its framework structure during chloromethane treatment; (iii) HKUST-1 is a good model MOF that possesses a high concentration of OCSs (every Cu2+ center has one OCS); (iv) all OCSs face toward the internal open spaces of the large cages, so guest molecules present in the open spaces are readily accessible to the OCSs and the molecules dissociated from the OCSs can be readily removed through the large open spaces; and (v) the paddle wheel-like Cu–Cu node is Raman active, with a stretching vibration mode that appears in the range of approximately 165–230 cm−1 depending on the absence or presence and type of coordinating solvent.Raman spectra of H2O- and EtOH-coordinated pristine HKUST-1 (hereafter denoted as pristine-HK; HK is fully desolvated HKUST-1), thermally activated pristine-HK (TA-HK), DCM-treated pristine-HK (DCM-HK), TCM-treated pristine-HK (TCM-HK), HK crystals wetted in DCM (DCM-wet-HK), and HK crystals wetted in TCM (TCM-wet-HK) clearly showed the possible coordination of chloromethanes (see Fig. 2).68–70 The stretching vibration mode of Cu–Cu for pristine-HK appeared at a Raman shift of approximately 178 cm−1. In contrast, the same stretching vibration in TA-HK, where coordinated solvent was definitely excluded, appeared at approximately 228 cm−1. The stretching vibration frequencies of both DCM-HK and TCM-HK were identical to that of TA-HK. These observations indicate that pore-filling solvent and coordinating solvent in pristine-HK are completely removed using only DCM or TCM without further thermal treatment, demonstrating the chemical function of DCM and TCM (see below). It was notable that Raman spectra of DCM-wet-HK and TCM-wet-HK samples were strikingly different from those of dried DCM-HK and TCM-HK samples. The Cu–Cu mode of DCM-wet-HK and TCM-wet-HK appeared at approximately 213–217 cm−1, thus demonstrating the presence of DCM and TCM coordination bonds as an intermediate state of the chemical process. However, DCM-wet-HK and TCM-wet-HK samples exhibited different Raman shifts for the Cu–Cu vibration. Whereas that of DCM-wet-HK appeared at approximately 213 cm−1, the TCM-wet-HK sample displayed the same vibrational mode at approximately 217 cm−1. Although small, this difference suggests the different strengths of the coordination bonds formed by DCM and TCM with the CuII center. Considering that the H2O- and EtOH-coordinated pristine-HK exhibited signals from Cu–Cu peaks at lower Raman shift (178 cm−1) than those of coordinating solvent-free TA-HK, DCM-HK, and TCM-HK (228 cm−1), it can be estimated that stronger solvent coordination bonding associated with the Cu–Cu center shifts the Cu–Cu vibration to lower frequency. Therefore, the above observations suggest that the coordination bond between Cu and DCM (fCu–Cu–DCM = 213 cm−1) is stronger than that between Cu and TCM (fCu–Cu–TCM = 217 cm−1).
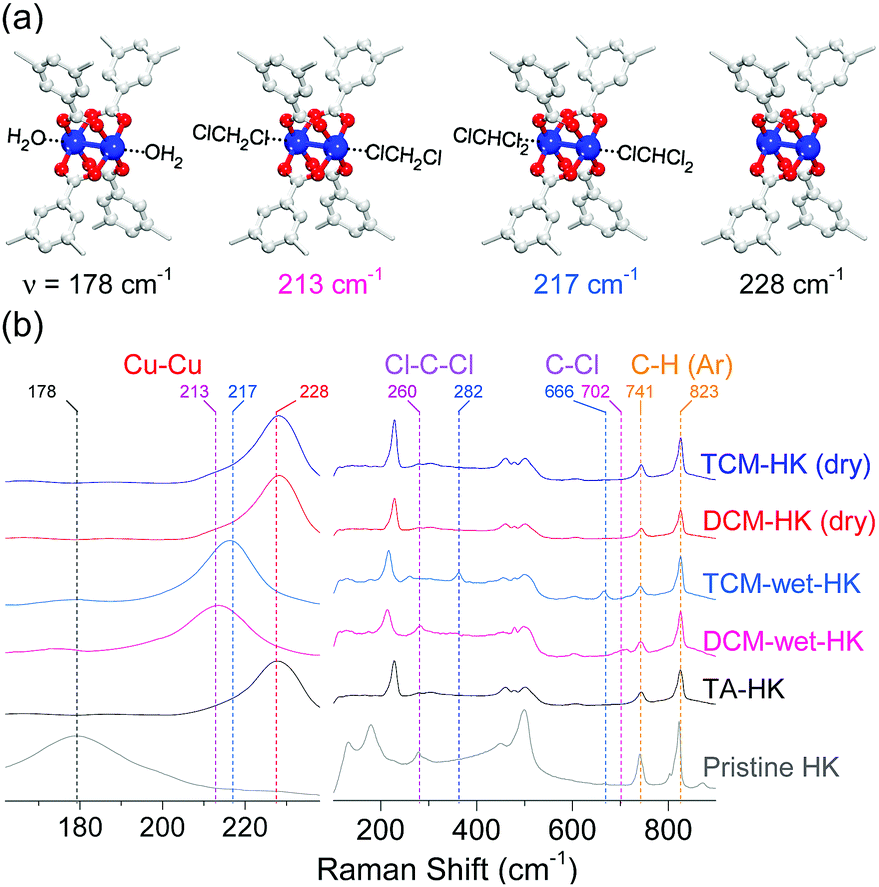 | ||
| Fig. 2 (a) Schematics of H2O-coordinated, DCM-coordinated, TCM-coordinated, and open-state (CuII)2 centers in HKUST-1 (from left to right). (b) Narrow (left) and wide (right) views of Raman spectra of pristine-HK (gray), TA-HK (black), DCM-wet-HK (pink), TCM-wet-HK (sky blue), dry DCM-HK (red), and dry TCM-HK (blue). Reprinted (adapted) with permission from ref. 70. Copyright (2018) American Chemical Society. | ||
3. Activation of open coordination sites by chloromethanes
The Raman spectra in Fig. 2 indicate that when pristine-HK was treated with either DCM or TCM, the coordinated EtOH dissociated from (CuII)2 metal center. The Raman spectra of DCM-wet-HK and TCM-wet-HK also indicate that the coordination bonds of DCM and TCM at the OCSs are formed as an intermediate state after replacing the EtOH molecules. These observations provide support for the feasibility of direct coordination exchange with DCM or TCM, which results in the removal of precoordinated solvents with moderate bond strength, such as EtOH. However, solvents that form strong coordination bonds such as N,N-dimethylformamide (DMF), N,N-diethylformamide (DEF), and dimethyl sulfoxide (DMSO) have proven difficult to remove by direct coordination exchange because the equilibrium weight of coordination exchange is substantially shifted to such strong coordination bonds. In these cases, multiple coordination exchange, which can be achieved by stepwise coordination exchanges, that is, replacement of a solvent that forms strong coordination bonds with one that forms coordination bonds of moderate strength, such as EtOH, and subsequent replacement of the solvent with moderate coordination bonds by DCM coordination, can be conducted to completely remove strongly coordinated solvent molecules at OCSs.3.1. Activation by direct coordination exchange
The aforementioned coordination exchange behavior of chloromethanes and the formation of intermediate-state chloromethane coordination were first monitored in the DCM treatment of pristine-HK at room temperature.68 These observations enabled a mechanism for the chemical function of DCM involving two successive reactions to be proposed (see Fig. 3). The first step is the coordination exchange of EtOH with DCM. The second step is the spontaneous dissociation of the coordinated DCM at room temperature. Whether or not this behavior was general for solvent coordination bonds was addressed by examining DCM treatment with pure MeOH-, EtOH-, and MeCN-coordinated HK samples (denoted MeOH-HK, EtOH-HK, and MeCN-HK, respectively).68Fig. 4a shows the 1H NMR spectra of pristine-HK, EtOH-HK, MeOH-HK, and MeCN-HK samples, which were measured after the samples were vacuum-treated at room temperature to remove only pore-filling solvent and subsequently dissolved in D2SO4.68,69 The peak for the three identical protons in the 1,3,5-benzenetricarboxylate ligand appeared at 8.8 ppm, and those for the two identical CH2 protons and three CH3 protons in EtOH appeared at 4.1 and 1.1 ppm, respectively. In addition, the peaks for the three identical CH3 protons in MeOH and MeCN appeared at 3.7 and 2.1 ppm, respectively. The 1H NMR spectrum of TA-HK was also measured for comparison. As expected, the peaks originating from EtOH in pristine-HK disappeared after TA.68 Conversely, the 1H NMR spectrum of DCM-treated pristine-HK (DCM-pristine-HK) was identical to that of TA-HK (see Fig. 4b). This behavior was also observed for DCM-treated MeOH-HK, EtOH-HK, and MeCN-HK (denoted DCM-MeOH-HK, DCM-EtOH-HK, and DCM-MeCN-HK, respectively).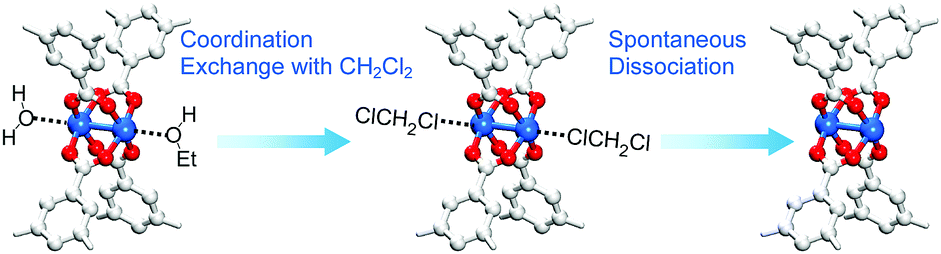 | ||
| Fig. 3 Schematics illustrating activation of the paddle wheel-like (CuII)2 node within HKUST-1 by DCM. Hydrogen atoms bound to carbon atoms in the benzene moieties are omitted for clarity. Reprinted (adapted) with permission from ref. 68. Copyright (2015) American Chemical Society. | ||
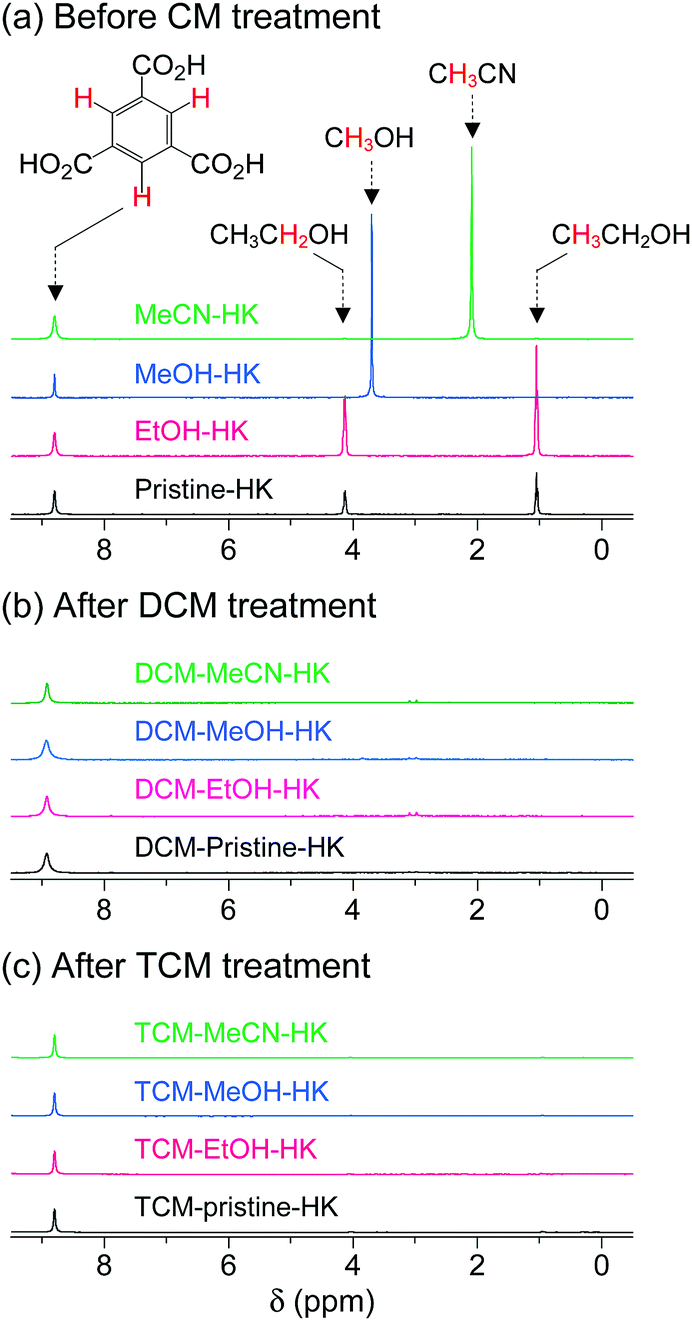 | ||
| Fig. 4 1H NMR spectra of pristine-, EtOH-, MeOH-, and MeCN-coordinated HKUST-1 (a) before chloromethane treatment, (b) after DCM treatment, and (c) after TCM treatment. NMR spectra were measured after completely dissolving the powder samples in D2SO4. Reprinted (adapted) with permission from ref. 68 and 70. Copyright (2015 and 2018) American Chemical Society. | ||
One might expect that TCM will behave similarly to DCM because TCM is a very similar chemical substance with neutral Cl atoms. The 1H NMR spectra of TCM-treated pristine-HK, MeOH-HK, EtOH-HK, and MeCN-HK (denoted TCM-pristine-HK, TCM-MeOH-HK, TCM-EtOH-HK, and TCM-MeCN-HK, respectively) proved that this was the case. TCM treatment resulted in similar patterns to those obtained following DCM treatment, indicating the complete removal of precoordinated solvent molecules in all pristine-HK, MeOH-HK, EtOH-HK, and MeCN-HK samples (see Fig. 4c). These results underscore the ability of chloromethanes to perform direct coordination exchange of previously coordinated solvents at OCSs and chemically activate the OCSs.
The phase purities of the pristine-HK, MeOH-HK, EtOH-HK, and MeCN-HK samples before and after DCM and TCM treatment were determined via powder X-ray diffraction (PXRD) measurements. The PXRD results indicated that the structural integrity of the MOF samples was well preserved after chloromethane treatment (Fig. 5). N2 adsorption isotherms of pristine-HK, DCM-pristine-HK, and TCM-pristine-HK offered further confirmation of their structural integrity. Pristine-HK was treated by TA at 150 °C under vacuum before the N2 adsorption measurements, whereas DCM-pristine-HK and TCM-pristine-HK were pretreated by applying vacuum at room temperature. The resulting internal surface areas of both DCM-pristine-HK and TCM-pristine-HK were comparable to that of thermally activated pristine-HK.68,70 Therefore, these results also underline the effectiveness of the direct coordination exchange process in terms of maintaining structural integrity.
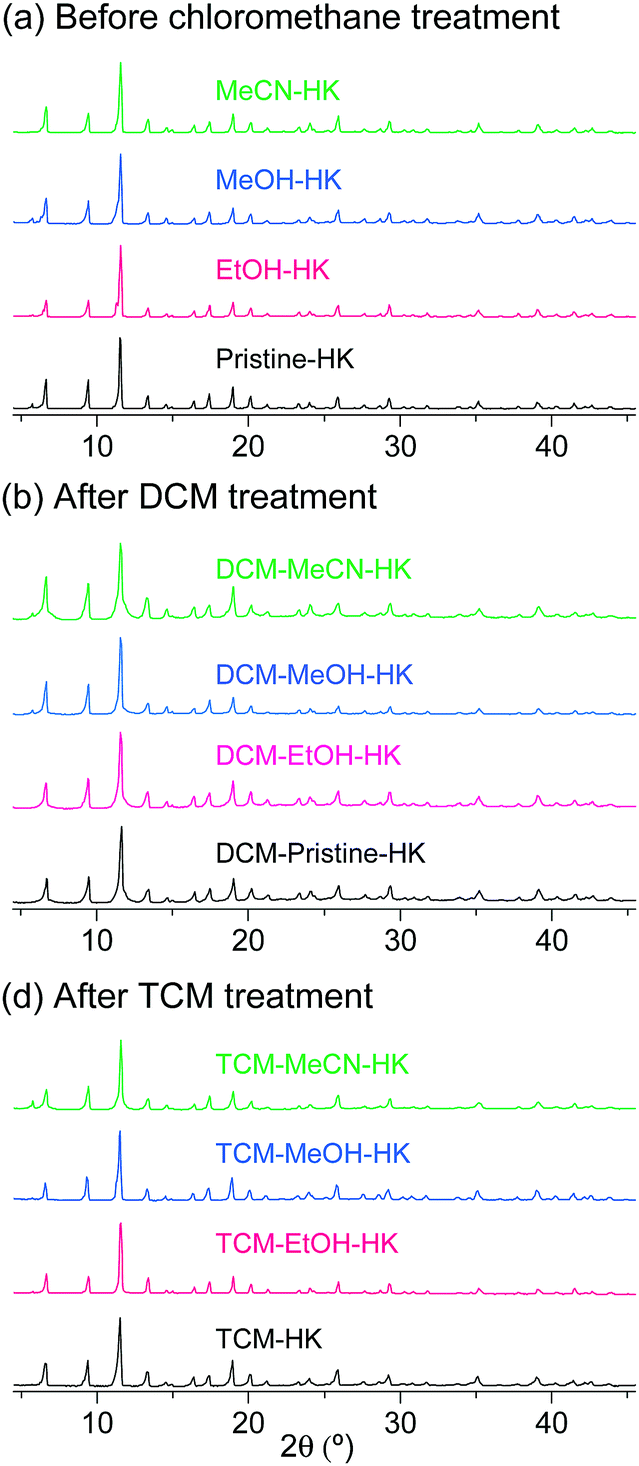 | ||
| Fig. 5 PXRD patterns of pristine-, EtOH-, MeOH-, and MeCN-coordinated HKUST-1 (a) before chloromethane treatment, (b) after DCM treatment, and (c) after TCM treatment. Reprinted (adapted) with permission from ref. 68 and 70. Copyright (2015 and 2018) American Chemical Society. | ||
As described above, TCM is an analogue of DCM in terms of not only chemical structure but also chemical functions. A unique difference between TCM and DCM is the number of Cl atoms, which gives rise to different molecular masses, polarities, and boiling points. The boiling point of DCM (40 °C) is fairly close to room temperature, whereas that of TCM (60 °C) is somewhat higher. Thus, it can be conceived that there will be some room to increase the temperature during TCM treatment. Thus, it is interesting to investigate whether the chemical function of TCM can be enhanced if a small amount of external thermal energy is supplied during TCM treatment. This issue was addressed by conducting TCM treatment of MeOH-HK at 55 °C (thermal energy at 55 °C, ET,55°C ∼ 28.3 meV) and simultaneously monitoring the 1H NMR peak of precoordinated MeOH. The TCM treatment was performed by soaking a MeOH-HK powder sample in pure TCM at 55 °C for 5 min. This process was repeated until the proton peak from MeOH completely disappeared from the 1H NMR spectrum of the sample. The TCM treatment was also performed at 25 °C (ET,25°C ∼ 25.7 meV) for comparison. The NMR results showed that while the treatment performed at 55 °C completely removed the precoordinated MeOH after only four cycles, that performed at 25 °C could remove only ∼80% of the precoordinated MeOH under the same conditions (see Fig. 6). The enhanced coordination exchange with TCM at higher temperature can be ascribed to the acceleration of translational motion of TCM molecules and the subsequent increase in the collision frequency of the TCM molecules with metal centers.
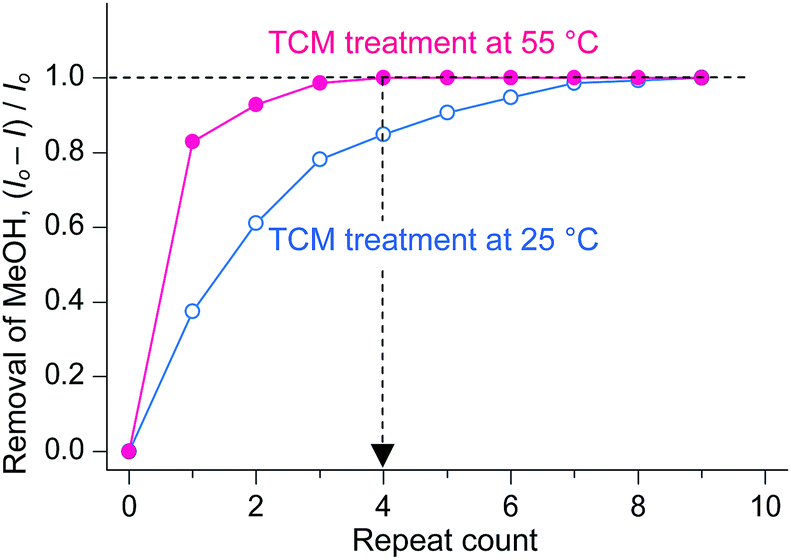 | ||
| Fig. 6 Changes in the amount of MeOH dissociated from MeOH-HK with respect to the number of TCM treatments performed at 25 °C (blue) and 55 °C (red). Reprinted (adapted) with permission from ref. 70. Copyright (2018) American Chemical Society. | ||
3.2. Multiple coordination exchange for activation
So far, we have discussed that OCSs can be chemically activated solely by DCM or TCM treatment via two stepwise reactions; that is, (i) direct coordination exchange of precoordinated MeOH, EtOH, and MeCN molecules with DCM or TCM and (ii) spontaneous dissociation of DCM or TCM at room temperature. Here, one might question whether solvents that form strong coordination bonds (i.e., DMF, DEF, and DMSO) can also be dissociated solely through chloromethane treatment. Direct coordination exchange of strong coordination bonds was examined by soaking a DMF-coordinated HKUST-1 (DMF-HK) powder sample in pure DCM at room temperature for 10 min. Under these conditions, DMF was not completely removed even when the DCM treatment was repeated 30 times (Fig. 7), as confirmed from 1H NMR spectra. From this observation, one might presume that only TA would be suitable to completely remove strongly coordinating solvent molecules and thereby fully activate the OCSs of MOFs. However, a stronger bond requires a higher temperature for its dissociation if TA is used. Heating an MOF at a temperature exceeding a critical point can be risky because high thermal energy can strongly influence metal–ligand coordination bonds and induce the structural damage of MOFs. For example, the complete removal of DMF from DMF-coordinated MOF-74(Ni) required thermal energy corresponding to a temperature of 240 °C, but the framework of MOF-74(Ni) completely collapsed above 180 °C.69 Thus, an alternative approach involving multiple coordination exchanges instead of direct coordination exchange could be conceived because the replacement of a strongly coordinating solvent with one with moderate coordination could eventually allow the removal of the solvent with moderate coordination strength by DCM treatment (Fig. 8). If the initial exchange of the strongly coordinating solvent is feasible, multiple coordination exchange should be an efficient chemical method to activate OCSs in the absence of an additional heat supply.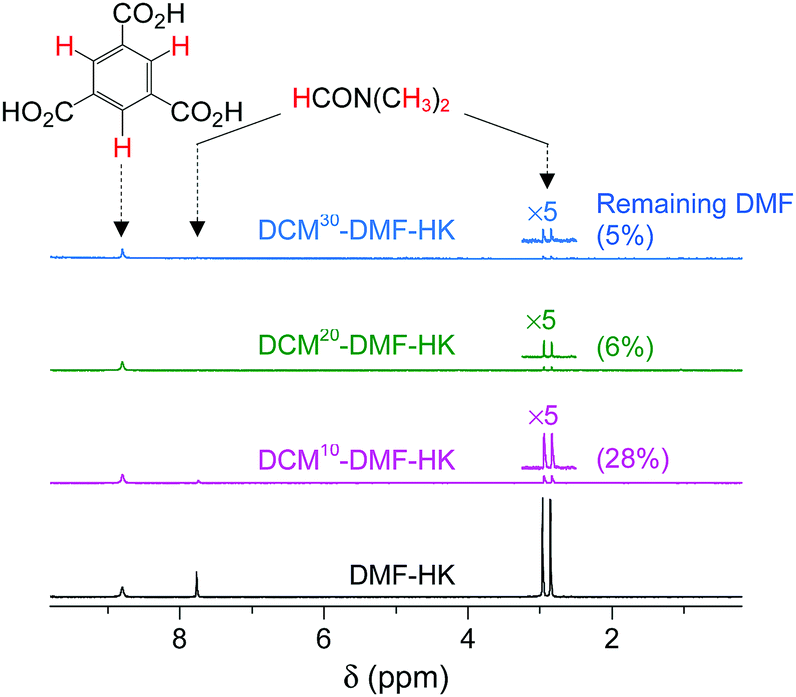 | ||
| Fig. 7 1H NMR spectra of DMF-coordinated HKUST-1 before and after DCM treatment 10, 20, and 30 times. NMR spectra were measured after the powder samples were completely dissolved in D2SO4. Reprinted (adapted) with permission from ref. 69. Copyright (2017) American Chemical Society. | ||
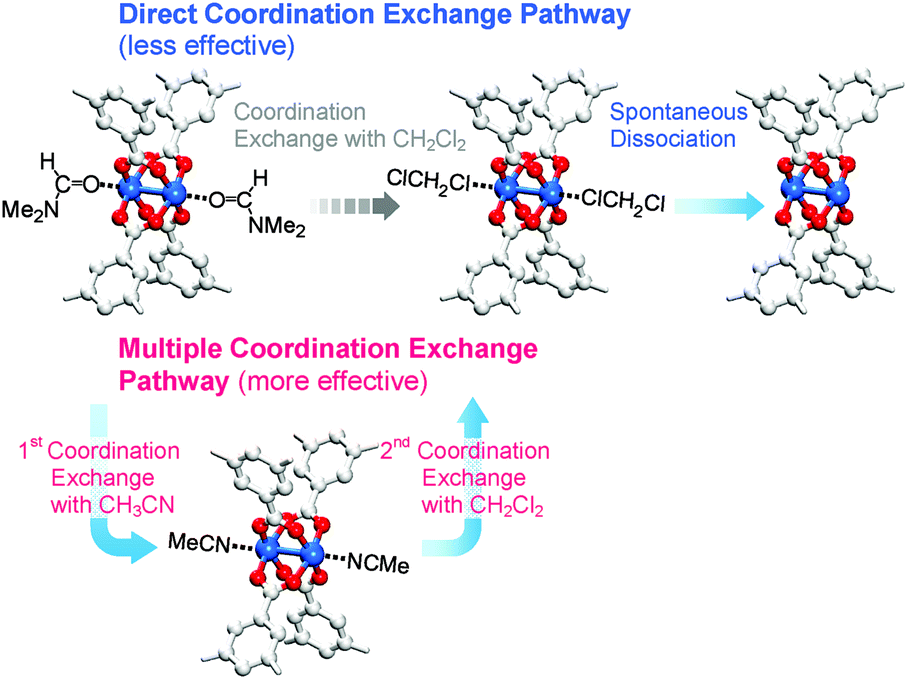 | ||
| Fig. 8 Schematics illustrating direct and multiple coordination exchange pathways for the activation of the paddle wheel-like (CuII)2 node of HKUST-1. Hydrogen atoms bound to the carbon atoms in the benzene moieties are omitted for clarity. Reprinted (adapted) with permission from ref. 69. Copyright (2017) American Chemical Society. | ||
The feasibility of multiple coordination exchange was systematically studied by monitoring 1H NMR spectra. A DMF-HK sample was initially treated with MeCN, MeOH, and EtOH (MeCN-DMF-HK, MeOH-DMF-HK, and EtOH-DMF-HK, respectively). The MeCN-DMF-HK, MeOH-DMF-HK, and EtOH-DMF-HK samples were subsequently treated with DCM (DCM-MeCN-DMF-HK, DCM-MeOH-DMF-HK, and DCM-EtOH-DMF-HK, respectively). Fig. 9 shows the 1H NMR spectra of these samples. While the peak from DMF in the DMF-HK sample disappeared after MeCN treatment, one from MeCN simultaneously appeared (Fig. 9a). The peak from MeCN then eventually disappeared during DCM treatment. It was also observed that this behavior was generic for the pathways involving initial MeOH and EtOH treatment (see the 1H NMR spectra in Fig. 9b and c).
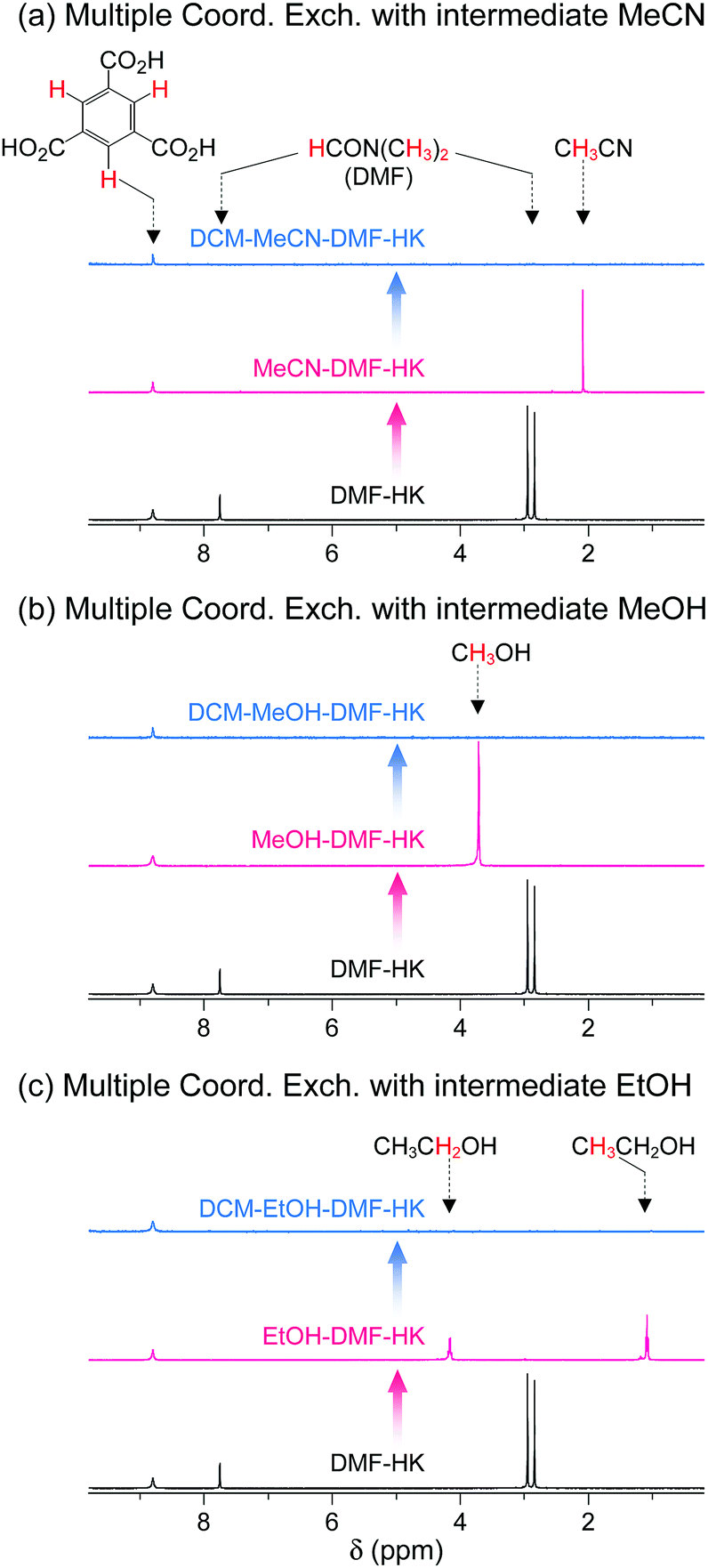 | ||
| Fig. 9 1H NMR spectra of DMF-coordinated HKUST-1 before and after initial solvent treatment with (a) MeCN, (b) MeOH, and (c) EtOH and after subsequent solvent treatment with DCM. NMR spectra were measured after the powder samples were completely dissolved in D2SO4. Reprinted (adapted) with permission from ref. 69. Copyright (2017) American Chemical Society. | ||
These observations were confirmed via in situ1H NMR experiments that were designed to monitor the progress of the coordination exchange process.69 The experiments were conducted in NMR tubes containing both target HKUST-1 crystals and deuterated solvent. More specifically, the DMF-HK crystals were immersed in deuterated MeCN (i.e., CD3CN) and then the amount of DMF that dissociated from the OCSs in DMF-HK by coordination exchange with CD3CN and subsequently dissolved in CD3CN was monitored over time, as illustrated in Fig. 10a. The amount of dissolved DMF was quantitatively analyzed by calibration with an internal standard, CHD2CN, which was present as an impurity in commercial CD3CN. DMF was initially absent in CD3CN, but its content increased upon exposure of DMF-HK crystals to CD3CN (see Fig. 10b). The integral area of the peak from DMF plateaued after 25 min, indicating the termination of the coordination exchange reaction (see Fig. 10c). The termination of the coordination exchange reaction was also confirmed by analyzing the amount of DMF remaining in the crystals (ca. 1.8 mol%), which was determined by taking a 1H NMR spectrum after the crystals were dissolved in D2SO4 (inset of Fig. 10c). A similar trend was observed in the second step, i.e., the coordination exchange of precoordinated MeCN in MeCN-HK with deuterated DCM. This experiment was performed by treating MeCN-HK crystals with CD2Cl2 (Fig. 10d and e). The proton peak from MeCN was initially absent, but rapidly evolved and reached a plateau after 15 min. The 1H NMR spectrum obtained for the MeCN-HK crystals after coordination exchange indicated that the amount of MeCN remaining in the crystals was only ca. 4.0 mol% (inset of Fig. 10e). Very similar trends were also observed for other in situ NMR experiments that were conducted with MeOD-d4 and EtOD-d6.69 Therefore, the in situ1H NMR spectroscopy results verify the ability of multiple coordination exchange to remove strongly coordinating solvent molecules from OCSs of MOFs.
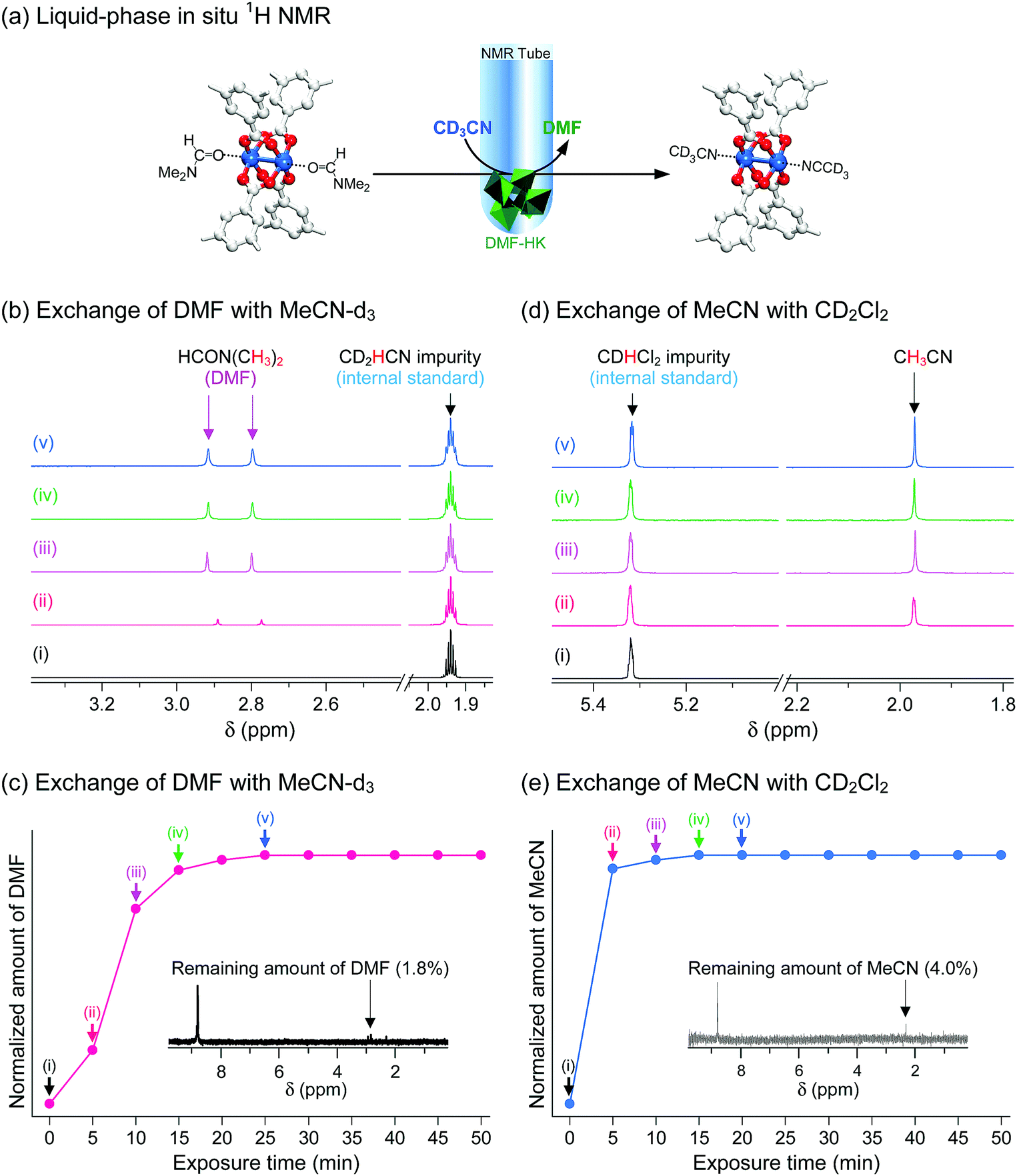 | ||
| Fig. 10 (a) Schematics illustrating the in situ1H NMR experiments designed to directly monitor coordination exchange. The coordination exchange of precoordinated DMF with postcoordinated deuterated acetonitrile (MeCN-d3, CD3CN) is shown. The in situ NMR setup was used to monitor the amount of DMF molecules that dissociated from the (CuII)2 node and thereby dissolved in MeCN-d3. Hydrogen atoms bound to the carbon atoms in the benzene moieties are omitted for clarity. In situ time-course 1H NMR spectra of (b) MeCN-d3-containing DMF-HK crystals and (c) deuterated dichloromethane (DCM-d2, CD2Cl2)-containing MeCN-DMF-HK crystals. The MeCN-d2 (CD2HCN) and DCM-d1 (CDHCl2) impurities present in the corresponding MeCN-d3 and DCM-d2 solvents were used as internal standards for the quantitative analysis of DMF and MeCN molecules, respectively. Plots of the amounts of (d) DMF and (e) MeCN dissociated from DMF-HK and MeCN-DMF-HK crystals, respectively, with respect to the exposure time to MeCN-d3 and DCM-d2, respectively. The insets display the 1H NMR spectra of the reaction-terminated (d) DMF-HK and (e) MeCN-DMF-HK crystals, which were measured after the crystals were completely dissolved in D2SO4. Reprinted (adapted) with permission from ref. 69. Copyright (2017) American Chemical Society. | ||
The absence or presence and type of coordinated solvent are reflected in the Raman vibrational mode of the (CuII)2 centers. For example, this mode for the solvent-free (CuII)2 node appears at approximately 229 cm−1, whereas those of the DMF- and EtOH-coordinated (CuII)2 nodes are found at approximately 175 and 185 cm−1, respectively. As also described above, the (CuII)2 vibrational mode of DCM-wet-HK lies at approximately 213 cm−1. On the basis of these observations, the function of multiple coordination exchange was further corroborated by monitoring successive changes in the Raman shifts of the Cu–Cu vibration.69 The successive changes were observed by in situ Raman spectroscopy during the sequential treatment of DMF-HK crystals with EtOH and then DCM (see Fig. 11). The resulting Raman spectra showed that the stretching vibration of the (CuII)2 node in DMF-HK appeared at 175 cm−1, and then shifted to approximately 185 cm−1 after the sample was exposed to EtOH. The (CuII)2 vibrational frequency further shifted to approximately 213 cm−1 when EtOH was completely supplanted by DCM. When the sample was exposed to ambient air, the peak shifted to approximately 228 cm−1, indicating the formation of open-state (CuII)2 centers. This shift implies the spontaneous dissociation of the coordinated DCM under ambient conditions, as described above. Upon continuous exposure to ambient air, the vibration mode of open-state (CuII)2 centers redshifted to 168 cm−1. This phenomenon occurred because of the coordination of H2O molecules present in the ambient atmosphere. Therefore, the peak at 213 cm−1 caused by DCM coordination and the observed sequential shifts (175 → 185 → 213 → 228 → 168 cm−1) provide strong support for the occurrence of multiple coordination exchanges at OCSs. Badger's rule states that the strength of a chemical bond affects the frequency of its vibration mode; that is, an increase in bond strength leads to an increase in the frequency of the bond vibration.75 Thus, Badger's rule indicates that the Cu–Cu bond strength increases in the order of H2O-HK < DMF-HK < EtOH-HK < DCM-HK < open-state-HK. Given that bond strength is generally inversely proportional to bond length, the Cu–Cu bond length should decrease in the order of H2O-HK > DMF-HK > EtOH-HK > DCM-HK > open-state-HK.
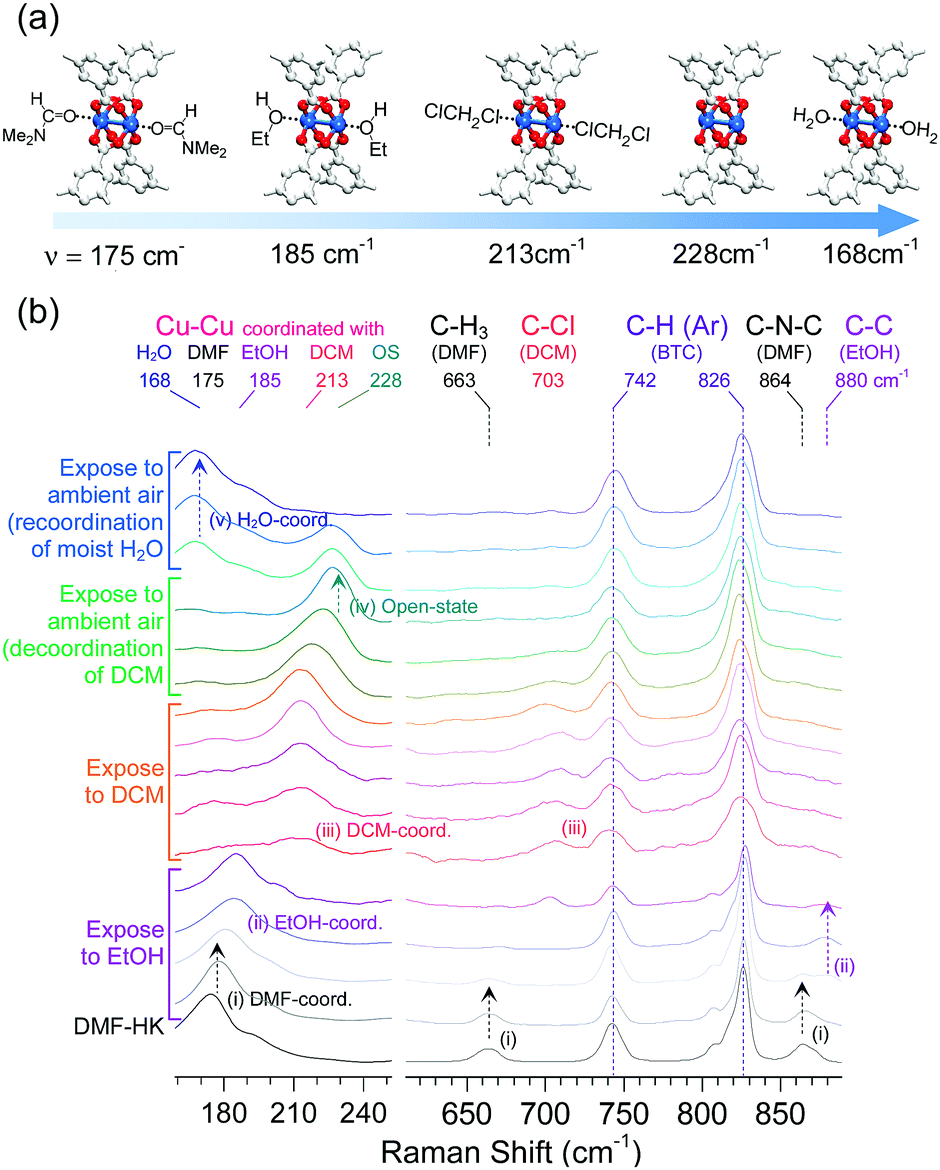 | ||
| Fig. 11 (a) Illustrations of DMF-coordinated, EtOH-coordinated, DCM-coordinated, open-state, and H2O-coordinated (CuII)2 centers in HKUST-1 (from left to right). (b) Successive changes of the in situ Raman spectra of DMF-coordinated HKUST-1 during sequential exposure to EtOH, DCM, and ambient air. Reprinted (adapted) with permission from ref. 69. Copyright (2017) American Chemical Society. | ||
The phase purities of MeCN-DMF-HK, MeOH-DMF-HK, and EtOH-DMF-HK samples before and after DCM treatment were determined by PXRD measurements (Fig. 12). As expected, the PXRD patterns indicated that the frameworks of the MOF samples remained intact even after strongly coordinating DMF was completely removed. The structural integrity of the DMF-HK, MeCN-DMF-HK, and DCM-MeCN-DMF-HK samples was also corroborated by examining their N2 adsorption–desorption isotherms. The DMF-HK and MeCN-DMF-HK samples were pretreated by TA at 150 °C prior to isotherm measurement and the DCM-MeCN-DMF-HK sample was pretreated only by applying vacuum at room temperature (see Fig. 13). The results showed that the internal surface area of MeCN-DMF-HK was approximately 1860 m2 g−1 and that of DMF-HK was approximately 1400 m2 g−1. This indicates that TA at 150 °C did not completely remove the strongly coordinating DMF molecules. Although the DCM-MeCN-DMF-HK sample was only vacuum-treated prior to isotherm measurement, its internal surface area of 1840 m2 g−1 was comparable to that of thermally activated MeCN-DMF-HK. Therefore, these results confirm that multiple coordination exchange is a promising strategy to remove strongly coordinating solvent molecules from OCSs and correspondingly, to fully activate OCSs.
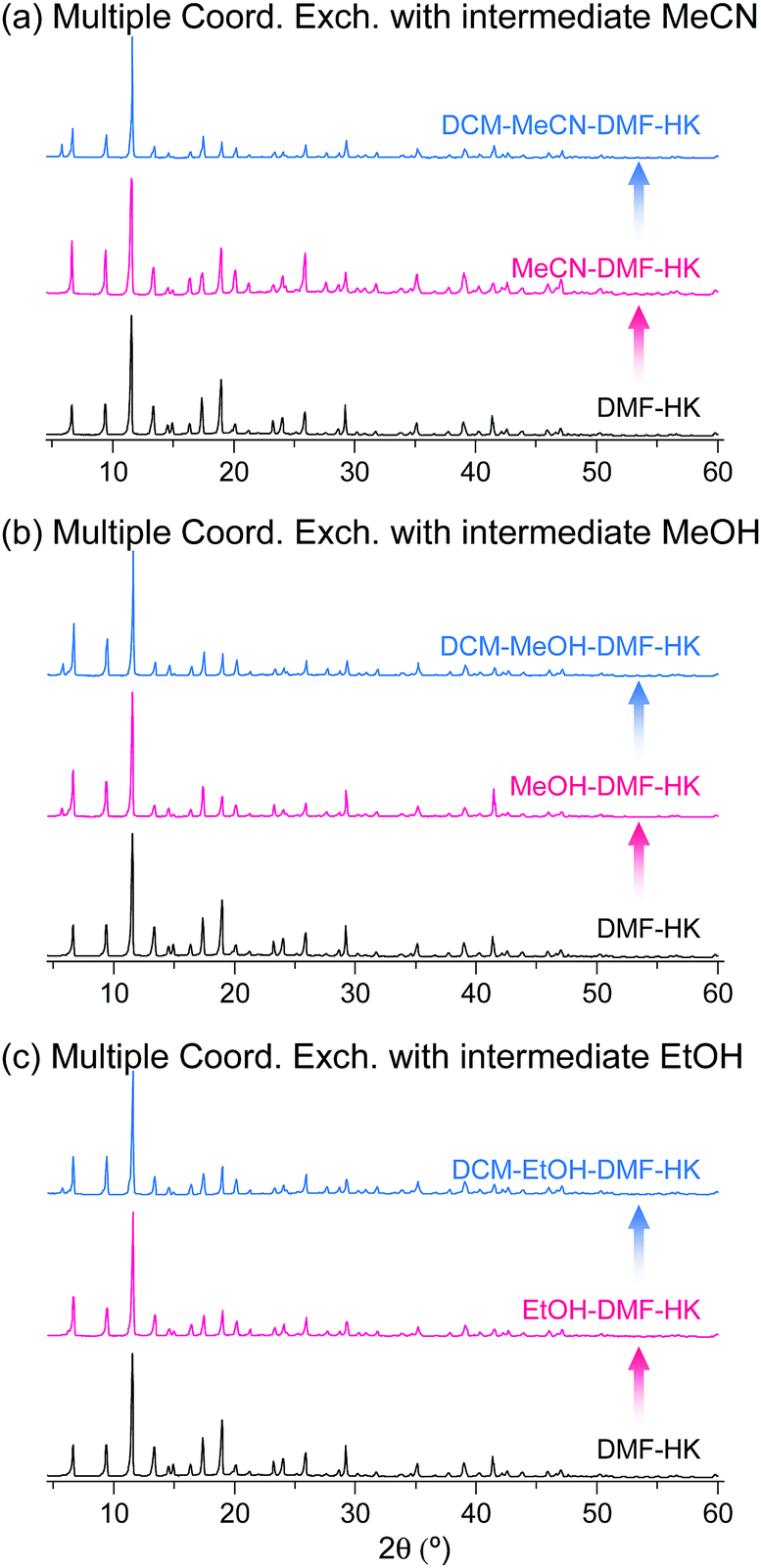 | ||
| Fig. 12 PXRD patterns of DMF-coordinated HKUST-1 before and after initial solvent treatment with (a) MeCN, (b) MeOH, and (c) EtOH and subsequent solvent treatment with DCM. Reprinted (adapted) with permission from ref. 69. Copyright (2017) American Chemical Society. | ||
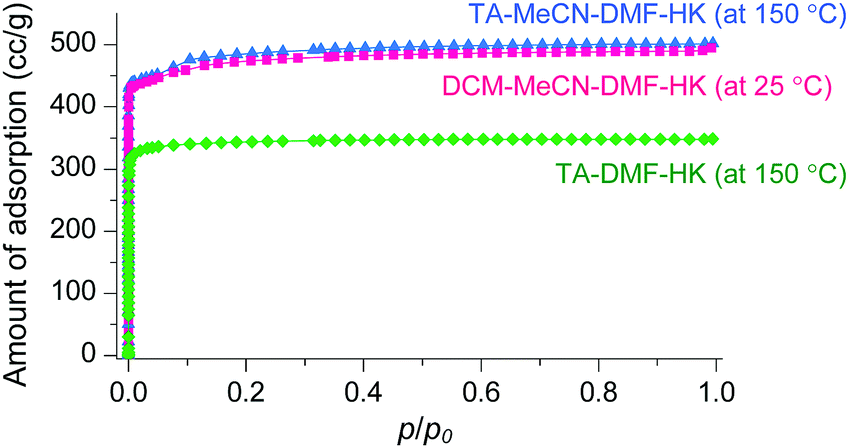 | ||
| Fig. 13 N2 adsorption isotherms of TA-DMF-HK (green), TA-MeCN-DMF-HK (blue), and DCM-MeCN-DMF-HK (pink). Reprinted (adapted) with permission from ref. 69. Copyright (2017) American Chemical Society. | ||
The wide applicability of multiple coordination exchange was verified by examining the coordination exchange behavior of DEF- and DMSO-coordinated HKUST-1 (hereafter denoted as DEF-HK and DMSO-HK, respectively). Prior to conducting multiple coordination exchange, direct coordination exchange was examined by performing DCM treatment of DEF-HK and DMSO-HK samples to determine if the coordinated DEF and DMSO molecules could be completely removed by DCM treatment alone. The results showed that 4% of DEF and 46% of DMSO remained in the DEF-HK and DMSO-HK samples, respectively, after DCM treatment 30 times. By contrast, MeCN treatment of DEF-HK and DMSO-HK completely replaced the coordinated DEF and DMSO with MeCN after only a few repetitions, and the subsequent DCM treatment completely removed the coordinated MeCN from the HKUST-1 samples (Fig. 14a and b). The crystallinities of both DEF-HK and DMSO-HK were well preserved after multiple coordination exchange using MeCN and DCM, as shown in Fig. 14c and d.
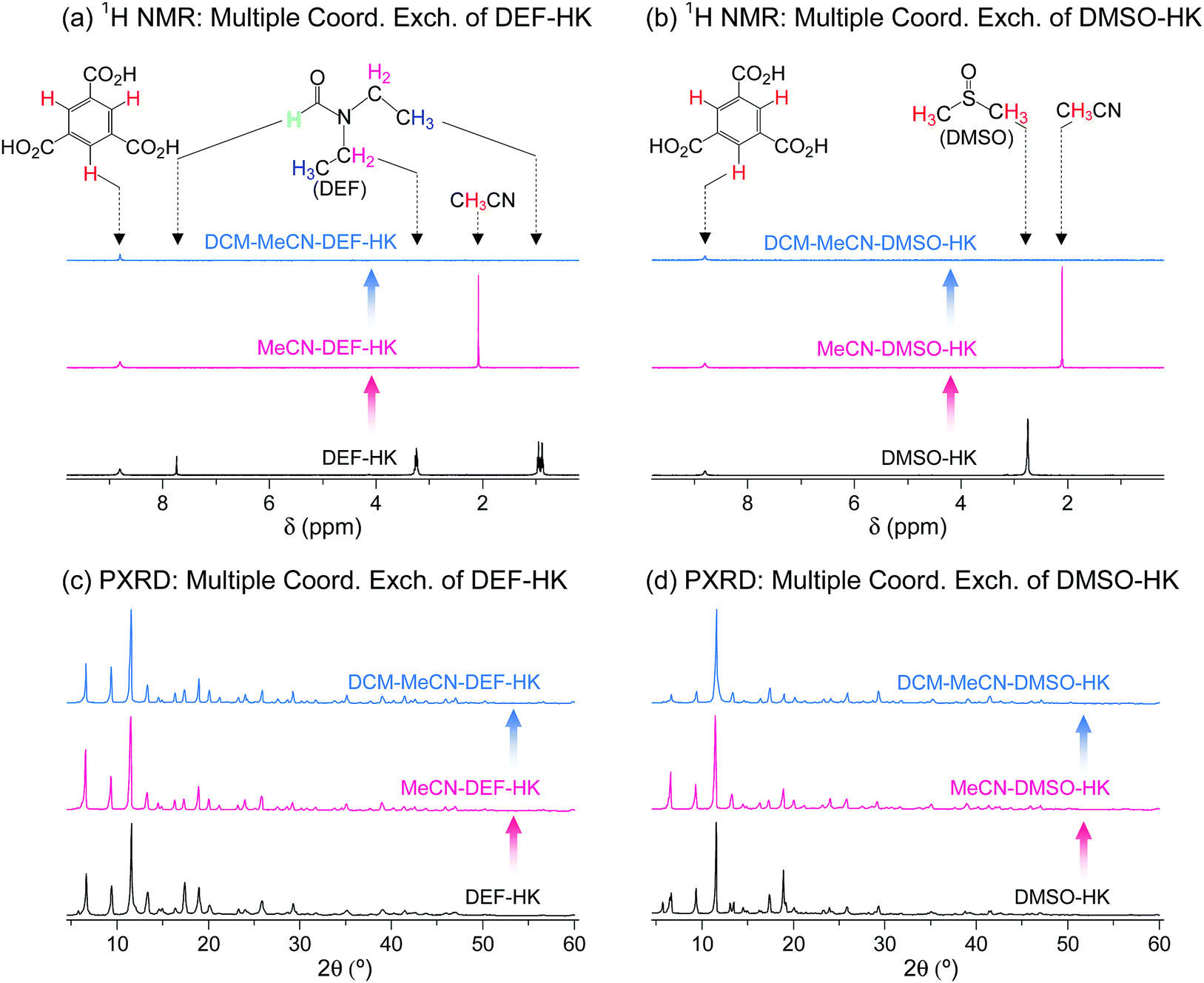 | ||
| Fig. 14 (a and b) 1H NMR spectra and (c and d) PXRD patterns of (a and c) DEF- and (b and d) DMSO-coordinated HKUST-1 before and after initial MeCN treatment and subsequent DCM treatment. NMR spectra were measured after the powder samples were completely dissolved in D2SO4. Reprinted (adapted) with permission from ref. 69. Copyright (2017) American Chemical Society. | ||
DMF-coordinated pristine MOF-74(Ni) is known as a thermally unstable MOF. For example, pristine-MOF-74(Ni) requires a temperature higher than 240 °C to completely remove coordinated DMF, but its framework completely collapses above 200 °C even though the coordinated DMF molecules have not fully dissociated.69 In addition, dimethylamine formed by the thermal decomposition of DMF during the TA process strongly coordinate to the OCSs of Ni2+ centers, suppressing the activation.69,76 Multiple coordination exchange was explored as an alternative method to completely remove coordinated DMF from pristine MOF-74(Ni) while maintaining the structural integrity of the MOF. The coordinated DMF molecules in pristine-MOF-74(Ni) were completely removed after sequential coordination exchanges with MeCN and then DCM (Fig. 15a). In terms of structural integrity, multiple coordination exchange gave results that differed from those of TA. That is, the framework of thermally unstable MOF-74(Ni) remained intact after multiple coordination exchange processes (Fig. 15b). Isostructural MOF-74(Cu) and MOF-74(Mg) exhibited similar behavior (Fig. 15c–f). Therefore, these results also support that multiple coordination exchange is a suitable strategy to activate thermally unstable or low-stability MOFs.
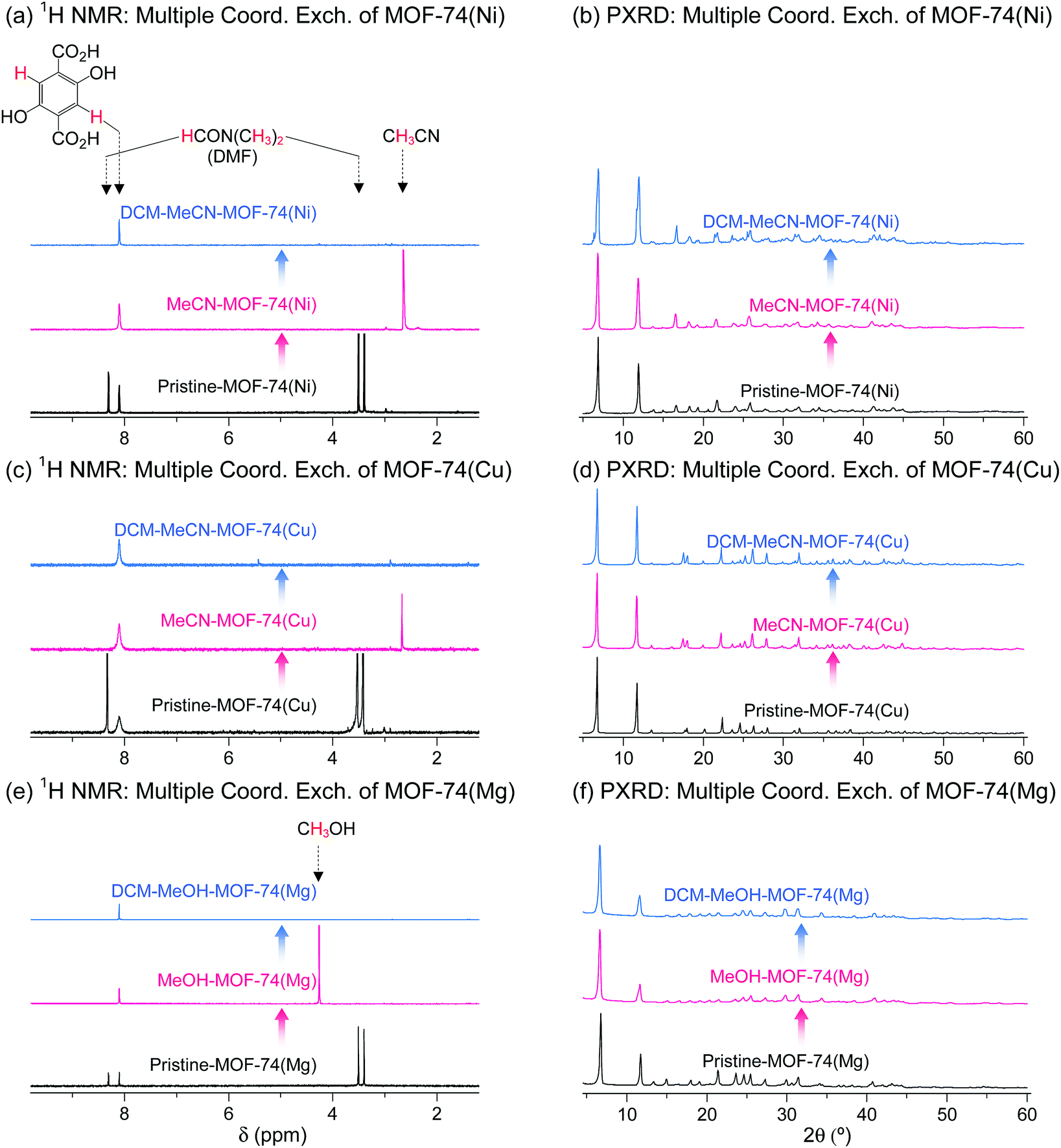 | ||
| Fig. 15 (a, c and e) 1H NMR spectra and (b, d and f) PXRD patterns of DMF-coordinated (a and b) MOF-74(Ni), (c and d) MOF-74(Cu), and (e and f) MOF-74(Mg) before and after initial (a–d) MeCN and (e and f) MeOH treatment and subsequent DCM treatment. NMR spectra were measured after the powder samples were completely dissolved in D2SO4. Reprinted (adapted) with permission from ref. 69. Copyright (2017) American Chemical Society. | ||
4. Possible applications of chloromethanes in activation of MOFs
TA requires effective facilities to make the temperature of surroundings higher and pressure lower relative to atmospheric conditions. Unlike TA, the chemical process performed by chloromethane treatment for the MOF activation is useful when the amount of a powder sample or size of an MOF film or membrane is large because it does not require such large facilities for heating and vacuum. In addition, conventional TA is sometimes unsuitable for MOF–polymer composite materials because thermally unstable polymers can be damaged or decomposed by the thermal energy supplied during TA. Therefore, the chemical process is attractive for application to large MOF films and MOF–polymer blends with low thermal stability.4.1. Activation of MOF films
The activation of large-area MOF films by DCM was examined using an HKUST-1 film fabricated on a metallic Cu substrate (Fig. 16a–c). The as-synthesized HKUST-1 film was sky blue, which changed to dark blue after DCM treatment (Fig. 16d). In agreement with the results obtained for powder samples, DCM treatment completely removed the coordinated EtOH from the HKUST-1 film (Fig. 16e and f). The framework structure of the HKUST-1 film remained intact after DCM treatment (Fig. 16g and h). Thus, these observations strongly support the suitability of DCM to chemically activate large-area MOF films.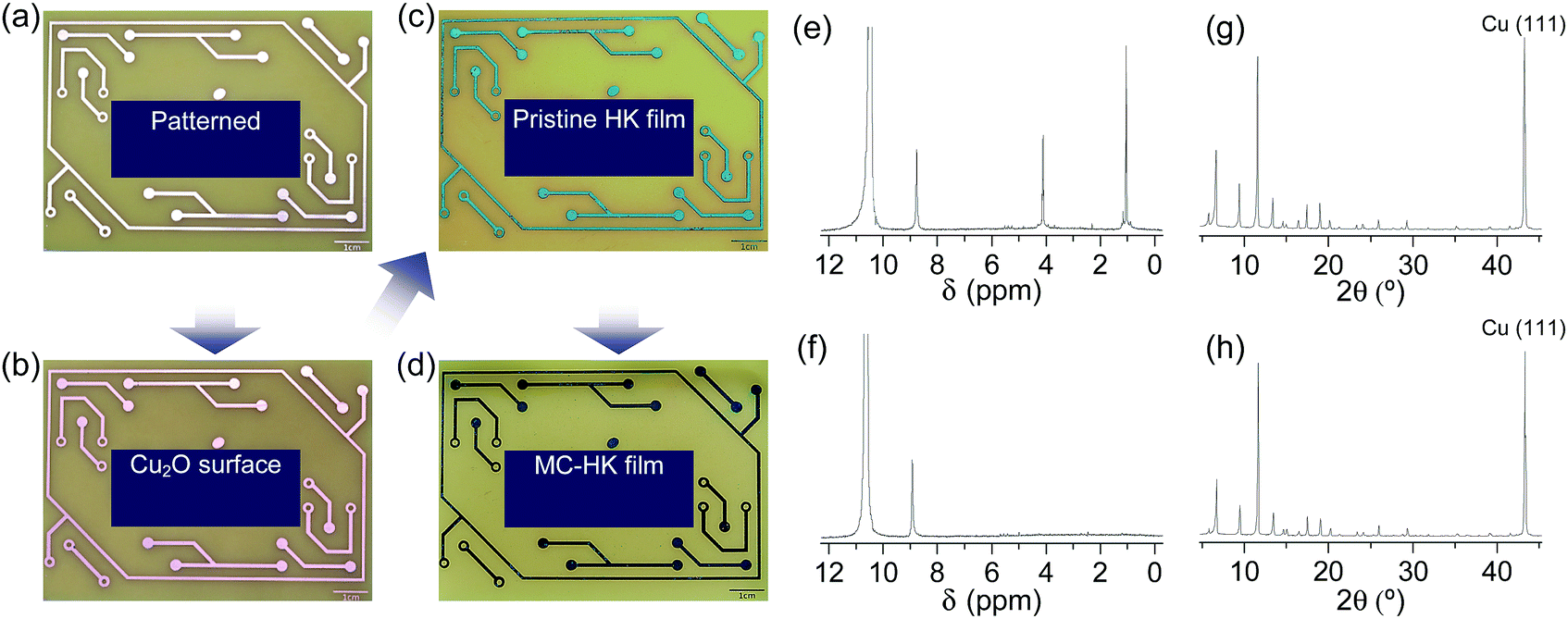 | ||
| Fig. 16 Photographs of the (a) patterned Cu substrate, (b) surface-oxidized Cu substrate (Cu2O/Cu), (c) pristine-HK film synthesized on the Cu substrate, and (d) DCM-treated pristine-HK film. (e and f) 1H NMR spectra and (g and h) XRD patterns of pristine-HK films (e and g) before and (f and h) after DCM treatment. Reprinted (adapted) with permission from ref. 68. Copyright (2015) American Chemical Society. | ||
4.2. Activation of MOF–polymer mixed matrices
MOF–polymer MMMs are an attractive platform to utilize MOFs in applications such as molecular sorption and separation.77–83 However, conventional TA is unsuitable for activating MOF–polymer MMMs when the polymer matrix has a low glass-transition or melting temperature.82–85 In this regard, moderate temperatures (of approximately 100–150 °C) have often been used for TA of MMMs to prevent the thermal damage of the polymer matrix, as illustrated in Fig. 17. However, moderate temperatures are not sufficient to completely remove solvents that form strong coordination bonds such as DMF (see below). By contrast, the activation of these MMMs by DCM or TCM treatment can be a promising alternative to fully activate OCSs of MOF–polymer MMMs, provided that the polymer matrix is chemically robust to DCM or TCM.70 As an example, a large-area HK-polyvinylidene fluoride MMM (ca. 10 × 10 cm2) was treated with TCM at room temperature (approximately 25 °C) (hereafter this sample is denoted as TCM25-MMM). TA of HK MMMs at 105, 150, and 200 °C under vacuum (hereafter these samples are denoted as TA105-MMM, TA150-MMM, and TA200-MMM, respectively) was also examined for comparison. The 1H NMR spectrum of TA105-MMM revealed that the thermal energy of 105 °C was not sufficient to remove the strongly coordinated DMF, although both the MOF and polymer matrix were well preserved (Fig. 18a and b). A considerable amount of DMF still remained in the TA150-MMM sample even though TA was conducted at 150 °C. A temperature of at least 200 °C was required to completely remove the coordinated DMF, as was observed for TA200-MMM. In this case, however, the polymer matrix hardened and eventually fragmented with serous deformation, as displayed in the photographs in Fig. 18c and d. In contrast, treatment with TCM completely removed the strongly coordinated DMF without deformation of the MMM (see Fig. 18). Notably, the Brunauer–Emmett–Teller (BET) surface area of TCM25-MMM (approximately 2000 m2 g−1) was much higher than those of TA105-MMM and TA150-MMM (both approximately 1400 m2 g−1), as indicated in Fig. 19. As predicted, TA200-MMM exhibited a very low surface area of 60 m2 g−1, which was ascribed to the polymer melting and then blocking the pores on the surface of the MOF crystals.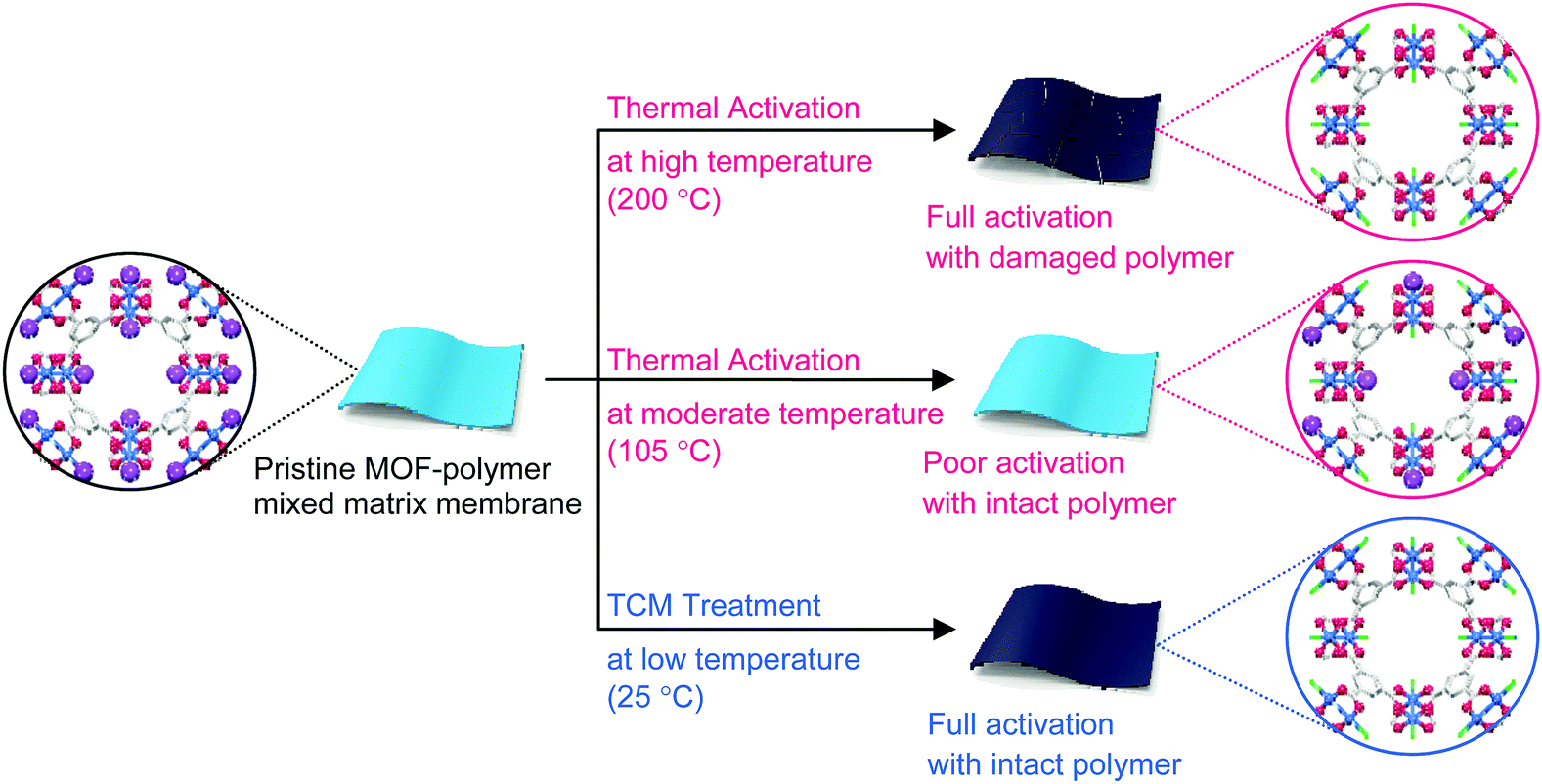 | ||
| Fig. 17 Schematics illustrating the activation of the paddle wheel-like (CuII)2 nodes within HKUST-1 by TCM. Hydrogen atoms bound to the carbon atoms in the benzene moieties are omitted for clarity. Reprinted (adapted) with permission from ref. 70. Copyright (2018) American Chemical Society. | ||
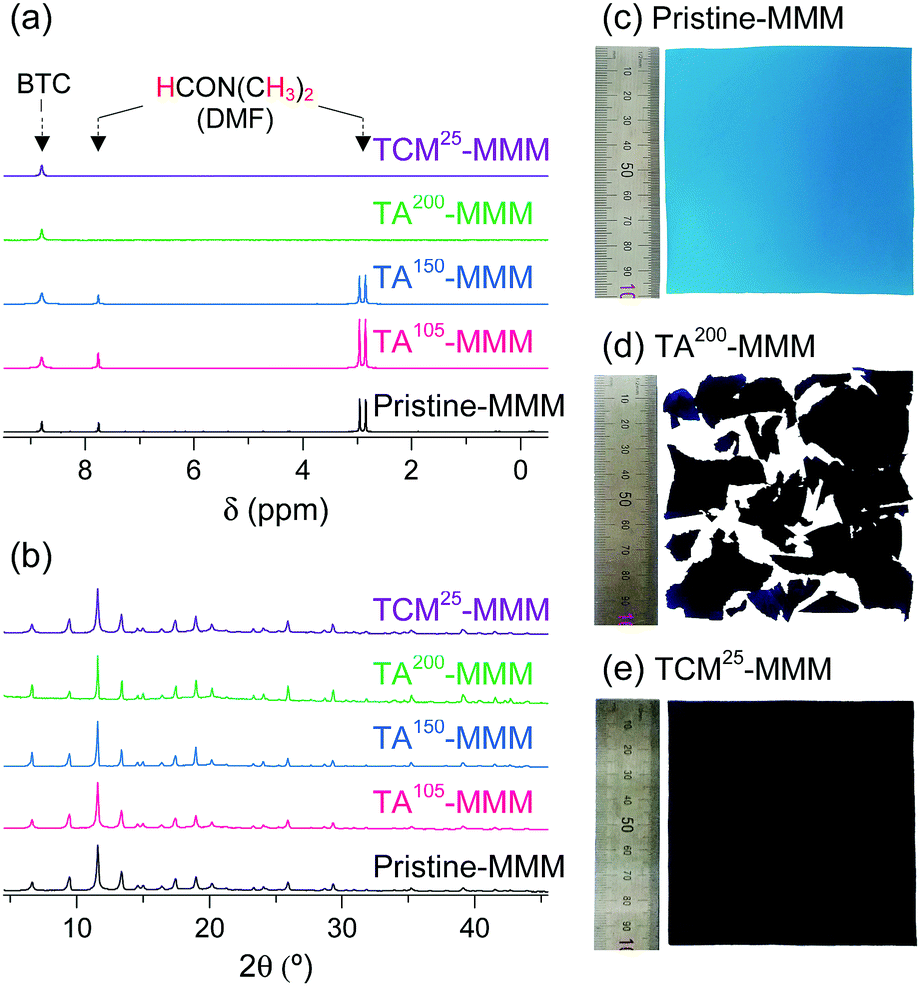 | ||
| Fig. 18 (a) 1H NMR spectra and (b) PXRD patterns of pristine-, TA105-, TA150-, TA200-, and TCM25-MMM. Photographs of (c) pristine-MMM, (d) TA200-MMM, and (e) TCM25-MMM. Reprinted (adapted) with permission from ref. 70. Copyright (2018) American Chemical Society. | ||
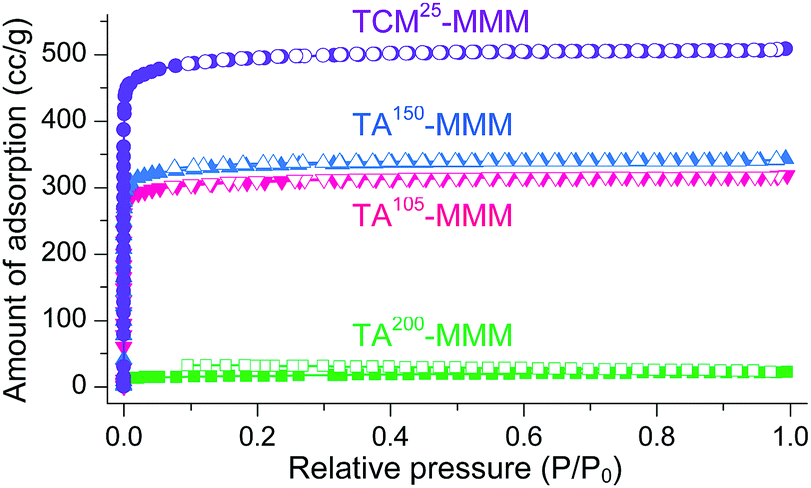 | ||
| Fig. 19 N2 adsorption/desorption isotherms of TA105-, TA150-, TA200-, and TCM25-MMM. The isotherms for the TCM25-MMM sample were measured after applying vacuum at room temperature for 4 h. Reprinted (adapted) with permission from ref. 70. Copyright (2018) American Chemical Society. | ||
The sorption efficiency of MOF–polymer MMMs following activation with chloromethanes was investigated. The sorption of H2O and NO by TCM25-MMM was examined; the former can be used in closed-cycle refrigerators as an environmentally friendly refrigerant and the latter is a harmful gas in flue emissions. The sorption performance of TA105-MMM was also examined for comparison. Consistent with the above BET results, TCM25-MMM displayed H2O and NO sorption capacities that were approximately twice those of TA105-MMM (Fig. 20). These results also demonstrate that activation by TCM treatment is an alternative strategy to TA for the effective activation of thermally unstable MOF–polymer composite materials.
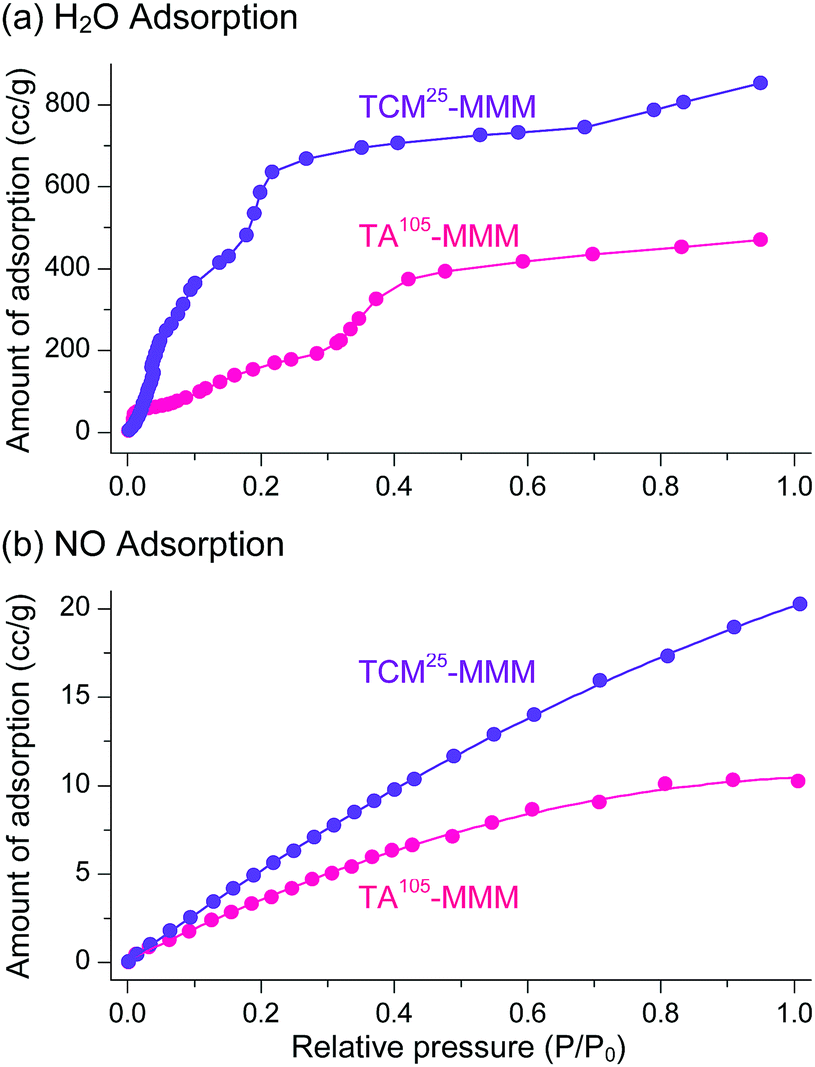 | ||
| Fig. 20 (a) H2O and (b) NO adsorption isotherms of TA105-MMM and TCM25-MMM. The isotherms for the TCM25-MMM sample were measured after applying vacuum at room temperature for 4 h. Reprinted (adapted) with permission from ref. 70. Copyright (2018) American Chemical Society. | ||
5. Conclusions
In this Feature Article, we discussed the possible formation of coordination bonds of chloromethanes with transition metal ions as well as the OCS-activation ability of chloromethanes to remove precoordinated solvent molecules from OCSs in MOFs. Chloromethanes are known as inert chemical substances and are widely used as solvents in a variety of chemical reactions even though they possess a lone pair of electrons that may be reactive or coordinate to transition metal ions. The chemical function of chloromethanes discussed in this article demonstrated their ability to form coordination bonds with metal ions even though their coordination strengths are relatively very weak. In the aspect of thermodynamics, the formation of DCM coordination bond and its subsequent coordination exchange behavior arise in equilibrium. Although the data are not displayed in this article, we have obviously observed an equilibrium between H2O and DCM coordination bonds, using a 1H NMR analytical technique. Thus, we infer that, in addition to the chloromethanes, molecules possibly to form weak coordination bond at metal center can engender a similar function for metal-node activation.In most cases, the coordination bonds in inorganic metal complexes are practically characterized by single-crystal X-ray diffraction measurements. However, weak coordination bonds are difficult to characterize by such X-ray diffraction measurements at room temperature because of their instantaneous dissociation. MOFs possessing both paddle wheel-type metal nodes and OCSs at the axial position, such as HKUST-1, are good model compounds to demonstrate the presence of weak coordination bonds because such bonds can be monitored by the vibrational modes of metal nodes. This article illustrated the presence of weak coordination bonds of Cl atoms using the intrinsic vibrational mode of the paddle wheel-type (CuII)2 node. For the examples discussed, the stretching vibration frequency of the (CuII)2 node was sensitive to the absence or presence and type of coordinated solvent molecules. The stretching vibration frequency of the EtOH-coordinated (CuII)2 node appeared at a Raman shift of approximately 185 cm−1, whereas that of the open-state (CuII)2 node appeared at approximately 228 cm−1. The presence of DCM or TCM coordination at (CuII)2 centers was evidenced by the (CuII)2 modes of DCM-wet-HK and TCM-wet-HK samples appearing at 213 and 217 cm−1, respectively. Considering the observed trend that a stronger coordination bond leads to a lower Raman shift of the (CuII)2 mode, it could be further supposed that the DCM coordination bond is stronger than the TCM coordination bond.
We envision that these observations represent the first step to revealing more fundamental scientific knowledge and developing more advanced applications for the limitless variety of inorganic complexes and MOFs. For instance, the presence of hydrogen bonding around metal centers can be directly monitored using Raman spectroscopy. We infer that stronger hydrogen bonding between a coordinated solvent molecule and non-coordinated (pore-filling) solvent molecules around the metal center can lead to weaker coordination strength of the coordinated solvent, and subsequently to stronger Cu–Cu binding strength, which consequently results in the higher Raman shift of the (CuII)2 node. In another example, the efficiency and turn-over number of a catalytic reaction may be raised if chloromethanes are used as the solvent in the catalytic reaction because chloromethanes that weakly coordinate and spontaneously dissociate can ensure that the metal centers are continuously in the open state and catalytically active.86–88 Altogether, it is remarkable how rapidly this field is developing at both fundamental and applied levels. Coupled with new directions to achieve advanced applications of these materials, progressive fundamental scientific understanding of unidentified weak chemical bonds will provide future potential to revolutionize a number of fields.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
NCJ gratefully acknowledges the support of a 2018 ChemComm Emerging Investigator Award and the financial support of Ministry of Science and ICT (MSIT) of Korea (NRF-2016R1A2B2014918).Notes and references
- M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782–1789 CrossRef PubMed.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- Y.-S. Bae, C. Y. Lee, K. C. Kim, O. K. Farha, P. Nickias, J. T. Hupp, S. T. Nguyen and R. Q. Snurr, Angew. Chem., Int. Ed., 2012, 51, 1857–1860 CrossRef CAS PubMed.
- J. B. DeCoste and G. W. Peterson, Chem. Rev., 2014, 114, 5695–5727 CrossRef CAS PubMed.
- D. Banerjee, A. J. Cairns, J. Liu, R. K. Motkuri, S. K. Nune, C. A. Fernandez, R. Krishna, D. M. Strachan and P. K. Thallapally, Acc. Chem. Res., 2015, 48, 211–219 CrossRef CAS PubMed.
- K. Adil, Y. Belmabkhout, R. S. Pillai, A. Cadiau, P. M. Bhatt, A. H. Assen, G. Maurin and M. Eddaoudi, Chem. Soc. Rev., 2017, 46, 3402–3430 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- Y. S. Bae and R. Q. Snurr, Angew. Chem., Int. Ed., 2011, 50, 11586–11596 CrossRef CAS PubMed.
- J. Yu, L. H. Xie, J. R. Li, Y. Ma, J. M. Seminario and P. B. Balbuena, Chem. Rev., 2017, 117, 9674–9754 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- M. Dinca and J. R. Long, Angew. Chem., Int. Ed., 2008, 47, 6766–6779 CrossRef CAS PubMed.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- S. Xiang, W. Zhou, Z. Zhang, M. A. Green, Y. Liu and B. Chen, Angew. Chem., Int. Ed., 2010, 49, 4615–4618 CrossRef CAS PubMed.
- Y. Peng, V. Krungleviciute, I. Eryazici, J. T. Hupp, O. K. Farha and T. Yildirim, J. Am. Chem. Soc., 2013, 135, 11887–11894 CrossRef CAS PubMed.
- L. C. Lin, J. Kim, X. Q. Kong, E. Scott, T. M. McDonald, J. R. Long, J. A. Reimer and B. Smit, Angew. Chem., Int. Ed., 2013, 52, 4410–4413 CrossRef CAS PubMed.
- Y. B. He, W. Zhou, G. D. Qian and B. L. Chen, Chem. Soc. Rev., 2014, 43, 5657–5678 RSC.
- A. J. Rieth, Y. Tulchinsky and M. Dinca, J. Am. Chem. Soc., 2016, 138, 9401–9404 CrossRef CAS PubMed.
- D. J. Levine, T. E. Runčevski, M. T. Kapelewski, B. K. Keitz, J. Oktawiec, D. A. Reed, J. A. Mason, H. Z. Jiang, K. A. Colwell and C. M. Legendre, J. Am. Chem. Soc., 2016, 138, 10143–10150 CrossRef CAS PubMed.
- E. D. Bloch, W. L. Queen, M. R. Hudson, J. A. Mason, D. J. Xiao, L. J. Murray, R. Flacau, C. M. Brown and J. R. Long, Angew. Chem., Int. Ed., 2016, 55, 8605–8609 CrossRef CAS PubMed.
- C.-Y. Gao, H.-R. Tian, J. Ai, L.-J. Li, S. Dang, Y.-Q. Lan and Z.-M. Sun, Chem. Commun., 2016, 52, 11147–11150 RSC.
- B. Liu, S. Yao, C. Shi, G. H. Li, Q. S. Huo and Y. L. Liu, Chem. Commun., 2016, 52, 3223–3226 RSC.
- M. S. Shah, M. Tsapatsis and J. I. Siepmann, Chem. Rev., 2017, 117, 9755–9803 CrossRef CAS PubMed.
- K. V. Kumar, K. Preuss, M. M. Titirici and F. Rodriguez-Reinoso, Chem. Rev., 2017, 117, 1796–1825 CrossRef CAS PubMed.
- N. S. Bobbitt, M. L. Mendonca, A. J. Howarth, T. Islamoglu, J. T. Hupp, O. K. Farha and R. Q. Snurr, Chem. Soc. Rev., 2017, 46, 3357–3385 RSC.
- E. Barea, C. Montoro and J. A. R. Navarro, Chem. Soc. Rev., 2014, 43, 5419–5430 RSC.
- P. Silva, S. M. F. Vilela, J. P. C. Tome and F. A. A. Paz, Chem. Soc. Rev., 2015, 44, 6774–6803 RSC.
- K. Schlichte, T. Kratzke and S. Kaskel, Microporous Mesoporous Mater., 2004, 73, 81–88 CrossRef CAS.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- D. Feng, Z. Y. Gu, J. R. Li, H. L. Jiang, Z. Wei and H. C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS PubMed.
- J. W. Liu, L. F. Chen, H. Cui, J. Y. Zhang, L. Zhang and C. Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC.
- S. Y. Moon, Y. Y. Liu, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2015, 54, 6795–6799 CrossRef CAS PubMed.
- H. Noh, Y. X. Cui, A. W. Peters, D. R. Pahls, M. A. Ortuno, N. A. Vermeulen, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 14720–14726 CrossRef CAS PubMed.
- P. Z. Li, X. J. Wang, J. Liu, J. S. Lim, R. Q. Zou and Y. L. Zhao, J. Am. Chem. Soc., 2016, 138, 2142–2145 CrossRef CAS PubMed.
- J. A. Johnson, B. M. Petersen, A. Kormos, E. Echeverria, Y. S. Chen and J. Zhang, J. Am. Chem. Soc., 2016, 138, 10293–10298 CrossRef CAS PubMed.
- E. D. Metzger, C. K. Brozek, R. J. Comito and M. Dinca, ACS Cent. Sci., 2016, 2, 148–161 CrossRef CAS PubMed.
- X. Liu, Y. Maegawa, Y. Goto, K. Hara and S. Inagaki, Angew. Chem., Int. Ed., 2016, 128, 8075–8079 CrossRef.
- L. Zhu, X. Q. Liu, H. L. Jiang and L. B. Sun, Chem. Rev., 2017, 117, 8129–8176 CrossRef CAS PubMed.
- Q. Yang, Q. Xu and H. L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- S. M. J. Rogge, A. Bavykina, J. Hajek, H. Garcia, A. I. Olivos-Suarez, A. Sepulveda-Escribano, A. Vimont, G. Clet, P. Bazin, F. Kapteijn, M. Daturi, E. V. Ramos-Fernandez, F. X. Llabresi Xamena, V. V. Speybroeck and J. Gascon, Chem. Soc. Rev., 2017, 46, 3134–3184 RSC.
- M. D. Korzynski and M. Dinca, ACS Cent. Sci., 2017, 3, 10–12 CrossRef CAS PubMed.
- C. D. Wu and M. Zhao, Adv. Mater., 2017, 1605446 CrossRef PubMed.
- J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 10294–10297 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- J. J. Gassensmith, J. Y. Kim, J. M. Holcroft, O. K. Farha, J. F. Stoddart, J. T. Hupp and N. C. Jeong, J. Am. Chem. Soc., 2014, 136, 8277–8282 CrossRef CAS PubMed.
- D. J. Wales, J. Grand, V. P. Ting, R. D. Burke, K. J. Edler, C. R. Bowen, S. Mintova and A. D. Burrows, Chem. Soc. Rev., 2015, 44, 4290–4321 RSC.
- M. G. Campbell, D. Sheberla, S. F. Liu, T. M. Swager and M. Dinca, Angew. Chem., Int. Ed., 2015, 54, 4349–4352 CrossRef CAS PubMed.
- Z. Y. Guo, X. Z. Song, H. P. Lei, H. L. Wang, S. Q. Su, H. Xu, G. D. Qian, H. J. Zhang and B. L. Chen, Chem. Commun., 2015, 51, 376–379 RSC.
- J. Wang, M. Jiang, L. Yan, R. Peng, M. J. Huangfu, X. X. Guo, Y. Li and P. Y. Wu, Inorg. Chem., 2016, 55, 12660–12668 CrossRef CAS PubMed.
- R. W. Huang, Y. S. Wei, X. Y. Dong, X. H. Wu, C. X. Du, S. Q. Zang and T. C. W. Mak, Nat. Chem., 2017, 9, 689–697 CrossRef CAS PubMed.
- N. C. Jeong, B. Samanta, C. Y. Lee, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 51–54 CrossRef CAS PubMed.
- P. Ramaswamy, N. E. Wong and G. K. Shimizu, Chem. Soc. Rev., 2014, 43, 5913–5932 RSC.
- L. Sun, M. G. Campbell and M. Dinca, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS PubMed.
- X. Meng, H. N. Wang, S. Y. Song and H. J. Zhang, Chem. Soc. Rev., 2017, 46, 464–480 RSC.
- Y. Yang, P. Shukla, S. Wang, V. Rudolph, X.-M. Chen and Z. Zhu, RSC Adv., 2013, 3, 17065–17072 RSC.
- J. E. Mondloch, O. Karagiaridi, O. K. Farha and J. T. Hupp, CrystEngComm, 2013, 15, 9258–9264 RSC.
- A. J. Howarth, A. W. Peters, N. A. Vermeulen, T. C. Wang, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 26–39 CrossRef CAS.
- B. F. Abrahams, B. F. Hoskins, D. M. Michail and R. Robson, Nature, 1994, 369, 727–729 CrossRef CAS.
- H. Li, M. Eddaoudi, T. L. Groy and O. Yaghi, J. Am. Chem. Soc., 1998, 120, 8571–8572 CrossRef CAS.
- C. J. Kepert and M. J. Rosseinsky, Chem. Commun., 1999, 375–376 RSC.
- G. Ferey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- A. P. Nelson, O. K. Farha, K. L. Mulfort and J. T. Hupp, J. Am. Chem. Soc., 2009, 131, 458–460 CrossRef CAS PubMed.
- M. R. Lohe, M. Rose and S. Kaskel, Chem. Commun., 2009, 6056–6058 RSC.
- W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443–6445 CrossRef CAS PubMed.
- H. K. Kim, W. S. Yun, M. B. Kim, J. Y. Kim, Y. S. Bae, J. Lee and N. C. Jeong, J. Am. Chem. Soc., 2015, 137, 10009–10015 CrossRef CAS PubMed.
- J. Bae, J. S. Choi, S. Hwang, W. S. Yun, D. Song, J. Lee and N. C. Jeong, ACS Appl. Mater. Interfaces, 2017, 9, 24743–24752 CAS.
- J. S. Choi, J. Bae, E. J. Lee and N. C. Jeong, Inorg. Chem., 2018 DOI:10.1021/acs.inorgchem.1028b00267 , accepted.
- M. R. Colsman, M. D. Noirot, M. M. Miller, O. P. Anderson and S. H. Strauss, J. Am. Chem. Soc., 1988, 110, 6886–6888 CrossRef CAS.
- T. D. Newbound, M. R. Colsman, M. M. Miller, G. P. Wulfsberg, O. P. Anderson and S. H. Strauss, J. Am. Chem. Soc., 1989, 111, 3762–3764 CrossRef CAS.
- M. R. Colsman, T. D. Newbound, L. J. Marshall, M. D. Noirot, M. M. Miller, G. P. Wulfsberg, J. S. Frye, O. P. Anderson and S. H. Strauss, J. Am. Chem. Soc., 1990, 112, 2349–2362 CrossRef CAS.
- D. M. Vanseggen, O. P. Anderson and S. H. Strauss, Inorg. Chem., 1992, 31, 2987–2990 CrossRef CAS.
- R. M. Badger, J. Chem. Phys., 1934, 2, 128–131 CrossRef CAS.
- G. P. Robertson, S. D. Mikhailenko, K. Wang, P. Xing, M. D. Guiver and S. Kaliaguine, J. Membr. Sci., 2003, 219, 113–121 CrossRef CAS.
- M. W. Anjum, F. Vermoortele, A. L. Khan, B. Bueken, D. E. De Vos and I. F. Vankelecom, ACS Appl. Mater. Interfaces, 2015, 7, 25193–25201 CAS.
- L. Cao, F. Lv, Y. Liu, W. Wang, Y. Huo, X. Fu, R. Sun and Z. Lu, Chem. Commun., 2015, 51, 4364–4367 RSC.
- R. Lin, L. Ge, S. Liu, V. Rudolph and Z. Zhu, ACS Appl. Mater. Interfaces, 2015, 7, 14750–14757 CAS.
- J. Dechnik, J. Gascon, C. J. Doonan, C. Janiak and C. J. Sumby, Angew. Chem., Int. Ed., 2017, 56, 9292–9310 CrossRef CAS PubMed.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- M. S. Denny, Jr. and S. M. Cohen, Angew. Chem., Int. Ed., 2015, 54, 9029–9032 CrossRef PubMed.
- J. Lim, E. J. Lee, J. S. Choi and N. C. Jeong, ACS Appl. Mater. Interfaces, 2018, 10, 3793–3800 CAS.
- J. C. Moreton, M. S. J. Denny and S. M. Cohen, Chem. Commun., 2016, 52, 14376–14379 RSC.
- J. B. DeCoste, M. S. J. Denny, G. W. Peterson, J. J. Mahle and S. M. Cohen, Chem. Sci., 2016, 7, 2711–2716 RSC.
- N. Pieper, C. Klaus-Mrestani, M. Schürmann, K. Jurkschat, M. Biesemans, I. Verbruggen, J. C. Martins and R. Willem, Organometallics, 1997, 16, 1043–1052 CrossRef CAS.
- J. Lewiński, W. Marciniak, Z. Ochal, J. Lipkowski and I. Justyniak, Eur. J. Inorg. Chem., 2003, 2753–2755 CrossRef.
- W. Bury, I. Justyniak, D. Prochowicz, Z. Wrobel and J. Lewinski, Chem. Commun., 2012, 48, 7362–7364 RSC.
| This journal is © The Royal Society of Chemistry 2018 |