
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dynamics of different steps of the photopyrolytic cycle of an eminent anticancer drug topotecan inside biocompatible lyotropic liquid crystalline systems†
Bibhisan Roy and
Partha Hazra*
Department of Chemistry, Indian Institute of Science Education and Research (IISER), Pune, 411008, Maharashtra, India. E-mail: p.hazra@iiserpune.ac.in; Fax: +91-20-2589-9790; Tel: +91-20-2590-8077
First published on 23rd December 2016
Abstract
In the present work, we have explored the dynamics of different steps of photopyrolytic processes of an eminent anticancer drug topotecan (TPT) inside the biocompatible reverse hexagonal (HII), gyroid (Ia3d) cubic and diamond (Pn3m) cubic lyotropic liquid crystalline (LLC) phases. Both steady state and time-resolved area normalized emission spectrum (TRANES) demonstrate the generation of an excited state zwitterionic (Z*) form of TPT in all LLC phases attributed to an excited state intermolecular proton transfer (ESPT) process from TPT (C* form) to the nearby water molecules. Using a kinetic model, we have calculated the dynamics of different steps, namely, proton transfer, recombination and dissociation of photopyrolytic processes of TPT inside different LLC phases. The calculated proton transfer dynamics of TPT are found to be severely retarded inside LLC phases compared to bulk water (32 ps), and they follows the order HII < Ia3d < Pn3m < H2O. The combined effect of slower solvation, hampered ‘Grotthuss’ proton transfer and the topological influence of LLC phases are believed to be responsible for the slower and different extent of ESPT dynamics. Geminate recombination dynamics are also found to be slower (nanosecond time scale) and follow the order HII < Ia3d < Pn3m. Slower geminate recombination dynamics mainly arise due to the lower dielectric constant and reduced water channel diameter of the LLC phases. The dissociation dynamics of TPT also get modulated inside LLC phases, and they are believed to be governed mainly by the topological influence and dielectric constant of the LLC phases.
Introduction
Biocompatible ‘smart materials’ exhibiting an organized host framework are at the heart of current material science revolution.1–7 Lipid based ‘soft’ and ‘wet’ lyotropic liquid crystalline (LLC) materials are recognized as the ‘smart materials’ owing to their remarkable topology,8–10 stimuli responsive phase change11,12 and multitude of applications in the biological,5,13 materials1–7 and pharmaceutical14,15 fields. These materials can be synthesized by the binary mixture of an amphiphilic surfactant (especially lipid) and water.16–19 Often, 1-linoleoyl-rac-glycerol (monolinolein; GML) lipids have been chosen to prepare the LLC materials due to their highly biocompatible nature owing to the presence of an ester group (Scheme 2) and their outstanding phase behaviour.16–19 Recently, lipid based reverse hexagonal (HII), gyroid (G) Ia3d and diamond (D) Pn3m phases (Scheme 1) received major attention due to their mimic with mitochondrial membrane,20,21 virally infected cells,22,23 eukaryotic cells,24 endoplasmic reticulum25 etc. Lipid based LLC phases have also established themselves as an efficient host for sustain release of bioactive guest molecules ranging from low molecular weight drugs26,27 to high molecular weight biomolecules (peptide, protein, nucleic acid)28 and dendrimer,29 due to their tuneable water nano-channel diameter.14 Topology of these LLC phase plays important role in biologically important chemical reactions; such as enzyme (HRP) kinetics,9 electrophoresis of oligonucleotides30 and hydrolysis of phosphate linkage.31 The topology of HII phase exhibits hexagonal array consisting one cylinder per corner in an unit cell.17 Each cylinder of HII phase is inverted type, where the aqueous channel is infinitely long straight rods enclosed by densely packed lipid (GML) surfactant (Scheme 1). However, Ia3d and Pn3m phases consist with minimal surface (mean area zero) arranged in ‘gyroid’ and ‘diamond’ fashion with the inverse bicontinuous cubic array.32 The prime difference between Ia3d and Pn3m phase lies in the orientation of water channel in the unit cell. In Ia3d phase lipid bilayer separates the water channels, which are intertwined, yet unconnected and joined in a three way junction at the angle of 120° (Scheme 1). Moreover, in Ia3d phase the two water channels are not identical to each other, but they are enantiomeric in nature.10,32 However, in Pn3m phase two autonomous water network are separated by the continuous layer of lipid molecules with four way junctions at the tetrahedral (109.5°) angle (Scheme 1), and in this phase both water channels are identical in nature.10,32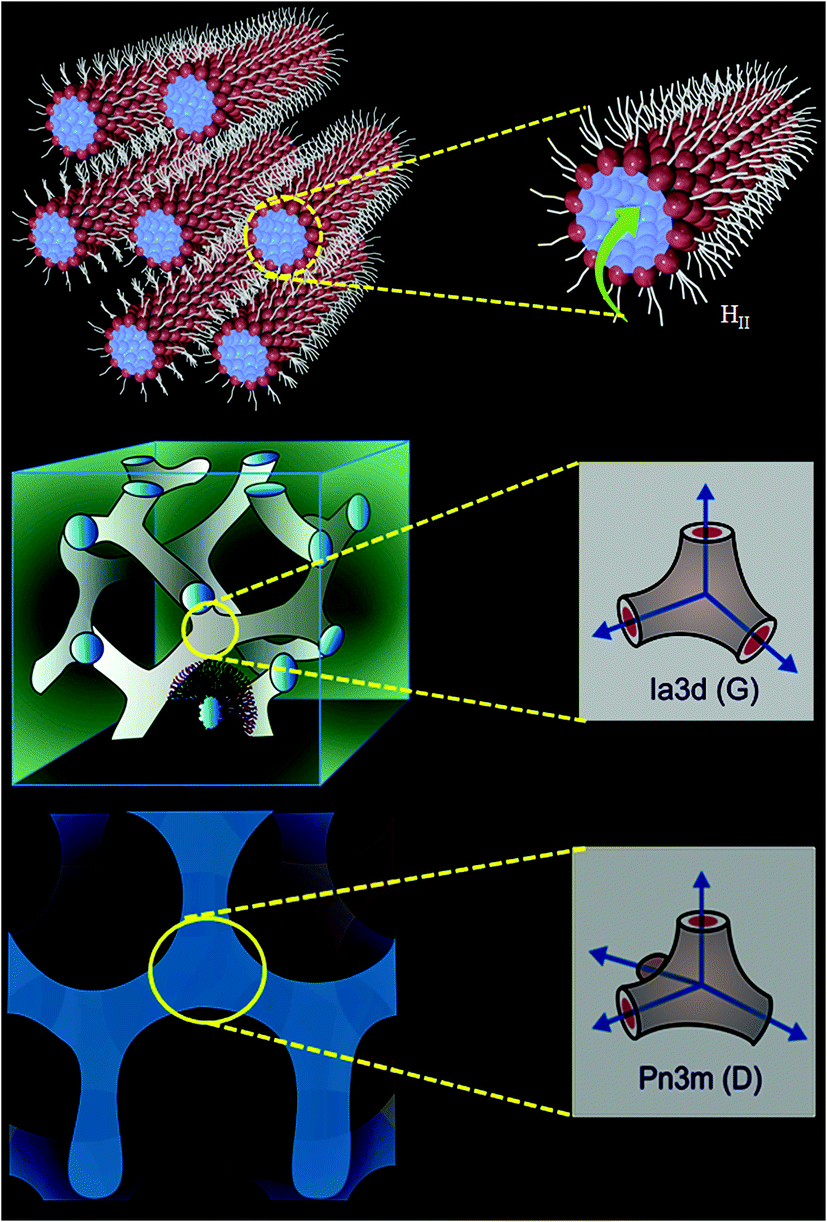 | ||
| Scheme 1 Schematic representation of HII, gyroid (G) Ia3d, diamond (D) Pn3m phases. The individual water nanochannel connection angle and diffusion path of ejected proton are shown right side. | ||
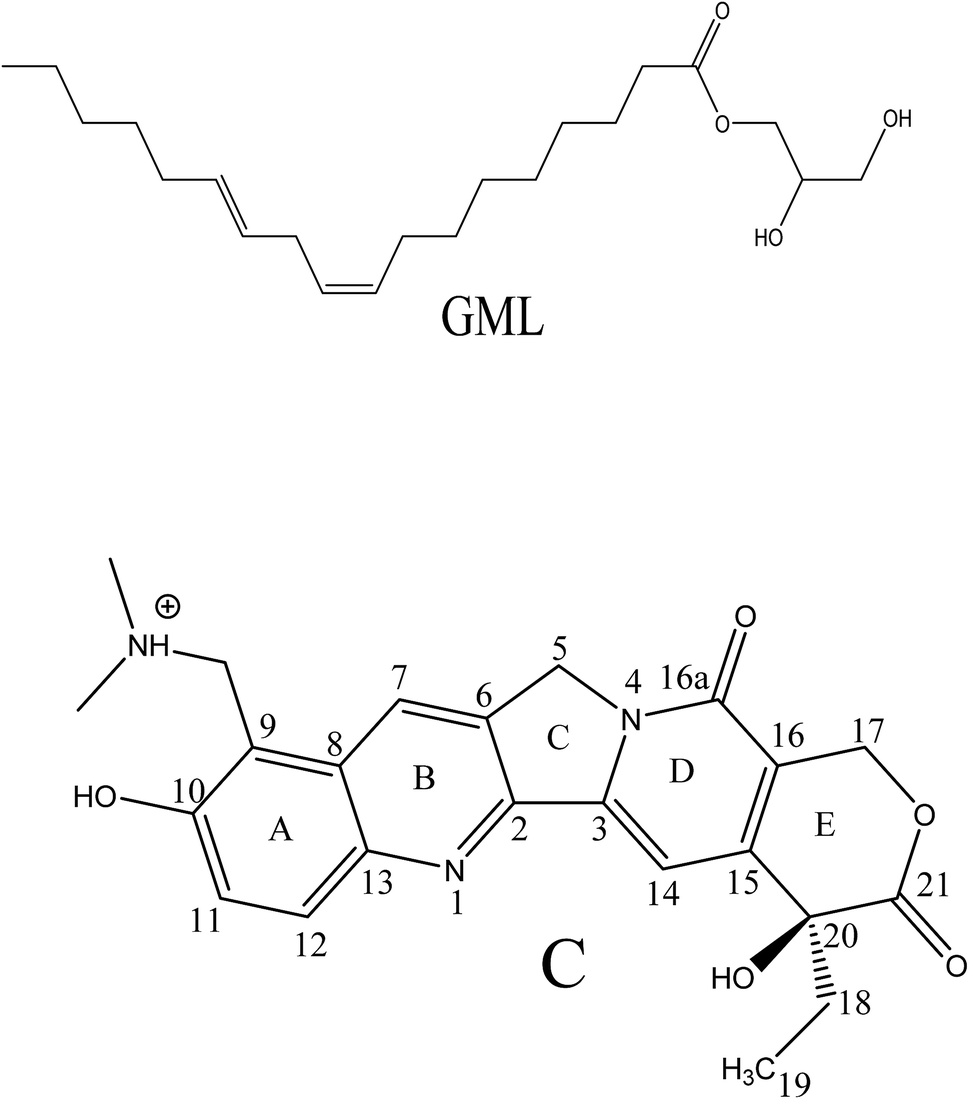 | ||
| Scheme 2 Chemical structures of monolinolein (GML) and cationic form (C) of topotecan (TPT). | ||
Chemotherapeutic agents received tremendous importance due to their potential pharmaceutical applications especially against the cancer threat.33 Camptothecin (CPT) and its derivative are found to be potential inhibitor for growth of tumour cells by the reduction of human topoisomerase I (Top1) activity through the Top1-DNA cleavable complex formation.34 Among CPT derivatives, TPT receives special importance owing to its high water solubility and minute toxic effect.34,35 TPT consists of five rings; among them four rings (A, B, D, E) are six-membered and one (C) is five-membered (Scheme 2). The 9 and 10 positions of ring A are occupied by dimethylaminomethylene and hydroxyl group, respectively. Interestingly, combination of 6-hydroxy quinoline (A and B rings) and 9-dimethylaminomethylene group gives rise to different protolytic forms of TPT in their ground as well as in excited state.36–38 In physiological conditions TPT exhibits equilibrium between enol (E), cationic (C) and zwitterionic (Z) forms in ground state (Scheme 2).36,38,39 Although TPT exists in different forms in ground state, the drug exhibits a single emission peak at ∼530 nm in the excited state responsible for Z* form in aqueous solution.36,38,39 The formation of Z* form in excited state is believed to be an outcome of the ‘excited state proton transfer (ESPT)’ from –OH group of C* form to the nearby water molecule.36,38,39 As ESPT process of TPT is sensitive to polarity,36,38,39 the ESPT process can be modulated inside confined media, where polarity is significantly different compared to bulk water. Hence, ESPT dynamics have been probed inside confined media, such as, calixarenes,40 cucurbituril,37 reverse micelle,39 etc. Although few works on ESPT dynamics of TPT inside some bio-mimicking and confined media have been reported,37,39,40 till date no efforts have been invested to explore the details photopyrolytic process of TPT inside the LLC phases, which are considered to be potential, biocompatible organized media for pharmaceutical and drug delivery applications.
Photophysics of potential anticancer drug inside confined environment has always been a budding topic of research. Considering the importance of this field, during last two decades several extensive efforts have been made to understand the photophysics of anticancer drug inside restricted environments, such as, micelle,41 reverse micelle,39 proteins environment,42 DNA43 etc. However, till now investigation of photophysics of the anticancer drugs inside lipid based lyotropic liquid crystalline materials are almost untouched. Owing to the biocompatible nature of LLC phases, few efforts have been made on drug delivery in recent.44,45 However, most of them are related with the release kinetics and maximizing the efficacy of the drug towards the specific site. Notably, incorporated drug molecules may undergo physical or chemical conversion in ground as well as in the excited state; hence, it is uttermost necessary to monitor the photophysical modulation of the drug inside LLC phases. Herein, we have probed photophysics of TPT inside liquid crystalline materials. To the best of our knowledge this is the first ever report on the details spectroscopic investigation of photopyrolytic processes and acid–base equilibria of an eminent anticancer drug, topotecan, inside the LLC materials. Besides that, for the first time we have investigated individual dynamics of different steps (initial proton transfer, geminate recombination and dissociation) involved in the photopyrolytic process of TPT in excited state. We strongly believe that our study will make significant contribution towards understandings of pharmacodynamic behavior of topotecan inside the different phases of LLC materials, which may help to promote the LLC materials as an efficient carrier for topotecan and other anti-cancer drugs.
Experimental section
Materials
1-Linoleoyl-rac-glycerol or monolinolein (GML; molecular weight 354.52 g mol−1, purity ≥ 97%) and topotecan hydrochloride (TPT; molecular weight 421.45 g mol−1, purity ≥ 98%; HPLC and enzymatic grade) purchased from Sigma Aldrich. Both of the chemicals were used as received. Milli-Q water (pH 6.5) was used for loading of TPT inside the water channels of monolinolein based HII, Ia3d, and Pn3m phases. Concentration of the TPT in each LLC phase have been calculated using the reported molar extinction co-efficient; ε380 nm = 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 at pH 6.5.46
000 M−1 cm−1 at pH 6.5.46
Synthesis of different liquid crystalline phases
We have synthesized monolinolein based type II reverse hexagonal (HII), gyroid (G) Ia3d and diamond (D) Pn3m phases by encapsulating 22 wt% of water (containing TPT) according to the phase diagram reported by Mezzenga and co-workers.16,19 For each phase, initially we have heated GML for few minutes at a particular temperature required for the respective phases. The required temperature for synthesizing HII, Ia3d and Pn3m phases are 358 K, 318 K and 325 K, respectively. TPT was dissolved in water and the concentration of the drug in each LLC phase was ∼23 μM. Before loading TPT containing water, it was slightly heated in order to remove dissolved O2 from the water. Then, the preheated water (containing TPT) was added to the liquefied GML with constant stirring. To avoid immediate solidification of the reaction mixture, we have added the drug containing water solution very slowly with extreme precaution. After water addition (containing TPT), we stirred the mixture for 15 minutes in order to get homogeneous mixture. The whole synthesis process for each phase was done under nitrogen atmosphere for avoiding the oxidation of GML as GML undergoes slow oxidation in presence of O2. After completion of the reaction, each sample was cooled to 298 K and it was allowed to equilibrate for 48 hours before running of the experiment. Once a particular phase is formed, then that particular phase is stable even at room temperature. The synthesized liquid crystalline phases are optically transparent, gel-like material.Methods
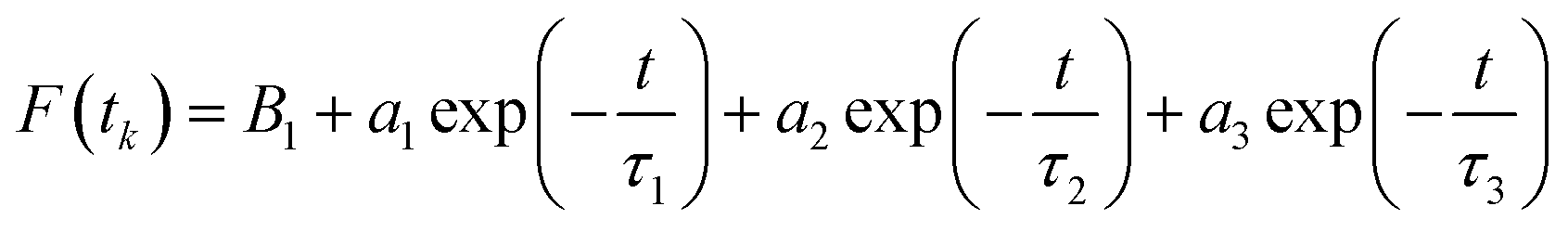 | (1) |
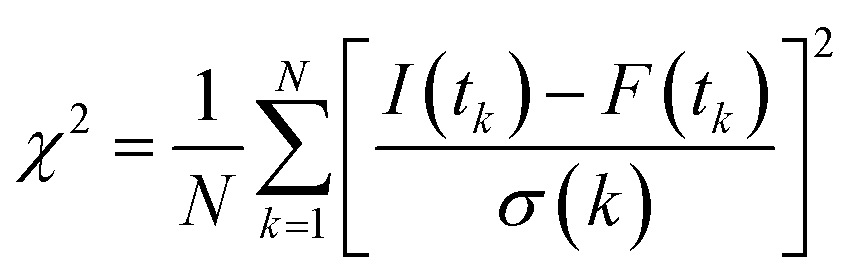 | (2) |
| Phases | λcoll (nm) | a1 | τ1 (ns) | a2 | τ2 (ns) | a3 | τ3 (ns) | τav (ns) | χ2 |
|---|---|---|---|---|---|---|---|---|---|
| Water | 570 | — | — | 0.13 | 0.633 | 0.87 | 5.93 | 5.24 | 1.01 |
| Pn3m | 570 | −0.28 | 0.129 | 0.23 | 2.8 | 0.5 | 5.35 | 3.32 | 0.99 |
| Ia3d | 570 | −0.28 | 0.199 | 0.2 | 2.79 | 0.52 | 5.38 | 3.42 | 0.95 |
| HII | 570 | −0.18 | 0.320 | 0.32 | 1.94 | 0.5 | 5.09 | 3.23 | 1.11 |
| Pn3m | 420 | 0.66 | 0.118 | 0.3 | 0.666 | 0.04 | 1.96 | 0.354 | 1.06 |
| Ia3d | 420 | 0.69 | 0.202 | 0.29 | 0.878 | 0.02 | 2.62 | 0.441 | 1.06 |
| HII | 420 | 0.73 | 0.305 | 0.24 | 1.22 | 0.03 | 3.01 | 0.605 | 1.18 |
Results and discussion
Characterization of liquid crystalline phases
Different LLC phases (containing TPT) can be distinguished by the visual inspection of the characteristics texture through the crossed polarizer in polarized light microscopy (PLM). Notably, the HII phase shows birefringence and colourful typical ‘fan’ like ‘cone’ textures (Fig. 1) in PLM image. The appearance of cone like typical ‘fan’ type texture in the PLM image corresponds to the reverse hexagonal liquid crystalline phase.17,47 However, Ia3d (G) and Pn3m (D) phases do not exhibit any texture (Fig. S1 in ESI†), as they cannot show any birefringence property due to the presence of cubic symmetry of those phases.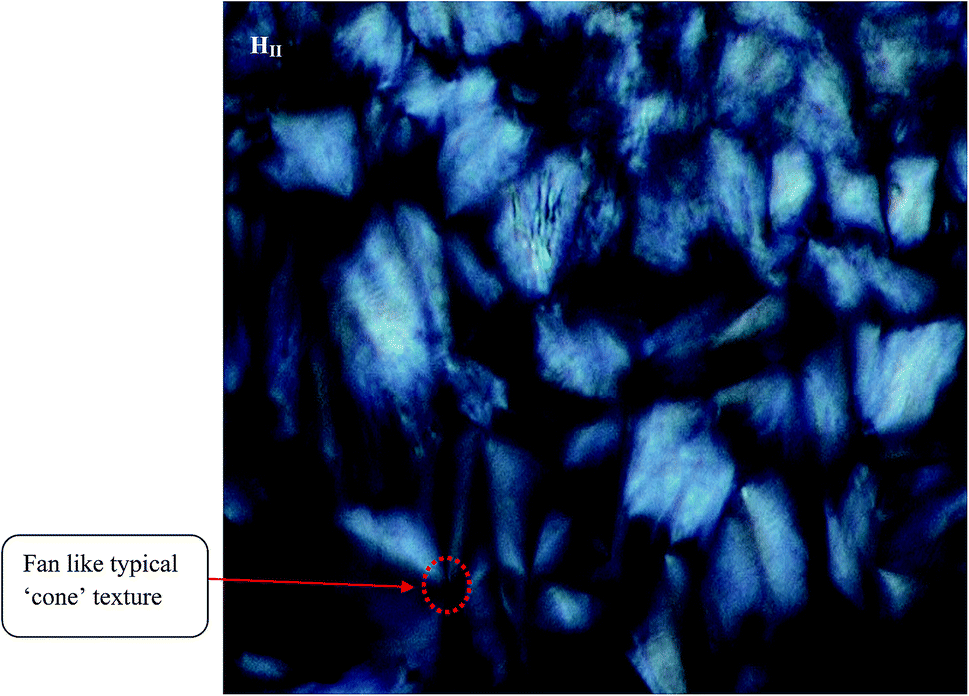 | ||
| Fig. 1 Polarized light microscopy (PLM) images of TPT loaded HII phase. The image has been taken at room (298 K) temperature. | ||
Steady state emission results
Emission spectrum of TPT in bulk water exhibits a single unstructured band at ∼532 nm attributed to the zwitterionic (Z*) form of the drug (Fig. 2).36–38 Here it is pertinent to mention that although the cationic (C) form of the drug is selectively excited at 380 nm (Fig. S2 in ESI†), the fluorescence appears from Z* form (532 nm) of TPT. This has been attributed to the excited state intermolecular proton transfer (ESPT) process by which water-assisted deprotonation of the 10-hydroxyl group takes place, and leads to the conversion of C* to Z* form of the drug.36–38 Thus, it is believe that the excited state conversion of C* to Z* is the dominating photophysics of TPT in aqueous solutions of neutral pH.36–38 However, in bulk monolinolein (GML) the emission spectrum exhibits a single emission peak at ∼425 nm (Fig. 2) corresponding to the cationic (C*) form of the drug.36–38 Since GML cannot act as a proton acceptor, TPT molecule does not undergo C* to Z* conversion following the ESPT process. Astonishingly, dual emission peak appears at 420 nm and 525 nm inside GML based LLC phases, which corresponds to C* and Z* form, respectively (Fig. 2). As the conversion from C* to Z* is the result of ESPT from the 10-hydroxyl group of TPT to nearby water molecule, this process will depend on the extent and nature of aqueous environments inside the LLC phases. Notably, inside each LLC phase individual water channel network and nature of the water molecules is distinctly different (Scheme 1), thereby, the efficiency for conversion from C* to Z*-TPT will be also different in each LLC phase. The quantitative idea of the efficiency of C* to Z* conversion can be obtained from the intensity ratio (IZ*/IC*) between Z* and C* forms of TPT. The most fascinating observation is the appearance of decreasing trend of ESPT efficiency going from water to HII, i.e. HII < Ia3d < Pn3m < H2O (Fig. 2). Since all the LLC phases are equally hydrated (22 wt% water), the observed slower ESPT efficiency of TPT may be due to the slower solvation, topological influence and lower dielectric constant of LLC phases, which will be discussed in the later part of manuscript.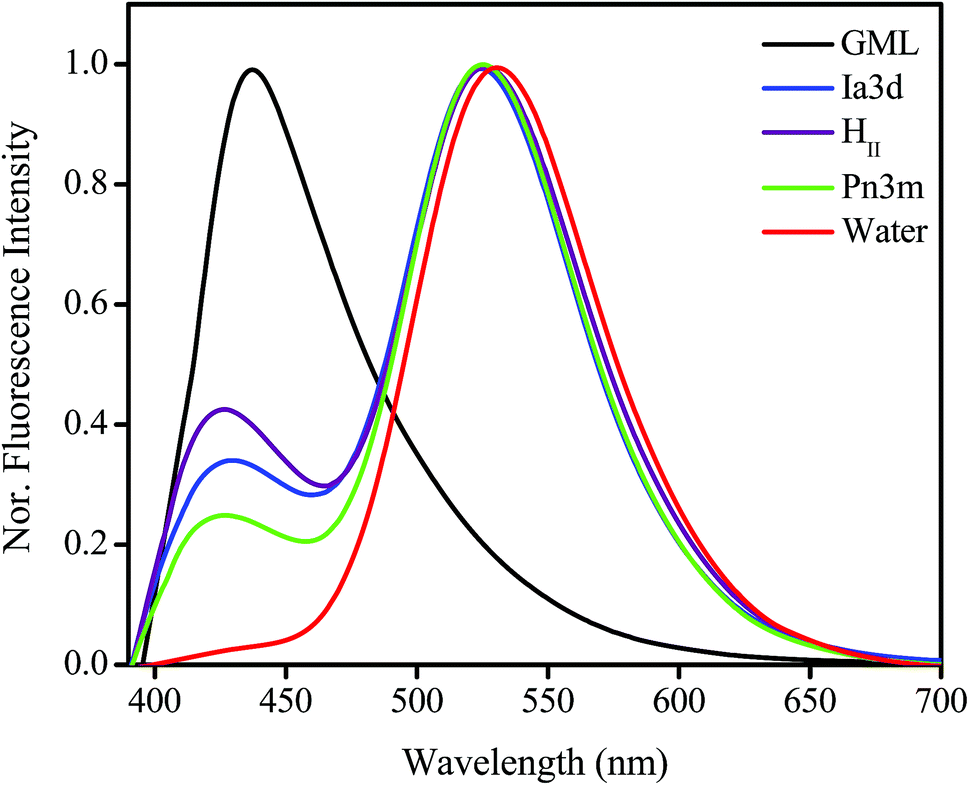 | ||
| Fig. 2 Normalized (normalized with respect to Z* emission maximum) steady state emission spectra of TPT in bulk water, GML and inside Pn3m, Ia3d and HII LLC phases (λex = 380 nm). | ||
Another intriguing observation in our result is the hypsochromic shift of Z* form of the drug inside LLC phases. The emission peak of Z*-TPT inside LLC phases is 7 nm blue shifted compared to bulk water (Fig. 2), suggesting that the drug molecules are sensing less polar environment inside LLC phases compared to bulk water. To get clear insight into the polarity inside the LLC phases, we have taken the emission spectra of Coumarin 343 (C-343), a polarity sensitive probe, by encapsulating inside LLC phases. The emission maxima of C-343 in HII, Ia3d and Pn3m phases closely lie at ∼480 nm, which is ∼10 nm blue-shifted compared to bulk water (Fig. S3 in ESI†). The comparison of the emission maxima of C-343 with deferent solvents48 suggests that polarity inside HII and cubic (Ia3d and Pn3m) phases are ethylene glycol type.
Time-resolved studies
Although steady state results provide a notion about the efficiency of ESPT process, the time-resolved fluorescence study can provide deep insight into the dynamics of ESPT process of TPT inside different LLC phases. Emission decay profiles of cationic (C*) and zwitterionic (Z*) form of TPT in water and inside the different LLC phases are shown in Fig. 3 and all the decay parameters are summarized in Table 1. The decay profile of Z* form (λcoll = 570 nm) of the drug in bulk water exhibits bi-exponential decay with an average lifetime of 5.24 ns (Table 1). Notably, the decay profile of TPT in bulk water is devoid of any rise component (Table 1 and Fig. 3), indicating that the ESPT dynamics is too fast (∼32 ps)36–38 to be detected by our TCSPC set up (IRF < 100 ps). The most intriguing observation of our work is the appearance of a rise component of Z* form of TPT (λcoll = 570 nm) inside different LLC phases (Table 1 and Fig. 3). For instance, the Z* form of TPT exhibits a rise component with a time scale of ∼320 ps in HII phase, which is ∼10 times slower than the reported rise component in bulk water (32 ps). Besides the HII phase, we have also observed rise component in Ia3d and Pn3m phases, which are also found to be considerably several times slower than that in bulk water (Table 1). Interestingly, the observed rise component of Z* form of TPT exactly matches with the decay component of C* (λcoll = 420 nm) form of TPT (Table 1) inferring that Z* is getting generated from C*-TPT by the ESPT process. This observation clearly indicates that the ESPT dynamics of TPT is getting modulated inside these lyotropic liquid crystalline systems.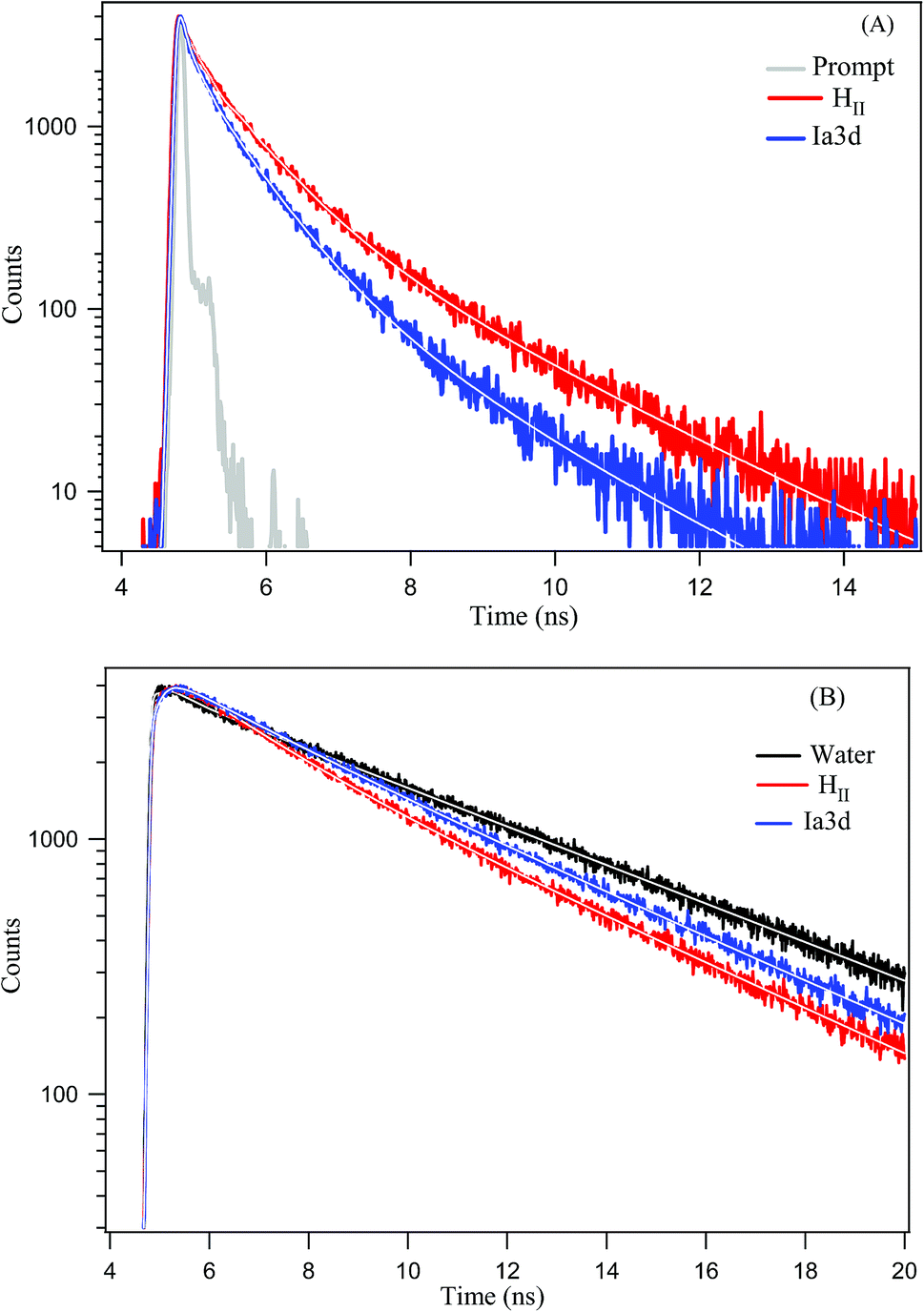 | ||
| Fig. 3 Overlay of fluorescence decays (λex = 375 nm) of TPT for (A) C* form (λcoll = 420 nm) and (B) Z* form (λcoll = 570 nm) in water and inside different LLC phases. In both the figures the horizontal axis is in linear scale and vertical axis is in logarithmic scale. While solid line indicates the fitting of experimental data by DAS6 analysis software. | ||
Although lifetime data furnishes an idea about the existence of multiple emissive species (C* and Z*) in all the LLC phases, time-resolved area normalized emission spectra (TRANES), a recently developed technique to explore the excited state species,49,50 provides further confirmation for the existence of two different emissive species in the excited state. The constructed TRANES of TPT inside different LLC phases are illustrated in the Fig. 4. It is clear from the figure that the intensity of C* form gradually decreases with concomitant rise of Z* emission intensity inside each LLC phase. This observation again reinforces our claim that the Z* form of TPT is generated at the cost of C* form. The time scale at which the Z* form starts appearing are 320 ps, 200 ps, 130 ps for HII, Ia3d and Pn3m phases, respectively (Fig. 4). Notably, the observed time scale are in excellent agreement with the rise time (Table 1, λcoll = 570 nm) of Z*-TPT inside different LLC phases. Moreover, we have observed an iso-emissive point at ∼490 nm inside each LLC phase, confirming the existence of two emitting species (C* and Z*) in the excited state of TPT.
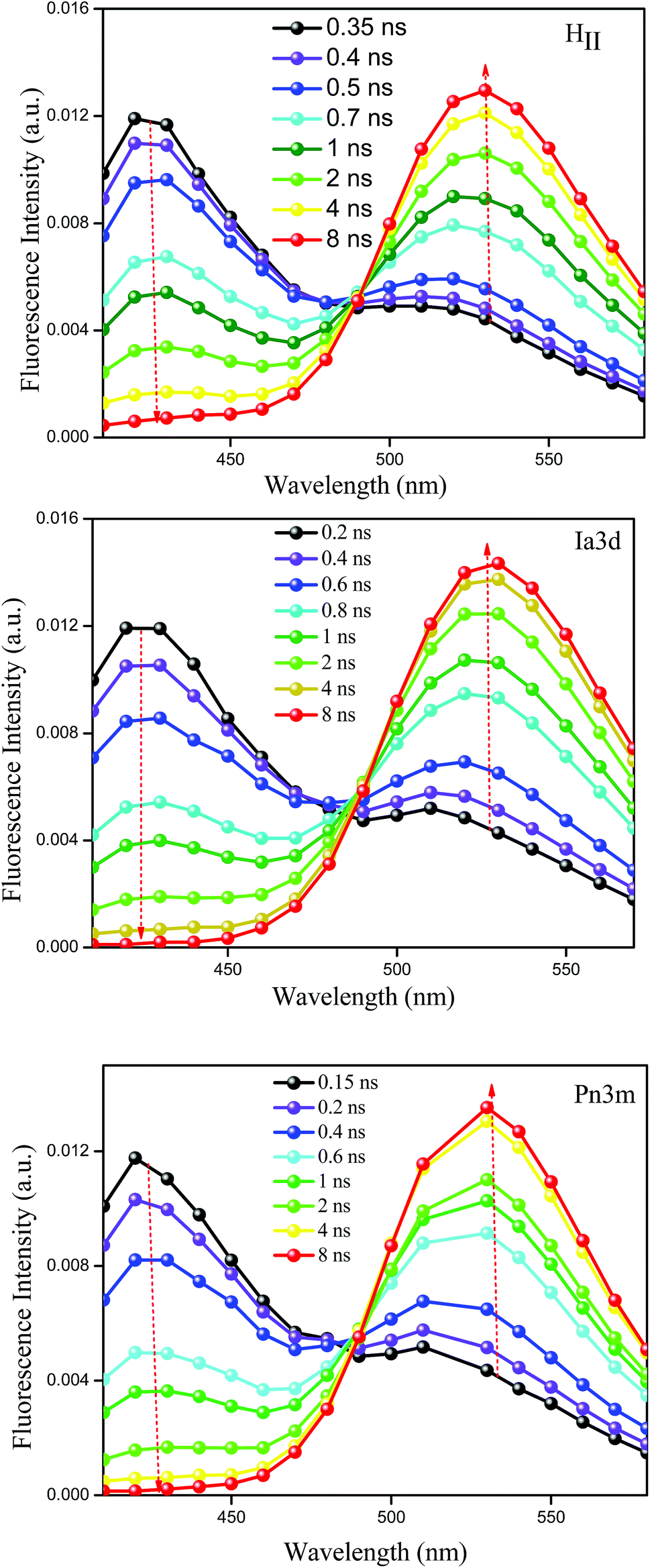 | ||
| Fig. 4 Time-resolved area normalized emission spectra (TRANES) of TPT encapsulated inside HII, Ia3d and Pn3m phases. Here the solid lines are not indicating the actual fittings, but they are used for visualization purpose. | ||
We have also focused on the dynamics of geminate ion-pair formation, because the ejected proton may come closer towards the conjugate base of the drug. For an effective achievement of geminate ion-pair the probe molecule must exhibit some important criteria. First of all, the dielectric constant (polarity) around the probe and ejected proton should be sufficiently low, so that they can come closer and correlate their movements.51 Secondly, the excited state life time of the conjugate base should be sufficiently long, so that the solvent separated geminate ion-pair can re-encounter.52 Interestingly, dielectric constant inside LLC phases is lower (ethylene glycol type) as evidenced from steady state and literature results,53 and the excited state lifetime of the conjugate base (Z*) is ∼6 ns inside LLC phases (τ3 in Table 1), which is long enough to re-encounter the solvent separated geminate ion-pair. Generally, geminate recombination process gives rise to a non-exponential long time decay tail in the time-resolved decay profile of the probe.54 However in the present scenario, non-exponential feature in the long time decay tail for C* form (λcoll = 420 nm) is not so prominent. Notably, the non-exponential long time decay tail is majorly dictated by the anionic charge of the conjugate base. This is prominent where conjugate base exhibits higher anionic charges (such as conjugate base of hydroxypyrene-1,3,6-trisulfonic acid (HPTS),55 which contains four negative charges), so that they can drag the ejected proton easily by the Coulomb attraction. But, singly anionic charged conjugate base of TPT can't drag the ejected proton as efficiently as that of HPTS.
Excited state kinetics of TPT inside LLC phases
Now, we focus on the kinetics of excited state proton transfer reaction of TPT inside different LLC phases. Here, it is necessary to mention that the time-resolved studies can provide an idea about the number of kinetically coupled species in excited state. For example, bi-exponential and tri-exponential decay profiles infer the presence of two and three kinetically coupled species in the excited state during ESPT process, respectively.51,56 Interestingly in the present scenario, the decay profiles of C* (λcoll = 420 nm) and Z* (λcoll = 570 nm) forms are found to be best fitted by tri-exponential decay function inside each LLC phase (Table 1). To justify the use of tri-exponential fitting function, we have provided the parameters of bi-exponential fitting for C* (λcoll = 420 nm) and Z* (λcoll = 570 nm) forms of TPT (Table S1 in ESI†). It is clear from the fittings parameters (Tables 1 and S1†) that the χ2 (defined in Experimental section) values are superior for tri-exponential case. Tri-exponential fittings of the decay profiles clearly suggest the existence of three kinetically coupled species in excited state of TPT inside liquid crystalline systems. Out of the three kinetically coupled species, one is solvent separated conjugate base (Z*⋯H3O+) responsible for the geminate recombination process. Other two species are C* and Z* form of TPT affirmed from the life time components of C* form (τ2 of λcoll = 420 nm) and Z* form (τ3 of λcoll = 570 nm), respectively (Table 1). Based on this observation, the kinetics of ESPT process of TPT inside LLC phases can be represented by Scheme 3. The rate constants involved in the kinetic scheme (Scheme 3) have been determined using the corresponding decay components and amplitudes of C* and Z* form of TPT, and has been discussed in detail in note S2 in ESI.† Using the calculated rate constants, we have estimated the time constants for different processes (Table 2), such as, the time constants for excited state proton transfer (τPT = 1/kPT), recombination (τrec = 1/krec) and dissociation (τdis = 1/kdis) processes.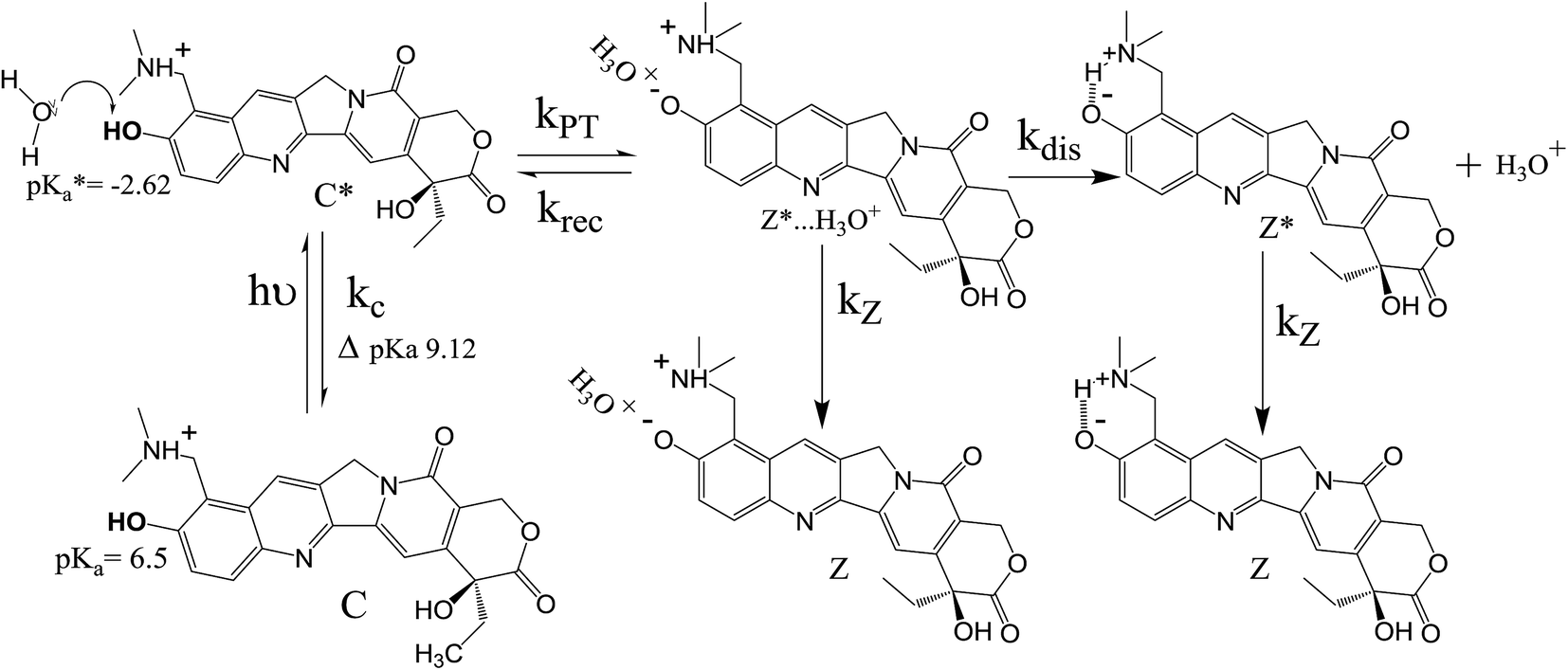 | ||
| Scheme 3 The photopyrolytic processes of topotecan depicting the proton transfer, geminate recombination and dissociation processes. | ||
| Phases | kPT (ps−1) | krec (ps−1) | kdis (ps−1) | τPT (ps) | τrec (ps) | τdis (ps) |
|---|---|---|---|---|---|---|
| Water | — | — | — | 32 | — | — |
| Pn3m | 6.29 × 10−3 | 16.6 × 10−4 | 1.83 × 10−3 | 158 | 603 | 547 |
| Ia3d | 3.82 × 10−3 | 7.93 × 10−4 | 1.29 × 10−3 | 262 | 1261 | 775 |
| HII | 2.66 × 10−3 | 3.3 × 10−4 | 0.68 × 10−3 | 375 | 2408 | 1234 |
The calculated time constants of initial proton transfer (τPT) inside Pn3m, Ia3d and HII phases are found to be ∼158 ps, 262 ps and 375 ps (Table 2), respectively, which are about 5 fold, 8 fold and 12 fold slower than that in bulk water (32 ps). An efficient ESPT process is mainly dictated by the solvation of ejected proton and conjugate base, dielectric constant of the surrounding media and availability of an adequate number of free water molecules around the probe.57–59 Out of these parameters, the most important one is solvation, which is mainly responsible for the stabilization of ejected proton and corresponding conjugate base (Z*) created just after the proton ejection.57–59 In one of our previous works, we have shown that the average solvation dynamics inside lipid base HII phase is ∼6674 ps, which is several order magnitude slower than that of bulk water.53 Bhattacharyya and co-workers also observed very slow solvation dynamics of ∼4500 ps inside Pluoronic-123 based cubic phase (Ia3d space group).60 Kim et al. reported slow dynamics of encapsulated water molecules inside lipidic cubic (Pn3m space group) phases.61 Thus, water dynamics inside LLC phases are slower compared to bulk water. As proton transfer activity heavily depends on the solvation of the water molecules, therefore, the retarded solvation dynamics inside different LLC phases plays a significant role for the appearance of slow proton transfer dynamics. Secondly, an efficient ESPT process also requires large number of available free water molecules (as proton acceptor and for solvation) around the probe molecules.57–59 However, the number of available free water molecules vastly reduces inside the lipid based LLC phases due to the presence of ‘bound water’ molecules as a result of H-bonding with the –OH groups of GML.53 Hence, the reduction of free water molecules inside liquid crystalline phases leads to slowing down of the ESPT dynamics of TPT. Thirdly, the decreased polarity (dielectric constant) inside the LLC phases slows down the ESPT dynamics of TPT.
In addition to these factors, proton transfer dynamics of TPT may also depend on the topology and the water channel diameters of the lyotropic liquid crystalline systems. As ESPT dynamics of TPT is highly sensitive to water content, the ESPT dynamics should be identical in each LLC phase, since they contain equal amount of water (22 wt%) molecules. However, the observed proton transfer dynamics inside different LLC phases follows the order as HII < Ia3d < Pn3m < H2O (Table 2). This means that either topology or water channel diameter or probably combination of both are playing significant role during ESPT process of the drug. Topology of HII phase exhibits inverted hexagonal cylinder (P6mm space group), where the aqueous channel is enclosed by densely packed infinitely long straight rods (Scheme 1).17 However, Pn3m and Ia3d cubic phases exhibit one set of bicontinuous lipid bilayer separating two sets of 3D water channels with tetra fold (109.5°) and tri-fold (120°) connectivity respectively (Scheme 1).10,32 The water channel in both cubic phases is unconnected, which is recently also visualized by Cryo-Electron Tomography (CET) technique.10 Notably, the water channels in Ia3d phase are associated with chirality and the two regions are enantiomeric with the non-identical nature; whereas, the water nano-channels in Pn3m phase are identical.10,32 Thus, from the architecture of different LLC phases, it is clear that HII phase provides most restricted pathway during ‘Grotthuss’ proton diffusion process owing to its cylindrical topology (Scheme 1). Interestingly, Pn3m phase can provide the faster pathway for proton diffusion due to tetra fold (109.5°) water channel connectivity than the tri-fold connectivity (120°) in Ia3d phase (Scheme 1, right handed images). Hence, ‘Grotthuss’ proton diffusion should follow the order as HII < Ia3d < Pn3m based on the topology of the LLC phases. At this stage, the influence of water channel diameter can't be ignored, as ESPT process proceeds following the proton diffusion pathway. The water channel diameter of HII, Ia3d and Pn3m phases varies from ∼2 nm to 3.5 nm, and follows the order as Pn3m > Ia3d > HII.9 This means, among all the LLC phases, the diffusion of the proton would be more preferable in Pn3m phase, followed by Ia3d and HII phases. However, we believe that in the present scenario the influence of channel diameter is not significant, as the size of TPT (0.13 nm) is significantly smaller compared to water channel diameter of any of the LLC phase. Moreover at this point, one may argue that viscosity may play a significant role, as proton transfer process is diffusion controlled. It is true that the macroscopic viscosity of this kind of gel material is large; however, the local viscosity inside the water filled pores is very low. As for example, local viscosity of water pores in polyacrylamide (PAA) gel is very low with respect to the bulk viscosity of the PAA gel.62 Even, we observed that the microviscosity of “quasi free” water region inside HII mesophase is 0.94 cP.38 Thus, we believe that the local viscosity inside the water pores of all LLC phases are very low compared to the bulk viscosity of the gel and will not play significant role in the proton transfer reaction dynamics.
Another intriguing observation of our results is the appearance of geminate recombination dynamics of TPT inside the liquid crystalline systems, which is absent in bulk water. The absence of proton recombination dynamics of TPT in bulk water is probably due to less efficiency 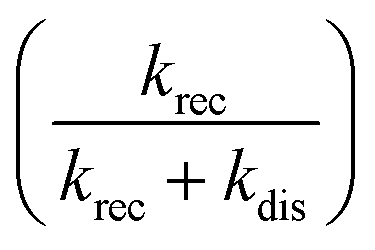 of ion-pair recombination process in bulk water. The calculated recombination dynamics (τrec) inside Pn3m, Ia3d and HII phases are about ∼600 ps, 1.26 ns and 2.4 ns (Table 2), respectively. As proton recombination repopulates the C* form in excited state, it enhances the fluorescence intensity at longer time-scale, and lead to the long non-exponential decay component of C* form.55,63 Interestingly, we have observed non-exponential long time fluorescence tail in the decay pattern of C* form (λcoll = 420 nm) of TPT, which is a fingerprint for proton recombination process in LLC phases. The recombination mainly arises due to the close proximity of the ion pair, which is mainly controlled by the reduced dielectric constant inside the liquid crystalline systems. Lower dielectric constant (polarity) favours the electrostatic interaction between ejected proton and the conjugate base; hence, accelerates the recombination process. It may be recalled that the dielectric constant of lipid based lyotropic liquid crystalline systems are ethylene glycol type. Hence, the lower dielectric constant (or polarity) favours the electrostatic interaction between the ejected proton and the conjugate base, resulting in proton recombination inside LLC phases. Channel diameter of the LLC phases may also influence the recombination dynamics. We have already mentioned that the channel diameter of LLC phases follows the order as Pn3m > Ia3d > HII.9 Because of the smallest channel diameter in HII phase, the ejected proton and conjugate base (Z*) stay close and they are hemmed tightly by the water molecules in HII phase. However, the relative distance between ejected proton and the conjugate base (Z*) will be long enough owing to the wider channel diameter in Ia3d and Pn3m phases. Hence, based on the channel diameter the recombination dynamics should obey the order as Pn3m < Ia3d < HII. However, the observed recombination dynamics follow the order as HII < Ia3d < Pn3m, which is probably because of the outcome of non-covalent interaction between ejected proton and the hydroxyl groups of surfactant (GML). Smallest channel diameter of HII phase favours the non-covalent interaction between the ejected proton and the –OH groups of GML, and hence, reduces the recombination dynamic. Such type of interactions is less favourable in Ia3d phase and least favourable in Pn3m phase owing to their larger channel diameter. Thus, combination of dielectric constant and channel diameter are believed to be responsible for the observed order of recombination dynamics.
of ion-pair recombination process in bulk water. The calculated recombination dynamics (τrec) inside Pn3m, Ia3d and HII phases are about ∼600 ps, 1.26 ns and 2.4 ns (Table 2), respectively. As proton recombination repopulates the C* form in excited state, it enhances the fluorescence intensity at longer time-scale, and lead to the long non-exponential decay component of C* form.55,63 Interestingly, we have observed non-exponential long time fluorescence tail in the decay pattern of C* form (λcoll = 420 nm) of TPT, which is a fingerprint for proton recombination process in LLC phases. The recombination mainly arises due to the close proximity of the ion pair, which is mainly controlled by the reduced dielectric constant inside the liquid crystalline systems. Lower dielectric constant (polarity) favours the electrostatic interaction between ejected proton and the conjugate base; hence, accelerates the recombination process. It may be recalled that the dielectric constant of lipid based lyotropic liquid crystalline systems are ethylene glycol type. Hence, the lower dielectric constant (or polarity) favours the electrostatic interaction between the ejected proton and the conjugate base, resulting in proton recombination inside LLC phases. Channel diameter of the LLC phases may also influence the recombination dynamics. We have already mentioned that the channel diameter of LLC phases follows the order as Pn3m > Ia3d > HII.9 Because of the smallest channel diameter in HII phase, the ejected proton and conjugate base (Z*) stay close and they are hemmed tightly by the water molecules in HII phase. However, the relative distance between ejected proton and the conjugate base (Z*) will be long enough owing to the wider channel diameter in Ia3d and Pn3m phases. Hence, based on the channel diameter the recombination dynamics should obey the order as Pn3m < Ia3d < HII. However, the observed recombination dynamics follow the order as HII < Ia3d < Pn3m, which is probably because of the outcome of non-covalent interaction between ejected proton and the hydroxyl groups of surfactant (GML). Smallest channel diameter of HII phase favours the non-covalent interaction between the ejected proton and the –OH groups of GML, and hence, reduces the recombination dynamic. Such type of interactions is less favourable in Ia3d phase and least favourable in Pn3m phase owing to their larger channel diameter. Thus, combination of dielectric constant and channel diameter are believed to be responsible for the observed order of recombination dynamics.
The dissociation time constants are found to be 547 ps, 775 ps and 1234 ps inside Pn3m, Ia3d and HII phase (Table 2), respectively. Normally, dissociation dynamics in bulk water is very fast due to the ‘Grotthuss’ proton diffusion process.57,64 Here, in bulk water the dissociation dynamics of TPT can't be calculated, since the kinetic scheme is only valid for tri-exponential decay profile. Notably, the efficiency of ‘Grotthuss’ diffusion mainly depends on the number of available ‘free’ water molecules in confined medium.57,64 Inside LLC phases, the available ‘free’ water molecules reduces severely due to binding (H-bonding) of water molecules with –OH group of GML, and thereby, proton can't escape through the ‘Grotthuss’ diffusion process. As a result, we have observed slower dissociation dynamics in LLC phases. In order to understand the trend of dissociation dynamics inside LLC phases, we have employed Debye–Smoluchowski equation (eqn (1)),65 which will provide a relationship between dissociation rate constant (kdis) with diffusion constant and dielectric constant inside LLC phases.
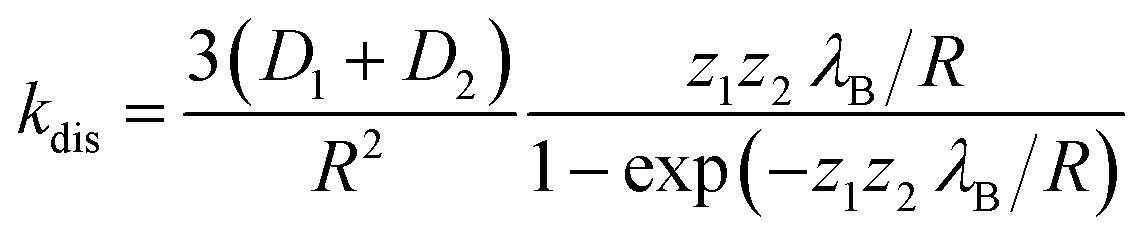 | (3) |
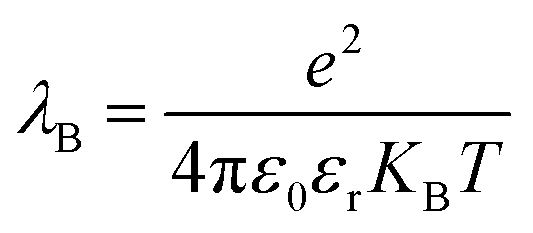 ; where e is the elementary charge, ε0 is the vacuum permittivity and εr is the dielectric constant. From the eqn (1) it is clear that lower dielectric constant and facile diffusion of ions (Z* and H+) favour the dissociation process. Notably, diffusion of ions entirely depends on topology of the LLC phases. As for example, inside HII phase both of the ions (Z* and H+) can diffuse throughout the cylinder (Scheme 1) as topology of HII phase is cylindrical.66 However, in Ia3d and Pn3m phase the ions diffuse through the two sets of water channels in non-communicating pathways owing to their intertwined autonomous water channel topology. Although, ions diffuses in non-communicating pathways in both the phases, ions get more space to diffuse in Pn3m phase owing to tetra-fold channel connectivity rather than tri-fold connectivity in Ia3d phase (different water channel connectivity have been shown in Scheme 1).10,32 Thus, the efficiency of proton diffusion is highest in Pn3m phase due to fascinating topology, followed by Ia3d and HII phase. In addition to topology, modulated dielectric constant inside LLC phases also takes significant role in dissociation dynamics. Lower dielectric constant brings the ion pair close enough, which hampers the dissociation process. Notably, dielectric constant significantly reduces inside nanochannel of LLC phases. Hence, dissociation process is more preferable in Pn3m phase and least preferable in HII phase. In summary, the fascinating topological feature along with dielectric constant play important roles during the proton dissociation dynamics through the ‘Grotthuss’ proton transfer process, and follows the dissociation dynamics as HII < Ia3d < Pn3m.
; where e is the elementary charge, ε0 is the vacuum permittivity and εr is the dielectric constant. From the eqn (1) it is clear that lower dielectric constant and facile diffusion of ions (Z* and H+) favour the dissociation process. Notably, diffusion of ions entirely depends on topology of the LLC phases. As for example, inside HII phase both of the ions (Z* and H+) can diffuse throughout the cylinder (Scheme 1) as topology of HII phase is cylindrical.66 However, in Ia3d and Pn3m phase the ions diffuse through the two sets of water channels in non-communicating pathways owing to their intertwined autonomous water channel topology. Although, ions diffuses in non-communicating pathways in both the phases, ions get more space to diffuse in Pn3m phase owing to tetra-fold channel connectivity rather than tri-fold connectivity in Ia3d phase (different water channel connectivity have been shown in Scheme 1).10,32 Thus, the efficiency of proton diffusion is highest in Pn3m phase due to fascinating topology, followed by Ia3d and HII phase. In addition to topology, modulated dielectric constant inside LLC phases also takes significant role in dissociation dynamics. Lower dielectric constant brings the ion pair close enough, which hampers the dissociation process. Notably, dielectric constant significantly reduces inside nanochannel of LLC phases. Hence, dissociation process is more preferable in Pn3m phase and least preferable in HII phase. In summary, the fascinating topological feature along with dielectric constant play important roles during the proton dissociation dynamics through the ‘Grotthuss’ proton transfer process, and follows the dissociation dynamics as HII < Ia3d < Pn3m.
Congregate literature reports revealed that there are studies on the encapsulation of anticancer and NSAID drugs inside the LLC phases. However, most of the focuses are invested on the release kinetics and maximising the efficacy of the drug towards the specific site.11,14,27,67–69 Notably, encapsulated drug molecules may undergo physical or chemical conversion in ground as well as in the excited state due to drastically different environments inside the LLC phases. Hence, it is of uttermost necessity to monitor the photophysical modulation of the encapsulated drug inside LLC phases, which has not yet been addressed so far. In the present study, we have investigated photopyrolytic process of an eminent anticancer drug topotecan inside LLC phases using fluorescence spectroscopic techniques. We have also consider the topological influence of LLC phases on different steps of photopyrolytic process such as initial proton transfer, recombination and dissociation processes of TPT. Until now there are no reports on recombination and dissociation dynamics of TPT in any bulk solvent or confined media. Therefore, to the best of our knowledge this is the first ever report on details spectroscopic investigation of photopyrolytic process of an eminent anticancer drug topotecan inside the LLC materials. We strongly believe that our study will make significant contribution towards understanding of pharmacodynamic behaviour of TPT inside the different phases of LLC materials, and it may help to promote the LLC materials as an efficient carrier for topotecan and other anti-cancer drugs.
Conclusion
In the current work, we have investigated the details of photopyrolytic processes of an eminent anticancer drug, topotecan inside different lyotropic liquid crystalline (LLC) systems. To the best of our knowledge this is the first ever report focusing detail investigation of photophysics of an anticancer drug (topotecan) inside biocompatible lyotropic liquid crystalline systems. The major and significant findings of the present study are summarized as follows. Steady state emission study infer the quantitative idea about the efficiency of ESPT process of TPT; following the order as HII < Ia3d < Pn3m < H2O. From the time-resolved measurement, we have observed a growth component (λcoll = 570 nm) in each LLC phase, which is not found in the bulk water due to fast (32 ps) ESPT dynamics of TPT. In addition, the time-resolved area normalized emission (TRANES) provides the direct spectroscopic evidence of for the conversion of Z*-TPT at the expense of C*-TPT form in excited state. Based on the time-resolved studies, we have proposed a kinetic scheme to analyse the different photopyrolytic processes of TPT inside different LLC phases. Calculated proton transfer dynamics is found to be several order slower than that of the bulk water (32 ps), and it follows the order as HII < Ia3d < Pn3m < H2O. Slower ESPT dynamics of TPT inside LLC phases is believed to be an outcome of the slower solvation dynamics, hampered ‘Grotthuss’ proton transfer and topological influence of the liquid crystalline phases. Interestingly, we observed the proton recombination (nano second time scale) of TPT inside the different LLC systems, which is not found in the bulk water. Recombination dynamics is found to pursue the order, as HII < Ia3d < Pn3m, which is mainly governed by dielectric constant and reduced water channel size inside LLC phases. The dissociation dynamics of TPT inside the LLC phases follows the order as HII < Ia3d < Pn3m. Modulation of dissociation dynamics inside the LLC phases may be attributed to topological influence along with reduced dielectric constant of the LLC phases.Acknowledgements
The authors are thankful to IISER-Pune for providing instrumental facilities. Authors are indebted to the reviewers for their valuable comments and suggestions.References
- E. Paineau, M.-E. M. Krapf, M.-S. Amara, N. V. Matskova, I. Dozov, S. Rouziere, A. Thill, P. Launois and P. Davidson, Nat. Commun., 2016, 7, 10271 CrossRef CAS PubMed.
- E. W. Kaler, Nat. Mater., 2005, 4, 11–12 CrossRef CAS.
- J. H. Ding and D. L. Gin, Chem. Mater., 2000, 12, 22–24 CrossRef CAS.
- G. S. Attard, P. N. Bartlett, N. R. B. Coleman, J. M. Elliott, J. R. Owen and J. H. Wang, Science, 1997, 278, 838–840 CrossRef CAS.
- J. J. Vallooran, S. Handschin, S. M. Pillai, B. N. Vetter, S. Rusch, H.-P. Beck and R. Mezzenga, Adv. Funct. Mater., 2016, 26, 181–190 CrossRef CAS.
- E. Pebay-Peyroula, G. Rummel, J. P. Rosenbusch and E. M. Landau, Science, 1997, 277, 1676–1681 CrossRef CAS PubMed.
- T. Kijima, T. Yoshimura, M. Uota, T. Ikeda, D. Fujikawa, S. Mouri and S. Uoyama, Angew. Chem., Int. Ed., 2004, 43, 228–232 CrossRef CAS PubMed.
- A. Zabara, R. Negrini, O. Onaca-Fischer and R. Mezzenga, Small, 2013, 9, 3602–3609 CrossRef CAS PubMed.
- W. Sun, J. J. Vallooran and R. Mezzenga, Langmuir, 2015, 31, 4558–4565 CrossRef CAS PubMed.
- D. Demurtas, P. Guichard, I. Martiel, R. Mezzenga, C. Hebert and L. Sagalowicz, Nat. Commun., 2015, 6, 8915 CrossRef CAS PubMed.
- R. Negrini and R. Mezzenga, Langmuir, 2011, 27, 5296–5303 CrossRef CAS PubMed.
- E. Nazaruk, M. Szlęzak, E. Górecka, R. Bilewicz, Y. M. Osornio, P. Uebelhart and E. M. Landau, Langmuir, 2014, 30, 1383–1390 CrossRef CAS PubMed.
- P. G. Hartley, M. R. Alderton, R. M. Dawson and D. Wells, Bioconjugate Chem., 2007, 18, 152–159 CrossRef CAS PubMed.
- R. Negrini and R. Mezzenga, Langmuir, 2012, 28, 16455–16462 CrossRef CAS PubMed.
- T. C. Le, X. Mulet, F. R. Burden and D. A. Winkler, Mol. Pharm., 2013, 10, 1368–1377 CrossRef CAS PubMed.
- R. Mezzenga, P. Schurtenberger, A. Burbidge and M. Michel, Nat. Mater., 2005, 4, 729–740 CrossRef CAS PubMed.
- N. Garti, D. Libster and A. Aserin, Food Funct., 2012, 3, 700–713 CAS.
- C. V. Kulkarni, W. Wachter, G. Iglesias-Salto, S. Engelskirchen and S. Ahualli, Phys. Chem. Chem. Phys., 2011, 13, 3004–3021 RSC.
- R. Mezzenga, C. Meyer, C. Servais, A. I. Romoscanu, L. Sagalowicz and R. C. Hayward, Langmuir, 2005, 21, 3322–3333 CrossRef CAS PubMed.
- Y. Deng, M. Marko, K. F. Buttle, A. Leith, M. Mieczkowski and C. A. Mannella, J. Struct. Biol., 1999, 127, 231–239 CrossRef CAS PubMed.
- C. V. Kulkarni, Nanoscale, 2012, 4, 5779–5791 RSC.
- Z. A. Almsherqi, S. D. Kohlwein and Y. Deng, J. Cell Biol., 2006, 173, 839–844 CrossRef CAS PubMed.
- B. G. Tenchov, R. C. MacDonald and D. P. Siegel, Biophys. J., 2006, 91, 2508–2516 CrossRef CAS PubMed.
- K. Larsson, K. Fontell and N. Krog, Chem. Phys. Lipids, 1980, 27, 321–328 CrossRef CAS.
- T. Landh, FEBS Lett., 1995, 369, 13–17 CrossRef CAS PubMed.
- E. Nazaruk, P. Miszta, S. Filipek, E. Górecka, E. M. Landau and R. Bilewicz, Langmuir, 2015, 31, 12753–12761 CrossRef CAS PubMed.
- R. Negrini, W.-K. Fong, B. J. Boyd and R. Mezzenga, Chem. Commun., 2015, 51, 6671–6674 RSC.
- A. Angelova, B. Angelov, R. Mutafchieva, S. Lesieur and P. Couvreur, Acc. Chem. Res., 2011, 44, 147–156 CrossRef CAS PubMed.
- L. Bitan-Cherbakovsky, D. Libster, A. Aserin and N. Garti, J. Phys. Chem. B, 2011, 115, 11984–11992 CrossRef CAS PubMed.
- N. Carlsson, A.-S. Winge, S. Engström and B. Åkerman, J. Phys. Chem. B, 2005, 109, 18628–18636 CrossRef CAS PubMed.
- S. Murgia, S. Lampis, P. Zucca, E. Sanjust and M. Monduzzi, J. Am. Chem. Soc., 2010, 132, 16176–16184 CrossRef CAS PubMed.
- A. L. Mackay, Nature, 1985, 314, 604–606 CrossRef CAS.
- G. M. Cragg, P. G. Grothaus and D. J. Newman, Chem. Rev., 2009, 109, 3012–3043 CrossRef CAS PubMed.
- A. G. Schultz, Chem. Rev., 1973, 73, 385–405 CrossRef CAS PubMed.
- I. K. Nordgren and A. Tavassoli, Chem. Soc. Rev., 2011, 40, 4307–4317 RSC.
- M. R. di Nunzio, Y. Wang and A. Douhal, J. Phys. Chem. B, 2012, 116, 8182–8190 CrossRef CAS PubMed.
- K. Gavvala, A. Sengupta, R. K. Koninti and P. Hazra, ChemPhysChem, 2013, 14, 3375–3383 CrossRef CAS PubMed.
- M. R. di Nunzio, Y. Wang and A. Douhal, J. Phys. Chem. B, 2012, 116, 7522–7530 CrossRef CAS PubMed.
- R. K. Koninti, K. Gavvala, A. Sengupta and P. Hazra, J. Phys. Chem. B, 2015, 119, 2363–2371 CrossRef CAS PubMed.
- G.-S. Wang, H.-Y. Zhang, F. Ding and Y. Liu, J. Inclusion Phenom. Macrocyclic Chem., 2010, 69, 85–89 CrossRef.
- M. El-Kemary, M. Gil and A. Douhal, J. Med. Chem., 2007, 50, 2896–2902 CrossRef CAS PubMed.
- M. Novaira, M. A. Biasutti, J. J. Silber and N. M. Correa, J. Phys. Chem. B, 2007, 111, 748–759 CrossRef CAS PubMed.
- K. M. Solntsev, E. N. Sullivan, L. M. Tolbert, S. Ashkenazi, P. Leiderman and D. Huppert, J. Am. Chem. Soc., 2004, 126, 12701–12708 CrossRef CAS PubMed.
- N. M. Correa and N. E. Levinger, J. Phys. Chem. B, 2006, 110, 13050–13061 CrossRef CAS PubMed.
- I. V. Gopich and N. Agmon, Phys. Rev. Lett., 2000, 84, 2730–2733 CrossRef CAS PubMed.
- N. Sanna, G. Chillemi, A. Grandi, S. Castelli, A. Desideri and V. Barone, J. Am. Chem. Soc., 2005, 127, 15429–15436 CrossRef CAS PubMed.
- J. Borné, T. Nylander and A. Khan, Langmuir, 2001, 17, 7742–7751 CrossRef.
- J. Kim, W. Lu, W. Qiu, L. Wang, M. Caffrey and D. Zhong, J. Phys. Chem. B, 2006, 110, 21994–22000 CrossRef CAS PubMed.
- A. S. R. Koti, M. M. G. Krishna and N. Periasamy, J. Phys. Chem. A, 2001, 105, 1767–1771 CrossRef CAS.
- A. S. R. Koti and N. Periasamy, J. Chem. Phys., 2001, 115, 7094–7099 CrossRef CAS.
- A. A. Freitas, F. H. Quina and A. A. L. Maçanita, J. Phys. Chem. A, 2011, 115, 10988–10995 CrossRef CAS PubMed.
- R. Chowdhury, A. Saha, A. K. Mandal, B. Jana, S. Ghosh and K. Bhattacharyya, J. Phys. Chem. B, 2015, 119, 2149–2156 CrossRef CAS PubMed.
- B. Roy, S. Satpathi, K. Gavvala, R. K. Koninti and P. Hazra, J. Phys. Chem. B, 2015, 119, 11721–11731 CrossRef CAS PubMed.
- A. Datta, S. Das, D. Mandal, S. K. Pal and K. Bhattacharyya, Langmuir, 1997, 13, 6922–6926 CrossRef CAS.
- N. Amdursky, R. Simkovitch and D. Huppert, J. Phys. Chem. B, 2014, 118, 13859–13869 CrossRef CAS PubMed.
- L. Giestas, C. Yihwa, J. C. Lima, C. Vautier-Giongo, A. Lopes, A. L. Maçanita and F. H. Quina, J. Phys. Chem. A, 2003, 107, 3263–3269 CrossRef CAS.
- K. Bhattacharyya, Acc. Chem. Res., 2003, 36, 95–101 CrossRef CAS PubMed.
- N. Nandi, K. Bhattacharyya and B. Bagchi, Chem. Rev., 2000, 100, 2013–2046 CrossRef CAS PubMed.
- L. M. Tolbert and K. M. Solntsev, Acc. Chem. Res., 2002, 35, 19–27 CrossRef CAS PubMed.
- U. Mandal, A. Adhikari, S. Dey, S. Ghosh, S. K. Mondal and K. Bhattacharyya, J. Phys. Chem. B, 2007, 111, 5896–5902 CrossRef CAS PubMed.
- J. Kim, W. Lu, W. Qiu, L. Wang, M. Caffrey and D. Zhong, J. Phys. Chem. B, 2006, 110, 21994–22000 CrossRef CAS PubMed.
- C. Caltagirone, A. M. Falchi, S. Lampis, V. Lippolis, V. Meli, M. Monduzzi, L. Prodi, J. Schmidt, M. Sgarzi, Y. Talmon, R. Bizzarri and S. Murgia, Langmuir, 2014, 30, 6228–6236 CrossRef CAS PubMed.
- S.-Y. Park, H. Jeong, H. Kim, J. Y. Lee and D.-J. Jang, J. Phys. Chem. C, 2011, 115, 24763–24770 CAS.
- O. F. Mohammed, D. Pines, J. Dreyer, E. Pines and E. T. J. Nibbering, Science, 2005, 310, 83–86 CrossRef CAS PubMed.
- M. Eigen, Angew. Chem., Int. Ed., 1964, 3, 1–19 CrossRef.
- I. Amar-Yuli, E. Wachtel, D. E. Shalev, H. Moshe, A. Aserin and N. Garti, J. Phys. Chem. B, 2007, 111, 13544–13553 CrossRef CAS PubMed.
- M. Cohen-Avrahami, A. I. Shames, M. F. Ottaviani, A. Aserin and N. Garti, J. Phys. Chem. B, 2014, 118, 6277–6287 CrossRef CAS PubMed.
- M. Cohen-Avrahami, D. Libster, A. Aserin and N. Garti, J. Phys. Chem. B, 2011, 115, 10189–10197 CrossRef CAS PubMed.
- S. Aleandri, C. Speziale, R. Mezzenga and E. M. Landau, Langmuir, 2015, 31, 6981–6987 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25200a |
| This journal is © The Royal Society of Chemistry 2017 |