
Sample pre-treatment techniques for use with ICP-MS hyphenated techniques for elemental speciation in biological samples
Han
Wang
,
Xiaolan
Liu
,
Kai
Nan
,
Beibei
Chen
,
Man
He
and
Bin
Hu
*
Key Laboratory of Analytical Chemistry for Biology and Medicine (Ministry of Education), Department of Chemistry, Wuhan University, Wuhan 430072, China. E-mail: binhu@whu.edu.cn; Fax: +86-27-68754067; Tel: +86-27-68752162
First published on 11th August 2016
Abstract
The biochemical reactions involving trace elements depend on the nature and form of their chemical species in a biological system. Consequently, the study of the chemical speciation of a given element in biological samples has become essential for understanding their bioavailability and toxicity. Inductively coupled plasma-mass spectrometry (ICP-MS) hyphenated techniques are the most powerful and widely used for elemental speciation combining both high sensitivity and good selectivity for the determination of ultra-trace elements in biological samples. However, direct analysis of biological samples can suffer from matrix interferences, and both the low concentrations of chemical species, and in some cases, the limited amount of sample available can present analytical challenges. Therefore, there is a need to develop sample pre-treatment methods that can be used to remove the matrix and pre-concentrate the species of interest prior to analysis. This review will focus specifically on the state-of-the-art sample pre-treatment methods published in the last ten years including liquid and solid phase micro-extraction, and chip-based manifolds that can be coupled to ICP-MS hyphenated techniques. The different methods are evaluated for their ability to be deployed for the pre-treatment of actual biological samples. The development trends in this area of research are discussed, and integrated automated miniaturized systems are also reviewed.
 Han Wang | Han Wang received her BSc degree from Wuhan University (China) in 2012. She is currently working as a PhD student with Professor Bin Hu in Wuhan University. Her current research is on the combination of chip-based microextraction and ICP-MS based analytical techniques for elements and their species analysis. |
 Xiaolan Liu | Xiaolan Liu received her BSc degree from Hubei University (China) in 2012. She is currently pursuing her PhD in Analytical Chemistry with Professor Bin Hu, from Wuhan University. Her PhD research is focused on monolithic capillary microextraction for the analysis of elements and their species. |
 Kai Nan | Kai Nan received his BSc degree in Chemistry (2014) from Wuhan University (China). He is currently pursuing his PhD in Analytical Chemistry at Wuhan University. His PhD research is focused on arsenic speciation in biological and environmental samples. |
 Beibei Chen | Beibei Chen is currently an Associate Professor in the College of Chemistry and Molecular Sciences, Wuhan University (China). She received her BSc degree in Chemistry (2005) and her PhD degree in Analytical Chemistry (2010) with Professor Bin Hu from Wuhan University (China). She worked as a Postdoctoral Fellow (2011–2014), Lecturer (2012–2015), and Associate Professor (2015–) at Wuhan University. She was a Visiting Postdoctoral Fellow with Professors Xing-Fang Li and X. Chris Le at the University of Alberta in Nov. 2013–Nov. 2014. Her research focuses on the development of new hyphenated techniques, involving mass spectrometry and chromatography, for elemental speciation and metallomics. |
 Man He | Man He has been working as an associate professor in Analytical Chemistry in Wuhan University (China) since 2007. She received her masters degree in Analytical Chemistry (2001) with Prof. Soulin Lin from the Chinese University of Geosciences (Wuhan, China) and her PhD degree in Analytical Chemistry (2004) with Prof. Zucheng Jiang from Wuhan University (China). She worked as a visiting scholar with Prof. Ryszard Lobinski and Prof. Joanna Szpunar for a year (2011–2012) in CNRS (Le Centre national de la recherche scientifique)-UPPA UMR 5254, France. Her research focuses on the development of miniaturized sample pretreatment techniques and ICP-MS based hyphenated techniques. |
 Bin Hu | Bin Hu is a full Professor of chemistry at Wuhan University, Vice-Dean of the College of Chemistry and Molecular Sciences at Wuhan University, China, and Vice-director of the Key Laboratory of Analytical Chemistry for Biology and Medicine of the Ministry of Education of China. His research interests concern hyphenated techniques for metallomics, electrothermal vaporization as a sample introduction technique for plasma spectrometry (ICP-OES/MS), novel microextraction techniques and advanced functional materials for spectrometric analysis, and novel ICP-MS based strategies for bioanalysis. Professor Hu is an Editorial Advisory Board member for J. Anal. At. Spectrom., Spectrochim. Acta Part B, and Appl. Spectrosc. Rev. |
1. Introduction
Inductively coupled plasma mass spectrometry (ICP-MS) as well as ICP-MS hyphenated techniques have become powerful tools for the analysis of trace elements and their species due to their high sensitivity and good selectivity. However, several challenges are encountered when ICP-MS hyphenated techniques are used to study the chemical species in biological samples and these include matrix effects, extremely low concentrations of target species, and the limited amount of available samples (e.g. cells). As a result, sample pre-treatment techniques can be deployed before ICP-MS hyphenated techniques. This review will focus specifically on the different state of the art sample pre-treatment methods that have been deployed before the use of ICP-MS hyphenated techniques for elemental speciation in biological samples in the last ten years.1.1. Elemental speciation: definition, importance and necessity
According to IUPAC, a chemical species is defined as the “specific form of an element defined as to isotopic composition, electronic or oxidation state, and/or complex or molecular structure” and speciation analysis is defined as “analytical activities of identifying and/or measuring the quantities of one or more individual chemical species in a sample”. However, there are variations in the definition of speciation analysis: Professor Caroli defined speciation as “identifying and quantifying the risks posed to human health and environment by the various forms under which an inorganic analyte may occur”1; Professor Lobinski defined it as “an excellent term to describe the state of distribution of an element among different chemical species in a real-world system (sample)”.2 Professor Hieftje defined it as “the process of obtaining information about the oxidation state, bonding characteristics, charge state and atomic partners of target analyte atoms”.3Elements and their species are constituents of complex biological systems, and play important roles in the maintenance/interference of normal physiological activities. It has been elucidated that the bioavailability and toxicity of elements in physiological systems depend on their different chemical forms (species)4–7 and their concentrations. For example, Cr(III) is essential for the metabolism of insulin, but Cr(VI) is highly toxic and it is a carcinogen.8,9 Arsenic toxicity study in fish is another example in which arsenobetaine (AsB), the main arsenic species in fish, is not toxic and therefore the comparison of the total arsenic contents in different fish may not be a good indicator of the toxicity of the fish. These examples indicate that the qualitative and quantitative analyses of elemental species especially the low abundant species are of great significance in the study of the biochemical behavior of the elements.
In 2004, Professor Haraguchi proposed “Metallomics”10 as a new scientific field of biometallic science, and raised it to be a scientific field in symbiosis with genomics and proteomics. In metallomics, metalloproteins, metalloenzymes and other metal-containing biomolecules are defined as “metallomes”. The most important research target of metallomics is the study of the physiological roles and functions of biomolecules binding with metallic ions in the biological systems. The establishment of metallomics further demonstrates the importance and necessity of elemental speciation in biological samples.
1.2. Elemental speciation in biological samples: difficulty and requirement
Various elemental species exist in biological systems in the form of free metal ions, organometallic species, coordination complexes and so on. Professor Szpunar11 classified elemental species in biological samples into six classes: (1) biosynthesized molecules with chemical bonds such as selenoamino acids, selenoglutathione, selenoproteins and methylarsonic acids, AsB, arsenocholine (AsC) and arseno sugars; (2) complexes with biosynthesized macrocyclic chelating agents such as chlorophyll, cobalamins, hemoglobin, myoglobin, cytochromes and peroxidases; (3) complexes with nucleobases, oligo- and polynucleotides and nucleosides (the interaction of Pt, Au, and Ru with protein and DNA is the key to understand their activities as components of many therapeutic drugs); (4) complexes with amino acids, oligopeptides and polypeptides (proteins) such as ferritin (Fe, Cu, Zn), β-amylase (Cu), alcohol dehydrogenase (Zn), carbonic anhydrase (Cu, Zn) and metallothioneins; (5) complexes with other biomacromolecules such as polysaccharides and glycoproteins; and (6) metallodrugs such as cisplatin and carboplatin.As can be seen, the elemental species in biological samples are complex and cover a wide range of chemical bonds. Factors to be considered in chemical analysis include:12 (1) lability of the target elemental species; since ambient conditions disturb the existing physicochemical equilibria, the species determined may not fully represent those which really occur in the biological sample analyzed (e.g. Al species are labile because they are weakly bound, and easy to transform with the change of pH and matrix13); (2) the contents of some elemental species in biological samples are extremely low and therefore sensitive and selective analytical methods are required (the concentration of most metallic elements in urine samples is about a few micrograms per litre14); (3) the determination of low abundance species may be affected by matrix interference even when sensitive and selective high performance liquid chromatography (HPLC)-ICP-MS techniques are used; (4) the quantities of some biological samples such as cells and blood are limited and therefore it is essential to apply miniaturized analytical methods.
As a result, the development of simple, fast, sensitive, miniaturized and reliable analytical methods for elemental speciation in biological samples is urgently required. The requirements that have to be met in order to limit the changes in elemental speciation include the following: (1) the original forms of target species should be maintained during the whole analytical process; (2) analytical techniques with high sensitivity, selectivity and excellent matrix resistance ability are urgently needed for the analysis of low abundant species; (3) matrix removal as well as simultaneous enrichment of target species should be achieved, and in some cases, derivatization is necessary; (4) miniaturized pre-treatment methods and sample introduction system (such as chip-based systems) are needed for the analysis of samples of limited amounts.
1.3. ICP-MS hyphenated techniques
ICP-MS is proved to be one of the most powerful detection techniques for trace elements due to its unique advantages such as high sensitivity, low mass interference, wide linear dynamic range, and multi-elemental and isotopic analysis ability.15–17 Coupling of efficient separation techniques (such as HPLC, gas chromatography (GC), and capillary electrophoresis (CE)) with ICP-MS has opened up possibilities for elemental speciation with high sensitivity and selectivity. Indeed, organic MS techniques can also be applied in elemental speciation for both qualitative and quantitative analyses.Among all the separation techniques, HPLC is the most frequently employed separation technique used in elemental speciation18 because of the following: (1) HPLC is relatively easy to be coupled with ICP-MS, the different flow rates (from nL min−1 to mL min−1) of the mobile phase are compatible with the different commercial interfaces and nebulizers19–21 of ICP-MS; (2) although the normal operation of the ICP does not tolerate high levels of organic solvents (>10%), the use of organic mode sample introduction systems (adding oxygen in the carrier gas) or the employment of microconcentric nebulizers (<10 μL min−1) can greatly improve the tolerance limit of organic solvents in the mobile phase; (3) species with properties ranging from nonpolar to ionic can be separated by applying different chromatographic modes such as reverse phase (RP)-HPLC,22 ion pair RP-HPLC,23 hydrophilic interaction liquid chromatography (HILIC),24 ion chromatography (IC),25 and macromolecules can be separated by size exclusion chromatography (SEC).26 In the case of volatile hydride-forming elements27–32 (such as Sb, As, Sn, Pb, Se) and Hg, the limit of detection (LOD) of HPLC-ICP-MS could be improved by coupling vapor generation (VG)33,34 to ICP-MS.
GC-ICP-MS is a suitable technique for the separation of volatile elemental species with high sample transport efficiency of nearly 100%. Compared with HPLC-ICP-MS, higher separation resolution, better stability and less interference would be achieved in GC-ICP-MS as a consequence of dry plasma conditions.35 The main requirement of GC-ICP-MS is to avoid the condensation of analytes to assure quantitative analyte transport and peak sharpness. In addition, as typical effluent flow rates (a few mL per minute) applied in GC are too low to obtain stable plasma, the addition of a make-up (aerosol carrier) gas is required.36 However, since GC separation is based on the volatility of analytes and the derivatization is complicated for biological samples, GC-based methods for elemental speciation are limited, therefore only a few elements such as Hg,37 Sn38 and Se39 have been analyzed by GC-ICP-MS.
Better separation efficiency can be obtained when CE is used instead of HPLC. CE is based on electrophoretic separation of the chemical species.40 CE is preferred for the analysis of ionic species and macromolecule speciation (metalloproteins) when limited sample volume is available. The coupling of CE with ICP-MS is not as straightforward as with HPLC and GC.41,42 The construction of the interface of CE-ICP-MS involves maintaining an effective electrical connection to the exit of the capillary, countering the suction-effect of the operating nebulizer (preventing laminar flow in the capillary), and minimizing band broadening and obtaining high transport efficiency.
In addition to chromatographic separation, there are also non-chromatographic methods that can be used for elemental speciation such as the combination of liquid phase microextraction (LPME),15,16 capillary microextraction (CME),43 or magnetic solid phase extraction (MSPE)17 with ICP-MS. Although non-chromatographic methods are simpler and cheaper to run, only a limited number of species can be analysed simultaneously.
Biological samples encompass urine, saliva, sweat, exhaled breath, whole blood, serum, plasma, cerebrospinal fluid, gastric fluid, tissue samples44 (such as hair, nail, skin, bone, muscle), cells and so on. Although some biological samples could be analysed without sample pre-treatment,45–49 for others, partial or complete removal of sample matrix is essential in order to limit possible interference effects.50–54
Sample pre-treatment methods that have been discussed in several reviews for elemental speciation are needed for the matrix removal as well as enrichment of target species.55–64 Sample pre-treatment methods include liquid phase extraction (LLE),56 LPME (e.g. single drop microextraction (SDME) and hollow fiber (HF)-LPME),57,58 solid phase extraction (SPE)59 (e.g. MSPE60), solid phase microextraction (SPME)61,62 (e.g. stir bar sorptive extraction (SBSE)63 and CME64), chip-based65–70 pre-treatment techniques and hybrid methods such as the combination of LLE with SPME.71 ICP-MS hyphenated techniques are subsequently used for sample analysis.
In this review, we will focus on these novel pre-treatment methods for elemental speciation in biological samples by ICP-MS hyphenated techniques. The merits, shortcomings and developing trends of the pre-treatment methods are discussed. The elemental species and combination characteristics for these pre-treatment methods as well as the applicability of the pre-treatment methods when coupled with ICP-MS hyphenated techniques will be evaluated.
2. Liquid phase microextraction for elemental speciation in biological samples
LLE is a widely used separation method based on the differences in the relative solubility of analytes in two/three types of immiscible liquids and has been used for elemental speciation in biological samples. Zachariadis et al.72 developed a method for the speciation of organotin compounds in urine based on LLE. Seven organotin species (monobutyltin (MBT), dibutyltin (DBT), tributyltin (TBT), tetrabutyltin (TTBT), monophenyltin (MPT), diphenyltin (DPT) and triphenyltin (TPT)) were in situ derivatized by sodium tetraethylborate (NaBEt4) to form less polar ethylated derivatives in the urine matrix. Then the derivatives were extracted using hexane and detected by using a GC-microwave induced plasma (MIP)-atomic emission detector (AED). The LODs were in the range of 0.42–0.67 μg L−1 (as Sn) and the relative standard deviations (RSDs) were in the range of 4.2–11.7%. A similar method was employed for speciation analysis of triethyl-lead and tributyl-tin in human urine73 and the LODs were 0.05 and 0.48 μg L−1 for lead and tin, respectively.However, the operation of conventional LLE is time-consuming and complex; large volumes of solvent are needed; the direct injection of large amounts of organic solvents can affect the performance of ICP, as a result tedious evaporation and resolubilization steps after extraction are required.74 Thus, the application of LLE in elemental speciation is limited.
LPME57,58,75–77 was developed as a new and environmentally friendly sample pre-treatment method because of the reduced amount of organic solvents. Generally speaking, LPME could be divided into two modes: two-phase LPME and three-phase LPME. In two-phase LPME, the analytes are extracted from the aqueous solution into the organic solvent, and in three-phase LPME, the analytes are extracted from the aqueous solution through the organic solvent into another aqueous solution. Compared with two-phase LPME, three-phase LPME is more compatible with HPLC and CE as the subsequent instrumental analyses. In addition, based on the applied extraction devices, LPME could be divided into SDME,78–80 HF-LPME,81,82 and dispersive liquid–liquid microextraction (DLLME).83 These LPME modes have been applied for elemental speciation in biological samples.57 The combination of LPME and ICP-MS hyphenated techniques for elemental speciation will be discussed below.
2.1. SDME
SDME is a simple, cheap, environmentally friendly pre-treatment technique and possesses a high enrichment factor (EF) based on a great reduction of the extractant phase to sample volume ratio. SDME can be operated in two different modes: headspace (HS)-SDME (the extractant drop is exposed to the headspace of the sample) which is suitable for the extraction of volatile elemental species, and direct-SDME (the extractant drop is immersed in sample solution). The most important parameter in SDME is the choice of the extractant which should be based on the comparison of selectivity, extraction efficiency, incidence of drop loss, rate of drop dissolution, toxicity and the compatibility with the analytical technique ultimately employed.Xiao et al.84 combined HS-SDME with GC-ICP-MS for the speciation analysis of butyltin compounds in environmental and biological samples. Sodium tetraethylborate was chosen as the derivatizing reagent for in situ derivatization of the butyltin species. For the derivatization, HS-SDME parameters such as the organic solvent, drop volume, sample pH, stirring rate, temperature, extraction time and ionic strength were examined systematically. The LODs were 1.4 ng L−1 for MBT, 1.8 ng L−1 for DBT and 0.8 ng L−1 for TBT. The RSDs were in the range of 1.1–5.3% (c = 1 μg L−1, n = 3). With tripropyltin as the internal standard, this HS-SDME-GC-ICP-MS system was applied for the speciation analysis of butyltins in shellfish samples, and 11.6–30.4 ng g−1 MBT, 11.8–8.9 ng g−1 DBT and 12.8–52.6 ng g−1 TBT were found in different shellfish samples. For validation, the developed method was also employed for the speciation analysis of butyltins in the certified reference material PACS-2 sediment, and the determined values were in good agreement with the certified values.
Pena-Pereira et al.85 combined ionic liquid-based SDME with HPLC-photodiode array detection for the analysis of mercury species. Dithizone was used for derivatisation of four mercury species in the samples prior to the extraction of mercury dithizonates onto an ionic liquid drop. With 1-hexyl-3-methylimidazolium hexafluorophosphate used as the microdrop for the SDME, the extractant was then analysed by HPLC with a photodiode array detector.
Although SDME is easy to carry out and possesses high EF, the main shortcoming is that the micro-drop on the tip of the micro-syringe is not very stable at high stirring rates and long extraction times. Therefore, the reproducibility of SDME is often poor.
2.2. HF-LPME
To solve the unstability problem, Pedersen-Bjergaard and Rasmussen86 found that reproducibility can be improved when a hollow fiber in LPME (called HF-LPME) in which the microliter extractant phase was filled in the lumen and membrane pores of the hollow fiber was used. Besides, the small pore size prevents large molecules and particles present in the donor phase from entering the acceptor phase, thus potential interferences are reduced. In addition, the disposable nature of HF eliminates possible carryover. HF-LPME is suitable for extracting the analytes from biological samples with simultaneous clean-up of the extracts. Furthermore, HF-LPME can also be operated in HS and direct modes for the extraction of volatile and non-volatile elemental species.Ghasemi et al.87 developed a method for the determination of volatile organic compounds dimethylselenide (DMSe) and dimethyldiselenide (DMDSe) based on HS-HF-LPME. 1-Decanol was used as the extracting solvent with 2 mg L−1 of ethyl benzene as the internal standard. The syringe pump was pre-programmed to refresh the surface of the organic phase in order to achieve maximum absorption of the volatile analytes. After extraction, the acceptor solution was collected and 1 μL of the extractant was injected into the GC-MS for analysis. Under optimum conditions, the EFs were up to 1250 and 1170-fold for DMSe and DMDSe, respectively. The method has been successfully applied to the quantification of both DMSe and DMDSe in urine samples.
Duan et al.88 developed a new derivatization-extraction method for analysis of selenoamino acids (Se-methyl-selenocysteine (SeMeCys), selenomethionine (SeMet) and selenoethionine (SeEth)) based on HF-LPME in which 4 μL of organic solvent (toluene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :chloroform, 3:1) was used as the acceptor phase following the derivatization with 100 μL of ethyl chloroformate for 2.5 mL of aqueous standard or sample solution. After extraction for 5 min, the organic solvent was collected for GC-ICP-MS analysis. Under the optimal conditions, the LODs obtained for selenoamino acids ranged from 11 to 23 ng L−1 (as Se). The method was applied to determine the concentration of selenoamino acids in the extracts of garlic, cabbage and mushroom samples with spiked recoveries ranging from 96.8 to 108% when SeEth was used as the internal standard.
:chloroform, 3:1) was used as the acceptor phase following the derivatization with 100 μL of ethyl chloroformate for 2.5 mL of aqueous standard or sample solution. After extraction for 5 min, the organic solvent was collected for GC-ICP-MS analysis. Under the optimal conditions, the LODs obtained for selenoamino acids ranged from 11 to 23 ng L−1 (as Se). The method was applied to determine the concentration of selenoamino acids in the extracts of garlic, cabbage and mushroom samples with spiked recoveries ranging from 96.8 to 108% when SeEth was used as the internal standard.
Moreno et al.89 developed an HF-LPME method for simultaneous preconcentration of ultra-trace concentrations of Hg and Se species in environmental and biological samples in which 1 mg of o-phenylenediamine was added to the aqueous standard or sample solution to derivatise Se(IV) to form a piazselenol, and then extracted with bromobenzene filled in the U-shape HF between two syringes, and kept for 20 min in a water bath. The organic phase was injected into the HPLC system for subsequent analysis. The LODs of the method were 110–230 ng L−1 with RSDs below 15% for all the analytes and the averaged recoveries were in the range of 71–99%. The method was applied to selenium speciation in human blood plasma.
Xiao et al.90 developed a method for the analysis of four polybrominated diphenyl ethers in human serum samples based on HF-LPME followed by GC-ICP-MS detection. The organic solvent in the porous hollow fiber was first dipped into the sample for a given time and the retracted organic phase was introduced into the GC-ICP-MS system for analysis. Parameters affecting the extraction efficiency of HF-LPME were studied. Under the optimized conditions, the LODs of the proposed method varied between 15.2 and 40.5 ng L−1. The developed process of HF-LPME with GC-ICP-MS was applied successfully for the determination of polybrominated diphenyl ethers in spiked human serum samples.
Compared with two phase HF-LPME, three phase HF-LPME (also called HF-LLLME) is compatible with HPLC and CE analyses because an aqueous solution is used as the acceptor phase. In HF-LLLME, the analytes are extracted from the aqueous sample, through the organic solvent in HF membrane pores, and then transferred into the aqueous acceptor solution that is present inside the lumen of HF.
A novel method based on HF-LLLME was proposed by Guo et al.91 for the speciation of phenylarsenic compounds in chicken and feed samples in which the phenylarsenic compounds were extracted into the organic phase (20% (v/v) [MTOA]+[Cl]− in toluene) and then extracted into the acceptor phase (0.3 mol L−1 NaBr) from aqueous samples at pH 10.2. After extraction, 9.0 μL of the acceptor was collected for HPLC-ICP-MS analysis. The LODs for the five phenylarsenic compounds and arsenate were in the range of 1.4–16.0 ng L−1 with RSD and EFs ranging from 3.6 to 10% (C = 0.05 μg L−1 except Cp-ASA = 0.10 μg L−1) and 86 to 372-fold, respectively. The method was successfully applied to the analysis of five phenylarsenic compounds and arsenate in the spiked chicken and spiked feed samples with recoveries of 79.2–105.4% and 81.1–117.8%, respectively. A similar method was proposed for the analysis of mercury species in environmental and biological samples from the East Lake by the same group,92 focusing on the investigation of biotransformation and biomagnification of mercury species through the food chain in a micro-ecosystem. Under the optimized conditions, the LODs were at the ng L−1 level with RSDs between 5.6 and 10.6%.
Li et al.93 also proposed a dual preconcentration method based on the combination of HF-LLLME and anion selective exhaustive injection (ASEI)-CE/ultraviolet (UV) detection for sensitive determination of phenylarsenic compounds in pig feed, pig litter, soil and lake water samples. The phenylarsenic compounds in the sample solution were finally extracted into the acceptor phase of 18 μL 0.8 mmol L−1 Tris solution in the lumen of the hollow fiber through a thin phase of tributyl phosphate inside the pores of the hollow fiber. Under the optimized conditions, EFs were in the range of 155–1780-fold by combining HF-LLLME with ASEI-CE/UV with LODs ranging from 0.68 to 6.90 μg L−1 and RSDs varying in the range of 5.6–11.8%. An ion pair HF-LLLME system combined with the CE-UV method94 was also developed for the simultaneous determination of six thyroid hormones (3,5-diiodothyronine (T2), 3,3′,5-triiodothyronine (T3), 3,3′,5,5′-tetraiodothyronine (T4), reversed-3,3′,5-triiodothyronine (rT3), monoiodotyrosine (MIT) and diiodotyrosine (DIT)) in human serum samples. By the addition of a low concentration of sodium dodecyl sulfate into the donor phase as an ion pair reagent, octanol as the organic extraction solvent and 30 mmol L−1 Na2CO3 as the acceptor phase, the six analytes with different polarities and water solubilities were successfully extracted simultaneously using HF-LLLME. CE-UV was then used for the determination of target analytes under the optimized conditions, EFs ranging from 183- to 366-fold were obtained and the LODs were at the sub μg L−1 level. The established ion pair HF-LLLME-CE-UV method was successfully applied to the simultaneous determination of thyroid hormones and relevant compounds in human serum samples with good recoveries for the spiked samples.
The automatic system was also developed based on HF-LLLME mode. Li et al.95 constructed an LLLME device based on a simple home-made automatic dynamic (AD)-HF-LLLME for the simultaneous extraction of organomercury and inorganic mercury with the help of a programmable flow injection analyzer. Mercury species complexed with 18-crown-6 were extracted into the acceptor phase of 0.1% (m/v) 3-mercapto-L-propanesulfonic acid aqueous solution through the organic phase (chlorobenzene). Compared with the automatic static (AS)-HF-LLLME system, the extraction equilibrium of AD-HF-LLLME was achieved in a shorter time with higher extraction efficiency. A 2195-fold enrichment of the mercury species was obtained with the LODs at sub μg L−1.
Both HF-LPME and HF-LLLME exhibit high EF, improved reproducibility and good matrix tolerance, as a result they have become one of the most widely used LPME techniques for trace analysis in biological samples. However, they also have some inherent shortcomings and these include the following: the existence of a membrane barrier increases the time at which extraction equilibrium is attained; the presence of air bubbles on the surface of HF influences the extraction; in real sample analysis, adsorption of hydrophobic substances on the HF surface may block the pores thus affecting the extraction efficiency.
2.3. Other LPME formats
As we discussed above, LLLME96 is more compatible than two phase LPME when coupled with HPLC and CE. Besides HF-LLLME, a novel LPME system called membrane-assisted phase transfer (PT)-LLLME was proposed by Li et al.97 for the simultaneous extraction of inorganic and organic mercury species in a certified reference material DORM-2 dogfish muscle (NRCC, Ottawa, Canada). A schematic diagram of the membrane-assisted PT-LLLME system is shown in Fig. 1. The donor phase is the sample solution containing an appropriate amount of acetonitrile and dodecylamine at pH 4.0; the organic phase is 200 μL toluene and the acceptor phase is 15 μL L-cysteine. The addition of acetonitrile to the sample solution promoted the dissolution of dodecylamine, accelerated the formation of the complexes between mercury species and dodecylamine, and thus facilitated extraction of the complexes into the organic phase. Besides, application of the membrane-assisted system improved the stability and increased the volume of the acceptor phase for subsequent large volume sample stacking (LVSS)-CE/UV analysis. The method of membrane-assisted PT-LLLME-LVSS-CE/UV provided EFs of up to 12138-fold with LODs at the sub μg L−1 level. The PT-LLLME system is more stable for high stirring rates, possesses better extraction efficiency (90.3%) and higher EF (478-fold) compared with HF-LLLME for MeHg+. PT-LLLME provides a new approach for the application of LPME in elemental speciation in biological samples.
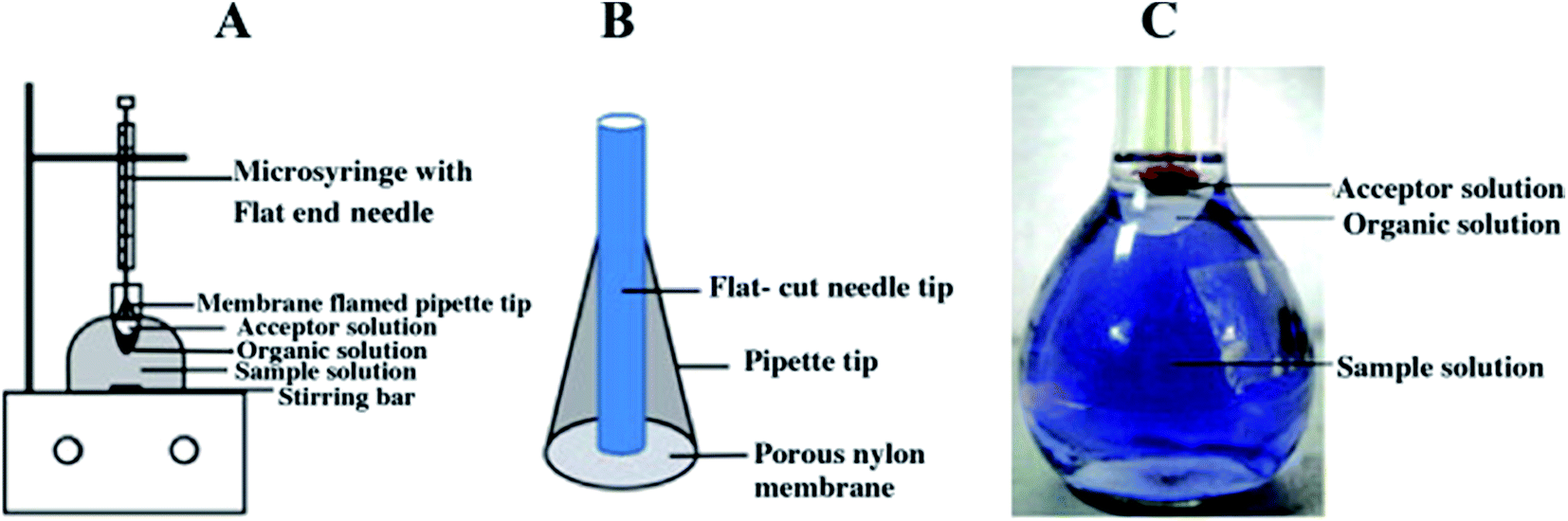 | ||
| Fig. 1 (A) Schematic diagrams of membrane-assisted PT-LLLME and (B) the home-made porous nylon membrane supported extraction tip. (C) Photographs of membrane-assisted PT-LLLME. | ||
Dispersive liquid–liquid microextraction (DLLME) is another novel LPME method proposed by Assadi and co-workers98 in 2006. In this method, a cloudy solution is formed when an appropriate mixture of extraction and dispersive solvents is injected into an aqueous sample, and the hydrophobic solutes are enriched in the extraction solvent. After centrifugation, the analytes in the settled phase can be determined by ICP-MS hyphenated techniques. Zhou et al.99 developed a simple and sensitive method for the speciation of selenium in tea samples by coupling DLLME with HPLC. After derivatization with 2,3-diaminonaphthalene, a mixed solution of acetonitrile and chlorobenzene was injected into the sample solution to form a cloudy solution. The supernatant was removed and the organic phase was redissolved with methanol for HPLC analysis. The LOD was 0.11 μg L−1 for Se(IV) with a linearity range of 0.4–40 μg L−1. The spiked recoveries were between 91.3 and 100%. The concentrations of total selenium, Se(VI) and total organoselenium were indirectly obtained by the determination of Se(IV) because only Se(IV) reacts with 2,3-diaminonaphthalene. DLLME is simple to operate, fast, cheap with high recovery, high EF and very short extraction times (a few seconds). However, its main drawback is poor matrix tolerance, which greatly limits its use in biological samples.
The advantages of using the LPME techniques include high efficiency, environmentally friendly operation (minimal solvent consumption) and high selectivity (systems designed for particular applications). These properties are essential for the extraction of target elemental species in various biological samples (mostly biological fluids and tissue extracts). A comparison of the various LPME formats is instructive. SDME is simple and has high EF, but suffers from the instability of the microdrop during the extraction; DLLME is easy to operate and has very fast extraction kinetics, however, it is not a suitable technique for biological sample analysis because of the matrix effect. Membrane supported LPME techniques (HF-LPME, HF-LLLME and PT-LLLME) provide better stability and greater tolerance to biological matrices compared to SDME and DLLME and as a result they are the most suitable LPME techniques for the enrichment of target elemental species in biological matrices.
Besides the selection of LPME modes, extraction conditions such as sample pH, organic solvents, complex reagents, and choice of acceptor would determine the extraction performance. Since most of the elemental species are polar compounds, choosing an appropriate chelating reagent to increase the hydrophobicity of the target species in order to facilitate their transfer into the organic phase is critical to the success of the extraction or otherwise of the method. For example, mercury species can complex with reagents such as APDC containing the thiol group which have high affinity for mercury. For selenoamino acids, ion-pairing reagents such as sodium 1-octanesulfonate are suitable for the extraction. Currently the choice of extraction systems is limited, however this situation is bound to change as new extraction systems are developed for known elemental species.
3. Solid phase extraction for elemental speciation in biological samples
SPE is the most common and widespread sample pre-treatment method currently used.59 Its applications in environmental, pharmaceutical, food and biological analyses have been reviewed.100–103 The main aims of SPE are analyte enrichment, matrix simplification and medium exchange. SPE is based on the selective distribution of analytes between the solid extraction material and liquid mobile phase. The analytes are transferred to the solid phase since they possess greater affinity for the solid phase than for the sample matrix, then they are recovered by elution using a suitable eluent or by thermal desorption into the gas phase. When in use, SPE can be used to process large volumes of liquid samples, it is less time consuming and the results of the analyses are reproducible.61,104Generally, there are several formats of SPE: SPE cartridges,105 SPE disks,106 SPE pipette tip,107 SPE microcolumn,108 magnetic SPE109 and so on. The commonly used SPE cartridges consist of a sorbent held in place by a frit at the top and bottom of the syringe. Commercial SPE cartridges are widely used, but often operated in an off-line mode. In contrast, the SPE microcolumn can easily be operated on-line coupled to compatible analytical techniques.
Traditionally, a variety of sorbents including reversed phase sorbents, normal phase sorbents, ion exchange sorbents to biological sorbents, carbon nanotubes, graphene, graphene oxide, metal–organic frameworks, mesoporous nanoparticles and functional magnetic nanoparticles, and so on have been used in SPE. As a result of the differences in the characteristics of the sorbents, SPE can be applied to a diverse range of analytes. The development of new sorbent materials is therefore an area of fruitful research.
In the following sections, different SPE techniques coupled to ICP-MS hyphenated techniques for elemental speciation will be discussed.
3.1. Commercial SPE cartridges
SPE cartridges are the most frequently used SPE mode.110 In different cartridges, different types and amounts of sorbents are packed between two frits which are made of polyethylene or stainless steel. Varied types of commercial SPE cartridges (packed with C18, cation exchange, anion exchange materials and so on) could be chosen for the extraction of different analytes, including inorganic analytes,106 organic pollutants111 and so on.There are several examples in which SPE cartridges have been coupled with ICP-MS hyphenated techniques for elemental speciation in biological samples. Wrobel et al.112 combined SPE with ion pair HPLC-ICP-MS for the detection/identification of selenium metabolites in human urine. C18 SPE cartridges modified with hexanesulfonic acid were used for the retention of cationic species in acidified urine. After elution with methanol, the eluate was introduced into HPLC-ICP-MS. Hernandez-Nataren et al.113 compared the extraction efficiency of Sb(III), Sb(V) and TMSb on several SPE cartridges after the determination of the species in the eluate by HPLC-ICP-MS. In the study, four commercial SPE cartridges: aminopropyl DCN-NH2 (ion exchange), amino DCN-SAX (ion exchange), polystyrene divinylbenzene (reversed phase) and SiO2 (normal phase) were used as sorbents and the retained species were eluted with the HPLC mobile phase. The results showed that anion exchange was the main separation mechanism for inorganic antimony species and partition mechanism was involved in the extraction of TMSb. Moreda-Pineiro et al.114 used ENVI-Carb™ SPE cartridges for the clean-up procedure after the enzymatic hydrolysis of seafood materials. This pre-treatment was then combined with HPLC-ICP-MS for the analysis of As(III), As(V), MMA, DMA, AsB and AsC. Shang et al.115 used SPE cartridges (Oasis MCX) for the clean-up of seafood samples and the eluate was injected into HPLC-ICP-MS for the detection of Al3+.
Commercial SPE cartridges could be used for elemental speciation based on hydrophobicity, hydrophilicity and ion exchange interaction. As the interactions between sorbents and target species are not specific, most of the commercial SPE cartridges are used for sample clean-up rather than preconcentration of the species. The development of sorbents designed to interact with a given group of species will undoubtedly improve the efficiency of extraction. In general, commercial SPE cartridges are simple and efficient but mostly suitable for the clean-up of biological fluids and tissue extracts.
3.2. Dispersive/magnetic/microcolumn SPE
Dispersive/magnetic and microcolumn SPEs containing nanoparticles are widely used as sorbents for the analysis of chemical species. The advantage of using nanoparticles is the ease with which surface functionalization can be performed in order to improve analyte selectivity.Duan et al.116 combined dispersive SPE with ligand-exchange micellar electrokinetic capillary chromatography (MEKC) for the chiral separation of D,L-SeMet in SELM-1 selenized yeast. The analytes were first retained on Cu(II) loaded mesoporous TiO2, then eluted by ammonia and finally separated by MEKC determination. A new phenylalanine derivative (L-N-(2-hydroxy-propyl)-phenylalanine) was synthesized and chelated with Cu(II) as the chiral selector for MEKC. With the EFs of 1400 and 1378, LODs of 0.44 and 0.60 ng mL−1 for L-SeMet and D-SeMet were obtained, respectively.
In recent years, with the development of functional magnetic materials, MSPE became a new and preferable method in the sample pre-treatment for biological samples. The operations of magnetic SPE are similar to dispersive SPE except that the sorbents could be collected and separated by an externally applied magnetic field which renders the whole extraction process easier, faster, more powerful and efficient especially for large scale operations.60,117 The application of MSPE shows great potential for elemental speciation. Huang et al.118 prepared Fe3O4@SiO2@γ-MPTMS magnetic nanoparticles for the analysis of Te species. They also prepared methacrylic acid (MAA) modified magnetic nanoparticles as sorbents and combined with HPLC-ICP-MS for the determination of carboplatin in urine samples.119 Liu et al.120 developed a microwave-assisted extraction procedure coupled with a magnetic nanoparticle-based pre-treatment method followed by HPLC-ICP-MS for the analysis of organotins in seafood. The LODs were 0.49, 0.35, 0.42 and 0.23 ng mL−1 for TET, TPT, TBT and azocyclotin, respectively.
Nanoparticles were also packed in an SPE microcolumn for elemental speciation in biological samples. Duan et al.121 prepared a micro-column packed with Cu(II) loaded nanometer-sized Al2O3 and coupled with ICP-MS for the separation and determination of SeMet and SeCys2 in selenized yeast, human urine and serum samples.
There are currently only a few published applications of functionalised magnetic materials used for MSPE in the elemental speciation of biological samples especially for biological fluids and extracts. However, with the development of new magnetic materials, the application of MSPE could be extended.
3.3. Matrix solid phase dispersion extraction (MSPD)
Matrix solid phase dispersion extraction (MSPD) was first introduced in 1989 by Barker et al.122 for disruption and extraction of solid samples. Solid samples such as animal tissues have to be broken down by dividing the sample into smaller and smaller pieces in order to provide greater surface area that can be exposed to extraction (water or organic solvents).123 MSPD combines aspects of several techniques for sample disruption and chromatographic separation of the extracted analytes. MSPD involves blending a viscous, solid or semi-solid sample with a solid support (derivatized to produce a bound coating on the surface, such as C18) in a mortar. The solid support serves as an abrasive to break samples into smaller pieces and also provides a further function: the analytes will dissolve and disperse into the bound coating on the surface of the particles. The solid support124,125 is essential for pre-treatment of solid samples with its unique ability to directly handle solid samples, disrupt and disperse the samples. As a result, it was widely used in biological126–128 and food samples, and there are several papers in which MSPD is coupled with ICP-MS hyphenated techniques for elemental speciation in biological samples.Moreda-Pineiro et al.129 combined MSPD with HPLC-ICP-MS for speciation of arsenic in seafood. The sample was first blended thoroughly with diatomaceous earth, then transferred into a syringe containing the C18 sorbent, and finally the analytes were eluted and introduced into an HPLC-ICP-MS system for the analysis of As(III), As(V), MMA, DMA, AsB and AsC, and the LODs ranged from 6.4 to 23.3 ng g−1.
Duarte et al.130 combined MSPD with GC-MS for the analysis of MeHg+ and Hg2+ in fish samples. The sample was blended with silica in a ceramic pestle and mortar to obtain a homogeneous mixture and was transferred into a polypropylene vessel. The treated sample was mixed by vortex and subjected to centrifugation, followed by injection of the derivatized analytes into a GC-MS. The LODs of this method were 0.06 and 0.12 μg g−1 for MeHg+ and Hg2+, respectively.
The MSPD on-line system was also reported. Deng et al.131 developed a multi-wall carbon nanotubes (MWCNTs) assisted on-line MSPD pre-treatment method, which was used in combination with HPLC-ICP-MS for the analysis of mercury species in fish. The fish sample and MWCNTs were blended and transferred to a stainless steel column which had been packed with C18 materials on the bottom of the column before. Then the columns were pumped to remove air and assembled on the on-line system as shown in Fig. 2a. The on-line system consists of two sequential injection valves and six MSPD columns between a six-port valve and an LC column. The eluent was injected to a 100 μL loop through the six-port valve by a syringe and then a mobile phase flushed the eluent to the MSPD column for the extraction of mercury species, and then the eluent was introduced into reversed-phase chromatography and detected by ICP-MS. The LODs were 9.9 and 8.4 ng g−1 for Hg2+ and MeHg+, respectively.
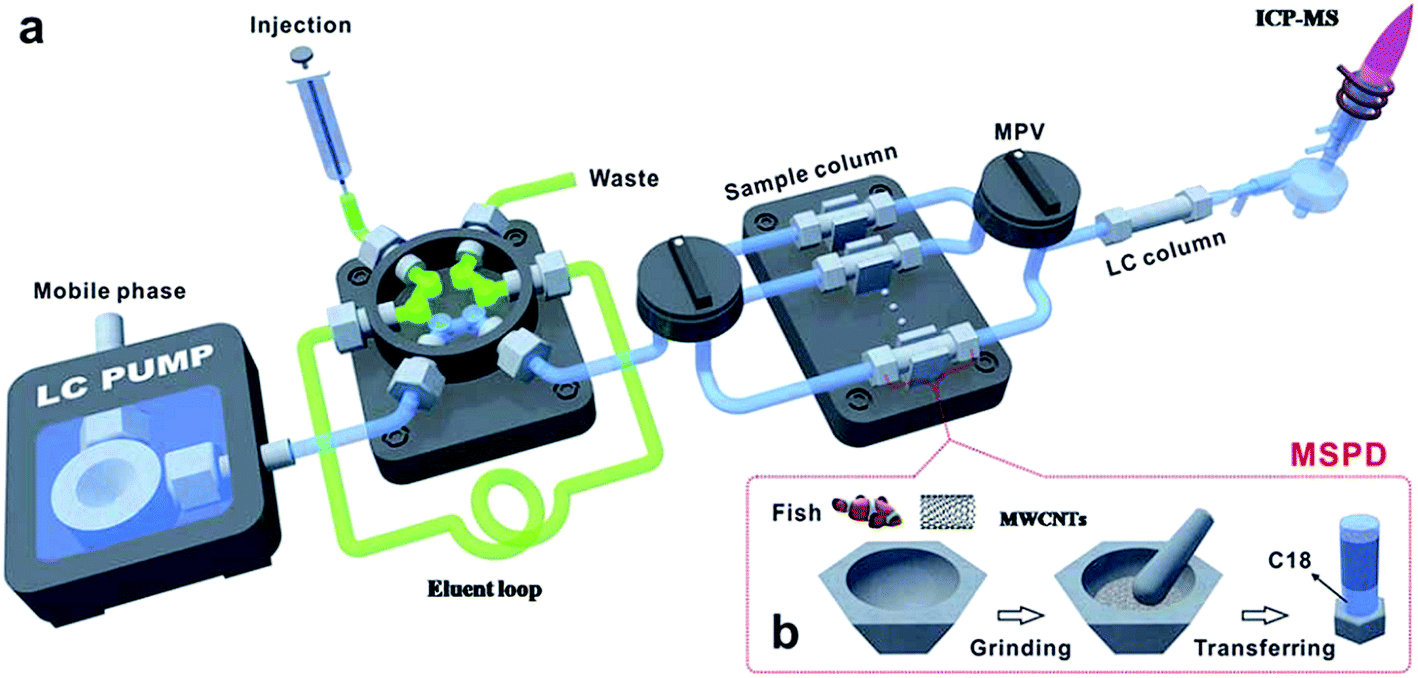 | ||
| Fig. 2 Schematic on-line MSPD platform coupled to an HPLC-ICP-MS system. | ||
MSPD is a pre-treatment method suitable for the analysis of solid and semi-solid biological samples, in which biological tissues can be directly prepared making the pre-treatment process simpler and faster. However, there are few reported applications of MSPD systems for elemental speciation in biological samples.
Off-line SPE has been widely used for the pre-treatment of biological samples, but it is not much suitable for use with cells and other sample types for which sample quantity is limited. In order to reduce the possibility of sample contamination the development of on-line SPE systems is essential. SPE systems with features of miniaturization and automation (i.e. SPME) are needed for biological samples, and these developments will be discussed in the following sections.
4. Solid phase microextraction for elemental speciation in biological samples
SPME was introduced by Arthur and Pawliszyn in the early 1990s132 and has played an important role in the field of sample preparation. SPME integrates sampling, sample preparation, preconcentration and sample introduction into a single step prior to instrumental analysis.133,134 Compared to conventional preparation methods, such as SPE and LLE, SPME features many advantages such as simplicity, rapidity, convenience, and low sample/reagent consumption. Significant advances in the development of SPME for elemental speciation have been made in recent years. The common formats of SPME include fiber SPME,135–138 in-tube SPME (also termed as CME),139,140 and SBSE in which a different extraction system is used. Recent advances in the above mentioned SPME modes used in the elemental speciation in biological samples are reviewed below.4.1. Fiber SPME
Fiber SPME is an extraction format with the stationary phase coated on a small end segment of silica fused fiber. It can be operated in three modes: direct SPME, headspace SPME and membrane protected SPME. Table 1 lists recent applications of fiber SPME combined with separation techniques for elemental speciation in biological samples.| Analytes | Sample | Coating/extraction mode | Detection | LOD (ng mL−1) | Ref. |
|---|---|---|---|---|---|
| MeHg, EtHg | Human urine | PDMS–DVB/HS-SPME | LC-ICP-MS | 0.06 | 141 |
| MeHg, EtHg | Waterfowl egg and fish | PDMS/HS-SPME | GC-AFS | 0.04, 0.13 ng g−1 | 142 |
| MeHg, Hg(II) | Fish, bivalves, prawns, crabs | PDMS/direct-SPME | GC-MS | 0.02, 0.05 ng | 143 |
| MeHg, EtHg, Hg(II) | Blood samples | Carboxen-PDMS/HS-SPME | GC-ICP-MS | 30 pg g−1 | 144 |
| MeHg, Hg(II) | Seabird eggs | PDMS–silica/HS-SPME | GC-ICP-MS | 4.2 g g−1 | 145 |
| MeHg, Hg(II) | Fish tissues | PDMS/HS-SPME | GC-AES | 1.5, 0.7 ng g−1 | 146 |
| MeHg, EtHg, Hg(II) | Blood samples | PDMS/HS-SPME | GC-ICP-DRC-MS | 0.12, 0.16, 0.27 | 147 |
| DMSe, DEtSe, DMDSe | Plants (Indian mustard, garlic and lupine) and yeast | Carboxen–PDMS/HS-SPME | GC-AES | 0.57, 0.47, 0.19 | 148 |
| SeMeCys, SeMet, γ-GluSeMeCys | Human B-cell lymphoma line | Carboxen–PDMS/HS-SPME | HPLC/GC-MS | 25.1, 35.5, 40.8 ng kg−1 | 149 |
| MeHg, Hg(II); TML, DML; TMT, DMT, MMT, MBT, DBT, TBT | Dogfish muscle, oyster tissue, mussel tissue | PDMS–DVB/HS-SPME | GC-ICP-MS | 1.3, 1.97; 0.03, 0.03; 0.09, 0.30, 0.51, 0.77, 0.99, 0.62 pg g−1 | 150 |
| MeHg, EtHg, Hg(II) | Dogfish liver, hepatopancreas, dogfish muscle | PDMS/HS-SPME | GC-AES | 0.55, 0.34, 0.23 ng g−1 | 151 |
Despite its commercialization and diverse applications, fiber SPME possesses some inherent shortcomings including fiber breakage, mechanical damage of the coating, inefficient adsorption capacity and difficulty for on-line analysis. In addition, the fiber-SPME-GC hyphenated technique is not suitable for the analysis of thermally unstable or non-volatile analytes. The extraction and determination steps in the manual use of fiber-SPME-HPLC are discontinuous processes, which impose restrictions on the analysis speed and automation. Recently, in vivo SPME152,153 for metabolomics study in living animals has been developed. A fiber/needle with better mechanical properties was used and directly introduced into biological tissues for the enrichment of target metabolites. This approach can be extended to the study of elemental speciation in biological samples.
4.2. Capillary microextraction (CME)
CME was first introduced by Bigham et al.154 as a viable extraction technique for preconcentration of a wide variety of polar and nonpolar analytes. The capillary can be prepared in a single-step procedure and allows for in situ creation of chemically bonded sorbents which are featured with high thermal and solvent stabilities. CME is a mode of SPME, it is featured with low cost, simple operation, high speed, high selectivity, low consumption of sample and solvent as well as easy automation; as a result, it has been widely used for on-line analysis of trace metals in biological and environmental samples. So far, three types of capillary columns: packed, open tubular and monolithic have been used for CME.CME has been hyphenated with different separation instruments including GC,155 HPLC156 and CE,157 and also with some detection instruments such as electrospray ionization (ESI)-MS158 and ICP-MS.159,160 However, it should be stressed that until now, most of the published studies on CME applications are mainly focused on the analysis of organic substances, the application of CME to inorganic elements analysis is much less common and fewer still are aimed at elemental speciation.
Wu et al.161 prepared a polypyrrole (PPY) coated capillary for the extraction of MMAs, DMAs, AsB and AsC followed by LC-ESI-MS detection. Compared with several commercially available capillaries, the PPY coated capillary was found to have higher extraction efficiency. Chen et al.162 developed an in-tube HF-SPME on-line coupled to HPLC-ICP-MS for arsenic speciation (As(III), As(V), MMA, DMA, AsB and AsC). Partial sulfonated poly(styrene) (PSP) and a mixed-sol of mercaptopropyl trimethoxysilane (MPTS) and N-(2-aminoethyl)-3-aminopropyltrimethoxysilane (AAPTS) were prepared and immobilized in the pores and the inner surface of polypropylene HF. The addition of MPTS with the mercapto group greatly improved the adsorption of neutral As(III), and an improvement of the adsorption of the positively charged AsC was observed due to the cation exchange interaction with PSP, while negatively charged As(V) and MMA as well as amphoteric DMA and AsB were adsorbed on the amino group of the AAPTS. The homemade in-tube HF-SPME-HPLC-ICP-MS system offered high efficiency and good stability, and the proposed method was sensitive and suitable for simultaneous speciation of organic and inorganic arsenic species (including anions and cations) in biological samples.
Hu's group has published several investigations that have focused on elemental speciation using a single CME step without the need for further separation. A dual-column CME system was developed for sequential separation/preconcentration of As(III), As(V), MMA and DMA in the extracts of human hair followed by electrothermal vaporization (ETV)-ICP-MS detection with iridium as a matrix modifier.163 A TiO2 nanoparticles functionalized monolithic column was also prepared and used in CME coupled on-line to an ICP-MS system for the sequential determination of Gd species in human urine samples43 exposed to Gd-based contrast agents.
CME is a simple, quick and easy-to-automate sample preparation technique that is suitable for the pre-treatment of mL and sub mL volumes of samples such as blood, serum and cerebrospinal fluid.
4.3. Stir bar sorptive extraction (SBSE)
SBSE was developed in 1999 as an environmentally friendly microextraction technique164 derived from SPME. SBSE possesses many merits, such as high EF, good reproducibility, high adsorption capacity and excellent recoveries.Most applications of SBSE are for the analysis of organic compounds. There are however applications of SBSE to trace element analysis and their speciation, which will be discussed below. In this part, studies that applied SBSE to elemental speciation in biological samples are discussed.
Highly polar TiO2 was used as a stir bar coating by immobilizing on polypropylene hollow fiber (PPHF) through sol–gel immersion and a low temperature hydrothermal process.165 The obtained TiO2–PPHF inherits both the adsorption properties of TiO2 and the toughness of PPHF, and the method of SBSE combined with HPLC-ICP-MS was successfully applied for the speciation of phenyl arsenic compounds and their possible transformation products in chicken tissues.
By using a PSP–TiO2 organic–inorganic hybrid material as the stir bar coating, Mao et al.166 combined SBSE with HPLC and ICP-MS for the determination of seleno-amino acids (including SeCys2, MeSeCys, SeMet and SeEth) and seleno-oligopeptides (including γ-GluMeSeCys and GS-Se-SG). The hybridization of PSP with TiO2 can avoid the swelling of PSP and cracking of TiO2 because of the good film-forming property of PSP with the high mechanical strength of TiO2. The developed method was applied to the analysis of certified reference materials prepared from selenium enriched yeast, human urine and garlic samples.
Fan et al.167 prepared polyethyleneglycol (PEG)/hydroxyl polydimethylsiloxane (OH-PDMS)/γ-MPTS coated stir bars and used for the SBSE of amphoteric thyroxines (T4, T3, rT3) and their metabolite (T2) followed by HPLC-UV and HPLC-ICP-MS analysis in human urine samples. The linear range obtained by SBSE-HPLC-UV was 2–500 μg L−1 for T2 and 5–500 μg L−1 for rT3, T3 and T4, while the linear range obtained by SBSE-HPLC-ICP-MS was 0.05–500 μg L−1 for T2 and rT3, 0.10–200 μg L−1 for T3 and 0.05–200 μg L−1 for T4. The LODs for the target thyroxines were 0.60–2.20 μg L−1 for SBSE-HPLC-UV and 0.0071–0.0355 μg L−1 for SBSE-HPLC-ICP-MS. The developed method was then applied for the determination of target thyroxines in urine samples, and the recovery for the spiked samples obtained by SBSE-HPLC-UV was in the range of 81.6–137.6% for human urine, while the recovery for the spiked urine samples obtained by SBSE-HPLC-ICP-MS was in the range of 72.0–121.5%.
Mao et al.168 also developed a new approach of C18-coated SBSE coupled with HPLC-ICP-MS for the speciation of butyltins. The butyltin compounds (MBT, DBT and TBT) were first extracted with a C18-coated stir bar, and then desorbed with 40% (v/v) methanol for subsequent HPLC-ICP-MS analysis. The analytical operation conditions of the C18-SBSE have been systematically studied so that the percentage of organic solvents in the mobile phase is reduced and separation resolution is improved. A CN column with carboxylic acids as the mobile phase additives was used for butyltin separation. Quick separation (<8 min) was accomplished using a methanol–formic acid–water (16:8:76, v/v/v) mobile phase containing 5 mmol L−1 mercaptoacetic acid, and the mobile phase was compatible with the conventional quadrupole ICP-MS detector without addition of oxygen gas or desolvation and post-column dilution systems. Under the optimal conditions, the limits of detection for the three target butyltins ranged from 15.6 to 29.4 ng L−1, with a linear range of 0.05–50 μg L−1. The EFs were 85–127-fold. The developed method is simple and rapid, with no derivatization involved.
In headspace mode, SBSE was applied in the simultaneous extraction of mercury and organometallic tin compounds present in oyster tissue, combined with thermal desorption and GC-MS detection.169 Before the SBSE process, all solid samples were subjected to extraction by diluted HCl or KOH/methanol and then derivatization by NaBEt4. Mellano et al.170 developed a method for the determination of MeSeCys and SeMet in yeast, algae and clam samples by SBSE-GC-MS. The samples were subjected to an ultrasonic-assisted enzymatic digestion before extraction by a 10 mm × 1 mm PDMS stir bar.
A method of HS-SBSE-GC-ICP-MS for the determination of volatile organoseleno compounds (DMSe and DMDSe) in garlic, onion and their juices has been developed.39 In this work, the extraction performance of various head space sorptive bar coatings, including PDMS, PDMS–poly(vinyl alcohol) (PVA), carbowax–PDMS–PVA, and PDMS–β-CD, has been studied, and the best extraction efficiencies for DMSe and DMDSe were obtained for the PDMS–PVA sorptive bar coating. The LODs were 33 and 7.1 ng L−1 for DMSe and DMDSe, respectively. Giraaldez et al.171 adopted a dual SBSE and single thermal desorption combined with capillary GC-MS for the speciation of Se(IV) and SeMet in clams. Before the SBSE step, samples are extracted by an ultrasonic probe assisted enzymatic hydrolysis, Se(IV) and selenoamino acids are derivatized to piazselenol and N-isobutoxycarbonylmethyl ester derivatives respectively, and then diluted with water.
The applications of SPME techniques including fiber-SPME, CME and SBSE have a number of advantages and these are rapidity, simplicity, low sample/solvent consumption and a convenient format. With the help of various microextraction formats, SPME has been successfully used for elemental speciation in biological fluids and extracts.
SPE/SPME has been widely used in elemental speciation of biological samples because of their excellent matrix tolerance and high EF, and they are less-time consuming. SPE is more suited for the analysis of large volume biological samples, while SPME, CME and SBSE are used with smaller volumes, less than 1 mL. Chip-based methods are the best choice for the pre-treatment of sample volumes less than 1 mL. It is noteworthy that MSPD can handle solid and semi-solid samples directly. Indeed SPME also possesses this potential when the in vivo SPME mode is used. In SPE/SPME, appropriate adsorbent selection is the key to successful method development. The extraction of elemental species is mostly based on the following types of interactions: hydrophobic, ion-exchange and complexation; as a result, sorbents such as C18, ion-exchange materials and thiol/amino/carboxyl group modified materials are widely used in SPE/SPME. Take mercury species for example, thiol group materials such as Fe3O4@SiO2@MPTS are suitable sorbents. For selenoamino acids, ion-exchange materials might be more suitable. C18 is a universal sorbent which is widely used for clean-up and extraction in biological samples.
5. Microfluidic chip for elemental speciation in biological samples
The micro-total analysis system (μTAS) was first proposed by Manz and Widmer172 and has been widely used in chemical and biological analysis over the past 25 years. The microfluidic chip presents a functional tool-set for chemical analysis offering numerous advantages including: reduced sample and reagent consumption; reduced analysis time; high sensitivity; integrated system with little human intervention which reduces the odds of human error and sample contamination.173,174Chip-based pre-treatment methods utilize the properties of μTAS, which integrate force (motion),175 heat,176 light,177 sound,178,179 electricity,180,181 and magnetism182,183 on a chip to optimize the conditions for the pre-treatment of real samples. Despite many examples of the application of the microfluidic chip in gene, protein, and cell research,184–190 there are few investigations that have focused on the development of chip-based analysis methods for elemental speciation in biological samples. Indeed the microfluidic chip is an excellent platform for the manipulation and analysis of biological samples especially cells and blood (serum), on account of its ability to handle micro-amounts of samples in an integrated and automated process. Several research groups have attempted to develop analytical (pre-treatment) systems for the determination of trace elements and elemental speciation.
5.1. Chip-based liquid phase microextraction
Microscale analytical technology is recognized as a new way to miniaturize liquid phase microextraction.191–193 Laminar flow and droplets between the aqueous phase and organic phase are easy to form in a microchannel, so chip-based liquid phase microextraction possesses larger contact area and higher extraction efficiency. Therefore, several LPME systems have been developed and combined with ICP-MS for the analysis of elements. Zhang et al.194 used modified channels on a chip to achieve the laminar flow extraction process of uranium. Nichols et al.195 described a method to achieve liquid–liquid extraction of lanthanides and actinides by using droplet-based microfluidics combined with ICP-MS. Besides, a chip-based liquid phase microextraction system196 (Fig. 3C) was developed and combined with ETV-ICP-MS for the determination of trace metals in cell samples by Hu and his colleagues. On the chip, laminar flow LPME was developed with DDTC as both the chemical modifier for low-temperature ETV-ICP-MS and the extracting reagent for chip-based LPME. Even though there are many advantages of chip-based LPME, the collection of extractants is difficult thus limiting its application in elemental speciation. There are currently no chip-based LPME systems that have been coupled to ICP-MS techniques.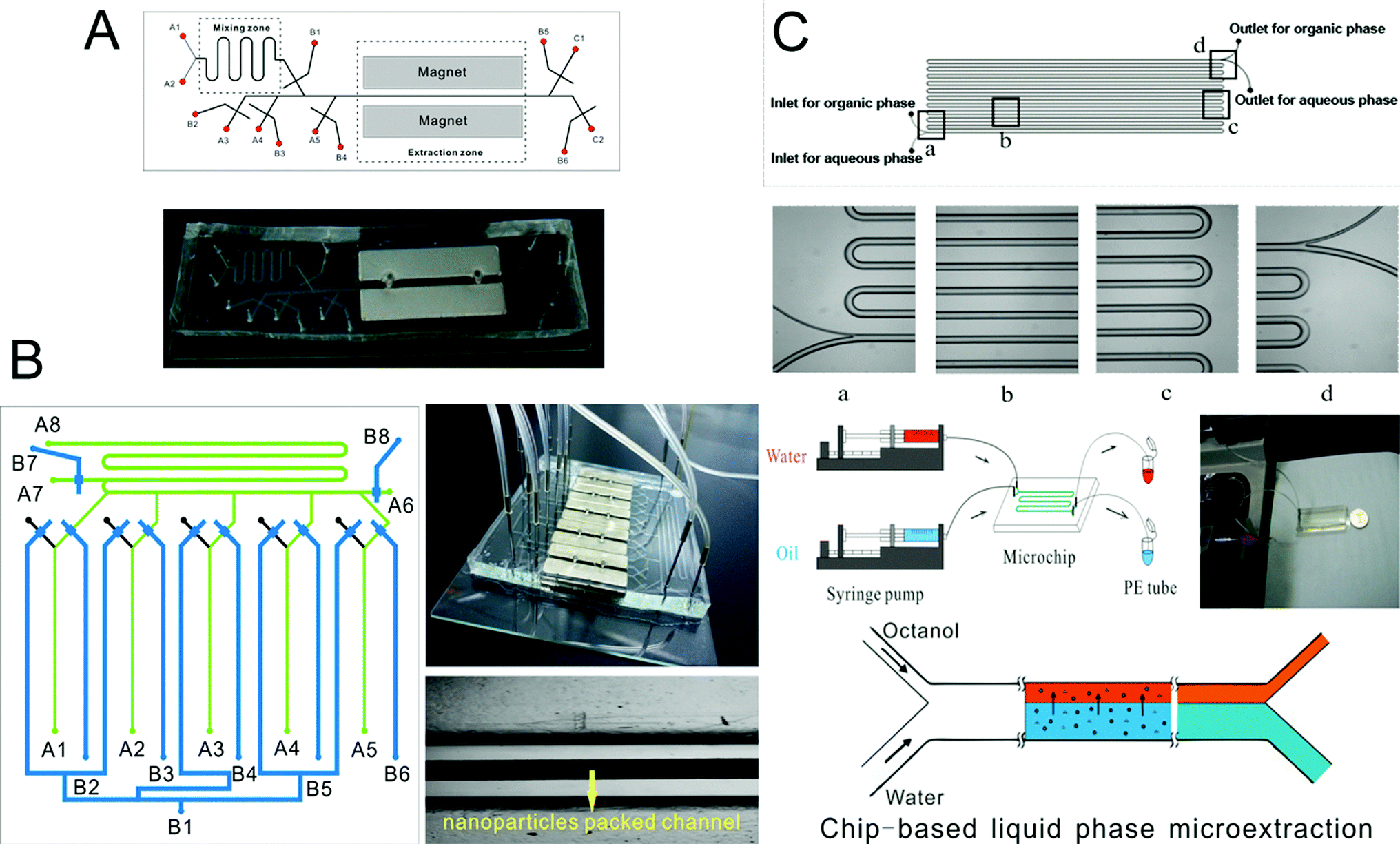 | ||
| Fig. 3 Schematic of chip-based MSPME (A), on-line array MSPME (B) and LPME (C) analysis systems for the determination of trace elements in cell samples. | ||
5.2. Chip-based solid phase microextraction
Zhang et al.197 prepared an ethanediamine modified polyglycidyl methacrylate trimethylol propane triacrylate monolithic column (poly(GMA-co-TRIM-NH2)) in the microfluidic chip channel, and established a novel method by combining chip-based poly(GMA-co-TRIM-NH2) monolithic microextraction with ICP-MS for the determination of bismuth in cell samples. Shih et al. developed an open-channel198 and dipole-assisted199 SPME chip for the determination of trace heavy metals. Hsu et al.200 developed a gold nanoparticle-coated chip for the determination of Hg2+ ions in aqueous samples. Besides ICP-MS, other techniques have been used with the chip for the determination of metals (e.g. Pb and Cd201). Park et al.202 fabricated a cation exchange resin packed microchamber combined with a DNA aptamer linked photoluminescent graphene oxide quantum dot sensor for the detection of lead ions. Date et al.203 developed an on-chip detection method for mercury ions which combined solid phase extraction (ion exchange resin) with Hg2+ immunoassay and labeled with Au nanoparticles.Magnetic nanoparticles204–206 which have a high surface-to-volume ratio and good biocompatibility have been integrated into a chip making it possible to control their behavior with the aid of a magnet. Hu's group207–210 has developed several chip-based magnetic solid phase microextraction (MSPME) pre-treatment systems and combined them with ICP-MS for the analysis of trace elements in cell samples. In the first work,207 a chip-based MSPME system was developed (Fig. 3A) and combined with ETV-ICP-MS for the determination of Cd, Hg and Pb in HepG2 cells. A packed column was fabricated in a PDMS chip channel in which γ-MPTS modified silica coated magnetic nanoparticles were self-assembled under the influence of a magnetic field. The microextraction column was integrated with a cell rupture section and several microvalves for the manipulation of cells on a chip. After the optimization of the main factors influencing extraction, the analytical performance was investigated and the LODs were 0.72, 0.86 and 1.12 ng L−1 for Cd, Hg and Pb, respectively. Quantitative analyses of trace Cd, Hg and Pb in only 5000 HepG2 cells were achieved by applying the microfluidic system combined with ETV-ICP-MS. However, not every laboratory has access to an ETV sample introduction system. In addition, there are drawbacks associated with these off-line systems, such as elaborate manual operations, possible analyte loss and contamination from reagents or vials. In order to limit the effects of these factors, an array MSPME chip208 was fabricated (Fig. 3B) by the same group and on-line combined with ICP-MS for the determination of trace heavy metals in three kinds of cells (HepG2, Jurkat T and MCF7 cells). Five microextraction channels were integrated with several microvalves on the chip which improved the sample throughput of this system; as a result, the microextraction was performed automatically with excellent reproducibility.
Chip-based MSPME could also be combined with ICP-MS hyphenated techniques for elemental speciation in cell samples. Chen et al.209 developed an on-chip MSPME system and combined with HPLC-ICP-MS for the speciation of selenium in selenium-enriched yeast cells. The chip design (Fig. 4A) was similar to that of previous reports. Sulfonated polystyrene-coated magnetic nanoparticles were prepared and self-assembled in microextraction channels.209 The LODs were 0.057, 0.117, 0.116, 0.149 and 0.095 μg L−1 for SeCys2, MeSeCys, SeMet, GluMeSeCys and SeEth, respectively. This on-chip MSPME-HPLC-ICP-MS method has its own unique features of low sample consumption and high integration, and capability of detecting trace levels of selenoamino acids and selenopeptide in a small amount (∼800) of selenium-enriched yeast cells. In another study,210 an on-line chip-based MSPME-HPLC-ICP-MS analysis platform was developed (Fig. 4B) for mercury speciation in HepG2 cells. Two microextraction channels packed with sulfhydryl modified magnetic nanoparticles were integrated with two cell lysis unit and on-line combined with microHPLC-ICP-MS through a commercial six-way valve. The LODs of this method were 18.8, 12.8, 17.4 and 41.8 ng L−1 for Hg2+, MeHg+, EtHg+ and PhHg+, respectively. Thanks to the advantages of integration, miniaturization and automatization, the analysis results of this on-line chip-based MSPME-microHPLC-ICP-MS method are of good consistency even with consumption of 800 HepG2 cells. This method was applied to the analysis of mercury species in HepG2 cells in order to investigate the protection mechanism of the cells against mercury toxicity. Shih et al.211 developed a hyphenated system employing a microfluidic-based photocatalyst-assisted vapor generation system combined with HPLC-ICP-MS for the analysis of inorganic selenium species in water samples.
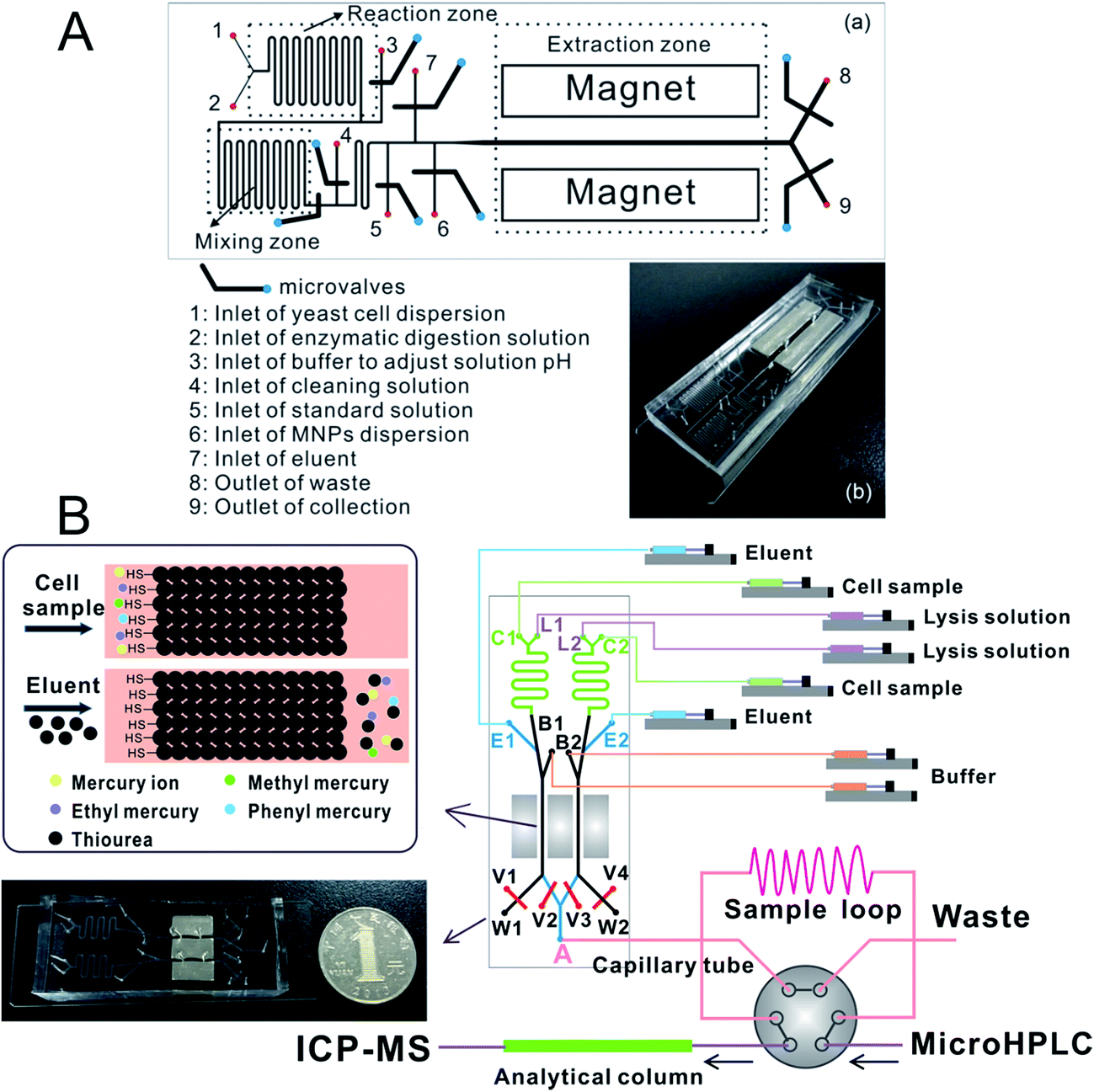 | ||
| Fig. 4 Schematic of chip-based MSPME-HPLC-ICP-MS (A) and on-line chip-based MSPME-HPLC-ICP-MS (B) systems for the analysis of cell samples for selenium and mercury species, respectively. | ||
In a number of studies, microfluidic chips have been coupled to ICP-MS for elemental speciation without the need for separation techniques. Xi et al.212 fabricated a centrifugal microfluidic chip that was integrated into an eight SPME column system packed with commercial silica gel for chromium speciation analysis in water samples. Hsu et al.213 developed a polyoxometalate cluster immobilized filter paper packed into a chamber on a chip, and coupled this to an ICP-MS for the study of chromium speciation in water samples. In another development, Li et al.214 fabricated a centrifugal microfluidic chip integrated with eight monolithic capillary SPME columns for the analysis of chromium speciation in water samples.
There are currently a few studies in which SPME/MSPME microfluidic chips are coupled to ICP-MS techniques, however, this combination of a miniaturized sample pre-treatment platform, highly efficient separation techniques and high sensitivity detection instrument exhibits excellent analytical performance.
5.3. Microfluidic electrophoresis chip
CE is a highly efficient separation technique, which can be used for chemical and biological analyses. Conventional CE is performed in fused silica capillaries, but microfabrication techniques make it possible to design an electrophoresis system on a microchip.215 A typical microchip CE device216 has a straight separation channel, buffer, sample and waste reservoirs and an outlet to the detection system. Literature on the application of microchip-electrophoresis to elemental speciation has been published.217 Indeed diverse detection techniques such as laser induced fluorescence, LED absorbance detection, chemiluminescence, and atomic fluorescence spectrometry have been coupled with microchip electrophoresis218,219 for elemental speciation.Although ICP-MS is the most powerful technique for the determination of elemental speciation, the interface between the ICP-MS and microchip electrophoresis presents technical challenges. Song et al.220 developed the interfaces between microchip electrophoresis and ICP-MS, and applied this method for the separation of Cr(III) and Cr(VI), Cu(II) and Cu(EDTA)2−, As(III) and As(V), but no real samples were analyzed. Cheng et al.221 developed a suction-free interface between microchip-electrophoresis and ICP-MS (Fig. 5). In this design, two porous polymer plugs were fabricated to eliminate the nebulizer suction and ensure the injection of makeup solution. The separation of I− and IO3− was achieved within 25 s with a separation length of 15 mm. Cheng et al.222 combined microchip electrophoresis-ICP-MS with the field-amplified sample injection technique for the determination of Br− and BrO3− in bread and the LODs were 0.18 and 0.22 μg L−1, respectively.
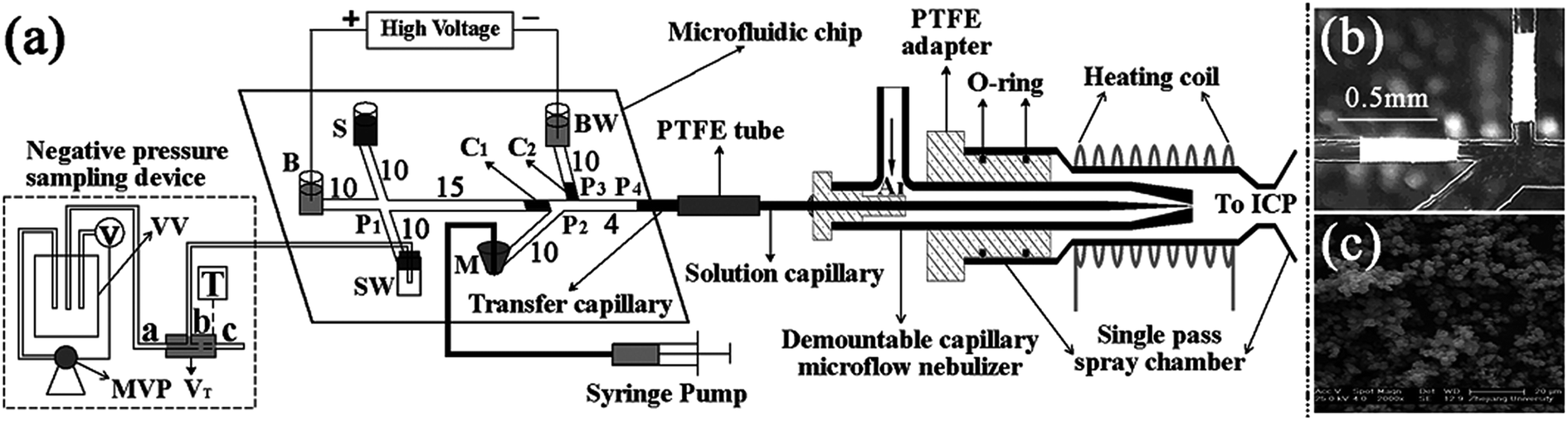 | ||
| Fig. 5 (a) Schematic diagram of the MCE-ICP-MS system, (b) photograph of the microchannels with the in situ polymerized plugs and (c) the scanning electron microscope micrograph of the macroporous plugs. | ||
There are currently no known applications of microchip CE-ICP-MS to the analysis of biological samples. Microchip CE is an efficient separation method, its application to the analysis of complex biological samples is not trivial. An integrated chip-based system which consists of a suitable pre-treatment method (such as SPME) and microchip electrophoresis may offer solution to the potential technical problems which are encountered with biological samples. Although there are no examples of their use for elemental speciation, applications in other areas provide evidence of the proof of concept in which miniaturized sample treatment platforms are combined with highly sensitive detection.
6. Conclusion/future prospects
In order to address the problems including elemental species lability, low concentration of the target species, matrix interference and limited sample quantities, a variety of sample pre-treatment methods have been developed and combined with highly sensitive ICP-MS hyphenated techniques. Indeed, their application potential has been demonstrated with less complex sample matrices.LLE/LPME offers high EF and can be deployed in multiple formats, but suffers from potential interferences and is usually used for the pre-treatment of biological fluids after appropriate dilution. SPE/SPME possesses excellent matrix tolerance and it is probable that with the development of new functional materials, different kinds of analytes could be extracted and enriched. More importantly, the extraction process can be performed in diversified operation modes making possible on-line and array analysis. In addition, these pre-treatment methods can be used for the analysis of elemental species in large to sub mL volumes, as well as in solid and semi-solid samples. Chip-based systems can be miniaturized and integrated for the pre-treatment and manipulation of cell samples. Although preliminary results are promising, several obstacles remain in the development of appropriate sample pre-treatment methods coupled with ICP-MS for elemental speciation in biological samples.
(1) There is currently a shortage of calibration and reference standards for use in elemental speciation. In the papers cited herein the focus has been mainly on the analysis of mercury, selenium and arsenic species. And only a few species of these three elements were determined due to the unavailability of appropriate standards. The characterization of such standards will depend on the use of organic mass spectrometry. The relatively low concentrations of the elemental species present analytical challenges. Even with the help of analyte enrichment, and the use of sensitive detection, the levels of some of the species are still lower than the limits of detection of current analytical methods. As a result, pre-treatment methods with higher EF and selectivity are still urgently needed. The use of multiple extractions could improve method sensitivity and reduce matrix interferences.
(2) Difficulty of low abundant species analysis. Even with the help of analyte enrichment in pre-treatment methods and highly sensitive analytical instruments, the amount of some low abundant species was still lower than the LODs of developed analytical systems. As a result, pre-treatment methods with higher EF and selectivity are still in urgent need. The combination of multiple extraction methods is a good choice to improve the method sensitivity and matrix resistance ability, and would play important roles in the elemental speciation in biological samples.
(3) It is essential that the chemical species of the elements are not altered during sample pre-treatment. Exposure to strong chelating reagents, long extraction times and high temperatures are all factors which affect the stability of elemental species. As a result, the development of fast, low temperature and mild pre-treatment methods are urgently needed especially for the analysis of labile elemental species.
(4) Going forward, miniaturization, integration and automation will be required for the analysis of minute sample quantities. Automation and integration will decrease manual operation errors and reduce potential contamination and thus the accuracy and reproducibility of the process will be improved. A few on-line, automated and miniaturized systems have been developed but much more needs to be done to improve the overall analytical performance of such systems.
List of abbreviations
| AAPTS | N-(2-Aminoethyl)-3-aminopropyltrimethoxysilane |
| AED | Atomic emission detector |
| AES | Atomic emission spectrometry |
| AFS | Atomic fluorescence spectroscopy |
| AS | Automatic static |
| As(III) | Arsenite |
| As(V) | Arsenate |
| AsB | Arsenobetaine |
| AsC | Arsenocholine |
| ASEI | Anion selective exhaustive injection |
| β-CD | β-Cyclodextrin |
| CE | Capillary electrophoresis |
| CME | Capillary microextraction |
| DBT | Dibutyltin |
| DDTC | Diethyldithiocarbamate |
| DEtSe | Diethylselenide |
| DLLME | Dispersive liquid–liquid microextraction |
| DMAs(V) | Dimethylarsenic |
| DMDSe | Dimethyldiselenide |
| DMSe | Dimethylselenide |
| DMT | Dimethyltin |
| DIT | Diiodotyrosine |
| DPT | Diphenyltin |
| DVB | Divinylbenzene |
| EF | Enrichment factor |
| ESI | Electrospray ionization |
| EtHg | Ethylmercury |
| ETV | Electrothermal vaporization |
| GC | Gas chromatography |
| γ-GluSeMeCys | γ-Glutamyl-Se-methyl-selenocysteine |
| GS-Se-SG | Selenodiglutathione |
| HF | Hollow fiber |
| HPLC | High performance liquid chromatography |
| HS | Headspace |
| ICP | Inductively coupled plasma |
| LC | Liquid chromatography |
| LLE | Liquid liquid extraction |
| LLLME | Liquid liquid liquid microextraction |
| LOD | Limit of detection |
| LPME | Liquid phase microextraction |
| LVSS | Large volume sample stacking |
| MAA | Methacrylic acid |
| MBT | Monobutyltin |
| MIP | Microwave induced plasma |
| MIT | Monoiodotyrosine |
| MeHg | Methylmercury |
| MEKC | Micellar electrokinetic capillary chromatography |
| MMAs(V) | Monomethylarsenic |
| MMT | Monomethyltin |
| MPT | Monophenyltin |
| γ-MPTS | γ-Mercaptopropyl trimethoxysilane |
| MS | Mass spectrometry |
| MSPD | Matrix solid phase dispersion extraction |
| MSPE | Magnetic solid phase extraction |
| MSPME | Magnetic solid phase microextraction |
| MWCNTs | Multi-wall carbon nanotubes |
| OH-PDMS | Hydroxyl polydimethylsiloxane |
| PDMS | Polydimethylsiloxane |
| PEG | Polyethyleneglycol |
| PhHg | Phenylmercury |
| PPHF | Polypropylene hollow fiber |
| PPY | Polypyrrole |
| PSP | Partial sulfonated poly(styrene) |
| PT | Phase transfer |
| PVA | Poly(vinyl alcohol) |
| RSD | Relative standard deviation |
| Sb(III) | Antimonite |
| Sb(V) | Antimonate |
| SBSE | Stir bar sorptive extraction |
| SDME | Single drop microextraction |
| Se(IV) | Selenite |
| Se(VI) | Selenate |
| SeEth | Selenoethionine |
| SeMeCys | Se-methyl-selenocysteine |
| SeMet | Selenomethionine |
| SPE | Solid phase extraction |
| SPME | Solid phase microextraction |
| T2 | 3,5-Diiodothyronine |
| T3 | 3,3′,5-Triiodothyronine |
| rT3 | Reversed-3,3′,5-triiodothyronine |
| T4 | 3,3′,5,5′-Tetraiodothyronine |
| μTAS | Micro-total analysis systems |
| TBT | Tributyltin |
| TET | Triethyltin |
| TML | Trimethyllead |
| TMT | Trimethyltin |
| TTBT | Tetrabutyltin |
| TPT | Triphenyltin |
| UV | Ultraviolet |
Acknowledgements
This work is financially supported by the National Nature Science Foundation of China (Nos. 21575107, 21375097, 21175102), the Science Fund for Creative Research Groups of NSFC (No. 20621502, 20921062), and the Fundamental Research Funds for the Central Universities (2015203020203), the MOE of China.References
- S. Caroli, Microchem. J., 1995, 51, 64–72 CrossRef CAS.
- R. Lobinski, Spectrochim. Acta, Part B, 1998, 53, 177–185 CrossRef.
- G. M. Hieftje, Spectrochim. Acta, Part B, 1998, 53, 165–167 CrossRef.
- A. M. Florea and D. Busselberg, BioMetals, 2006, 19, 419–427 CrossRef CAS PubMed.
- I. L. Heras, M. Palomo and Y. Madrid, Anal. Bioanal. Chem., 2011, 400, 1717–1727 CrossRef CAS PubMed.
- C. T. Chasapis, A. C. Loutsidou, C. A. Spiliopoulou and M. E. Stefanidou, Arch. Toxicol., 2012, 86, 521–534 CrossRef CAS PubMed.
- I. F. Scheiber, J. F. B. Mercer and R. Dringen, Prog. Neurobiol., 2014, 116, 33–57 CrossRef CAS PubMed.
- D. G. Barceloux, J. Toxicol., Clin. Toxicol., 1999, 37, 173–194 CrossRef CAS PubMed.
- D. M. Stearns, BioFactors, 2000, 11, 149–162 CrossRef CAS PubMed.
- H. Haraguchi, J. Anal. At. Spectrom., 2004, 19, 5–14 RSC.
- J. Szpunar, Analyst, 2000, 125, 963–988 RSC.
- A. Sanz-Medel, Spectrochim. Acta, Part B, 1998, 53, 197–211 CrossRef.
- E. Devoto and R. A. Yokel, Environ. Health Perspect., 1994, 102, 940–951 CrossRef CAS PubMed.
- S. Caroli, A. Alimonti, E. Coni, F. Petrucci, O. Senofonte and N. Violante, Crit. Rev. Anal. Chem., 1994, 24, 363–398 CrossRef CAS.
- Y. Liu, M. He, B. B. Chen and B. Hu, Talanta, 2015, 142, 213–220 CrossRef CAS PubMed.
- Y. Zhang, J. K. Duan, M. He, B. B. Chen and B. Hu, Talanta, 2013, 115, 730–736 CrossRef CAS PubMed.
- S. W. Su, B. B. Chen, M. He, Z. W. Xiao and B. Hu, J. Anal. At. Spectrom., 2014, 29, 444–453 RSC.
- V. L. Dressler, F. G. Antes, C. M. Moreira, D. Pozebon and F. A. Duarte, Int. J. Mass Spectrom., 2011, 307, 149–162 CrossRef CAS.
- B. B. Chen, M. He, X. J. Mao, R. Cui, D. W. Pang and B. Hu, Talanta, 2011, 83, 724–731 CrossRef CAS PubMed.
- F. Vanhaecke, M. VanHolderbeke, L. Moens and R. Dams, J. Anal. At. Spectrom., 1996, 11, 543–548 RSC.
- D. Schaumloffel, J. R. Encinar and R. Lobinski, Anal. Chem., 2003, 75, 6837–6842 CrossRef PubMed.
- B. Chen, Y. Wu, X. Guo, M. He and B. Hu, J. Anal. At. Spectrom., 2015, 30, 875–881 RSC.
- X. J. Mao, B. Hu, M. He and B. B. Chen, J. Chromatogr. A, 2012, 1256, 32–39 CrossRef CAS PubMed.
- J. Kunnemeyer, L. Terborg, B. Meermann, C. Brauckmann, I. Moller, A. Scheffer and U. Karst, Environ. Sci. Technol., 2009, 43, 2884–2890 CrossRef PubMed.
- S. Imanaka and H. Hayashi, Water Sci. Technol.: Water Supply, 2013, 13, 96–103 CrossRef CAS.
- P. Pacheco, T. Hanley and J. A. Landero Figueroa, Metallomics, 2014, 6, 560–571 RSC.
- M. Slachcinski, Appl. Spectrosc. Rev., 2014, 49, 271–321 CrossRef CAS.
- S. L. C. Ferreira, W. N. L. dos Santos, I. F. dos Santos, M. M. S. Junior, L. O. B. Silva, U. A. Barbosa, F. A. de Santana and A. F. D. Queiroz, Microchem. J., 2014, 114, 22–31 CrossRef CAS.
- Y. Gao, R. Liu and L. Yang, Chin. Sci. Bull., 2013, 58, 1980–1991 CrossRef CAS.
- W. Maher, F. Krikowa, M. Ellwood, S. Foster, R. Jagtap and G. Raber, Microchem. J., 2012, 105, 15–31 CrossRef CAS.
- Y. Arslan, E. Yildirim, M. Gholami and S. Bakirdere, TrAC, Trends Anal. Chem., 2011, 30, 569–585 CrossRef CAS.
- P. Wu, L. A. He, C. B. Zheng, X. D. Hou and R. E. Sturgeon, J. Anal. At. Spectrom., 2010, 25, 1217–1246 RSC.
- A. H. Petursdottir, H. Gunnlaugsdottir, H. Jorundsdottir, A. Mestrot, E. M. Krupp and J. Feldmann, Anal. Bioanal. Chem., 2012, 404, 2185–2191 CrossRef CAS PubMed.
- S. S. de Souza, A. D. Campiglia and F. Barbosa, Anal. Chim. Acta, 2013, 761, 11–17 CrossRef CAS PubMed.
- M. Popp, S. Hann and G. Koellensperger, Anal. Chim. Acta, 2010, 668, 114–129 CrossRef CAS PubMed.
- M. M. Bayon, M. G. Camblor, J. I. G. Alonso and A. Sanz-Medel, J. Anal. At. Spectrom., 1999, 14, 1317–1322 RSC.
- D. Esteban-Fernandez, M. Mirat, M. I. M. de la Hinojosa and J. I. G. Alonso, J. Agric. Food Chem., 2012, 60, 8333–8339 CrossRef CAS PubMed.
- R. Wahlen and C. Wolff-Briche, Anal. Bioanal. Chem., 2003, 377, 140–148 CrossRef CAS PubMed.
- J. Duan, X. Li, C. Yu and B. Hu, J. Anal. At. Spectrom., 2009, 24, 297–303 RSC.
- B. Meermann and M. Sperling, Anal. Bioanal. Chem., 2012, 403, 1501–1522 CrossRef CAS PubMed.
- S. S. Kannamkumarath, K. Wrobel, K. Wrobel, C. B'Hymer and J. A. Caruso, J. Chromatogr. A, 2002, 975, 245–266 CrossRef CAS PubMed.
- A. Prange and D. Profrock, Anal. Bioanal. Chem., 2005, 383, 372–389 CrossRef CAS PubMed.
- X. L. Liu, B. B. Chen, L. Zhang, S. Y. Song, Y. B. Cai, M. He and B. Hu, Anal. Chem., 2015, 87, 8949–8956 CrossRef CAS PubMed.
- Y. J. Xue, H. Gao, Q. C. Ji, Z. Lam, X. P. Fang, Z. P. Lin, M. Hoffman, D. Schulz-Jander and N. D. Weng, Bioanalysis, 2012, 4, 2637–2653 CrossRef CAS PubMed.
- P. Kawczak and T. Baczek, Cent. Eur. J. Chem., 2012, 10, 570–584 Search PubMed.
- M. R. Brunetto, M. A. Obando, A. Fernandez, M. Gallignani, J. L. Burguera and M. Burguera, Talanta, 2002, 58, 535–542 CrossRef CAS PubMed.
- W. M. Mullett, J. Biochem. Biophys. Methods, 2007, 70, 263–273 CrossRef CAS PubMed.
- M. Rambla-Alegre, R. Marti-Centelles, J. Esteve-Romero and S. Carda-Broch, J. Chromatogr. A, 2011, 1218, 4972–4981 CrossRef CAS PubMed.
- W. Zhang, F. T. Han, H. Zhao, Z. P. Lin, Q. T. Huang and N. D. Weng, Biomed. Chromatogr., 2012, 26, 1163–1169 CrossRef CAS PubMed.
- N. C. Hughes, N. Bajaj, J. A. Fan and E. Y. K. Wong, Bioanalysis, 2009, 1, 1057–1066 CrossRef CAS PubMed.
- M. L. Chiu, W. Lawi, S. T. Snyder, P. K. Wong, J. C. Liao and V. Gau, JALA, 2010, 15, 233–242 CAS.
- E. Chambers, D. M. Wagrowski-Diehl, Z. L. Lu and J. R. Mazzeo, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 852, 22–34 CrossRef CAS PubMed.
- Y. Huang, R. Shi, W. Gee and R. Bonderud, Bioanalysis, 2012, 4, 271–279 CrossRef CAS PubMed.
- S. Soltani and A. Jouyban, Bioanalysis, 2014, 6, 1691–1710 CrossRef CAS PubMed.
- B. B. Chen, M. He, C. Zhong and B. Hu, J. Chromatogr. A, 2014, 1363, 62–70 CrossRef CAS PubMed.
- F. Pena-Pereira, I. Lavilla and C. Bendicho, Spectrochim. Acta, Part B, 2009, 64, 1–15 CrossRef.
- B. Hu, M. He, B. B. Chen and L. B. Xia, Spectrochim. Acta, Part B, 2013, 86, 14–30 CrossRef CAS.
- A. Sarafraz-Yazdi and A. Amiri, TrAC, Trends Anal. Chem., 2010, 29, 1–14 CrossRef CAS.
- B. Hu, M. He and B. B. Chen, Anal. Bioanal. Chem., 2015, 407, 2685–2710 CrossRef CAS PubMed.
- M. Wierucka and M. Biziuk, TrAC, Trends Anal. Chem., 2014, 59, 50–58 CrossRef CAS.
- Z. Mester, R. Sturgeon and J. Pawliszyn, Spectrochim. Acta, Part B, 2001, 56, 233–260 CrossRef.
- Z. Mester and R. Sturgeon, Spectrochim. Acta, Part B, 2005, 60, 1243–1269 CrossRef.
- M. He, B. B. Chen and B. Hu, Anal. Bioanal. Chem., 2014, 406, 2001–2026 CrossRef CAS PubMed.
- B. Hu, F. Zheng, M. He and N. Zhang, Anal. Chim. Acta, 2009, 650, 23–32 CrossRef CAS PubMed.
- T. C. Chao and A. Ros, J. R. Soc., Interface, 2008, 5, S139–S150 CrossRef CAS PubMed.
- M. C. Peoples and H. T. Karnes, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 866, 14–25 CrossRef CAS PubMed.
- X. J. Feng, W. Du, Q. M. Luo and B. F. Liu, Anal. Chim. Acta, 2009, 650, 83–97 CrossRef CAS PubMed.
- E. W. K. Young and D. J. Beebe, Chem. Soc. Rev., 2010, 39, 1036–1048 RSC.
- V. Lecault, A. K. White, A. Singhal and C. L. Hansen, Curr. Opin. Chem. Biol., 2012, 16, 381–390 CrossRef CAS PubMed.
- T. C. Chao and N. Hansmeier, Proteomics, 2013, 13, 467–479 CrossRef CAS PubMed.
- X. Q. Guo, X. T. Tang, M. He, B. B. Chen, K. Nan, Q. Y. Zhang and B. Hu, RSC Adv., 2014, 4, 19960–19969 RSC.
- G. A. Zachariadis and E. Rosenberg, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 1140–1144 CrossRef CAS PubMed.
- G. A. Zachariadis and E. Rosenberg, J. Sep. Sci., 2012, 35, 1132–1137 CrossRef CAS PubMed.
- S. Udrescu, I. D. Sora, F. Albu, V. David and A. Medvedovici, J. Pharm. Biomed. Anal., 2011, 54, 1163–1172 CrossRef CAS PubMed.
- A. Spietelun, L. Marcinkowski, M. de la Guardia and J. Namiesnik, Talanta, 2014, 119, 34–45 CrossRef CAS PubMed.
- S. Dadfarnia and A. M. H. Shabani, Anal. Chim. Acta, 2010, 658, 107–119 CrossRef CAS PubMed.
- E. Psillakis and N. Kalogerakis, TrAC, Trends Anal. Chem., 2003, 22, 565–574 CrossRef CAS.
- J. M. Kokosa, TrAC, Trends Anal. Chem., 2015, 71, 194–204 CrossRef CAS.
- L. Xu, C. Basheer and H. K. Lee, J. Chromatogr. A, 2007, 1152, 184–192 CrossRef CAS PubMed.
- E. Psillakis and N. Kalogerakis, TrAC, Trends Anal. Chem., 2002, 21, 53–63 CrossRef CAS.
- S. Pedersen-Bjergaard and K. E. Rasmussen, J. Chromatogr. A, 2008, 1184, 132–142 CrossRef CAS PubMed.
- J. Y. Lee, H. K. Lee, K. E. Rasmussen and S. Pedersen-Bjergaard, Anal. Chim. Acta, 2008, 624, 253–268 CrossRef CAS PubMed.
- M. Rezaee, Y. Yamini and M. Faraji, J. Chromatogr. A, 2010, 1217, 2342–2357 CrossRef CAS PubMed.
- Q. Xiao, B. Hu and M. He, J. Chromatogr. A, 2008, 1211, 135–141 CrossRef CAS PubMed.
- F. Pena-Pereira, I. Lavilla, C. Bendicho, L. Vidal and A. Canals, Talanta, 2009, 78, 537–541 CrossRef CAS PubMed.
- S. Pedersen-Bjergaard and K. E. Rasmussen, Anal. Chem., 1999, 71, 2650–2656 CrossRef CAS PubMed.
- E. Ghasemi, M. Sillanpaa and N. M. Najafi, J. Chromatogr. A, 2011, 1218, 380–386 CrossRef CAS PubMed.
- J. Duan and B. Hu, J. Mass Spectrom., 2009, 44, 605–612 CrossRef CAS PubMed.
- F. Moreno, T. Garcia-Barrera and J. L. Gomez-Ariza, J. Chromatogr. A, 2013, 1300, 43–50 CrossRef CAS PubMed.
- Q. Xiao, B. Hu, J. K. Duan, M. He and W. Q. Zu, J. Am. Soc. Mass Spectrom., 2007, 18, 1740–1748 CrossRef CAS PubMed.
- X. Q. Guo, B. B. Chen, M. He, B. Hu and X. Q. Zhou, J. Anal. At. Spectrom., 2013, 28, 1638–1647 RSC.
- B. B. Chen, Y. L. Wu, X. Q. Guo, M. He and B. Hu, J. Anal. At. Spectrom., 2015, 30, 875–881 RSC.
- P. J. Li and B. Hu, J. Chromatogr. A, 2011, 1218, 4779–4787 CrossRef CAS PubMed.
- P. J. Li, B. Hu, M. He and B. B. Chen, J. Chromatogr. A, 2014, 1356, 23–31 CrossRef CAS PubMed.
- P. J. Li, M. He, B. B. Chen and B. Hu, J. Chromatogr. A, 2015, 1415, 48–56 CrossRef CAS PubMed.
- M. Ma and F. F. Cantwell, Anal. Chem., 1998, 70, 3912–3919 CrossRef CAS.
- P. J. Li, X. Zhang and B. Hu, J. Chromatogr. A, 2011, 1218, 9414–9421 CrossRef CAS PubMed.
- M. Rezaee, Y. Assadi, M.-R. Milani Hosseini, E. Aghaee, F. Ahmadi and S. Berijani, J. Chromatogr. A, 2006, 1116, 1–9 CrossRef CAS PubMed.
- Q. X. Zhou, M. Lei, J. Li, M. Y. Wang, D. C. Zhao, A. Xing and K. F. Zhao, J. Sep. Sci., 2015, 38, 1577–1583 CrossRef CAS PubMed.
- H. Kataoka, TrAC, Trends Anal. Chem., 2003, 22, 232–244 CrossRef CAS.
- C. F. Poole, TrAC, Trends Anal. Chem., 2003, 22, 362–373 CrossRef CAS.
- C. Baggiani, L. Anfossi and C. Giovannoli, Anal. Chim. Acta, 2007, 591, 29–39 CrossRef CAS PubMed.
- F. G. Tamayo, E. Turiel and A. Martín-Esteban, J. Chromatogr. A, 2007, 1152, 32–40 CrossRef CAS PubMed.
- M. C. Hennion, J. Chromatogr. A, 1999, 856, 3–54 CrossRef CAS PubMed.
- G. Tangen, T. Wickstrom, S. Lierhagen, R. Vogt and W. Lund, Environ. Sci. Technol., 2002, 36, 5421–5425 CrossRef CAS PubMed.
- E. M. Thurman and K. Snavely, TrAC, Trends Anal. Chem., 2000, 19, 18–26 CrossRef CAS.
- C. Yang, T. W. Lv, H. Y. Yan, G. C. Wu and H. N. Li, J. Agric. Food Chem., 2015, 63, 9650–9656 CrossRef CAS PubMed.
- C. Z. Huang, Z. C. Jiang and B. Hu, Talanta, 2007, 73, 274–281 CrossRef CAS PubMed.
- J. C. He, M. Y. Huang, D. M. Wang, Z. M. Zhang and G. K. Li, J. Pharm. Biomed. Anal., 2014, 101, 84–101 CrossRef CAS PubMed.
- B. Buszewski and M. Szultka, Crit. Rev. Anal. Chem., 2012, 42, 198–213 CrossRef CAS.
- J. C. Carlson, J. K. Challis, M. L. Hanson and C. S. Wong, Environ. Toxicol. Chem., 2013, 32, 337–344 CrossRef CAS PubMed.
- K. Wrobel, S. S. Kannamkumarath and J. A. Caruso, Anal. Bioanal. Chem., 2003, 377, 670–674 CrossRef CAS PubMed.
- E. Hernandez-Nataren, A. Sahuquillo, R. Rubio and J. F. Lopez-Sanchez, Microchem. J., 2011, 97, 74–77 CrossRef CAS.
- A. Moreda-Pineiro, J. Moreda-Pineiro, P. Herbello-Hermelo, P. Bermejo-Barrera, S. Muniategui-Lorenzo, P. Lopez-Mahia and D. Prada-Rodriguez, J. Chromatogr. A, 2011, 1218, 6970–6980 CrossRef CAS PubMed.
- D. R. Shang, Y. F. Zhao, Y. X. Zhai, J. S. Ning, H. Y. Ding and X. F. Sheng, Chin. Sci. Bull., 2013, 58, 4437–4442 CrossRef CAS.
- J. Duan, M. He and B. Hu, J. Chromatogr. A, 2012, 1268, 173–179 CrossRef CAS PubMed.
- L. J. Xie, R. F. Jiang, F. Zhu, H. Liu and G. F. Ouyang, Anal. Bioanal. Chem., 2014, 406, 377–399 CrossRef CAS PubMed.
- C. Huang and B. Hu, J. Sep. Sci., 2008, 31, 760–767 CrossRef CAS PubMed.
- J. Chen, M. He, B. Chen and B. Hu, J. Anal. Sci., 2014, 30, 607–612 CAS.
- Y. X. Liu, L. Guo, X. F. Wan, Y. H. Xiong and Y. Q. Wan, Anal. Lett., 2015, 48, 966–981 CrossRef CAS.
- J. K. Duan and B. Hu, Talanta, 2009, 79, 734–738 CrossRef CAS PubMed.
- S. A. Barker, A. R. Long and C. R. Short, J. Chromatogr., 1989, 475, 353–361 CrossRef CAS PubMed.
- S. A. Barker, J. Chromatogr. A, 2000, 885, 115–127 CrossRef CAS PubMed.
- S. A. Barker, J. Biochem. Biophys. Methods, 2007, 70, 151–162 CrossRef CAS PubMed.
- A. L. Capriotti, C. Cavaliere, P. Giansanti, R. Gubbiotti, R. Samperi and A. Lagana, J. Chromatogr. A, 2010, 1217, 2521–2532 CrossRef CAS PubMed.
- M. Sergi, A. Gentili, D. Perret, S. Marchese, S. Materazzi and R. Curini, Chromatographia, 2007, 65, 757–761 CAS.
- L. Y. Guo, M. Guan, C. D. Zhao and H. X. Zhang, Anal. Bioanal. Chem., 2008, 392, 1431–1438 CrossRef CAS PubMed.
- Y. Moliner-Martinez, P. Campins-Falco, C. Molins-Legua, L. Segovia-Martinez and A. Seco-Torrecillas, J. Chromatogr. A, 2009, 1216, 6741–6745 CrossRef CAS PubMed.
- A. Moreda-Pineiro, E. Pena-Vazquez, P. Hermelo-Herbello, P. Bermejo-Barrera, J. Moreda-Pineiro, E. Alonso-Rodriguez, S. Muniategui-Lorenzo, P. Lopez-Mahia and D. Prada-Rodriguez, Anal. Chem., 2008, 80, 9272–9278 CrossRef CAS PubMed.
- F. A. Duarte, B. M. Soares, A. A. Vieira, E. R. Pereira, J. V. Maciel, S. S. Caldas and E. G. Primel, Anal. Chem., 2013, 85, 5015–5022 CrossRef CAS PubMed.
- D. Y. Deng, S. Zhang, H. Chen, L. Yang, H. Yin, X. D. Hou and C. B. Zheng, J. Anal. At. Spectrom., 2015, 30, 882–887 RSC.
- C. L. Arthur and J. Pawliszyn, Anal. Chem., 1990, 62, 2145–2148 CrossRef CAS.
- F. M. Musteata and J. Pawliszyn, TrAC, Trends Anal. Chem., 2007, 26, 36–45 CrossRef CAS.
- H. Kataoka, H. L. Lord and J. Pawliszyn, J. Chromatogr. A, 2000, 880, 35–62 CrossRef CAS PubMed.
- D. C. Kapsimali and G. A. Zachariadis, Talanta, 2010, 80, 1311–1317 CrossRef CAS PubMed.
- D. C. Kapsimali and G. A. Zachariadis, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 3210–3214 CrossRef CAS PubMed.
- L. Carrasco, S. Diez and J. M. Bayona, J. Chromatogr. A, 2007, 1174, 2–6 CrossRef CAS PubMed.
- P. Das, M. Gupta, A. Jain and K. K. Verma, J. Chromatogr. A, 2004, 1023, 33–39 CrossRef CAS PubMed.
- X. Liu, M. He, B. Chen and B. Hu, Spectrochim. Acta, Part B, 2014, 101, 254–260 CrossRef CAS.
- H. Bagheri, P. Bayat and H. Piri-Moghadam, J. Chromatogr. A, 2013, 1318, 58–64 CrossRef CAS PubMed.
- Y. K. Tsoi, S. Tam and K. S. Y. Leung, J. Anal. At. Spectrom., 2010, 25, 1758–1762 RSC.
- L. Carrasco, S. Diez and J. M. Bayona, J. Chromatogr. A, 2009, 1216, 8828–8834 CrossRef CAS PubMed.
- S. Mishra, R. M. Tripathi, S. Bhalke, V. K. Shukla and V. D. Puranik, Anal. Chim. Acta, 2005, 551, 192–198 CrossRef CAS.
- G. M. M. Rahman, M. M. Wolle, T. Fahrenholz, H. M. Kingston and M. Pamuku, Anal. Chem., 2014, 86, 6130–6137 CrossRef CAS PubMed.
- W. C. Davis, S. S. Vander Pol, M. M. Schantz, S. E. Long, R. D. Day and S. J. Christopher, J. Anal. At. Spectrom., 2004, 19, 1546–1551 RSC.
- P. Grinberg, R. C. Campos, Z. Mester and R. E. Sturgeon, Spectrochim. Acta, Part B, 2003, 58, 427–441 CrossRef.
- Y. L. Sommer, C. P. Verdon, M. R. Fresquez, C. D. Ward, E. B. Wood, Y. Pan, K. L. Caldwell and R. L. Jones, Anal. Bioanal. Chem., 2014, 406, 5039–5047 CrossRef CAS PubMed.
- C. Dietz, J. S. Landaluze, P. Ximenez-Embun, Y. Madrid-Albarran and C. Camara, Anal. Chim. Acta, 2004, 501, 157–167 CrossRef CAS.
- H. G. Infante, S. P. Joel, E. Warburton, C. Hopley, R. Hearn and S. Juliger, J. Anal. At. Spectrom., 2007, 22, 888–896 RSC.
- P. Jitaru, H. G. Infante and F. C. Adams, J. Anal. At. Spectrom., 2004, 19, 867–875 RSC.
- P. Grinberg, R. C. Campos, Z. Mester and R. E. Sturgeon, J. Anal. At. Spectrom., 2003, 18, 902–909 RSC.
- G. F. Ouyang, D. Vuckovic and J. Pawliszyn, Chem. Rev., 2011, 111, 2784–2814 CrossRef CAS PubMed.
- D. Vuckovic, S. Risticevic and J. Pawliszyn, Angew. Chem., Int. Ed., 2011, 50, 5618–5628 CrossRef CAS PubMed.
- S. Bigham, J. Medlar, A. Kabir, C. Shende, A. Alli and A. Malik, Anal. Chem., 2002, 74, 752–761 CrossRef CAS PubMed.
- L. Fang, S. Kulkarni, K. Alhooshani and A. Malik, Anal. Chem., 2007, 79, 9441–9451 CrossRef CAS PubMed.
- S. S. Segro, Y. Cabezas and A. Malik, J. Chromatogr. A, 2009, 1216, 4329–4338 CrossRef CAS PubMed.
- H. R. Lee, S. M. Cho, J. Kim and D. S. Chung, J. Chromatogr. A, 2014, 1346, 117–122 CrossRef CAS PubMed.
- J. C. Wu, W. Xie and J. Pawliszyn, Analyst, 2000, 125, 2216–2222 RSC.
- W. Hu, F. Zheng and B. Hu, J. Hazard. Mater., 2008, 151, 58–64 CrossRef CAS PubMed.
- B. Chen, H. Peng, F. Zheng, B. Hu, M. He, W. Zhao and D. Pang, J. Anal. At. Spectrom., 2010, 25, 1674–1681 RSC.
- J. C. Wu, Z. Mester and J. Pawliszyn, Anal. Chim. Acta, 2000, 424, 211–222 CrossRef CAS.
- B. Chen, B. Hu, M. He, X. Mao and W. Zu, J. Chromatogr. A, 2012, 1227, 19–28 CrossRef CAS PubMed.
- F. Zheng and B. Hu, J. Mass Spectrom., 2010, 45, 205–214 CrossRef CAS PubMed.
- E. Baltussen, P. Sandra, F. David and C. Cramers, J. Microcolumn Sep., 1999, 11, 737–747 CrossRef CAS.
- X. Mao, B. Chen, C. Huang, M. He and B. Hu, J. Chromatogr. A, 2011, 1218, 1–9 CrossRef CAS PubMed.
- X. Mao, B. Hu, M. He and B. Chen, J. Chromatogr. A, 2012, 1256, 32–39 CrossRef CAS PubMed.
- W. Fan, X. Mao, M. He, B. Chen and B. Hu, J. Chromatogr. A, 2013, 1318, 49–57 CrossRef CAS PubMed.
- X. J. Mao, W. Y. Fan, M. He, B. B. Chen and B. Hu, J. Anal. At. Spectrom., 2015, 30, 162–171 RSC.
- A. Prieto, O. Zuloaga, A. Usobiaga, N. Etxebarria, L. A. Fernández, C. Marcic and A. de Diego, J. Chromatogr. A, 2008, 1185, 130–138 CrossRef CAS PubMed.
- F. Mellano, M. Bujalance, I. Giraldez, P. Ruiz-Azcona, D. Sanchez-Rodas and E. Morales, J. Chromatogr. A, 2013, 1300, 151–158 CrossRef CAS PubMed.
- I. Giraaldez, P. Ruiz-Azcona, A. Vidal and E. Morales, Microchem. J., 2015, 122, 197–204 CrossRef.
- A. Manz, N. Graber and H. M. Widmer, Sens. Actuators, B, 1990, 1, 244–248 CrossRef CAS.
- A. A. S. Bhagat, H. Bow, H. W. Hou, S. J. Tan, J. Han and C. T. Lim, Med. Biol. Eng. Comput., 2010, 48, 999–1014 Search PubMed.
- L. Y. Yeo, H.-C. Chang, P. P. Y. Chan and J. R. Friend, Small, 2011, 7, 12–48 CrossRef CAS PubMed.
- Y. Luo, J. H. Qin and B. C. Lin, Front. Biosci., 2009, 14, 3913–3924 CrossRef CAS.
- Y. Wang and Z. G. Wang, Int. J. Therm. Sci., 2014, 86, 227–245 CrossRef CAS.
- M. Forbes, J. Gourlay and M. Desmulliez, Electron. Commun. Eng. J., 2001, 13, 221–232 Search PubMed.
- T. Laurell, F. Petersson and A. Nilsson, Chem. Soc. Rev., 2007, 36, 492–506 RSC.
- L. Y. Yeo and J. R. Friend, in Annual Review of Fluid Mechanics, Vol 46, ed. S. H. Davis and P. Moin, 2014, vol. 46, pp. 379–406 Search PubMed.
- L. M. Wang, X. Q. Gong and W. J. Wen, in Microfluidics: Technologies and Applications, ed. B. C. Lin, Springer-Verlag Berlin, Berlin, 2011, vol. 304, pp. 91–115 Search PubMed.
- P. Sheng and W. J. Wen, in Annual Review of Fluid Mechanics, Vol 44, ed. S. H. Davis and P. Moin, Annual Reviews, Palo Alto, 2012, vol. 44, pp. 143–174 Search PubMed.
- N. Pamme, Lab Chip, 2006, 6, 24–38 RSC.
- N. T. Nguyen, Microfluid. Nanofluid., 2012, 12, 1–16 CrossRef.
- E. K. Sackmann, A. L. Fulton and D. J. Beebe, Nature, 2014, 507, 181–189 CrossRef CAS PubMed.
- H. Yin and D. Marshall, Curr. Opin. Biotechnol., 2012, 23, 110–119 CrossRef CAS PubMed.
- R. Zhang, H. Q. Gong, X. Zeng, C. Lou and C. Sze, Anal. Chem., 2013, 85, 1484–1491 CrossRef CAS PubMed.
- C. T. Culbertson, T. G. Mickleburgh, S. A. Stewart-James, K. A. Sellens and M. Pressnall, Anal. Chem., 2014, 86, 95–118 CrossRef CAS PubMed.
- P. Ertl, D. Sticker, V. Charwat, C. Kasper and G. Lepperdinger, Trends Biotechnol., 2014, 32, 245–253 CrossRef CAS PubMed.
- N. T. Huang, H. L. Zhang, M. T. Chung, J. H. Seo and K. Kurabayashi, Lab Chip, 2014, 14, 1230–1245 RSC.
- D. K. Kang, M. M. Ali, K. X. Zhang, E. J. Pone and W. A. Zhao, TrAC, Trends Anal. Chem., 2014, 58, 145–153 CrossRef CAS.
- K. Sato, M. Tokeshi, T. Sawada and T. Kitamori, Anal. Sci., 2000, 16, 455–456 CrossRef CAS.
- A. Aota, M. Nonaka, A. Hibara and T. Kitamori, Angew. Chem., Int. Ed., 2007, 46, 878–880 CrossRef CAS PubMed.
- R. Hu, X. J. Feng, P. Chen, M. Fu, H. Chen, L. Guo and B. F. Liu, J. Chromatogr. A, 2011, 1218, 171–177 CrossRef CAS PubMed.
- J. Zhang, J. S. Liao, D. C. Deng, H. S. Wang and Z. L. Chen, Adv. Mater. Res., 2013, 709, 485–490 CrossRef.
- K. P. Nichlos, R. R. Pompano, L. Li, A. V. Gelis and R. F. Ismagilov, 15th International Conference on Miniaturized Systems for Chemistry and Life Sciences October 2-6, Seattle, Washington, USA, 2011 Search PubMed.
- H. Wang, Z. K. Wu, Y. Zhang, B. B. Chen, M. He and B. Hu, J. Anal. At. Spectrom., 2013, 28, 1660–1665 RSC.
- J. Zhang, B. B. Chen, H. Wang, X. Huang, M. He and B. Hu, J. Anal. At. Spectrom., 2016, 31, 1391–1399 RSC.
- T.-T. Shih, W.-Y. Chen and Y.-C. Sun, J. Chromatogr. A, 2011, 1218, 2342–2348 CrossRef CAS PubMed.
- T.-T. Shih, I. H. Hsu, S.-N. Chen, P.-H. Chen, M.-J. Deng, Y. Chen, Y.-W. Lin and Y.-C. Sun, Analyst, 2015, 140, 600–608 RSC.
- K.-C. Hsu, C.-F. Lee, W.-C. Tseng, Y.-Y. Chao and Y.-L. Huang, Talanta, 2014, 128, 408–413 CrossRef CAS PubMed.
- X. S. Zhu, C. Gao, J. W. Choi, P. L. Bishop and C. H. Ahn, Lab Chip, 2005, 5, 212–217 RSC.
- M. Park, H. D. Ha, Y. T. Kim, J. H. Jung, S. H. Kim, D. H. Kim and T. S. Seo, Anal. Chem., 2015, 87, 10969–10975 CrossRef CAS PubMed.
- Y. Date, A. Aota, S. Terakado, K. Sasaki, N. Matsumoto, Y. Watanabe, T. Matsue and N. Ohmura, Anal. Chem., 2013, 85, 434–440 CrossRef CAS PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, 167–181 CrossRef.
- B. B. Chen, S. J. Heng, H. Y. Peng, B. Hu, X. Yu, Z. L. Zhang, D. W. Pang, X. Yue and Y. Zhu, J. Anal. At. Spectrom., 2010, 25, 1931–1938 RSC.
- H. Wang, Z. K. Wu, B. B. Chen, M. He and B. Hu, Analyst, 2015, 140, 5619–5626 RSC.
- B. Chen, B. Hu, M. He, Q. Huang, Y. Zhang and X. Zhang, J. Anal. At. Spectrom., 2013, 28, 334–343 RSC.
- H. Wang, B. Chen, S. Zhu, X. Yu, M. He and B. Hu, Anal. Chem., 2016, 88, 796–802 CrossRef PubMed.
- T. T. Shih, C. H. Lin, I. H. Hsu, J. Y. Chen and Y. C. Sun, Anal. Chem., 2013, 85, 10091–10098 CrossRef CAS PubMed.
- Y. Q. Xi and H. S. Zhuang, Chin. J. Anal. Chem., 2010, 38, 1523–1527 CrossRef CAS.
- K. C. Hsu, C. C. Sun, Y. C. Ling, S. J. Jiang and Y. L. Huang, J. Anal. At. Spectrom., 2013, 28, 1320–1326 RSC.
- P. Li, Y.-j. Chen, H.-z. Lian and X. Hu, J. Anal. At. Spectrom., 2014, 29, 1785–1790 RSC.
- N. A. Lacher, K. E. Garrison, R. S. Martin and S. M. Lunte, Electrophoresis, 2001, 22, 2526–2536 CrossRef CAS PubMed.
- R. S. Martin, A. J. Gawron, S. M. Lunte and C. S. Henry, Anal. Chem., 2000, 72, 3196–3202 CrossRef CAS PubMed.
- K. Faure, M. Pravda and J. D. Glennon, Anal. Lett., 2006, 39, 435–449 CrossRef CAS.
- F. Li, D. D. Wang, X. P. Yan, J. M. Lin and R. G. Su, Electrophoresis, 2005, 26, 2261–2268 CrossRef CAS PubMed.
- F. Li, D. D. Wang, X. P. Yan, R. G. Su and J. M. Lin, J. Chromatogr. A, 2005, 1081, 232–237 CrossRef CAS PubMed.
- Q. J. Song, G. M. Greenway and T. McCreedy, J. Anal. At. Spectrom., 2004, 19, 883–887 RSC.
- H. Y. Cheng, J. H. Liu, X. F. Yin, H. Shen and Z. G. Xu, Analyst, 2012, 137, 3111–3118 RSC.
- H. Cheng, C. Han, Z. Xu, J. Liu and Y. Wang, Food Anal. Methods, 2014, 7, 2153–2162 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |