
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Strategies for sensing neurotransmitters with responsive MRI contrast agents
Goran
Angelovski
 *a and
Éva
Tóth
b
*a and
Éva
Tóth
b
aMR Neuroimaging Agents, Max Planck Institute for Biological Cybernetics, Tübingen, Germany. E-mail: goran.angelovski@tuebingen.mpg.de
bCentre de Biophysique Moléculaire, UPR 4301 CNRS, Université d'Orléans, rue Charles Sadron, 45071 Orléans Cedex 2, France
First published on 6th January 2017
Abstract
A great deal of research involving multidisciplinary approaches is currently dedicated to the understanding of brain function. The complexity of physiological processes that underlie neural activity is the greatest hurdle to faster advances. Among imaging techniques, MRI has great potential to enable mapping of neural events with excellent specificity, spatiotemporal resolution and unlimited tissue penetration depth. To this end, molecular imaging approaches using neurotransmitter-sensitive MRI agents have appeared recently to study neuronal activity, along with the first successful in vivo MRI studies. Here, we review the pioneering steps in the development of molecular MRI methods that could allow functional imaging of the brain by sensing the neurotransmitter activity directly. We provide a brief overview of other imaging and analytical methods to detect neurotransmitter activity, and describe the approaches to sense neurotransmitters by means of molecular MRI agents. Based on these initial steps, further progress in probe chemistry and the emergence of innovative imaging methods to directly monitor neurotransmitters can be envisaged.
 Goran Angelovski | Goran Angelovski holds a diploma degree from the University of Belgrade, PhD degree from the University of Dortmund and venia legendi (Habilitation) from Tuebingen University, all in organic chemistry. He is currently leading a multidisciplinary research group at the Max Planck Institute for Biological Cybernetics in Tuebingen, merging diverse chemistry fields with other natural sciences to investigate brain function. In this research, Goran and his coworkers use an integrated approach, which includes design and preparation of bioresposnive MRI contrast agents, their in vitro characterization, and finally the in vivo validation in animal models. |
 Éva Tóth | Eva Toth is expert in the design and characterization of metal chelates for imaging applications. After a PhD from the University of Debrecen, Hungary in lanthanide chemistry, she occupied research positions at EPFL Lausanne, Switzerland. In 2005, she was appointed CNRS research director in Orléans, France and since 2012 she is head of the Centre of Molecular Biophysics. She edited “The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging” (Wiley) and chaired the European COST Network “Metal-Based Systems for Molecular Imaging Applications”. Her recent research focuses on detection of enzymatic activities, neurotransmitters or amyloid peptides and on theragnostic agents. |
Key learning points1. Principle of direct assessment of neurosignalling events by MRI using neurotransmitter-sensitive probes.2. Main mechanisms that can induce MRI signal change upon interaction between an MRI probe and a target analyte. 3. Design principles for preparing a genetically engineered protein with MRI response to monoamine neurotransmitters. 4. Design principles for preparing responsive paramagnetic complexes with artificial receptors for amino acid neurotransmitters. 5. Possible supramolecular chemistry approaches in the design of artificial hosts when creating neurotransmitter-sensitive MRI agents. |
Introduction
Numerous research efforts have been dedicated to the understanding of various aspects of brain function. Multidisciplinary approaches have been developed to study the complex phenomena underlying neural signalling, computing and plasticity of the brain. Formerly, characterization of signal propagation through electrical impulses and a variety of chemicals has been performed involving moderate to highly invasive electrophysiology and electrochemistry methods.1,2 The development of novel imaging techniques has brought a new momentum in neurosciences, allowing whole brain coverage and detailed non-invasive investigations.3 Amongst these, magnetic resonance imaging (MRI) is probably the most preferred choice as it enables mapping of neural events with excellent specificity, spatiotemporal resolution and unlimited tissue penetration. Its functional variant, fMRI, is currently the mainstay of neuroimaging. Although extremely common and useful, fMRI experiences drawbacks related to the physiological origin of the signal. Namely, it provides indirect measurement of neural activity, observing phenomena in the brain vasculature.4 During neural activity, blood oxygenation changes and causes the spin alteration of heme iron. Due to the increase in the ratio of paramagnetic iron, the local field homogeneity changes can be detected in an MRI experiment, thus revealing the active brain regions.The direct monitoring of neural activity by means of MRI is a clear ambition of neuroimaging. Converting action potential changes into an MRI signal would evidently be an amazing accomplishment, yet the field still lacks the approaches which may ensure such measurements in a robust manner.5 On the other hand, monitoring neurotransmitter release is becoming realistic and achievable; indeed a few approaches have been explored to date. Taking advantage of common NMR spectroscopy principles, neurotransmitters can be directly monitored by means of resolving their 1H or 13C spectra, resulting in magnetic resonance spectroscopy (MRS), a technique suitable for studies on living biological subjects including humans.6 Alternatively, the chemical exchange saturation transfer (CEST) effect between the amine group of glutamate (Glu) and bulk water can be detected to result in GluCEST MRI.7 Both of these approaches rely on the observation of NMR signals generated directly by neurotransmitters and experience limitations. MRS measurements need to be performed at high magnetic fields in order to increase sensitivity and resolution. On the other hand, utilization of the CEST MRI methodology is possible only for a limited group of neurotransmitter molecules which exhibit an observable CEST effect.
However, there are numerous possibilities to target neurotransmitters by means of MRI-detectable exogenous contrast agents, taking benefit of the knowledge and the tools of coordination chemistry and molecular biology. Such molecular MRI sensors will be capable of establishing a specific interaction with the desired neurotransmitter molecule. This recognition event, resulting in the formation of a host–guest complex between the MRI sensor and the neurotransmitter, alters the paramagnetic properties of the sensor which results in concomitant changes of the acquired MRI signal. The varying concentration of neurotransmitters during the neuronal signalling events can thus be detected through the activity of such bioresponsive or smart contrast agents (SCA). The consequence of their specific interaction with neurotransmitters is hence a change in MRI contrast that can be directly related to brain activity in a particular region.
In this review, we aim to survey approaches for direct MR imaging of neurotransmitters. We introduce the readers to the chemistry of SCA and discuss the major strategies so far explored in the design of neurotransmitter-sensitive SCA (NT-SCA), as well as the practical aspects related to their utilization in fMRI. Finally, we also indicate possible directions for further evolution to provide, hopefully in a close future, a very powerful tool to study brain function.
Neurotransmitters: function, chemical composition and detection
Neurotransmitters are chemical messengers involved in synaptic transmission. They are released by the neuron and bound to specific receptor proteins that are embedded in the postsynaptic neurons. This interaction with the receptors is typically transient and lasts from milliseconds to minutes. Although the reuptake of neurotransmitters is rapid, the consequence of their action on target cells can last considerably longer (i.e. several hours).8Substances involved in the synaptic transmission have very diverse chemical structures and properties. In fact, a molecule is classified as neurotransmitter if the following four criteria are met:
1. The molecule is synthesized in the presynaptic neuron.
2. It is present in the presynaptic terminal and is released in sufficient amounts to initiate a postsynaptic response.
3. When applied exogenously to the synapse in appropriate concentrations, the molecule yields an identical postsynaptic response as the one generated naturally by synaptic release of the endogenous neurotransmitter.
4. There exists a mechanism for terminating the postsynaptic interaction with the neurotransmitter.
Major classes of neurotransmitters involve (Fig. 1): (1) amino acids, such as glutamic acid, γ-aminobutyric acid, aspartic acid and glycine; (2) monoamines, such as dopamine, norepinephrine or noradrenaline and serotonin; and (3) peptides, such as vasopressin, oxytocin, leucine- or methionine-enkephalin. In addition to these three main classes, several small-molecule neurochemicals exist, such as acetylcholine, adenosine, adenosine triphosphate, arachidonic acid (AA), or nitric oxide, etc.
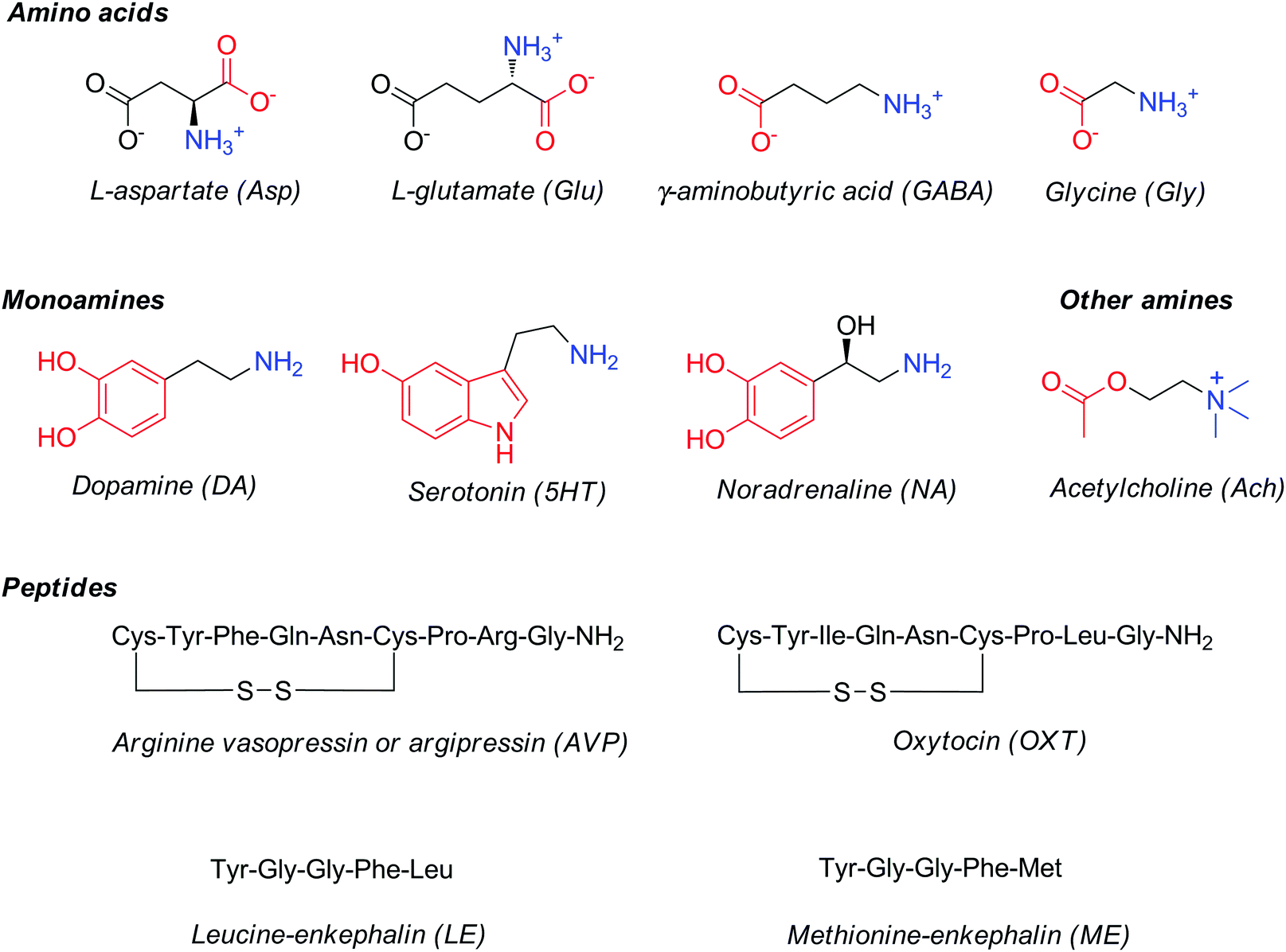 | ||
| Fig. 1 Structures and common abbreviations of selected amino acid, amine or peptide neurotransmitters. | ||
Numerous conventional analytical methods (mass spectrometry, voltammetry, liquid chromatography, electrophoresis, fluorescence, colorimetric assays) exist to detect neurotransmitters that provide selective and multi-analyte detection.9 Combination with sampling approaches has allowed in vitro studies on cultured cells, ex vivo measurements on brain slices, as well as in vivo monitoring of neurotransmitters and their dynamics. Extracellular concentrations for many neurotransmitters have been thus determined and fall in the range from pM to low μM.10,11 These conventional methods are highly invasive, often lack specificity and sensitivity, and provide poor spatiotemporal resolution and low biocompatibility.
Imaging of neurotransmitters
Imaging methodologies can circumvent many downsides of conventional analytical techniques used to detect neurotransmitters. Most importantly, imaging can provide noninvasive, repeated monitoring with a large spatial coverage and tissue penetration depth which makes it favourable for studying synaptic transmission. Clearly, each imaging technique has its own specificities with respect to resolution and sensitivity, as well as concerning the physical and technical requirements or the chemical nature of the imaging probes applied.Fluorescence microscopic imaging
Optical microscopic imaging is widely applied in neuroscience. Both small molecule and genetically encoded protein sensors have been developed to sense neurotransmitters.12,13 Fluorescence imaging is appealing as it can provide sufficient spatiotemporal resolution (∼1 μm and ms to s time scale) to assess activity at individual synapses in cell culture, brain tissue, and potentially in the intact brain. However, even with the development of multiphoton approaches and near-infrared dyes, tissue penetration does not exceed few millimetres. Among the most characteristic fluorescent probes to study neurotransmitter activity are fluorescent false neurotransmitters, small molecules capable of labelling synaptic vesicles and providing an optical readout of neurotransmitter release kinetics at individual synapses (Fig. 2). For instance, the fluorescent dye 1 targets the vesicular monoamine transporter 2 (VMAT2), which then transports 1 to synaptic sites. Its release during firing leads to a fluorescence change related to neural activity.14 In a different and more conventional strategy, the molecule 2 can selectively report on NA and DA. Due to its reactive aldehyde group on the cumarine derivative, 2 reacts irreversibly with these neurotransmitters providing an optical response, while remaining insensitive to secondary amines, such as epinephrine (adrenaline).15 The probe was successfully used for selective microscopic imaging of NA in secretory vesicles and for discrimination between norepinephrine- and epinephrine-enriched populations of chromaffin cells.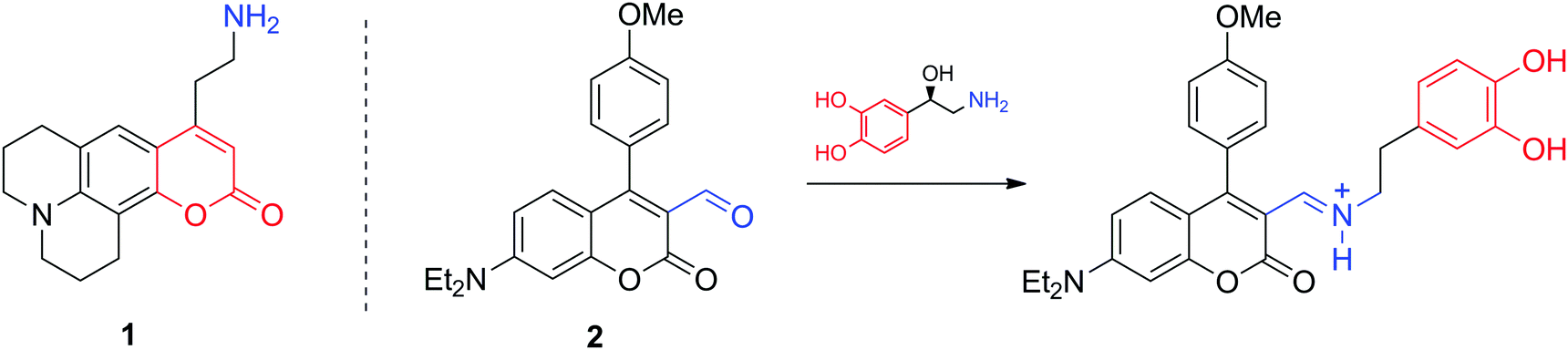 | ||
| Fig. 2 Structures of the false neurotransmitter 1 which targets the VMAT2 transporter to reveal the activity of monoamine neurotransmitters (left) and the turn-on fluorescent sensor 2 which reports on the presence of primary amine neurotransmitters (right). | ||
Indirect imaging methods
Among the macroscale imaging modalities, positron emission or single-photon emission computed tomography (PET and SPECT, respectively) using radiolabelled ligands allows for indirect monitoring of neurotransmitter activity. In this case, binding competition is used to image and study synaptic neurotransmission.16 It takes advantage of the interaction between radiolabelled metabolites or receptor agonists or antagonists and the target receptor. Subsequently, information can be obtained concerning active receptors, their dynamics and regions where specific neurotransmitters are released. Although extremely sensitive and quantitative, PET and SPECT imaging lack spatial and often sufficient temporal resolution, requiring alternative imaging methods.In a similar fashion, MRI visualization of neurotransmitter receptors has also been proposed to image neurotransmission in the brain. The general principles and examples of this approach were recently summarized in another tutorial review by Sim and Parker.17 Like the PET and SPECT tracers discussed above, these MRI contrast agents are targeted to neurotransmitter receptors, thus their molecular design is based on the conjugation of a receptor ligand to the MRI reporter. The specific binding of the conjugate to cell-surface or intracellular receptor targets yields in increased MR signal intensity. Evidently, signal intensity depends not only on the amount of the accumulated bound contrast agent, but also on its relaxivity and the magnetic field used (vide infra).
In this tutorial review, we intend to introduce the readers to a fundamentally different approach towards MR imaging of neurotransmission. It relies on a direct interaction between a bioresponsive MRI contrast agent and a neurotransmitter molecule and takes advantage of the capability of the contrast agent to alternate MRI signal via modulation of their contrast enhancing properties (proton relaxivity, CEST effect, etc.) in response to a given biomarker. Hence, this functional molecular imaging technique is distinct from those mentioned above in providing a more direct readout of the presence and concentration changes of neurotransmitters.
Responsive MR imaging
Mechanisms to modulate the contrast enhancing capability of MRI agents and design of responsive probes
Classical MRI provides images that are dependent on proton density and on the relaxation times of water proton nuclei. Paramagnetic substances can decrease, mainly via dipolar interactions, the nuclear relaxation times and this will enhance the image contrast. Among metal ions, mostly Gd3+ and Mn ions (in +2 and +3 redox state), and to some extent high spin Fe2+ and Fe3+ received attention in the context of MRI. High electron spin and slow electronic relaxation are the two main features required for a paramagnetic metal ion to provide efficient nuclear relaxation enhancement. In this respect, with seven unpaired electrons (S = 7/2) and a symmetric S state leading to remarkably slow electron spin relaxation (T1e ≈ 10−7 s), Gd3+ ensures the best efficiency. Despite its smaller electron spin (S = 5/2), Mn2+ remains a strong relaxing agent and has been under scrutiny for MRI use. High spin Fe2+ complexes (S = 2) were recently proposed as magnetogenic probes18 and importantly, BOLD MRI is also based on the relaxation effect of high spin heme iron of deoxyhemoglobin as endogenous contrast agent.4 However, the nuclear relaxation efficiency of Fe2+ complexes remains rather limited due to fast electronic relaxation (T1e ≈ 10−12 s).The majority of currently used clinical MRI agents are Gd3+ complexes. In order to create a smart (activatable or responsive) probe for T1 or T2-weighted MRI, the efficiency of the complex to accelerate water proton relaxation (relaxivity) needs to be selectively modulated by the biomarker of interest. Relaxivity is the term expressing the capacity of a paramagnetic substance to affect nuclear relaxation of surrounding water proton nuclei, and is defined as the paramagnetic relaxation enhancement of water proton relaxation rate per mM concentration of the paramagnetic ion. It is determined by various microscopic properties characterizing the paramagnetic metal complex as defined by the Solomon–Bloembergen–Morgan theory of paramagnetic relaxation.19 Briefly, the relaxation enhancement of a paramagnetic ion is the result of dipole–dipole interactions between the electron spin of the metal and the nuclear spin of the proton and originates from two distinct mechanisms. The metal electron spin interacts with the freely diffusing water molecules yielding the outer sphere relaxation mechanism as well as with the water protons situated on the coordinated water, called inner sphere mechanism. This latter is much more interesting since it can be modified by varying the structure of the complex. Among the most significant parameters that affect inner sphere relaxivity, we cite (i) the number of water molecules directly coordinated to the metal ion (hydration number, q), (ii) the exchange rate, kex, of these water molecules with the bulk water, (iii) the rotational correlation time, τR, of the complex and (iv) the electron spin relaxation times, T1e and T2e (Fig. 3). Additionally in some cases, water protons in the second hydration sphere of the complex, hydrogen-bound to the ligand or to the inner sphere water, can also contribute to the relaxation effect (second sphere relaxivity). Second sphere relaxivity is governed by the same theory as inner sphere relaxivity.
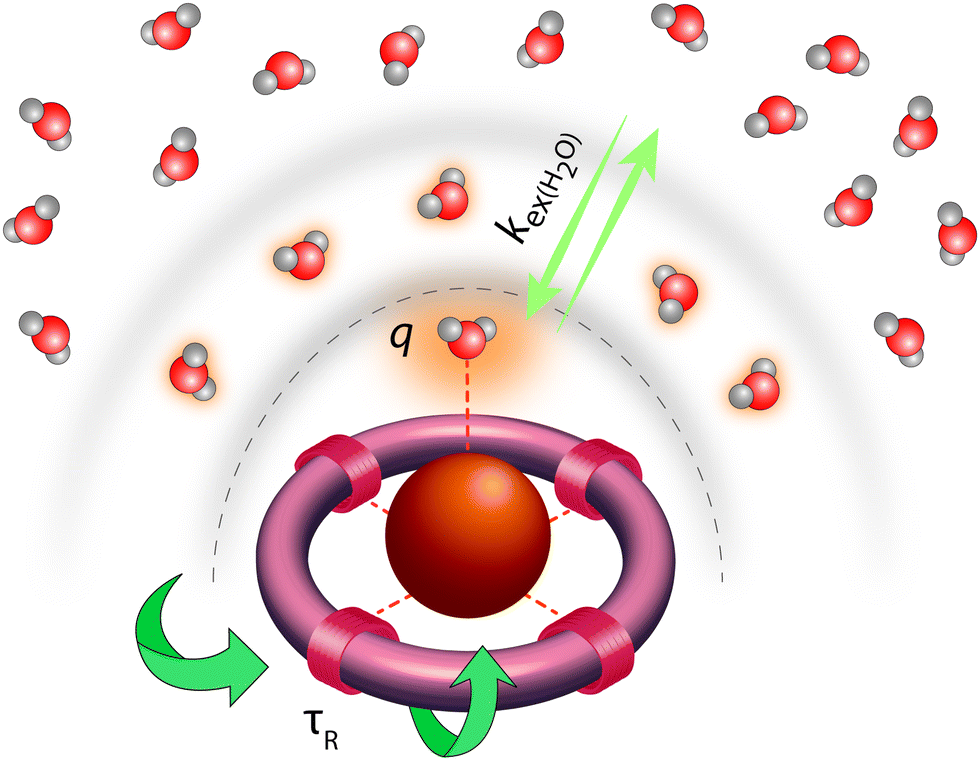 | ||
| Fig. 3 In activatable, T1-weighted MRI contrast agents, MR signal change can be generated via modulation of the main parameters that determine relaxivity: the number of water molecules directly coordinated to the paramagnetic ion (Gd3+), q; the exchange rate of the inner sphere water with bulk water, kex(H2O); the rotational motion of the complex, characterized by the rotational correlation time, τR. | ||
To create responsive probes, one or several of these factors has to be modulated by the interaction between the biomarker and the metal complex. Today, there is a vast literature on smart Gd3+-based agents that can detect metal ions, pH, enzymes, etc.19 Most of these agents are based on changes in the hydration state or in the rotational dynamics of the molecule which are the most predictable and straightforward to modulate. These two parameters affect proton relaxivity very differently and this difference should be considered when designing responsive probes. The inner sphere relaxivity contribution is linearly proportional to the hydration number and this proportionality is valid at any magnetic field. On the other hand, the effect of the reorientational correlation time of the agent is field-dependent. At magnetic fields typically between 0.5–1 T, slower rotation (larger size and reduced flexibility) translates to higher relaxivity of the chelate, and the relaxivity of slowly rotating nanosized complexes can attain 20-fold values with respect to small chelates. On the other hand, at higher magnetic fields, the effect of rotational dynamics vanishes and the relaxivity is the highest for medium-sized systems. Given the increasing preponderance of MRI scanners operating at fields 3 T and above, it is more sensible to design responsive probes operating via modulation of the hydration number.
Genetically encoded, neurotransmitter-sensitive MRI contrast agents
It can be anticipated that designing bioresponsive MRI agent for molecular targets such as neurotransmitters is much more challenging than designing an SCA for small ion targets. Numerous studies have identified host molecules for many ions, whereas the choice of host molecules which specifically and non-covalently interact with other molecules, charged or neutral, is definitely poorer. Two approaches can be envisaged to tackle this problem, one involving host molecules developed by nature (i.e. proteins), and the second one involving supramolecular chemistry and artificial host molecules.The first approach has been extensively explored by the Jasanoff lab in the last years. This group considered a naturally existing paramagnetic metalloprotein, the fatty acid hydroxylase flavocytochrome P450-BM3 which acts like a natural SCA. This protein contains a single heme-bound paramagnetic Fe3+ (mixed spin 1/2 and 5/2) embedded in the binding pocket and coordinated to a water molecule. The natural protein form has high affinity for AA. The binding of AA to the protein replaces the water molecule in the binding pocket, thus reducing the protein's longitudinal relaxivity due to modulation of the hydration number (see previous section).
This observation constituted the starting point for a very intelligent methodology of directed evolution to result in potent NT-SCAs based on P450-BM3.20 Namely, focusing on the heme domain of BM3 (BM3h), several mutation rounds by replacing one to two amino acids were performed, aiming to tune the affinity of the BM3h binding pocket for the monoamine neurotransmitter DA instead of AA. The screening procedure yielded two mutant proteins from the fourth and fifth evolution round denoted BM3h-8C8 and BM3h-B7. The mutations resulted in a 100–300-fold increase in DA affinity, in parallel to a ∼100-fold decrease in AA affinity. On the other hand, these proteins exhibited a certain affinity towards 5HT and NA, still 4–10 times lower than for DA (Table 1). Importantly, DA binding to mutant BM3h could be followed with MRI as the protein saturation with DA reduced the longitudinal proton relaxivity, r1, to 85% of the initial value in both cases, providing Kd values of 4.9 ± 2.7 and 2.7 ± 2.9 μM for BM3h-8C8 and BM3h-B7, respectively. Subsequently, the potential of BM3h mutants to sense DA was assessed in MRI experiments using a standard cellular model of dopaminergic function. Finally, the use of this approach in in vivo functional MRI (fMRI) was demonstrated utilizing the sensor BM3h-8C8, with higher DA affinity over 5HT and NA. Solutions of BM3h-8C8 and wild type BM3h were injected into the two hemispheres of rat brain. The stimulation of brain cells was performed by depolarization with elevated concentrations of K+. The resulting DA release was monitored by T1-weighted MRI scans, and these results were compared to those during injection of low K+ concentrations. The recorded results revealed a significant MRI signal difference between high- and low-K+ (translating to high- and low-DA release) periods, exclusively in the regions where BM3h-8C8 was injected (Fig. 4).
| Protein | r 1 (mM−1 s−1) at 21–23 °C and 4.7 T | −Δr1 (%) | K d (μM) | ||||
|---|---|---|---|---|---|---|---|
| Unbound | + Ligand | AA | DA | 5HT | NA | ||
| a Ref. 20. b Ref. 21. | |||||||
| BM3ha | 1.23 | 0.42 (AA), 0.76 (DA) | 66 (AA), 38 (DA) | 6.8 | 990 | — | — |
| BM3h-8C8a | 1.10 | 0.17 (DA) | 85 | 750 | 8.9 | 80 | 44 |
| BM3h-B7a | 0.96 | 0.14 (DA) | 85 | 660 | 3.3 | 11.8 | 18.6 |
| BM3h-9D7b | 1.31 | 0.07 (DA) | 95 | — | 1.3 | 70 | 37 |
| BM3h-3DB10b | 0.73 | 0.22 (5HT) | 70 | — | 46.1 | 1.1 | 190 |
| BM3h-2G9C6b | 1.89 | 0.14 (5HT) | 93 | — | 198 | 0.7 | 275 |
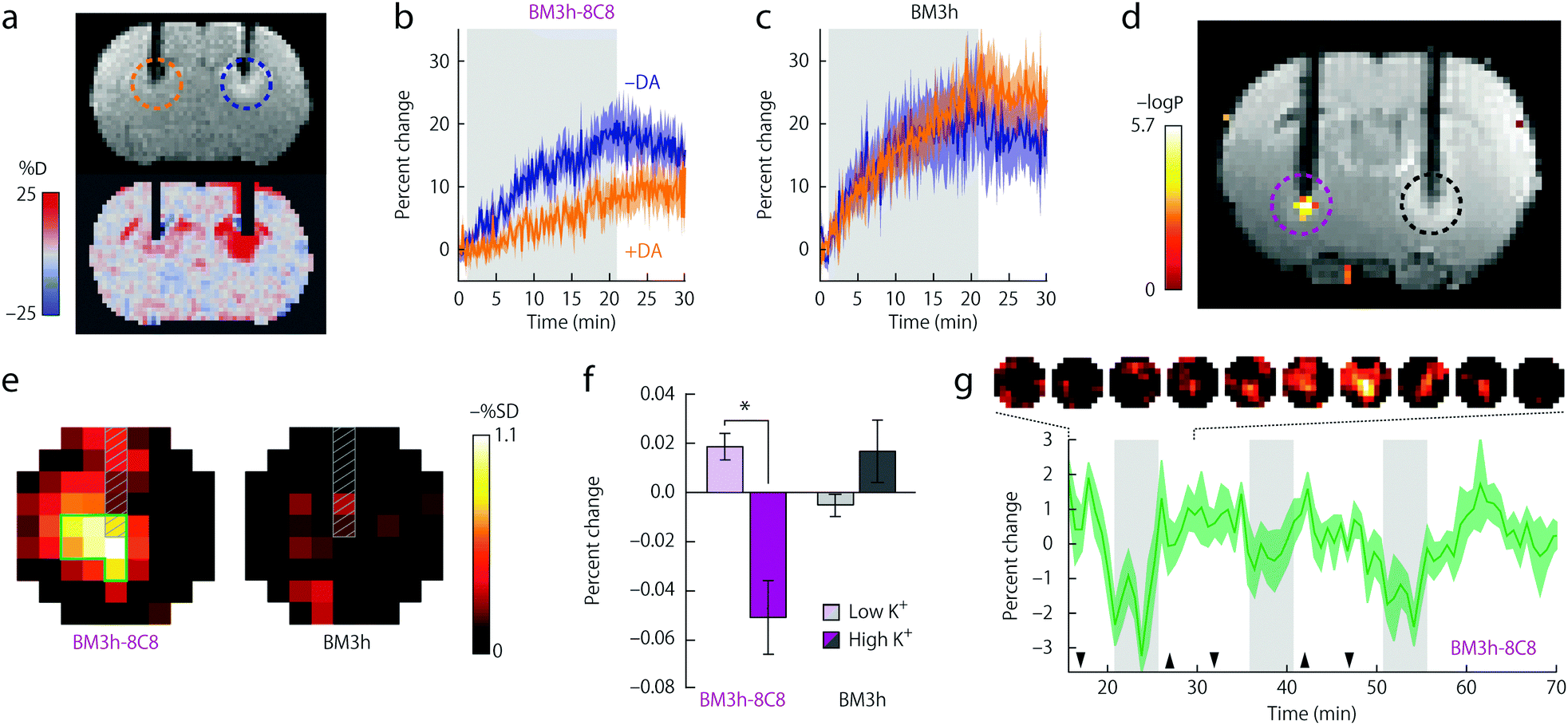 | ||
| Fig. 4 Functional MRI in vivo experiments of BM3h-8C8 with DA in injected rat brains. (a) Coronal MR image of injected BM3h-8C8 in the absence (blue dashed circle) and the presence (orange dashed circle) of DA (top) or map of percent signal change (SC) for the same animal (bottom). (b) Time courses of relative SC observed during injection of BM3h-8C8 in the absence (blue) and presence of DA (orange). (c) Corresponding time courses of a control injection with native BM3h. (d) Correlation of MRI intensity with low- and high-K+ conditions in an individual rat (coloured voxels inside purple dashed circle) presented on a corresponding T1-weighted coronal slice, showing injection cannulae used for BM3h-8C8 infusion (left, purple dashed circle) and native BM3h control infusion (right, black dashed circle). Statistical parametric map of t-test significance values is presented on the side colour scale. (e) Maps of percent signal difference between high- and low-K+ conditions around BM3h-8C8 sensor (left) and native BM3h control (right) injection sites. (f) Mean MRI signal change from baseline observed during high-K+ (dark bars) and low-K+ (light bars) periods in regions of interest (ROIs) centered around infusion sites for BM3h-8C8 (purple) and native BM3h (gray) proteins. (g) The mean time course of MRI signal in voxels within the BM3h-8C8 – infused ROI and identified as correlated (P < 0.05) with the stimulus, averaged over animals and binned over 1.5 min intervals. Gray vertical bars denote periods associated with highest K+ stimulation, hence the greatest MRI signal change. Adapted by permission from ref. 20. Copyright 2010, Macmillan Publishers Ltd. | ||
Encouraged by these initial results, the same research group applied another round of directed evolution of the protein.21 The further modification of the BM3h active site resulted in several NT-SCAs with improved selectivity and low micro- to submicromolar affinity towards DA and 5HT (Table 1). Furthermore, protein crystallization in the absence and in the presence of DA or 5HT enabled structural characterization of these sensors (Fig. 5). The X-ray crystallographic data revealed the existence of two mutants denoted BM3h-9D7 and BM3h-2G9C6 as very potent sensors for DA and 5HT, respectively. The former one, BM3h-9D7, exhibited high affinity towards DA even over 5HT and NA, while DA addition dramatically reduced its r1 leading to virtually no signal produced by this contrast agent (Table 1). Possessing such advantageous properties, this protein was used to demonstrate molecular fMRI in vivo.22
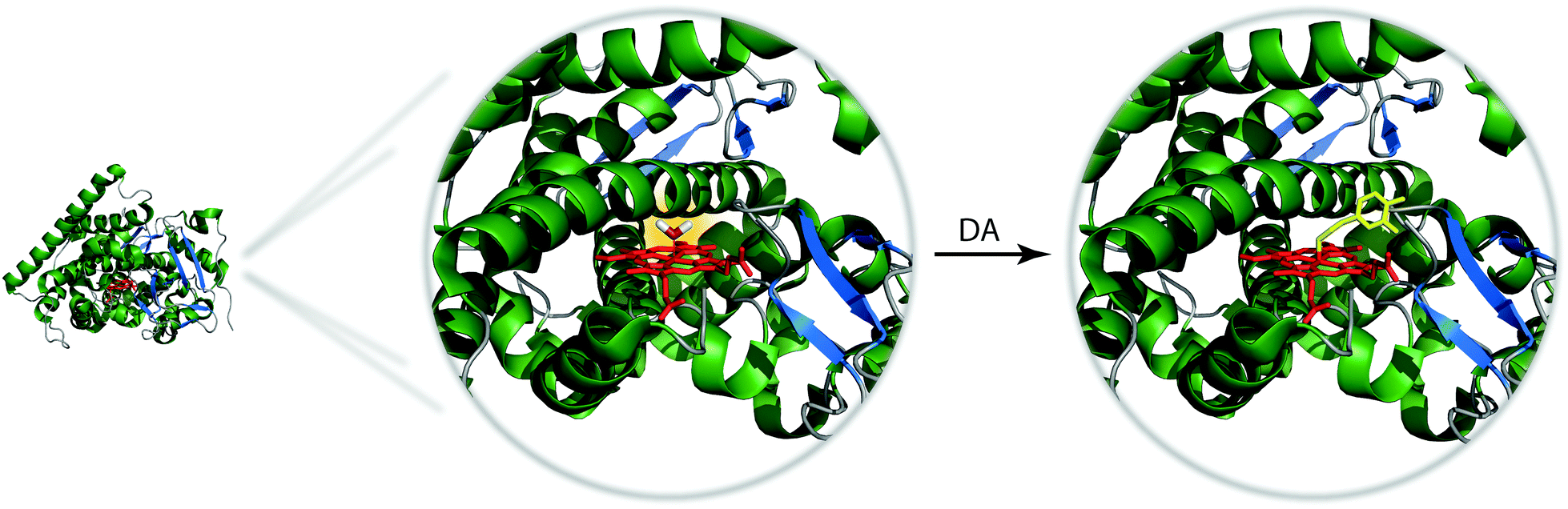 | ||
| Fig. 5 Active sites of BM3h-9D7 in the absence (left) and presence (right) of DA. The heme iron in BM3h-9D7 form without DA is coordinated to water molecule which gives rise to higher r1 and hence higher signal in the MRI experiment. Addition of DA decreases r1 and reduces the MRI signal. | ||
The NT-SCA was injected in the ventral striatum of the brain and the cells were electrically stimulated to release dopamine. T1-weighted MR images were collected every 8 s during blocks of 16 s stimulation and 5 min rest periods. Comparison of the MRI signal intensities allowed tracking of the dopamine release from neurons and then its reuptake into the cells. In addition to the dynamics of this process, this method allowed quantitative mapping of dopamine concentrations in regions where BM3h-9D7 was applied.
Very recently, a serotinin-sensitive mutant BM3h-2G9C6 was used in another type of molecular fMRI study. The MRI sensor was co-injected with 5HT in its ‘off’ state, namely when exhibiting low r1 relaxivity due to BM3h-2G9C6 association with 5HT (see Table 1). The activity of serotonin transporters was consequently followed by means of fMRI. 5HT reuptake could be imaged following the increase in the relaxivity of the sensor due to the replacement of the iron-coordinated 5HT by a water molecule. This switch to a ‘turn-on’ high relaxivity state is then detected as an increased MRI signal. The study revealed the three-dimensional mapping of serotonin transport and provided spatially resolved estimates of serotonin removal rates.23
These exemplary results demonstrated the full richness and potential of monitoring brain function with exceptional spatial and temporal resolution and with the molecular specificity of a NT-SCA. They also showed that imaging probes that are not able to cross the blood brain barrier, but are directly injected into the brain, can still be extremely useful to provide novel insights into fundamental neuroscience issues.
Synthetic, small molecule, neurotransmitter-sensitive MRI contrast agents
The other approach towards neurotransmitter sensing involves the use of artificial hosts applying concepts of supramolecular chemistry. While such synthetic molecular platforms might lack the specificity of engineered proteins for neurotransmitter recognition, their robustness, easier accessibility, and considerably higher relaxation enhancement efficiency might represent potential advantages. Although responsive Gd3+-based molecular imaging probes have been targeted to a wide variety of biologically relevant biomarkers, neurotransmitter detection has been only rarely addressed. As the single examples in this field, we have recently explored crown-ether appended Gd3+ complexes.24,25 Our choice was to target zwitterionic amino acid neurotransmitters which include the most prevalent ones such as Glu (millimolar concentration range), which is excitatory at well over 90% of the synapses in the human brain, or GABA, the major inhibitory neurotransmitter. In this first approach, we did not seek for selectivity towards individual neurotransmitters within zwitterionic molecules; the objective was rather to monitor brain activity via the detection of the cumulative effect of concentration variation of several neurotransmitters.When designing a host for zwitterionic neurotransmitters, one has to consider these molecules as bifunctional guests. The SCA has to provide specific binding sites for the protonated amine as well as for the deprotonated carboxylate function, which have to be separated at a distance suiting α- or γ-amino acids for Glu, Gly and Asp, or GABA, respectively. Only such divalent supramolecular interactions might enable sufficiently strong and specific binding of zwitterionic neurotransmitters with respect to endogenous competitors.
Various synthetic receptors have been examined for amine binding, including metal complexing agents, crown ethers, calixarenes, porphyrins, cyclodextrins, cyclopeptides, etc.26 Among these, crown ethers represent a versatile class of artificial hosts for ammonium binding.27 They typically bind primary and secondary amines via hydrogen bonds and size-discrimination has an important role in selectivity between primary and secondary amine and over other cations. Namely, 18-crown-6 has good affinity for primary amines, while larger crown-ethers favor secondary ammonium ions. Therefore, we used monoaza- and triaza-18-crown-6 ethers as amine recognition units. In the triazacrown, the position of the three nitrogen atoms corresponds to the symmetry axis of a primary ammonium cation which can promote the formation of three strong N⋯H–N hydrogen bonds to increase selectivity for ammonium over K+ ions.
The resulting smart contrast agents combined a coordinatively unsaturated and positively charged Gd3+ chelate that acts as a host for the carboxylate function and an azacrown ether to bind the amine function of the zwitterionic neurotransmitters. The two entities have been conjugated via linkers that differ in their length (i.e. ethylene and propylene linker). In the absence of the neurotransmitter, one inner sphere water molecule is coordinated to the Gd3+ ion, which will be replaced by the carboxylate function of the neurotransmitter, yielding a relaxivity decrease (“turn-off” response) (Fig. 6).
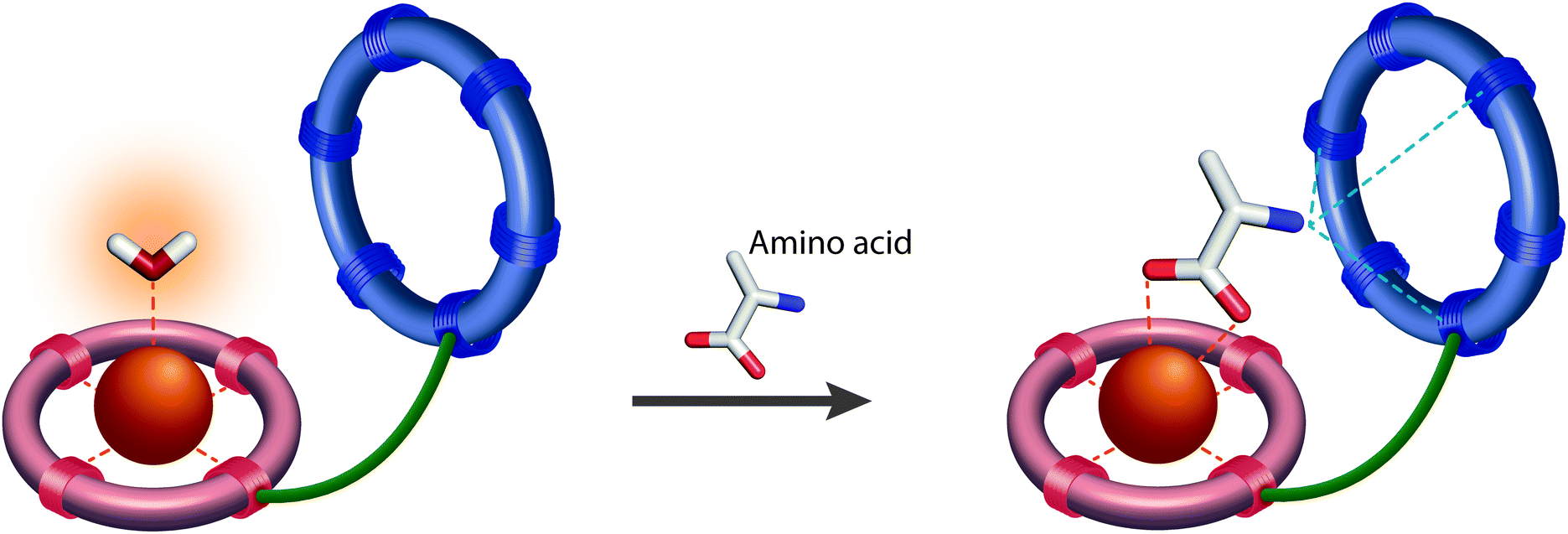 | ||
| Fig. 6 The mechanism responsible for change in r1 in crown ether-containing NT-SCA: ditopic binding of the amino acid neurotransmitter to the Gd3+ ion (complexed in the red ring) and to the crown ether (blue ring) causes replacement of the inner sphere coordinated water molecule, thus reducing the r1 of NT-SCA. | ||
As expected, a significant relaxivity decrease was observed, amounting to ∼80%, when zwitterionic neurotransmitters such as Glu, Asp and Gly are titrated into the contrast agent solutions (Fig. 7). This is the result of the decreasing hydration number which was evidenced by analogous luminescence titration data using the corresponding Eu3+ complex (q changes from 1.2 in the absence to 0.3 in the presence of 50 equivalents of Glu). On the other hand, the effect of non-zwitterionic neurotransmitters, like Ach, 5HT or NA is negligible. It is interesting to observe the size discrimination: GdL1 with the shorter ethylene linker has selectivity for α-amino acids with respect to the γ-amino acid GABA. GdL2 containing a propylene linker is more adapted to GABA as shown by the 3-fold increase in affinity with respect to GdL1. The dissociation constants, extracted from the relaxometric titration data, are in the millimolar range which can be adapted to the detection of Glu concentration during neural Glu release. The triaza-crown derivative GdL3 exhibited an improved affinity (three-fold) as compared to the monoaza-crown GdL2 with the same linker, in accordance with the fact that the triazacrown is a better receptor for amines. In order to assess the steric requirements of ditopic neurotransmitter binding to these agents, density functional theory (DFT) calculations have been performed for the GdL1–Gly complex.24 They showed not only that Gly can bind GdL1 in a bivalent manner but also suggested that the interaction between the amine and the crown ether moiety provides a substantial contribution to the overall stability of the GdL1–Gly system.
 | ||
| Fig. 7 Structures of NT-SCA GdL1–3 with crown ethers as artificial host molecules (top) and 1H NMR relaxometric titration of GdL3 with different neurotransmitters or bicarbonate (bottom). The lines represent the fit to the experimental data to yield Ka values presented in Table 2. | ||
However, selected endogenous anions appeared to be strong competitors binding to the metal ion in GdL1–3. The undesired interaction with bicarbonate (extracellular concentrations of about 25 mM) leads to a relaxivity decrease similar to that observed with neurotransmitters, while phosphate anions (extracellular concentrations of about 1 mM) did not interfere with GdL1–3. Achieving selectivity for amino acids over carbonate remains an important challenge. Logically, increasing the strength and thus the contribution of the amine binding to the overall affinity would be helpful. However, high affinity amine receptors are typically more elaborated synthetic structures and it can be very challenging to conjugate them to a Gd3+ chelate in a way that neurotransmitter binding has also an effect on the relaxation properties of the agent.
Most importantly, GdL3 could be used to image neurotransmitter release in acute mouse brain slices following neuronal stimulation.25 To avoid saturation of the agent by carbonate, normally present in the cerebrospinal fluid in much higher concentration than the neurotransmitters, we used an artificial cerebrospinal fluid which contains HEPES buffer instead of carbonate to maintain the mouse brain slices. Intense neural activity was promoted by adding high concentration KCl (40 mM) to the medium and MR images were recorded for a period of 12 min in different regions of interest. Signal intensity consistently decreased in the cortex, the striatum, and the hippocampus, without any significant change in the medium surrounding the brain slice. The hippocampus underwent the most important signal intensity-decrease, whereas signal change was delayed and less significant in the cortex and striatum (Fig. 8).
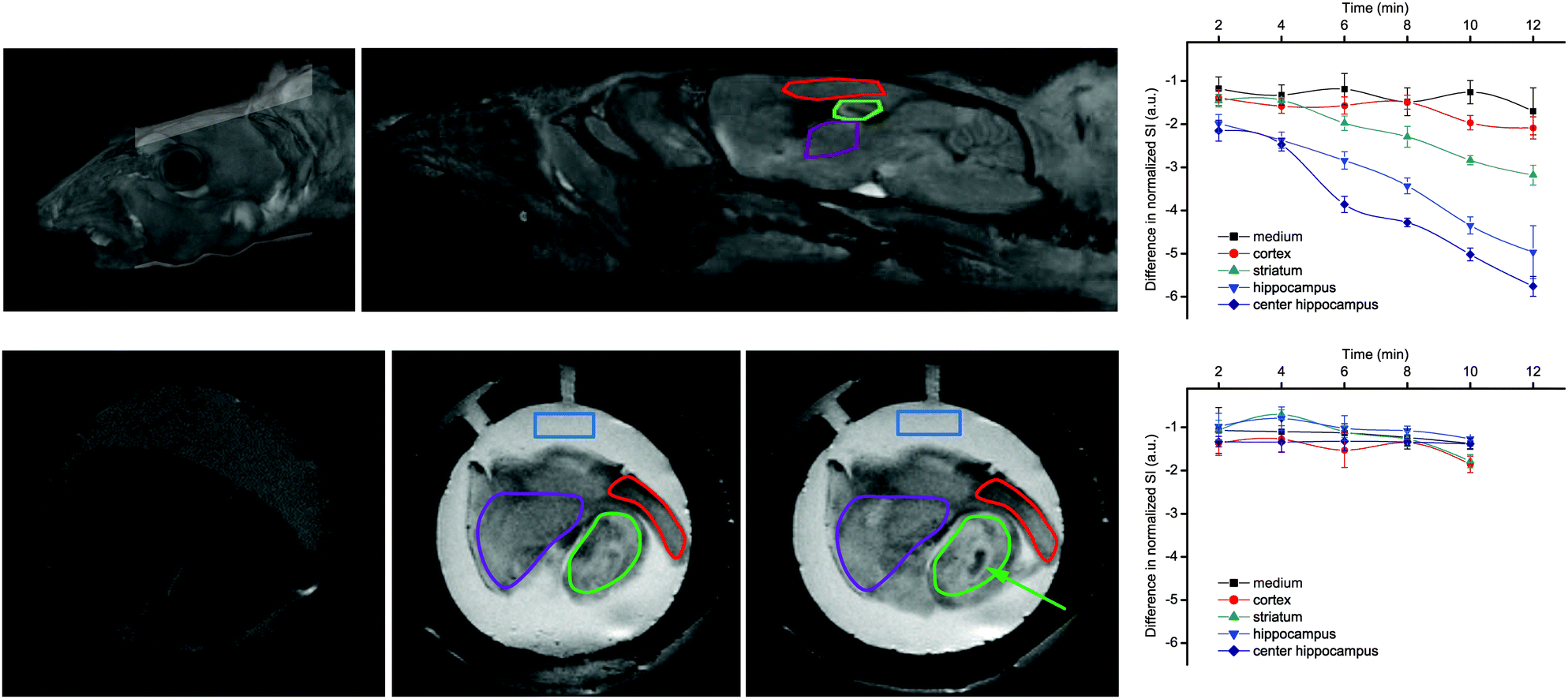 | ||
| Fig. 8 MRI experiments showing: (top) reconstituted 3D images of a mouse head with the orientation of the examined slice and the brain structures with the ROIs; (bottom) MR images of a mouse brain slice in the absence (left) or in the presence of 500 μM GdL3 before (middle) or 10 min after KCl addition (right). The ROIs indicate: medium (blue), striatum (purple), cortex (red), and hippocampus (green), while the arrow indicates the highest signal intensity decrease observed in the center of the hippocampus. Corresponding time courses of normalized signal intensity with respect to the first image taken immediately after KCl addition are presented on the graphs in the absence (top) and presence of TTX (bottom). The lines connecting SI values within the same ROI serve to guide eyes. Adapted with permission from ref. 25. Copyright 2015 American Chemical Society. | ||
The signal intensity decrease observed in the presence of GdL3 can be interpreted as a result of the decreasing relaxivity of the agent upon neurotransmitter binding. When the brain slices were pretreated with the sodium channel antagonist tetrodotoxin (TTX, 10 μM), no intensity change was observed. Indeed, TTX inhibits voltage-gated Na+ channels and the generation of action potential required for stimulus-evoked neurotransmitter release in most neurons. These findings showed that Gd3+-based MRI agents can provide an alternative towards monitoring neural activity under biologically relevant conditions.
Practical challenges in functional molecular imaging using neurotransmitter-sensitive MRI probes
Along with the encouraging results in the field of NT-SCA, the practical challenges and current barriers should be also addressed. First, MRI is typically characterized by low sensitivity, thus it is important to consider the detection limit that one can achieve by using molecular MRI agents based on paramagnetic metal chelates. It is generally admitted that the effect of typical MRI agents becomes detectable on the images above local in vivo [Gd3+] concentrations of 10–100 μM, which defines the lower limit of biomarkers that can be potentially visualized by typical molecular MRI probes. The concentration of neurotransmitters in the extracellular space (between neurons) covers several orders of magnitude. Although their normal concentration in the extracellular brain fluids is typically in the micromolar range,10,11 the concentration of synaptically released Glu, the major excitatory neurotransmitter in the mammalian central nervous system, was estimated to reach the low millimolar level,28 which is in the range that can be comfortably detected by MRI. We note that Glu concentration inside neurons of the brain is also in the 1–10 mM range; however, if the goal is to provide a functional molecular imaging method to monitor neural activity, neurotransmitter concentration changes occurring during neural signalling in the extracellular space represent the main target.The delivery of detectable amounts of NT-SCA to the brain is also a challenging task. Ideally, minimally invasive or non-invasive methods should be used; however, so far all approaches rely on intracranial probe injections and hence the local spreading of the agent into the tissue. The delivery of NT-SCA to large brain volumes to achieve full brain coverage would be very important. Preferentially, this could be done through blood–brain barrier penetration, albeit hypothetically, genetically encoded NT-SCA could be expressed directly in the tissue of interest, as much easier solutions exist for gene delivery. For synthetic probes such as Gd3+ complexes, the opening of the blood–brain-barrier by ultra-sound or by mannitol injection could be realized.29
Finally, the temporal resolution of molecular fMRI must also be considered. MRI has certainly the best potential compared to other imaging methods which allow investigations in deep tissues (i.e. PET or SPECT), with a capacity to reach submillimetre and subsecond spatial and temporal resolution, respectively.4,30 Although the initial proof-of-principle studies were performed with a temporal resolution on a minute scale,20,25 recent in vivo experiments using BM3h-9D7 were done with 8 s temporal resolution (vide supra).22 Principally, responsive T1-weighted MRI contrast agents allow monitoring dynamic events with subsecond resolution, provided changes in relaxivity (or T1 relaxation time) are at least a few per cent.30 On the other hand, recent work has shown that high temporal resolution is not always required to yield very important findings (e.g. to assess and quantify the reuptake rates of 5HT transporters).23
Perspectives for development of further NT-SCA classes
The previous chapters highlighted two distinct approaches in preparing bioresponsive MRI contrast agents which directly interact with neurotransmitters and can serve for reporting on their function in neuronal signal processing. Given the limited number of NT-SCAs developed so far, we want to explore possible directions for preparing more potent probes based on existing or entirely novel classes of agents. The following section will presume the main design principles which might arise from the currently established directions, while the emergence of entirely novel approaches to design NT-SCA would be welcome by the research community.Genetically encoded, protein-based SCAs proved to be very efficient and capable of providing essential information on DA or 5HT fluctuations with high specificity.20,22,23 They possess great selectivity and capacity for tuning the binding affinity for specific target molecules. However, the currently developed generation of mutants contains paramagnetic Fe3+, which gives rise to low longitudinal relaxivity of these proteins and hence low latitude for MRI signal change induction.21 Instead of iron, using better relaxing agents such as Mn2+/Mn3+ or Gd3+ would be a logical and preferable choice (vide supra); their influence on water proton relaxation times and on the concomitant MRI signal would be stronger. In this line, Jasanoff and colleagues attempted making more potent NT-SCA which should display higher r1 changes upon interaction with the desired neurotransmitter molecule. Using conventional protein engineering techniques, the porphyrin-bound ferric ion in BM3h, which is thought to exist primarily in a low-spin S = 1/2 state, was replaced with the manganese(III)–protoporphyrin IX complex containing a higher-spin (S = 2) Mn3+ ion to result in the metalloprotein Mn-BM3h.31 The applied modification improved protein stability and led to a two-fold increase in relaxivity (r1 = 2.6 mM−1 s−1 for Mn-BM3h in comparison to r1 = 1.3 mM−1 s−1 for the native ferric BM3h; 23 °C and 4.7 T). The addition of AA did not induce changes in r1, albeit optical titrations indicated AA binding to Mn-BM3h with high affinity (Table 3). After repeated mutation procedures similar to those previously made on native BM3h, a variant denoted Mn-C1634 exhibited AA-triggered changes in r1 which are greater by a factor of 2.4 than those for the native BM3h.
| Protein | r 1 (mM−1 s−1) | −Δr1 (%) | K d (μM) | |
|---|---|---|---|---|
| Unbound | + AA | |||
| Mn-BM3h | 2.60 | 2.57 | 1 | 0.7 |
| Mn-C1634 | 3.30 | 1.20 | 63 | 2.5 |
This work pointed to a promising direction towards protein-based NT-SCA which exhibit a robust MRI response to desired targets. It is obvious that the replacement of paramagnetic ions also changes the nature of the binding site in the protein; the consequence is usually a loss in the binding affinity for the desired target and thus the necessity to perform new rounds of directed evolution in order to re-refine the properties of the mutant. Even if tedious, this strategy could be potentially extended to the incorporation of other metals which are better relaxation agents in order to induce more robust MRI signal changes. In addition, techniques which involve de novo synthesis of metalloproteins sound promising and might bring exciting results in this direction in the near future.32
On the other hand, when discussing artificial host molecules for neurotransmitters, one can build on the great amount of work that has been performed in the field of supramolecular host–guest chemistry. Numerous books and review articles have summarized interactions of various host molecules with cationic, anionic or neutral guests.33–36 While these data are essential for the successful design of artificial MRI sensors for neurotransmitters, some specific aspects, such as the host/guest charge, solvent effects and the mechanism of MRI signal generation require further consideration.
The majority of abundant neurotransmitters are amino acids or amines (Fig. 1), hence one can identify a number of structures and functional groups as appropriate hosts. Receptors with amide/sulfonamide, urea/thiourea, pyrrol/imidazolium/pyridinium, guanidinium or ammonium groups incorporated in podands, crown ethers, cryptands, calixarenes, spherands, cyclodextrins, cyclophanes, cryptophanes, etc. are known to form supramolecular assemblies with neurotransmitters. For instance, cyclophane-type and cucurbit[n]uril molecules 3–6 are artificial biomimetic receptors that interact with amine neurotransmitters such as Ach, DA, NA (Fig. 9).37–39 The downsides of this interaction are the lack of selectivity for a particular guest molecule, and more importantly, the decrease of binding affinity in aqueous solution. Indeed, many of the supramolecular complexes are formed through non-ionic interactions (hydrogen bond, anion–, cation– or π–π interactions) which are strong in nonpolar or polar organic solvents. In water solvent, however, many hosts exhibit very low solubility, or the complex formation becomes weak.39–41
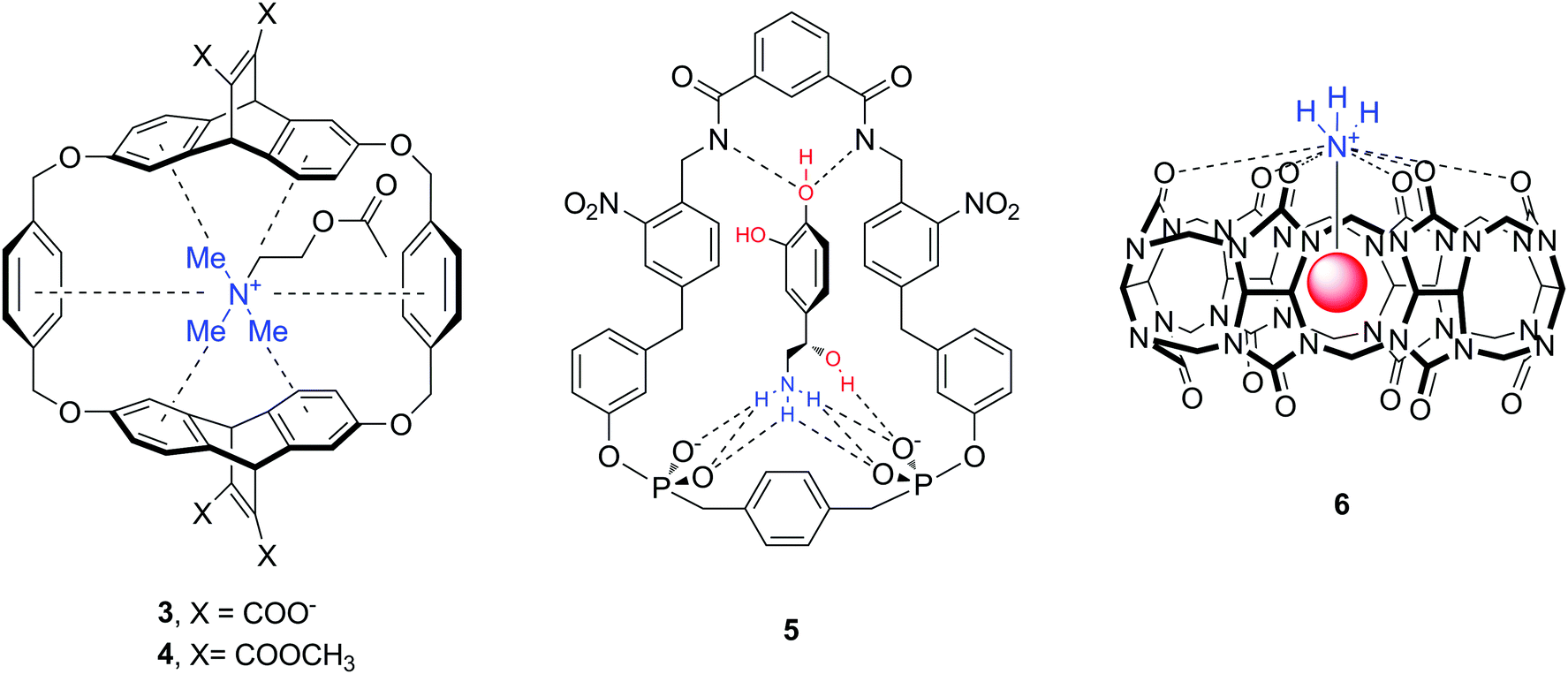 | ||
| Fig. 9 Structures of biomimetic receptors 3–6. | ||
An additional hurdle with amino acid neurotransmitters is their zwitterionic nature. Biological receptors (enzymes and proteins) that bind amino acids possess combined positively charged regions and carboxylate groups at an appropriate distance for binding anions and cations, respectively. The design of artificial amino acid receptors follows an analogous principle by mimicking these interactions. To date, only a limited number of hosts have been described for zwitterions, as compared to the total number of artificial supramolecular hosts. Here we highlight a few structures which are reported to bind natural amino acids Phe and Trp, but also neurotransmitters GABA, Glu or Asp (Fig. 10).42–44 All molecules 7–10 contain cationic and anionic or polar parts in order to accommodate the amino acid zwitterion, while the chemical nature of these moieties is quite diverse. For instance, the cation binding sites can be aza-crown ethers or ester carbonyl groups, while guanidinium, a cationic cage or terminal hydroxyl groups serve for anionic binding.
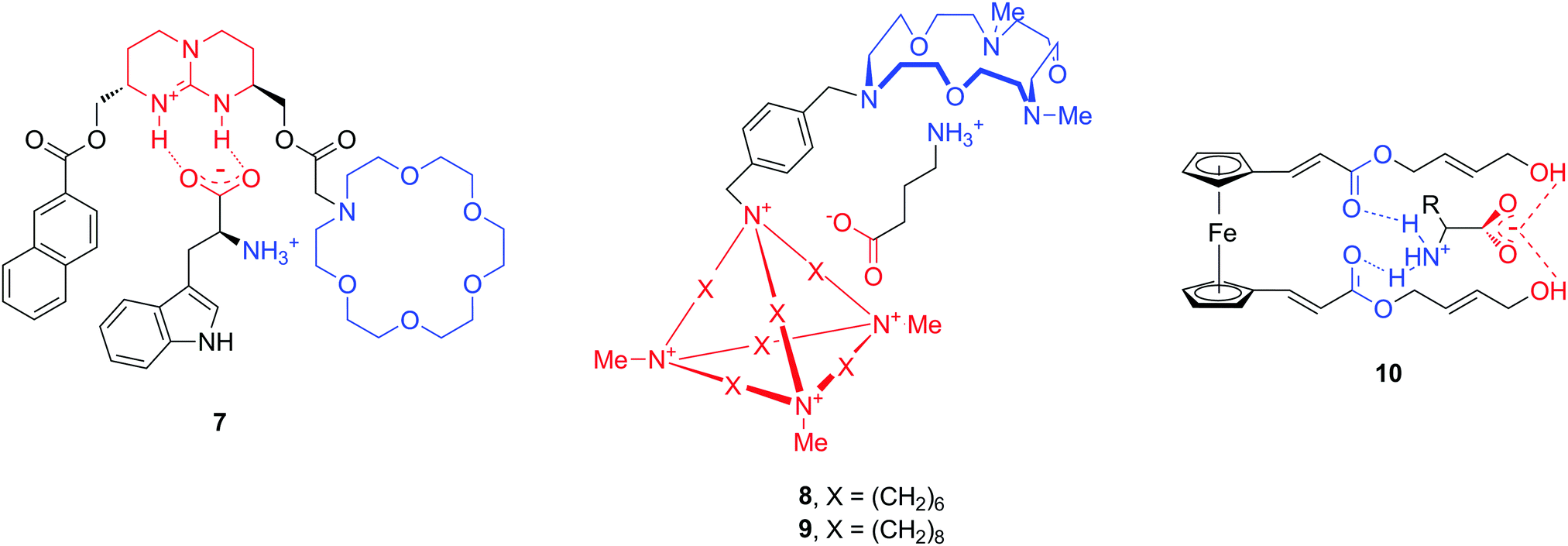 | ||
| Fig. 10 Structures of ditopic neurotransmitter hosts 7–10. | ||
There is an additional issue beyond the appropriate NT-SCAs design that complicates faster progress in the field of smart MRI probes and requires serious considerations. As already described above, an NT-SCA needs to report on the presence of neurotransmitters through a change in MRI signal intensity. After establishing an interaction with the neurotransmitter, the contrast agent has to generate an MRI signal which is dependent on the complex formation or dissociation. NT-SCAs described in this work have all achieved this through the modulation of the inner-sphere hydration of the paramagnetic metal upon neurotrasmitter binding. This is the most common, reliable and magnetic-field independent mechanism to design bioresponsive agents and should lead to further NT-SCA examples.
Other contrast mechanisms can be exploited too to create NT-dependent MR signal. The last decade has seen the emergence of a fundamentally different class of potential paramagnetic MRI contrast agents which operate on the basis of CEST. ParaCEST (paramagnetic CEST) agents are complexes of paramagnetic lanthanide or transition metal ions which possess paramagnetically shifted exchangeable protons but do not induce strong paramagnetic relaxation.19 Except for Gd3+ (strong relaxing agent) and the diamagnetic La3+ and Lu3+, complexes of all other lanthanides are potential candidates as paraCEST probes. Among transition metal ions, chelates of Fe2+, Co2+ and Ni2+ have been mostly investigated. The exchangeable protons of such paramagnetic chelates (typically amide-, amine-, alcoholic –OH or water protons) are in slow exchange with the bulk water (Fig. 11). During a CEST experiment, they are selectively saturated by radiofrequency pulses. Given their chemical exchange with bulk water protons, the saturation will lead to a decrease of the water proton signal intensity as well which can then be translated to MR contrast. When designing smart probes, one can play with the exchange rate, the resonance frequency and the concentration of mobile protons on the paraCEST agent to modulate the MRI signal as a response to a specific biomarker.45 Many responsive paraCEST probes have been explored and some proved their utility in vivo.46 Although sensitivity issues might be more important for CEST than for T1-weighted MRI, the millimolar concentration of Glu for instance, attained during neural firing, might be adapted to CEST detection.
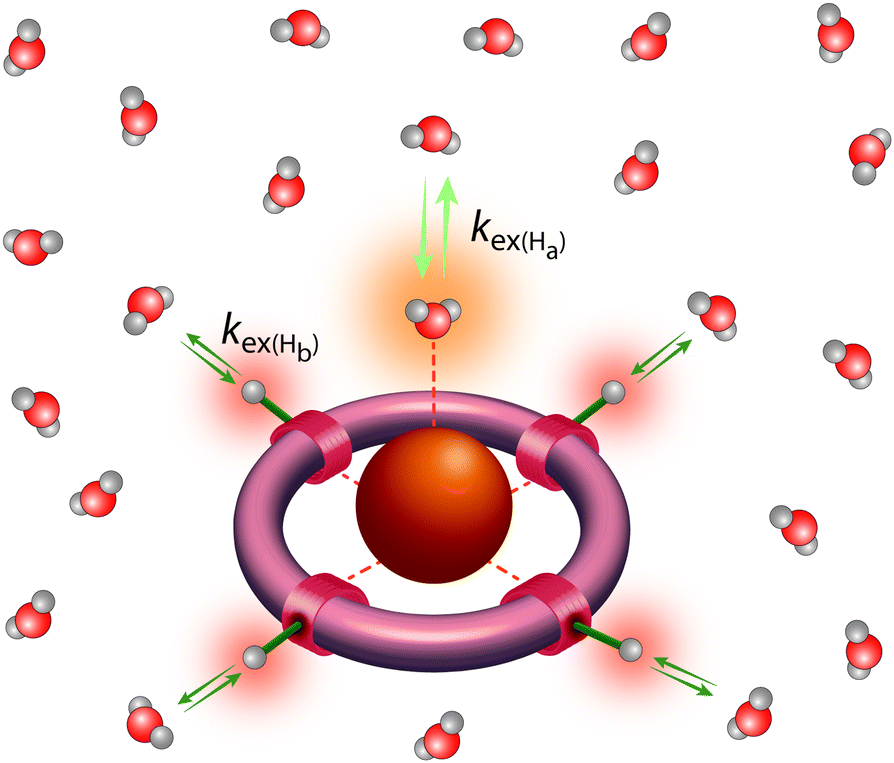 | ||
| Fig. 11 Metal complexes as paraCEST agents: the paraCEST effect originates from proton exchange between the bulk water and protons on the coordinated water molecules or on the ligand and is dependent on the proton exchange rate, kex(Ha) and kex(Hb), respectively; if the NMR chemical shifts of Ha- and Hb-type protons are different, their paraCEST effects will be observed at different frequencies. | ||
Recently, Parker et al. took a combined benefit of the relaxation and the shift inducing properties of lanthanides in a single paraSHIFT agent to achieve efficient in vivo imaging of highly shifted and fast relaxing protons.47 To this end, they simultaneously optimized, for given magnetic fields, the nature of the lanthanide and the distance of the reporter protons on the chelate from the paramagnetic centre. This shifted the observed proton resonance to a spectral region which is far from the water and fat signals and thus has no background signal, and simultaneously accelerated relaxation and thus allowed for rapid pulsing in the MRI sequences. As a result, the sensitivity of chemical shift imaging could be increased by two orders of magnitude with respect to previous chemical shift imaging studies using diamagnetic molecules. This novel concept will likely be extended to the design of responsive imaging probes for the detection of various biologically relevant molecules.
Aside from 1H, 19F is another nucleus which attracts considerable attention in MRI as its detection sensitivity is comparable to that of 1H. The absence of background signal in most tissues facilitates the observation of exogenous 19F probes in vivo, however relatively high concentrations are required as compared to classical relaxation agents.48 Both the 19F chemical shifts, which expand over a range of 300 ppm, and the relaxation rates are highly sensitive to changes in the microenvironment which can be exploited for the sensing of various biomarkers, such as pO2, metal ions or pH. 19F probes have been also conjugated to paramagnetic, mainly lanthanide complexes to enhance relaxation and therefore increase detection sensitivity. When the paramagnetic ion in the proximity of the 19F nucleus is a very strong relaxing agent such as Gd3+, the 19F signal is undetectable due to very fast relaxation and it becomes observable only upon cleavage of the Gd3+ moiety from the 19F label. Such a design was utilized for detection of bond-cleaving enzymes.48 Principally, 19F MRI has a good potential to produce NT-dependent signal changes in vitro, though in vivo translation seems difficult due to sensitivity limits.
Finally, the quickly growing field of hyperpolarized MRI can also offer additional means towards responsive imaging. Hyperpolarization of 13C, 129Xe, 3He, 15N or other nuclear spins results in signal enhancement by 4–6 orders of magnitude. While today it is mainly metabolic imaging that takes advantage of the use of hyperpolarized MRI based on 13C probes, various approaches to derive biosensors based on hyperpolarized 13C or 129Xe have been already reported and will be certainly followed by novel examples.49,50 Responsive imaging using hyperpolarized nuclei is starting to emerge and might play an important role in near future. It is interesting to note for instance that 129Xe and some amino acids can be considered as competitive binders to common hosts based on cucurbit[n]urils (example 6, Fig. 9).39,50
Conclusions
Routine direct visualization of neurotransmission processes by imaging in deep living tissues would be a big step forward in the understanding of brain function. Fully materializing such a methodology is still a great challenge for contemporary science. Nevertheless, the first advances have been made in molecular fMRI to provide molecular level insights into the physiology of neuronal processing. In this Tutorial Review, we summarize the recent exciting progress in the field of neurotransmitter-sensitive MRI contrast agents which hold promise to greatly improve functional MRI and neuroimaging in general. Based on the main principles which govern MRI signal modulation by responsive contrast agents, we present engineered proteins and artificial neurotransmitter receptors as two chemically distinct approaches to design NT-SCA. In addition, we give a survey of potential artificial hosts and biotechnology techniques which can be useful for the creation of NT-SCA. With this, the article aims to familiarize researchers with different topics which are merging into a concerted effort to study brain function. Ideally, it will stimulate new ideas, aid interaction between scientists involved in different facets of this kind of research and encourage new approaches arising from complementary areas. Given the young age of this interdisciplinary research field, we expect that novel and emerging chemistry and biotechnology techniques will give rise to rapid progress towards the final goal: direct monitoring of neuronal processes by means of molecular functional MRI.Acknowledgements
The authors thank Prof. Nikos K. Logothetis for inspirational discussions. The financial support from the Max Planck Society, CNRS, the Agence Nationale de Recherche, and academic research support program of Région Centre France (project ENZY-NEURO) is gratefully acknowledged.References
- R. P. Vertes and R. W. Stackman, Electrophysiological recording techniques, Humana Press, New York, 2011 Search PubMed.
- E. S. Bucher and R. M. Wightman, Annu. Rev. Anal. Chem., 2015, 8, 239–261 CrossRef CAS PubMed.
- M. Carter and J. C. Shieh, Guide to research techniques in neuroscience, Elsevier, Academic Press, Amsterdam, 2015 Search PubMed.
- N. K. Logothetis, Nature, 2008, 453, 869–878 CrossRef CAS PubMed.
- A. P. Koretsky, Neuroimage, 2012, 62, 1208–1215 CrossRef PubMed.
- E. J. Novotny, R. K. Fulbright, P. L. Pearl, K. M. Gibson and D. L. Rothman, Ann. Neurol., 2003, 54, S25–S31 CrossRef CAS PubMed.
- K. Cai, M. Haris, A. Singh, F. Kogan, J. H. Greenberg, H. Hariharan, J. A. Detre and R. Reddy, Nat. Med., 2012, 18, 302–306 CrossRef CAS PubMed.
- E. R. Kandel, J. H. Schwartz and T. M. Jessell, Principles of neural science, McGraw-Hill, New York, NY, London, 2000 Search PubMed.
- D. L. Robinson, A. Hermans, A. T. Seipel and R. M. Wightman, Chem. Rev., 2008, 108, 2554–2584 CrossRef CAS PubMed.
- M. Perry, Q. Li and R. T. Kennedy, Anal. Chim. Acta, 2009, 653, 1–22 CrossRef CAS PubMed.
- S. Fliegel, I. Brand, R. Spanagel and H. R. Noori, In Silico Pharmacol., 2013, 1, 7 CrossRef PubMed.
- T. Pradhan, H. S. Jung, J. H. Jang, T. W. Kim, C. Kang and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4684–4713 RSC.
- R. Q. Liang, G. J. Broussard and L. Tian, ACS Chem. Neurosci., 2015, 6, 84–93 CrossRef CAS PubMed.
- N. G. Gubernator, H. Zhang, R. G. W. Staal, E. V. Mosharov, D. B. Pereira, M. Yue, V. Balsanek, P. A. Vadola, B. Mukherjee, R. H. Edwards, D. Sulzer and D. Sames, Science, 2009, 324, 1441–1444 CrossRef CAS PubMed.
- K. S. Hettie, X. Liu, K. D. Gillis and T. E. Glass, ACS Chem. Neurosci., 2013, 4, 918–923 CrossRef CAS PubMed.
- M. Laruelle, J. Cereb. Blood Flow Metab., 2000, 20, 423–451 CrossRef CAS PubMed.
- N. Sim and D. Parker, Chem. Soc. Rev., 2015, 44, 2122–2134 RSC.
- J. Hasserodt, J. L. Kolanowski and F. Touti, Angew. Chem., Int. Ed., 2014, 53, 60–73 CrossRef CAS PubMed.
- A. E. Merbach, L. Helm and É. Tóth, The chemistry of contrast agents in medical magnetic resonance imaging, Wiley, Chichester, 2013 Search PubMed.
- M. G. Shapiro, G. G. Westmeyer, P. A. Romero, J. O. Szablowski, B. Kuster, A. Shah, C. R. Otey, R. Langer, F. H. Arnold and A. Jasanoff, Nat. Biotechnol., 2010, 28, 264–270 CrossRef CAS PubMed.
- E. M. Brustad, V. S. Lelyveld, C. D. Snow, N. Crook, S. T. Jung, F. M. Martinez, T. J. Scholl, A. Jasanoff and F. H. Arnold, J. Mol. Biol., 2012, 422, 245–262 CrossRef CAS PubMed.
- T. Lee, L. X. Cai, V. S. Lelyveld, A. Hai and A. Jasanoff, Science, 2014, 344, 533–535 CrossRef CAS PubMed.
- A. Hai, L. X. Cai, T. Lee, V. S. Lelyveld and A. Jasanoff, Neuron, 2016, 92, 754–765 CrossRef CAS PubMed.
- F. Oukhatar, H. Meudal, C. Landon, N. K. Logothetis, C. Platas-Iglesias, G. Angelovski and É. Tóth, Chem. – Eur. J., 2015, 21, 11226–11237 CrossRef CAS PubMed.
- F. Oukhatar, S. Même, W. Même, F. Szeremeta, N. K. Logothetis, G. Angelovski and É. Tóth, ACS Chem. Neurosci., 2015, 6, 219–225 CrossRef CAS PubMed.
- J. W. Steed and P. A. Gale, Supramolecular chemistry: from molecules to nanomaterials, Wiley, Chichester, 2012 Search PubMed.
- H. Tsukube and S. Shinoda, Chem. Rev., 2002, 102, 2389–2403 CrossRef CAS PubMed.
- T. Budisantoso, H. Harada, N. Kamasawa, Y. Fukazawa, R. Shigemoto and K. Matsui, J. Physiol., 2013, 591, 219–239 CrossRef CAS PubMed.
- Y. Chen and L. H. Liu, Adv. Drug Delivery Rev., 2012, 64, 640–665 CrossRef CAS PubMed.
- M. G. Shapiro, T. Atanasijevic, H. Faas, G. G. Westmeyer and A. Jasanoff, Magn. Reson. Imaging, 2006, 24, 449–462 CrossRef CAS PubMed.
- V. S. Lelyveld, E. Brustad, F. H. Arnold and A. Jasanoff, J. Am. Chem. Soc., 2011, 133, 649–651 CrossRef CAS PubMed.
- A. F. A. Peacock, Curr. Opin. Chem. Biol., 2016, 31, 160–165 CrossRef CAS PubMed.
- J. W. Steed and J. L. Atwood, Supramolecular chemistry, Wiley, Oxford, 2009 Search PubMed.
- P. A. Gale and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3595–3596 RSC.
- F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609–1646 CrossRef CAS PubMed.
- M. J. Langton, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 1974–1987 CrossRef CAS PubMed.
- D. A. Dougherty and D. A. Stauffer, Science, 1990, 250, 1558–1560 CrossRef CAS PubMed.
- M. Herm, O. Molt and T. Schrader, Angew. Chem., Int. Ed., 2001, 40, 3148–3151 CrossRef CAS.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS PubMed.
- H. J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924–3977 CrossRef CAS PubMed.
- G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366–2393 CrossRef CAS PubMed.
- A. Galan, D. Andreu, A. M. Echavarren, P. Prados and J. Demendoza, J. Am. Chem. Soc., 1992, 114, 1511–1512 CrossRef CAS.
- F. P. Schmidtchen, J. Org. Chem., 1986, 51, 5161–5168 CrossRef CAS.
- P. Debroy, M. Banerjee, M. Prasad, S. P. Moulik and S. Roy, Org. Lett., 2005, 7, 403–406 CrossRef CAS PubMed.
- D. V. Hingorani, A. S. Bernstein and M. D. Pagel, Contrast Media Mol. Imaging, 2015, 10, 245–265 CrossRef CAS PubMed.
- D. D. Castelli, G. Ferrauto, J. C. Cutrin, E. Terreno and S. Aime, Magn. Reson. Med., 2014, 71, 326–332 CrossRef PubMed.
- P. Harvey, A. M. Blamire, J. I. Wilson, K. L. N. A. Finney, A. M. Funk, P. K. Senanayake and D. Parker, Chem. Sci., 2013, 4, 4251–4258 RSC.
- J.-X. Yu, R. R. Hallac, S. Chiguru and R. P. Mason, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 70, 25–49 CrossRef CAS PubMed.
- A. R. Lippert, K. R. Keshari, J. Kurhanewicz and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 3776–3779 CrossRef CAS PubMed.
- Y. Wang and I. J. Dmochowski, Acc. Chem. Res., 2016, 49, 2179–2187 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |