
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rh(III)-catalyzed diastereoselective C–H bond addition/cyclization cascade of enone tethered aldehydes†
Jeffrey A.
Boerth
and
Jonathan A.
Ellman
*
Department of Chemistry, Yale University, Connecticut 06520, USA. E-mail: jonathan.ellman@yale.edu; Tel: +1-203-432-2647
First published on 1st December 2015
Abstract
The Rh(III)-catalyzed cascade addition of a C–H bond across alkene and carbonyl π-bonds is reported. The reaction proceeds under mild reaction conditions with low catalyst loading. A range of directing groups were shown to be effective as was the functionalization of alkenyl in addition to aromatic C(sp2)–H bonds. When the enone and aldehyde electrophile were tethered together, cyclic β-hydroxy ketones with three contiguous stereocenters were obtained with high diastereoselectivity. The intermolecular three-component cascade reaction was demonstrated for both aldehyde and imine electrophiles. Moreover, the first X-ray structure of a cationic Cp*Rh(III) enolate with interatomic distances consistent with an η3-bound enolate is reported.
Introduction
Recently, the design and implementation of Rh(III)-catalyzed C–H bond functionalization has led to a diverse array of structural motifs, including many that are present in drugs and natural products.1–3 In particular, Rh(III)-catalyzed C–H bond additions to polarized π-bonds3 provides convergent entry to drug relevant amines,4 amides,5 alcohols,6 and oxygen or nitrogen heterocycles.7 While a variety of methods have been developed for direct C(sp2)–H bond addition into polarized π-bonds, cascade addition sequences would offer an attractive strategy for rapidly building complexity into organic structures. To date, Rh(III)-catalyzed cascade C–H bond functionalization has primarily been reported for 5-membered ring synthesis by additions to an alkene or alkyne followed by cyclization upon the directing group for C–H bond activation.8 However, to the best of our knowledge, Lin and coworkers have reported the only example of Rh(III)-catalyzed C–H bond cascade addition to a π-bond and an electrophile other than the directing group.9 In their study, Rh(III)-catalyzed C–H bond addition to enones tethered to an alkyne proceeded to give substituted tetrahydrofurans (Fig. 1A). Herein, we demonstrate the Rh(III)-catalyzed cascade addition of a C–H bond across alkene and carbonyl π-bonds. When the two electrophiles are tethered together, cyclic β-hydroxy ketone products incorporating three contiguous stereocenters are produced with high diastereoselectivity (Fig. 1B).10,11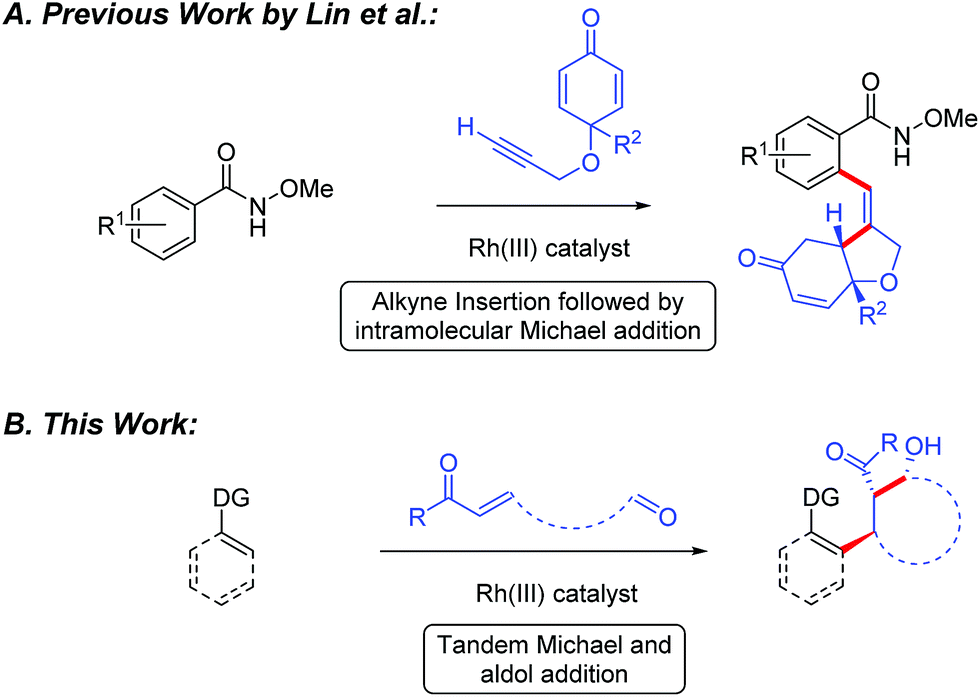 | ||
| Fig. 1 Rh(III)-catalyzed cascade C–H bond addition across two different π-bonds. | ||
Results and discussion
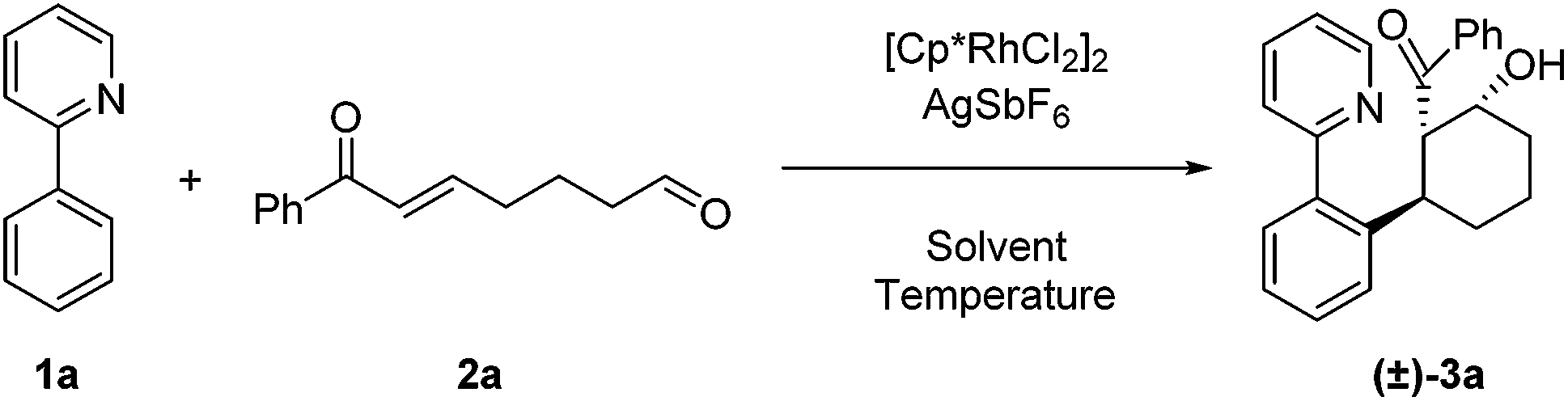
For our initial exploration of this reaction we employed 2-phenylpyridine and the enone tethered aldehyde 2a based on successful Rh(III)-catalyzed hydroarylation of enones (Table 1).12 After considerable optimization, we found that β-hydroxy ketone 3a could be obtained in high yield as a single diastereomer with only 2.5 mol% of Rh precatalyst and 10 mol% of AgSbF6 (entry 1). A 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 dioxane/H2O reaction solvent at 50 °C was found to be effective. Lower conversion was observed at 30 °C (entry 2). Solvents commonly used in Rh(III)-catalyzed C–H functionalization such as dichloroethane (entry 3) and dioxane (entry 4) provided lower yields, and reducing the amount of H2O also was detrimental (entry 5). In contrast, acetic acid as the solvent proved to be optimal,7a,12a giving a near quantitative yield of the desired β-hydroxy ketone 3a (entry 6). Additionally, 3a was obtained in 57% yield when employing only the rhodium dimer [Cp*RhCl2]2 in the absence of a silver halide abstractor demonstrating that a pre-formed cationic Rh catalyst is not required for this transformation (entry 7). However, when rhodium was excluded, the desired product 3a was not obtained (entries 8 and 9).
:2 dioxane/H2O reaction solvent at 50 °C was found to be effective. Lower conversion was observed at 30 °C (entry 2). Solvents commonly used in Rh(III)-catalyzed C–H functionalization such as dichloroethane (entry 3) and dioxane (entry 4) provided lower yields, and reducing the amount of H2O also was detrimental (entry 5). In contrast, acetic acid as the solvent proved to be optimal,7a,12a giving a near quantitative yield of the desired β-hydroxy ketone 3a (entry 6). Additionally, 3a was obtained in 57% yield when employing only the rhodium dimer [Cp*RhCl2]2 in the absence of a silver halide abstractor demonstrating that a pre-formed cationic Rh catalyst is not required for this transformation (entry 7). However, when rhodium was excluded, the desired product 3a was not obtained (entries 8 and 9).
|
|
||||
|---|---|---|---|---|
| Entry | Rh (mol%)/Ag (mol%) | Solvent | Temp (°C) | Yield 3ab (%) |
| a Conditions: 1a (2.0 equiv.), 2a (1.0 equiv.) using [Cp*RhCl2]2 and AgSbF6 for 20 h (0.2 M). b Determined by NMR analysis relative to 1,3,5-trimethoxybenzene as an external standard. | ||||
| 1 | (2.5)/(10) | 3:2 Dioxane/H2O |
50 | 92 |
| 2 | (2.5)/(10) | 3:2 Dioxane/H2O |
30 | 67 |
| 3 | (2.5)/(10) | DCE | 50 | 58 |
| 4 | (2.5)/(10) | Dioxane | 50 | 35 |
| 5 | (2.5)/(10) | 95:5 Dioxane/H2O |
50 | 61 |
| 6 | (2.5)/(10) | Acetic acid | 50 | 99 |
| 7 | (2.5)/(0) | Acetic acid | 50 | 57 |
| 8 | (0)/(10) | Acetic acid | 50 | 0 |
| 9 | None | Acetic acid | 50 | 0 |
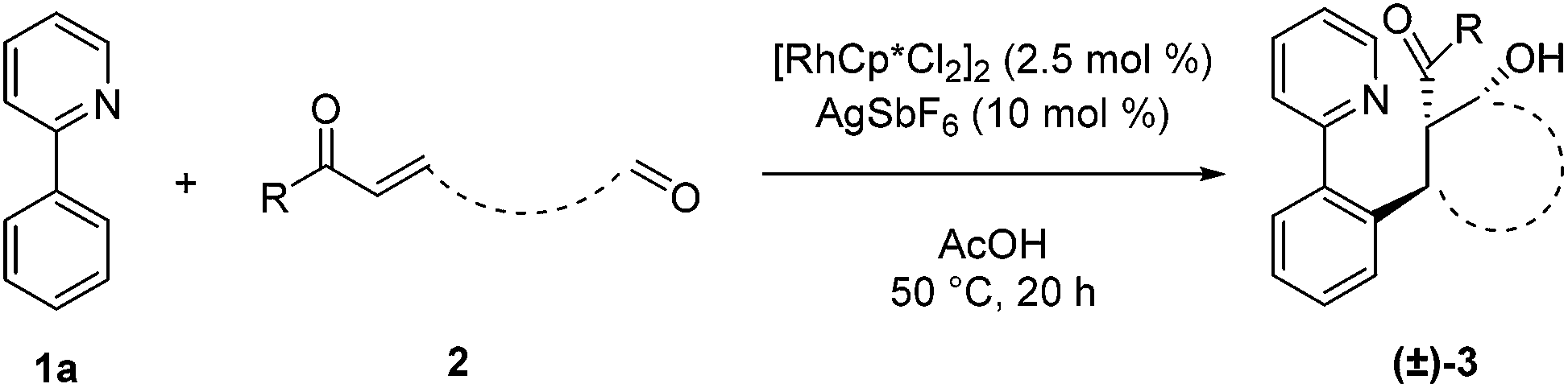
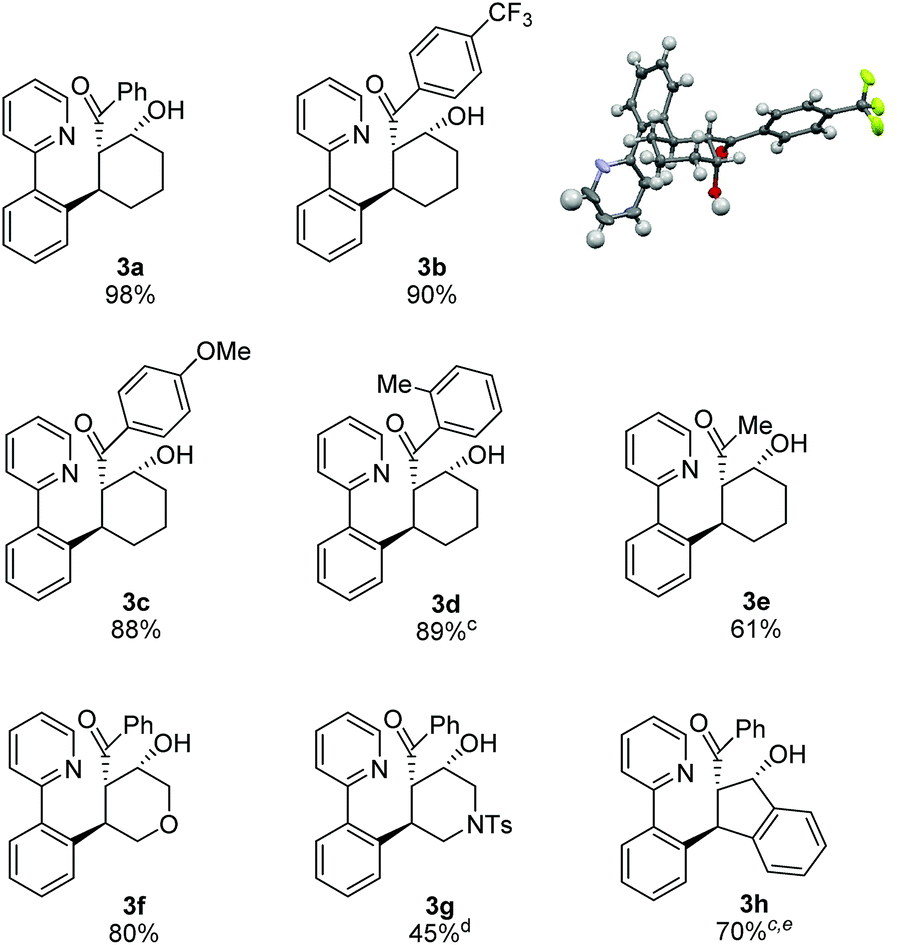
After identifying optimal conditions for the formation of β-hydroxy ketone 3a, we next explored the scope of the tethered electrophile substrate (Table 2). Pure 3a was isolated in near quantitative yield from the parent substrate 2a. Substrates with electron-donating and withdrawing substituents on the phenyl ring also afforded β-hydroxy ketones 3b, 3c, and 3d in high yield. X-ray structural analysis of 3b provided rigorous confirmation of product stereochemistry. An alkyl enone, which is a weaker acceptor than the corresponding aryl enone substrates, proved to be effective giving 3e with only a modest reduction in yield. Introduction of oxygen and nitrogen heteroatoms into the tether were also acceptable substitutions, and provided the substituted tetrahydropyran 3f and piperidine 3g, respectively. Additionally, the use of an aromatic tether was well-tolerated and gave indane 3h in good yield. However, for this substrate, 3:2 dioxane/H2O as solvent, a higher catalyst loading, and a lower reaction temperature were employed to minimize β-hydroxyl group elimination, which is particularly facile for β-hydroxy ketone 3h.
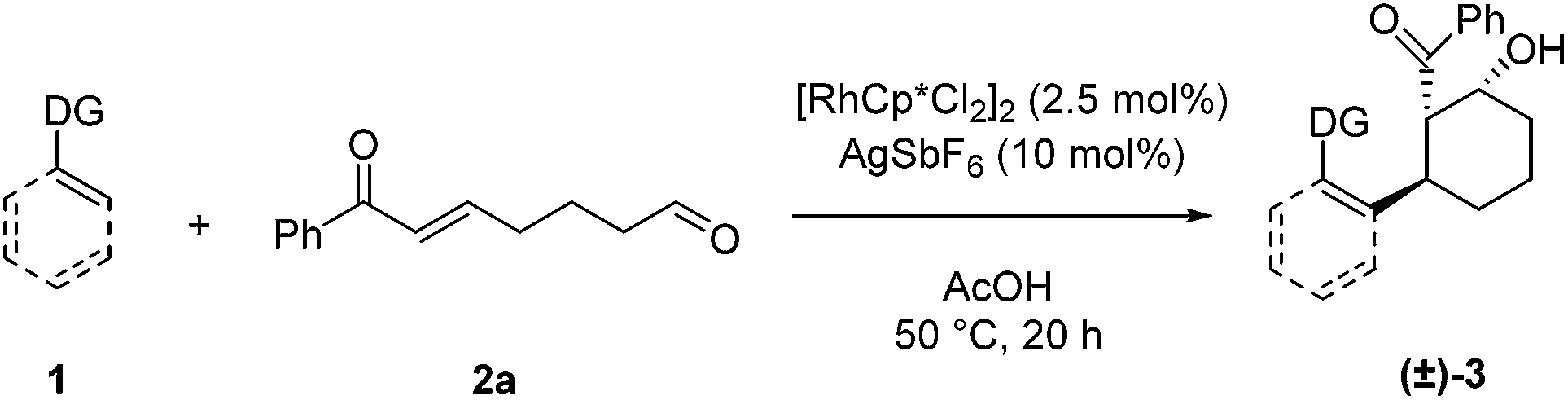
We next examined the scope for different C–H bond coupling partners (Table 3). Derivatives of 2-phenylpyridine with electron-donating and withdrawing substituents also provided high yields of the corresponding β-hydroxy ketones 3i, 3j, and 3k. Notably, the applicability of alkenyl C(sp2–H) functionalization to this transformation was demonstrated with 2-cyclohexenylpyridine, which gave 3l in good yield. Deviations from the pyridyl directing group are also noteworthy. The pyrazole and pyrimidine heterocycles were quite efficient in forming products 3m and 3n. N-Pyrimidylindole provided 3o, highlighting C–H functionalization on a heteroaryl ring. In addition, the N-methoxybenzamide and O-methyl oxime directing groups provided access to the products 3p and 3q in moderate yields. For these substrates, higher yields were obtained with 3:2 dioxane/H2O rather than acetic acid as solvent.
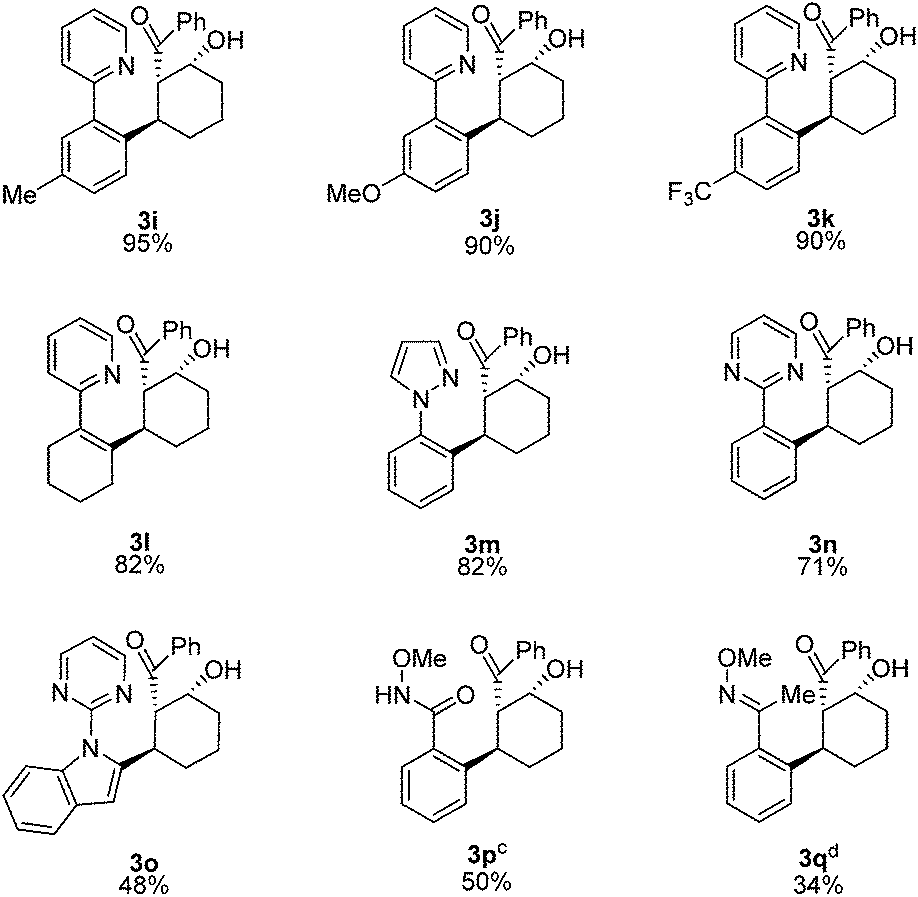
A mechanism for the Rh-catalyzed cascade C–H bond addition/aldol reaction is depicted in Scheme 1. The first step of this process proceeds via concerted metalation/deprotonation of 1 to generate rhodacycle 4, which has previously been proposed as an intermediate in other Rh-catalyzed C–H functionalization reactions.3,13 Coordination of the enone π-bond provides 5, which undergoes conjugate addition to give rhodium enolate 6. The rhodium enolate 6 can then undergo an intramolecular aldol reaction with the tethered aldehyde to form rhodium alkoxide 7.14 Coordination of another equivalent of 1 to the Rh–alkoxide complex then provides 8, which undergoes concerted metalation/deprotonation to release the alcohol product 3 and regenerate the active rhodium species 4.15
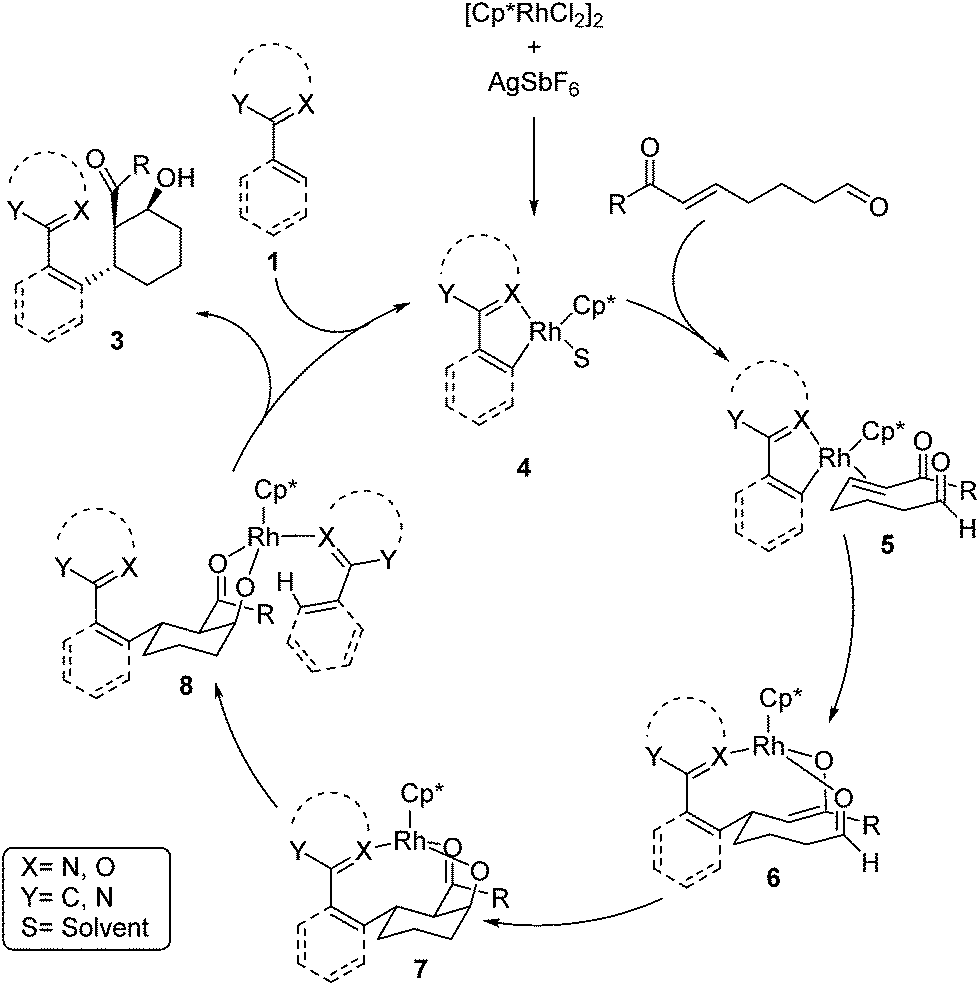 | ||
| Scheme 1 Proposed mechanism for transformation. | ||
To ascertain whether or not cyclization upon the aldehyde carbonyl after enone addition proceeds via the proposed Rh-enolate intermediate 6 (Scheme 1), the independently prepared acyclic enone addition product 9a was subjected to the reaction conditions with and without the cationic rhodium catalyst (Table 4). With dioxane/H2O as the solvent (entries 1 and 2), a low yield of β-hydroxy ketone 3a was obtained even in the presence of the Rh precatalyst and AgSbF6 (entry 1). In addition, whether or not Rh was added, complete consumption of 9a was observed, presumably as a result of selective formation of enolates or enols from the more acidic aldehyde functionality followed by unproductive side-reactions. These results are consistent with aldol cyclization proceeding via the Rh-enolate generated upon enone addition. When acetic acid was used as the solvent, a moderate yield of 3a was obtained suggesting that acetic acid can mediate this cyclization step, although once again decomposition pathways competed with the desired cyclization pathway (entries 3 and 4).

C–H bond addition to generate a rhodium-enolate intermediate was rigorously established using the simplified enone substrate phenyl vinyl ketone (Fig. 2). To facilitate crystallization of the Rh-enolate, tetrakis(pentafluorophenyl)borate was employed as the counterion with DCE as the solvent, which for the cascade reaction is only slightly less effective than acetic acid or dioxane/water as solvent, vide infra (see 3r in Table 5). After only 30 min at rt, rhodacycle 10 was isolated in very high yield. The X-ray structure of 10 shows a coordinatively saturated Rh(III) complex with interatomic distances consistent with an η3-bound enolate. Although rhodium enolates have been proposed as intermediates for a number of transformations,16 very few X-ray structures have been reported.17 To our knowledge, the only published example of a Cp*Rh(III) enolate is for a neutral complex with an η1 C-bound enolate.17a
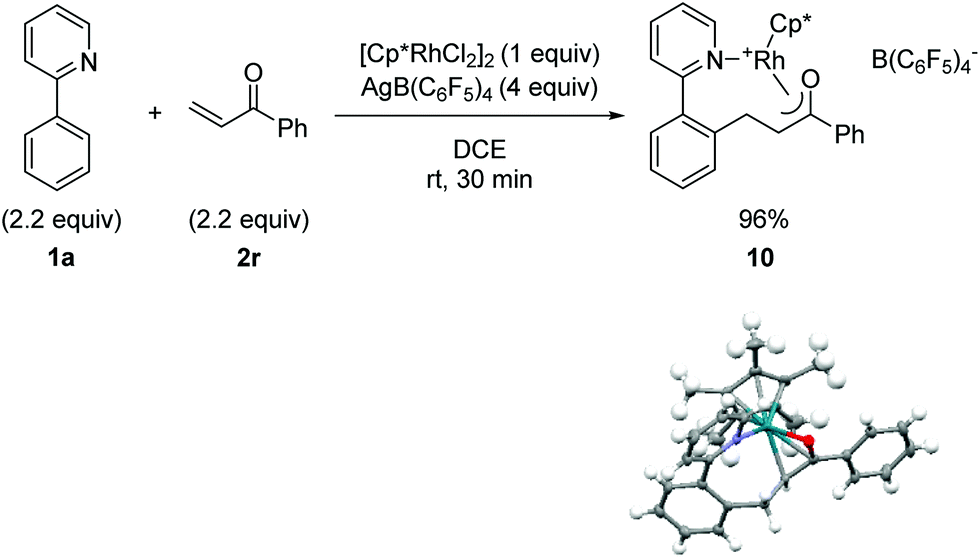 | ||
| Fig. 2 Preparation, isolation and X-ray structural characterization of a cationic Rh(III)-enolate. The B(C6F5)4 ion has been omitted from the X-ray structure for clarity. | ||
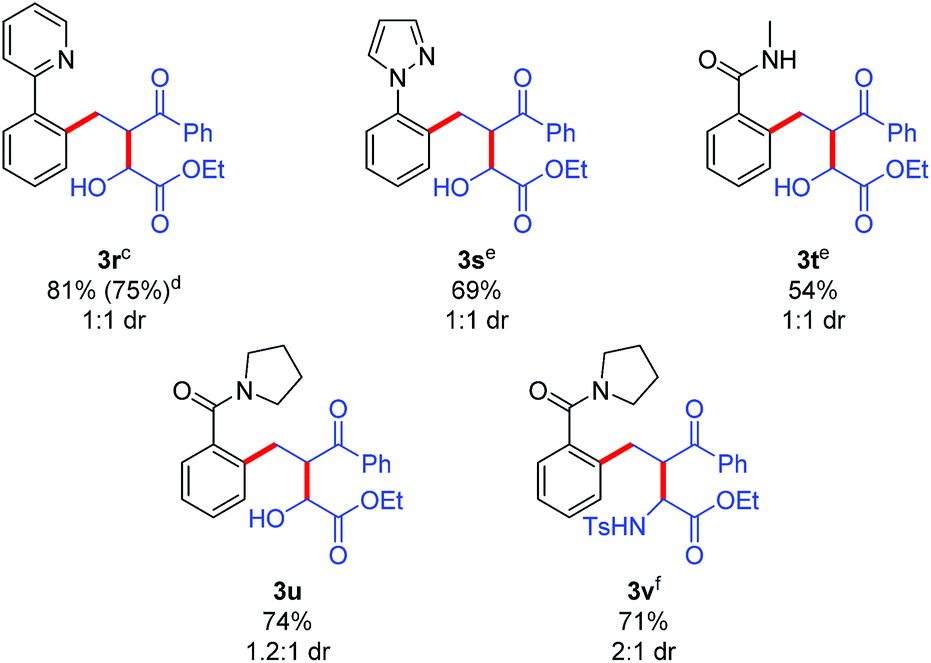
The potential for performing an intermolecular three-component C–H activation/addition/aldol reaction cascade was also evaluated (Table 5). Several different directing groups provided three-component cascade addition products using enone 2r along with either an activated aldehyde or imine. Coupling 2-phenylpyridine and N-phenyl pyrazole with enone 2r and ethyl glyoxylate gave products 3r and 3s in good yields, respectively. Moreover, the synthetically more versatile secondary and tertiary amide directing groups provided products 3t and 3u in moderate to good yields. In addition, three-component coupling with the N-tosyl imine derived from ethyl glyoxylate efficiently provided amine 3v, though the use of DCE as solvent was necessary to minimize the competitive imine hydrolysis that occurred in acetic acid.
The complete selectivity for initial C–H bond addition to the enone rather than the aldehyde is an interesting feature of this reaction. This outcome might result from kinetic control with C–H bond addition to the enone occurring much faster than to the aldehyde. Alternatively, thermodynamic control might be operative because C(sp2)–H bond addition to aldehydes is known to be reversible,18 although C–H bond addition products are favored for destabilized aldehydes such as ethyl glyoxylate.6f
Conclusions
In summary, a Rh(III)-catalyzed C–H bond addition/aldol cyclization cascade has been developed and represents the first C–H bond addition across an alkene π-bond and a carbonyl. This robust transformation can be carried out under mild conditions and for tethered substrates generates three contiguous stereocenters with high diastereoselectivity. An intermolecular three-component C–H bond addition/aldol reaction cascade has also been demonstrated. Preliminary mechanistic studies also provide the first X-ray structural characterization of a cationic Cp*Rh(III) enolate. In continuing efforts we are broadening the scope of this class of cascade reactions to include different tethered electrophilic species. We are also actively investigating stereoselective intermolecular three-component reactions with different carbon–carbon π-bonds and carbonyl/imine electrophiles.Acknowledgements
This work was supported by the NIH (GM069559). We gratefully acknowledge Dr Brandon Mercado (Yale University) for solving the crystal structure of 3b and 10.Notes and references
- For recent reviews on C−H functionalization, see: (a) L. Yang and H. Huang, Chem. Rev., 2015, 115, 3468 CrossRef CAS PubMed; (b) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (c) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (d) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC; (e) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (f) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (g) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (h) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (i) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (j) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed; (k) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (l) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC.
- For recent reviews on Rh(III)-catalyzed C−H functionalization, see: (a) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (b) F. W. Patureau, J. Wencel-Delord and F. Glorius, Aldrichimica Acta, 2012, 45, 31 CAS; (c) T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212 CrossRef CAS PubMed.
- For reviews on C−H bond additions to polarized π-bonds, see: (a) X.-S. Zhang, K. Chen and Z.-J. Shi, Chem. Sci., 2014, 5, 2146 RSC; (b) G. Yan, X. Wu and M. Yang, Org. Biomol. Chem., 2013, 11, 5558 RSC.
- For examples of Rh(III)-catalyzed C−H bond additions to imines, see: (a) A. Wangweerawong, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2014, 136, 8520 CrossRef CAS PubMed; (b) B. Zhou, Y. Yang, S. Lin and Y. Li, Adv. Synth. Catal., 2013, 355, 360 CAS; (c) Y. Li, X.-S. Zhang, H. Li, W.-H. Wang, K. Chen, B.-J. Li and Z.-J. Shi, Chem. Sci., 2012, 3, 1634 RSC; (d) Y. Li, X.-S. Zhang, Q.-L. Zhu and Z.-J. Shi, Org. Lett., 2012, 14, 4498 CrossRef CAS PubMed; (e) K. D. Hesp, R. G. Bergman and J. A. Ellman, Org. Lett., 2012, 14, 2304 CrossRef CAS PubMed; (f) Y. Li, B.-J. Li, W.-H. Wang, W.-P. Huang, X.-S. Zhang, K. Chen and Z.-J. Shi, Angew. Chem., Int. Ed., 2011, 50, 2115 CrossRef CAS PubMed; (g) A. S. Tsai, M. E. Tauchert, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2011, 133, 1248 CrossRef CAS PubMed.
- For examples of Rh(III)-catalyzed C−H bond amidation with isocyanates, see: (a) X.-Y. Shi, K.-Y. Liu, J. Fan, X.-F. Dong, J.-F. Wei and C.-J. Li, Chem.–Eur. J., 2015, 21, 1900 CrossRef CAS PubMed; (b) X.-Y. Shi, A. Renzetti, S. Kundu and C.-J. Li, Adv. Synth. Catal., 2014, 356, 723 CrossRef CAS; (c) K. Shin, J. Ryu and S. Chang, Org. Lett., 2014, 16, 2022 CrossRef CAS PubMed; (d) W. Hou, B. Zhou, Y. Yang, H. Feng and Y. Li, Org. Lett., 2013, 15, 1814 CrossRef CAS PubMed; (e) B. Zhou, W. Hou, Y. Yang and Y. Li, Chem.–Eur. J., 2013, 19, 4701 CrossRef CAS PubMed; (f) S. Takebayashi, T. Shizuno, T. Otani and T. Shibata, Beilstein J. Org. Chem., 2012, 8, 1844 CrossRef CAS PubMed; (g) K. D. Hesp, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2011, 133, 11430 CrossRef CAS PubMed.
- For examples of Rh(III)-catalyzed C−H bond additions to aldehydes, see: (a) Y. Li, X.-S. Zhang, K. Chen, K.-H. He, F. Pan, B.-J. Li and Z.-J. Shi, Org. Lett., 2012, 14, 636 CrossRef CAS PubMed; (b) See ref. 4d; (c) S. Sharma, E. Park, J. Park and I. S. Kim, Org. Lett., 2012, 14, 906 CrossRef CAS PubMed; (d) B. Zhou, Y. Yang and Y. Li, Chem. Commun., 2012, 48, 5163 RSC; (e) J. Park, E. Park, A. Kim, Y. Lee, K. W. Chi, J. H. Kwak, Y. H. Jung and I. S. Kim, Org. Lett., 2011, 13, 4390 CrossRef CAS PubMed; (f) L. Yang, C. A. Correia and C.-J. Li, Adv. Synth. Catal., 2011, 353, 1269 CrossRef CAS.
- (a) Y. Lian, R. G. Bergman, L. D. Lavis and J. A. Ellman, J. Am. Chem. Soc., 2013, 135, 7122 CrossRef CAS PubMed; (b) Y. Lian, T. Huber, K. D. Hesp, R. G. Bergman and J. A. Ellman, Angew. Chem., Int. Ed., 2013, 52, 629 CrossRef CAS PubMed; (c) X.-Y. Shi and C.-J. Li, Adv. Synth. Catal., 2012, 354, 2933 CrossRef CAS; (d) Y. Lian, R. G. Bergman and J. A. Ellman, Chem. Sci., 2012, 3, 3088 RSC.
- For examples of C–H bond additions to alkynes followed by cyclization upon an imine or carbonyl directing group, see: (a) Z. Shi, C. Grohmann and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 5393 CrossRef CAS PubMed; (b) J. Zhang, A. Ugrinov and P. Zhao, Angew. Chem., Int. Ed., 2013, 52, 6681 CrossRef CAS PubMed; (c) S. Chen, J. Yu, Y. Jiang, F. Chen and J. Cheng, Org. Lett., 2013, 15, 4754 CrossRef CAS PubMed; (d) Y. Chen, F. Wang, W. Zhen and X. Li, Adv. Synth. Catal., 2013, 355, 353 CAS; (e) Z. Qi, M. Wang and X. Li, Org. Lett., 2013, 15, 5440 CrossRef CAS PubMed; (f) L. Dong, C. H. Qu, J. R. Huang, W. Zhang, Q. R. Zhang and J. G. Deng, Chem.–Eur. J., 2013, 19, 16537 CrossRef CAS PubMed; (g) P. Zhao, F. Wang, K. Han and X. Li, Org. Lett., 2012, 14, 5506 CrossRef CAS PubMed; (h) B. J. Li, H. Y. Wang, Q. L. Zhu and Z. J. Shi, Angew. Chem., Int. Ed., 2012, 51, 3948 CrossRef CAS PubMed; (i) R. K. Chinnagolla and M. Jeganmohan, Eur. J. Org. Chem., 2012, 2012, 417 CrossRef CAS; (j) D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 11098 CrossRef CAS PubMed; (k) K. Muralirajan, K. Parthasarathy and C. H. Cheng, Angew. Chem., Int. Ed., 2011, 50, 4169 CrossRef CAS PubMed; (l) F. W. Patureau, T. Besset, N. Kuhl and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2154 CrossRef CAS PubMed; (m) Z. M. Sun, S. P. Chen and P. Zhao, Chem.–Eur. J., 2010, 16, 2619 CrossRef CAS PubMed; (n) T. Fukutani, N. Umeda, K. Hirano, T. Satoh and M. Miura, Chem. Commun., 2009, 5141 RSC; (o) Y. Kuninobu, Y. Tokunaga, A. Kawata and K. Takai, J. Am. Chem. Soc., 2006, 128, 202 CrossRef CAS PubMed ; for examples of C–H bond additions to alkenes followed by cyclization upon imine or carbonyl directing group, see:; (p) W. Liu, D. Zell, M. John and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 4092 CrossRef CAS PubMed; (q) T. Nishimura, Y. Ebe and T. Hayashi, J. Am. Chem. Soc., 2013, 135, 2092 CrossRef CAS PubMed; (r) X.-Y. Shi and C.-J. Li, Org. Lett., 2013, 15, 1476 CrossRef CAS PubMed; (s) T. Nishimura, M. Nagamoto, Y. Ebe and T. Hayashi, Chem. Sci., 2013, 4, 4499 RSC; (t) D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2010, 49, 8181 CrossRef CAS PubMed.
- Y. Fukui, P. Liu, Q. Liu, Z. He, N. Wu, P. Tian and G. Lin, J. Am. Chem. Soc., 2014, 136, 15607 CrossRef CAS PubMed.
- For a review on cascade Michael/aldol reactions, see: E. V. Gorobets, M. S. Miftakhov and F. A. Valeev, Russ. Chem. Rev., 2000, 69, 1001 CrossRef CAS.
- For relevant metal-catalyzed cascade Michael/aldol reactions, see: for Rh(I)-catalyzed examples, see: (a) D. F. Cauble, J. D. Gipson and M. J. Krische, J. Am. Chem. Soc., 2003, 125, 1110 CrossRef CAS PubMed; (b) R. R. Huddleston and M. J. Krische, Org. Lett., 2003, 5, 1143 CrossRef CAS PubMed ; for Cu(II)-catalyzed examples, see:; (c) A. Alexakis, G. P. Trevitt and G. Bernardinelli, J. Am. Chem. Soc., 2001, 123, 4358 CrossRef CAS PubMed; (d) L. A. Arnold, R. Naasz, A. J. Minnaard and B. L. Feringa, J. Am. Chem. Soc., 2001, 123, 5841 CrossRef CAS PubMed; (e) B. L. Feringa, M. Pineschi, L. A. Arnold, R. Imbos and A. H. M. de Vries, Angew. Chem., Int. Ed., 1997, 36, 2620 CrossRef CAS ; for a Co(II)-catalyzed example, see:; (f) T. G. Baik, A. L. Luis, J. C. Wang and M. J. Krische, J. Am. Chem. Soc., 2001, 123, 5112 CrossRef CAS PubMed ; for a Ni(0)-catalyzed example, see:; (g) J. Montgomery, E. Oblinger and A. V. Savchenko, J. Am. Chem. Soc., 1997, 119, 4911 CrossRef.
- (a) L. Yang, B. Qian and H. Huang, Chem.–Eur. J., 2012, 18, 9511 CrossRef CAS PubMed; (b) L. Yang, C. A. Correia and C.-J. Li, Org. Biomol. Chem., 2011, 9, 7176 RSC.
- For select relevant mechanistic studies for Rh(III)-catalysis, see: (a) A. P. Walsh and W. D. Jones, Organometallics, 2015, 34, 3400 CrossRef CAS; (b) M. E. Tauchert, C. D. Incarvito, A. L. Rheingold, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2012, 134, 1482 CrossRef CAS PubMed; (c) Y. Li, X.-S. Zhang, H. Li, W.-H. Wang, K. Chen, B.-J. Li and Z.-J. Shi, Chem. Sci., 2012, 3, 1634 RSC; (d) L. Li, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2008, 130, 12414 CrossRef CAS PubMed.
- For a stoichiometric aldol reaction of a neutral Rh(I)-enolate, see: G. A. Slough, R. G. Bergman and C. H. Heathcock, J. Am. Chem. Soc., 1989, 111, 938 CrossRef CAS.
- The Z-enone tethered aldehyde might be expected to provide a different enolate stereochemistry that could result in a different aldol stereoisomer. The Z-isomer of compound 2a was therefore prepared and subjected to the reaction conditions. Unfortunately, E/Z isomerization occurred more rapidly than the rate of C–H functionalization in all of the solvents investigated.
- For leading references, see: K. Fagnou and M. Lautens, Chem. Rev., 2003, 103, 169 CrossRef CAS PubMed.
- (a) J. Wu and R. G. Bergman, J. Am. Chem. Soc., 1989, 111, 7628 CrossRef CAS; (b) G. A. Slough, R. Hayashi, J. R. Ashbaugh, S. L. Shamblin and A. M. Aukamp, Organometallics, 1994, 13, 890 CrossRef CAS.
- H. Li, Y. Li, X.-S. Zhang, K. Chen, X. Wang and Z.-J. Shi, J. Am. Chem. Soc., 2011, 133, 15244 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1413993 for 3b and 1431932 for 10. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04138d |
| This journal is © The Royal Society of Chemistry 2016 |