
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigher-order structural interrogation of antibodies using middle-down hydrogen/deuterium exchange mass spectrometry†
Jingxi
Pan
a,
Suping
Zhang
b,
Albert
Chou
a and
Christoph H.
Borchers
*ac
aUniversity of Victoria-Genome British Columbia Proteomics Centre, Vancouver Island Technology Park, #3101-4464 Markham St., Victoria, BC V8Z 7X8, Canada. E-mail: christoph@proteincentre.com
bMRM Proteomics Inc., 4464 Markham Street, Suite #2108, Victoria, British Columbia V8Z 7X8, Canada
cDepartment of Biochemistry & Microbiology, University of Victoria, Petch Building Room 207, 3800 Finnerty Rd., Victoria, BC V8P 5C2, Canada
First published on 24th November 2015
Abstract
Although X-ray crystallography is the “gold standard” method for protein higher-order structure analysis, the challenges of antibody crystallization and the time-consuming data analysis involved make this technique unsuitable for routine structural studies of antibodies. In addition, crystallography cannot be used for the structural characterization of an antibody in solution, under conditions where antibody drugs are active. Intact antibodies are also too large and too complex for NMR. Top-down mass spectrometry coupled to hydrogen/deuterium exchange (HDX) is a powerful tool for high-resolution protein structural characterization, but its success declines rapidly as protein size increases. Here we report for the first time a new hybrid “middle-down” HDX approach that overcomes this limitation through enabling the nonspecific enzyme pepsin to perform specific restricted digestion at low pH prior to HPLC separation at subzero temperatures and online electron transfer dissociation (ETD). Three large specific peptic fragments (12 to 25 kDa) were observed from the heavy chain and light chain of a therapeutic antibody Herceptin, together with a few smaller fragments from the middle portion of the heavy chain. The average amino-acid resolutions obtained by ETD were around two residues, with single-residue resolution in many regions. This middle-down approach is also applicable to other antibodies. It provided HDX information on the entire light chain, and 95.3% of the heavy chain, representing 96.8% of the entire antibody (150 kDa). The structural effects of glycosylation on Herceptin were determined at close-to-single residue level by this method.
Introduction
The higher-order protein structure and protein dynamics basically determine the “personality” of a protein,1,2 therefore, comprehensive characterization of these structure features is essential for a better understanding of their functions. This is of particular importance for the development of antibody drugs and biosimilars, as even a small change in their higher-order structure may dramatically impact their therapeutic activities and produce unpredictable side effects in patients.3 X-ray crystallography is the “gold standard” method for protein higher-order structure analysis, but the challenges of antibody crystallization and the time-consuming data analysis involved make this technique unsuitable for routine structural studies of antibodies. In addition, crystallography cannot be used for the structural characterization of an antibody in solution, under conditions where antibody drugs are active. Intact antibodies are also too large and too complex for NMR.Hydrogen/deuterium exchange (HDX) monitored by mass spectrometry (MS) is a powerful tool for analyzing protein structures in solution.4–7 There are two commonly used analytical strategies: “bottom-up” and “top-down”. “Bottom-up” HDX-MS relies on enzymatic protein digestion followed by HPLC-MS analysis of the resulting peptides. The advantage of this approach is that there is effectively no limit on protein size, but it does have limitations such as significant deuterium label loss (typically 10–50%), limited spatial resolution, and incomplete sequence coverage.8 The “top-down” HDX approach overcomes these problems through the analysis of intact proteins by electron capture dissociation (ECD) or electron transfer dissociation (ETD).9–13 However, its success decreases as protein size increases, and its application has thus been limited to smaller proteins of <30 kDa.9–12 Therefore, a hybrid approach that combines the positive aspects of both bottom-up and top-down would be highly desirable.
In a previous publication, we demonstrated a top-down HDX approach to obtain amino acid-level structural information for antibodies by limiting back-exchange to 2% by the use of subzero temperature HPLC. Using this method, structural information was obtained for the entire sequence of an antibody light chain.13 However, for the antibody heavy chain (∼50 kDa) – the largest protein analyzed by top-down HDX-MS with high resolution to date – the use of this method covered only ca. 50% of the protein, leaving the middle portion uncharacterized. To circumvent the size-limit barrier, we developed a novel “middle-down” approach, in which large proteins such as antibodies are digested into a limited number of specific fragments under HDX quenching conditions, followed by HPLC separation and online gas-phase fragmentation. Compared to the traditional bottom-up HDX-MS approach, where proteins including antibodies are non-specifically digested into hundreds of small peptides (around 10 residues each),14–16 the main novelty of the current middle-down approach is that antibodies are digested prior to disulfide reduction, so the digestion is sterically hindered and leads to the generation of much larger fragments. Because each digestion event leads to the loss of deuteration information for the two amides at the newly generated peptide's N-terminus,9 the smaller number of digestion sites in our middle-down approach can potentially overcome the high back-exchange problem of the bottom-up approach. In addition, the smaller number of fragments makes it possible to separate by subzero temperature HPLC13 and subsequently perform ETD/ECD on each of them in an online fashion. This can not only further suppress deuterium back exchange, but can also provide residue level structural information.13
Experimental
Reagents
Recombinant Herceptin (HER) was purchased from Genscript USA Inc. (Piscataway, NJ). Porcine pepsin, ubiquitin, tris(2-carboxyethyl)phosphine hydrochloride (TCEP), calmodulin, and pepstatin were obtained from Sigma-Aldrich (St. Louis, MO), and deuterium oxide was from Cambridge Isotope Laboratories (Andover, MA). The peptide enfuvirtide was purchased from Thermo Scientific (Bremen, Germany). Peptides PEP1 (VAQVIIPSTYVPGTTNHDIALLR), PEP2 (AVPPNNSNAAEDDLPTVELQG-VVPR), PEP3 (YSSDPTGALTEDSIDDTFLPVPEYINQSVPK) were synthesized in-house. Deglycosylated HER (dHER) was prepared by incubating approximately 2 mg of HER with 40 mU of PNGase F (New England Biolabs, Whitby, ON) in 20 mM sodium phosphate pH 7.4 for 16 h at 37 °C. Complete deglycosylation was confirmed by intact mass measurements of the reduced antibodies before and after deglycosylation (ESI Fig. S5†). HER and dHER stock solutions were both buffer-exchanged into 20 mM sodium phosphate containing 100 mM NaCl (pH 7.4) using centrifugal filters (10K MWCO, Millipore, Billerica, MA).Hydrogen/deuterium exchange
HDX was carried out by mixing HER or dHER (100 μM, pH 7.4) with D2O buffer at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 (v/v). After incubation for a specific amount of time (20 s, 7 min, 1 h), 10 μL aliquots were removed and quickly quenched by reducing the pH to 2.5 with 3 μL of phosphate buffer at pH 2.0. Then 2 μL of pepsin (100 μM) was added and the solution was incubated for 1 min on ice, followed by the addition of 30 μL of 8 M urea solution containing 1 M TCEP and 35 μM pepstatin (pH 2.5). The samples without HDX were prepared similarly but without adding any D2O. The samples were flash-frozen using liquid nitrogen and stored at −80 °C. The fully deuterated peptides were prepared by incubating PEP1, PEP2 and PEP3 (100 μM) in 90% D2O for 24 hours, and were diluted by a factor of 10 into 0.1% formic acid immediately before injection for LC-MS.
:4 (v/v). After incubation for a specific amount of time (20 s, 7 min, 1 h), 10 μL aliquots were removed and quickly quenched by reducing the pH to 2.5 with 3 μL of phosphate buffer at pH 2.0. Then 2 μL of pepsin (100 μM) was added and the solution was incubated for 1 min on ice, followed by the addition of 30 μL of 8 M urea solution containing 1 M TCEP and 35 μM pepstatin (pH 2.5). The samples without HDX were prepared similarly but without adding any D2O. The samples were flash-frozen using liquid nitrogen and stored at −80 °C. The fully deuterated peptides were prepared by incubating PEP1, PEP2 and PEP3 (100 μM) in 90% D2O for 24 hours, and were diluted by a factor of 10 into 0.1% formic acid immediately before injection for LC-MS.
Liquid chromatography
LC-MS at −20 °C was carried out using a subzero setup described previously,13 while LC-MS at 0 °C was carried out by placing the column (C4, 5 μm, 30 × 2 mm, Phenomenex Inc., Torrance, CA) in the ice bath. The back-exchange level during LC-MS at −20 °C was determined to be ∼2%.13 The sample injector (Rheodyne Model 7125, sample loop volume 20 μL) was embedded in an ice bath beside the freezer. A 13 minute binary solvent gradient was used for protein elution, including a 1.5 min desalting time. Solvent A contained 35% methanol with 0.1% formic acid, while solvent B was 100% acetonitrile with 0.1% formic acid. The flow rate was set at 200 μL min−1. The eluent was diverted to waste for the first 2 min to prevent salts from entering the instrument.Both the samples with and without HDX were quickly thawed and kept on ice for 3 min in order to reduce the disulfide bonds in the antibody, then injected onto the column and analyzed by LC-MS. It was found that the H/D back exchange was negligible (<2%) during pepsin digestion and disulfide bond reduction under quench conditions.17,18 The peptides in the sample were separated using a linear gradient containing 20% to 60% solvent B, and the flow rate was decreased to 100 μL min−1 for ETD.
Mass spectrometry
All MS data were acquired on a Thermo Scientific Orbitrap Fusion mass spectrometer equipped with ETD (Thermo Scientific, Bremen, Germany). Basic instrumental parameters for the Orbitrap were set as follows: spray voltage 3500 V (positive), transfer tube temperature 300 °C, vaporizer temperature 275 °C, sheath gas 25, auxiliary gas 10, S-lens RF level 60. The Orbitrap detection was calibrated to within <5 ppm error by using Calmix. Detection of the intact protein fragments in the LC-MS experiments was performed in the Orbitrap mass analyzer over the m/z 600–1500 mass range, and the resolving power was set at 120000 at m/z 200. The AGC (automatic gain control) target was set at 2 × 105, and the maximum injection time was 100 ms. In the ETD experiments, fluoranthene radical anions were introduced into the ion trap over 50 ms with an ETD reagent target value of 3 × 105, and the ETD reaction time was 9 ms. Online ETD experiments were done by selecting one charge state for each peptide/fragment in a single HPLC run. The charge state selected for ETD was 23+ for intact light chain, 13+ for HC-C, 5+ for HC-m, and 24+ for HC-N, with an isolation window of 4 m/z units in the quadrupole. The accumulation time of ETD data was 2.5 min for light chain, 1 min for HC-C, 0.5 min for HC-m, and 2.5 min for HC-N. This corresponds to 50, 20, 10, and 50 scans, and 150, 60, 30, and 150 microscans, respectively. ETD fragment ions were detected in the Orbitrap using a scan range of 200–2500 m/z, with a resolution of 120000.
Data analysis
The raw MS data were processed using Xcalibur software (version 3.0.63, Thermo Scientific). The ETD product ions were identified through ProteinProspector (http://prospector.ucsf.edu) as described previously.9,13 Briefly, one middle-down fragment is analyzed at a time. On the “MS-product” webpage, the amino acid sequence of one protein fragment is pasted into the “Sequence” area, optional modifications to the N-terminal and C-terminal (e.g., “acetyl”) can be selected on the “N-term” and “C-term” pages, respectively. In our case, these areas were left blank as there was no modification. We analyzed c-, z-, b-, and y-type ions for ETD fragment ion identification because these are the major products of peptide/protein ETD, with c- and z-ions being predominant.19 Only c and z ions, however, were used for HDX analysis because they have been reported to be free of H/D scrambling.20,21 The fragment tolerance was set as 10 ppm for this analysis. The mass list generated from the ETD spectrum of a given protein was searched against the theoretical m/z values of the ions selected. The ETD cleavage maps in Fig. 2 were made using ProSight Lite (http://prosightlite.northwestern.edu), but the assignments were confirmed by comparing these to the match results from ProteinProspector and by manual inspection. The centroid m/z values for the unlabeled ETD ions were obtained from ProteinProspector, and the centroid m/z values after HDX were determined using an in-house written excel micro (Microsoft, Redmond, WA) spreadsheet. Only ETD fragment ions which had a signal-to-noise ratio (S/N) > 5 were used for deuteration content determination. The amino acid level deuteration information was obtained using the analytical strategy we described in detail previously,13 and the error was on the order of ±0.1. The global HDX data and ETD data shown in the figures represent experiments were made in triplicate. The error of the global HDX data was determined as ±0.1 Da for peptides including HC-m, and ±0.5 Da for the large fragments including HC-N, HC-C, and the intact light chain.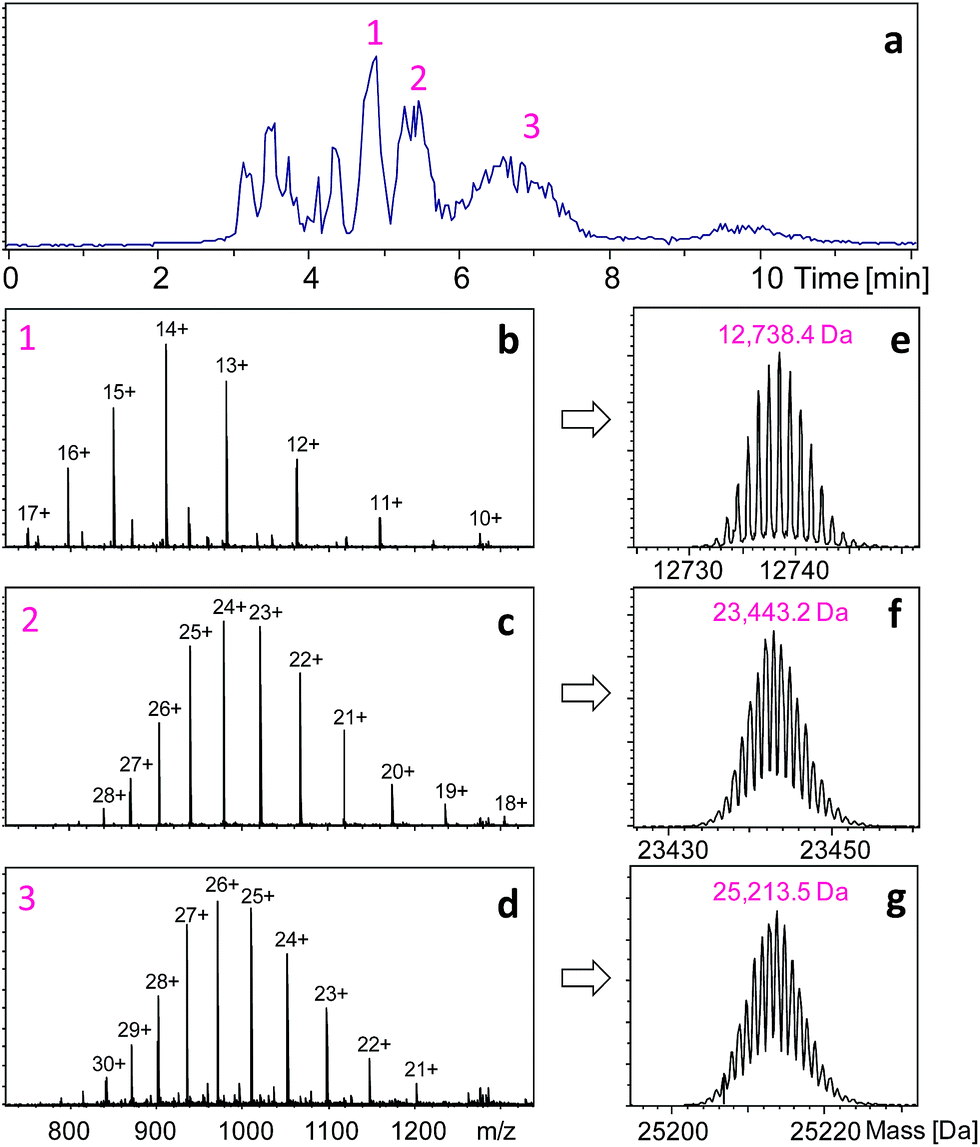 | ||
| Fig. 1 HPLC separation and intact mass measurements of HER fragments obtained after 1 min pepsin digestion and 3 min reduction at pH 2.5. The ESI-MS spectra of the three main HPLC peaks (labeled as 1, 2, and 3) are shown in (b–d), respectively. The corresponding masses after deconvolution are given in (e–g). | ||
 | ||
| Fig. 2 ETD cleavage sites mapped onto the amino acid sequence of representative peptic fragments from HER. | ||
Results and discussion
Restricted pepsin digestion
The key to the proposed middle-down approach is the selection of an appropriate enzyme system that works under HDX quench conditions (pH ∼ 2.5). There are several enzymes (e.g., OmpT22 and IdeS23) that are currently used to generate larger peptides for middle-down proteomics; however, none of them work at low pH. We achieved this goal by using the nonspecific enzyme pepsin to perform specific restricted digestion. Pepsin is a broad range protease with preferential cleavage for hydrophobic residues (e.g., Phe, Tyr, Trp and Leu). This usually leads to the production of tens to hundreds of small peptides from an unstructured protein, in only one minute at pH 2.5 and 0 °C.5–7 Although this characteristic is preferable in peptide-based bottom-up HDX-MS because it maximizes the protein coverage,14–16 it is unsuitable for middle-down proteomics. Pepsin has also been used for preparing F(ab)2 fragments from IgGs, but the digestion needs to be carried out at pH 4.0–4.5 and 37 °C for more than 8 hours24 – conditions that are not compatible with HDX. During the revision process of the current paper, an interesting study published which used pepsin-containing nylon membranes for controlled proteolysis of reduced antibodies, but it was not used for HDX studies.25 Direct ETD/ECD analysis of intact antibodies normally can only provide information for non-disulfide-linked regions.26,27 The digestion condition required by HDX experiments (pH 2.5) can lead to protein denaturation, which may result in non-specific cleavage. However, we hypothesized that specific digestion may be still achievable by using the disulfide bonds present in the target protein to limit the cleavage sites available to pepsin, and by carrying out the digestion for a very short period of time. Therefore, instead of performing digestion after disulfide reduction as is done in the bottom-up approach,15,16 we performed pepsin digestion of an intact IgG1 antibody Herceptin (HER) prior to disulfide reduction at pH 2.5 and 0 °C, with a reaction time of 1 min. Another prerequisite is the addition of a pepsin inhibitor, pepstatin in this case, during the subsequent reduction step to prevent further proteolysis of the resulting fragments. Shown in Fig. 1a is the HPLC separation profile of HER fragments obtained after a 1 min digestion.
The three main HPLC peaks represent three large protein fragments, with molecular weights of 12738.4, 23443.2, and 25213.5 Da, respectively (Fig. 1). The first peak corresponds to a C-terminal fragment from the heavy chain (HC-C, residues 337–449, theoretical mass = 12738.4 Da), and the third peak corresponds to a big N-terminal fragment (HC-N, residues 1–237, theoretical mass = 25213.6 Da). This indicates that the two pepsin digestion sites on HER heavy chains are between L237-L238 and E336-K337 (ESI Fig. S2†). The second mass (23443.2 Da) corresponded to the mass of intact light chain (214 residues, theoretical mass = 23443.3 Da), suggesting that no sites were available for pepsin under non-reduced conditions. In addition to the three large fragments, a few smaller fragments were also observed. These peptides originated from the middle portion of the heavy chain (Fig. S2†), and were identified as T310-E336 (HC-m), L238-F244, L245-M255, I256-D268, V269-W280, and T302-L309. In total, these peptic fragments covered 96.8% of the entire antibody. To see if the production of this restricted digestion pattern was true for antibodies in general, experiments were carried out on another antibody, Bevacizumab. Very similar results were observed – no digestion for the light chain (observed mass: 23451.2 Da), two large fragments from heavy chain (HC-N: 25776.5 Da, HC-C: 12738.5 Da) which were produced by cleavage at L240-L241 and E339-K340 (Fig. S3†). The different mass and residue numbering compared to that of HER is due to the different sequences of the two antibodies. Taken together, these results indicate that the disulfide bonds intrinsic to the structure of the antibody can be used as a tool for controlling the digestion pattern of pepsin at low pH.
Online ETD fragmentation
The assignment of the fragments based on intact mass measurements was also confirmed using online tandem MS. The low number of total fragments allowed the accumulation of high-quality MS/MS data during the HPLC separation. We used online ETD on an Orbitrap mass spectrometer to sequence each of the fragments within one HPLC run (12 min). These fragments were of perfect size for ETD fragmentation (ESI Fig. S4†), which resulted in extensive cleavage of the peptide bonds (Fig. 2). The average amino-acid resolutions obtained in this manner for the four biggest peptic fragments from HER were ∼1.7 aa for light chain, ∼2.3 aa for HC-N, ∼1 aa for HC-m, and ∼1.5 aa for HC-C, with single-residue resolution in many regions. Because proline does not have any amide hydrogens, this residue is usually not considered during the resolution calculation in HDX studies.Middle-down HDX-MS
Next, this middle-down method was combined with HDX for characterizing the higher-order structure of an antibody. HER is a humanized antibody that is highly effective for HER2-overexpressing breast cancer. A number of posttranslational modifications have been reported for this antibody, including 16 disulfide bridges, near 99% truncation of the C-terminal lysine of the heavy chain (−128 Da), methionine oxidation (+16 Da), and a single glycosylation site on residue N300 of the heavy chain (+1444 Da).28 Based on the measured mass of the HC-C fragment and its ETD fragment ions (Fig. 1 and results shown above), the residue K450 of the HER we are using is nearly 100% cleaved off. No methionine oxidation was observed on any of the peptic fragments, or on the intact light chain and heavy chain (Fig. S1†). The single glycosylation site on only the heavy chain was confirmed by intact mass measurements of both the light chain and heavy chain before and after deglycosylation (Fig. S1†), where deglycosylation induced a 1443.7 Da mass decrease on the heavy chain, but had no effect on the light chain.Removal of the glycans from HER is known to lead to a diminished receptor binding as well as immune response.29 The HDX behaviors of both native HER and deglycosylated HER (dHER) were investigated here to decipher the structural effect of glycosylation. HPLC separation was performed at −20 °C to reduce H/D back exchange to a level of 2%.13 The deuterium uptake of most peptic fragments was found to be similar for both HER and dHER, but a significant difference was observed for fragment HC-m (Fig. 3 and ESI Fig. S5†). To obtain structural information at amino acid resolution, these fragments were subjected to online ETD as described above. The H/D scrambling during ETD20 was tested using ubiquitin and was found to be negligible (data not shown). Because fragment HC-m is a peptide 27 residues long, we synthesized three similar peptides (23–31 residues) to determine if the subzero temperature LC-based middle down method could also be used to suppress the back exchange of relatively small peptides. The fully deuterated peptides prepared from 90% D2O were run under the same LC-MS conditions as for the antibody, at both 0 °C and −20 °C. The mass spectra obtained are provided as Fig. S7† and the back-exchange calculations are given as Table S1 in the ESI.† It was found that the back exchange level was reduced from 31.3% at 0 °C to 1.9% at −20 °C for PEP1, 27.3% to 2.1% for PEP2, and 37.2% to 6.8% for PEP3. The dramatic decrease in back exchange indicates that the middle-down method works not only for large protein fragments, but also for smaller peptides. Based on the deuteration levels of corresponding ETD fragment ions, the deuteration status of each residue was calculated.9,13 The amide deuteration results are shown in Fig. 4. No significant differences were observed for HC-N, HC-C, and the light chain (Fig. 4B and S6†), which is consistent with the HDX data at intact fragment level. For HC-m, where a significant difference was observed before and after deglycosylation, we can now see that the differences mainly occurred on two residues, namely L317 and N318 (Fig. 4A).
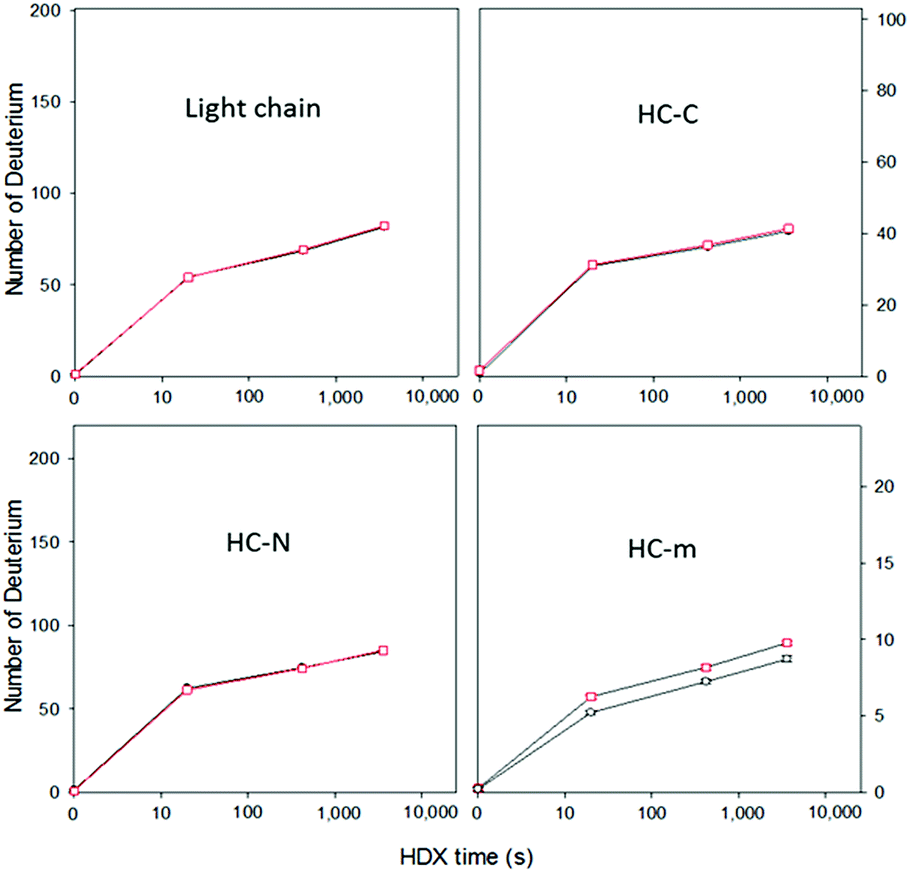 | ||
| Fig. 3 Time course of deuterium uptake for intact peptic fragments of HER (black) and dHER (red). The data represent an average of three replicates. The error bars for HC-m are shown. The error bars for the light chain, HC-N, and HC-C (<0.2 Da) are smaller than the symbols and are not shown. | ||
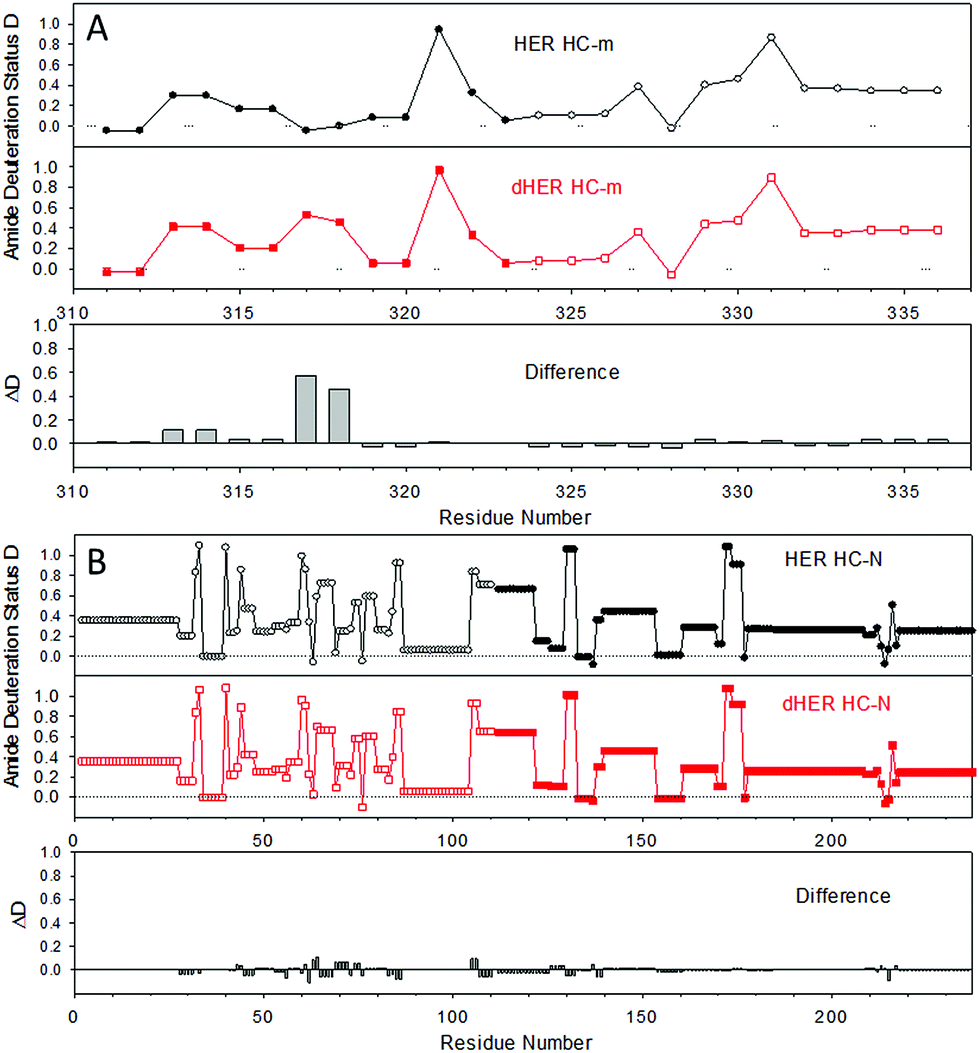 | ||
| Fig. 4 Amide deuteration levels of the middle-down fragments after HDX for 1 h, as obtained from c ions (open symbol) and z ions (filled symbol). (A) HC-m; (B) HC-N. The data represent an average of triplicate measurements, and the error on the deuterium incorporation of each amide was within ±0.1. | ||
To better visualize the locations of these residues, the results were mapped onto the crystal structure of an IgG1 Fc (fragment crystallizable) region (PDB entry 3D6G, Fig. 5). These two residues were found to form part of an α-helix at the CH2–CH3 (constant region of heavy chain) interface. Although Borrok and coworkers proposed (based on X-ray data for Fc) that deglycosylation induces opening of the two CH2 domains (pdb entry 3DNK),30 this seems unlikely for the whole antibody because the top portion of the two CH2 domains are held together by two disulfide bonds in the hinge region. Based on our in-solution HDX data and the fact that the glycans in native HER are too far away to provide much shielding of the two affected residues, we hypothesize that removal of the glycans may have increased the flexibility of the “joint” region. The resulting motion may destabilize the interaction between the connecting loop and the helix containing L317 and N318 (Fig. 5), possibly through a simplified mechanism as shown in Fig. 6. In other words, part of the role of N300 glycan in HER may be to limit the movement of the Fc and to restrict the movement of the CH2–CH3 interface. This mechanism is in line with earlier fluorescence data where IgG displayed an increase in the peak width of the energy transfer efficiency histogram upon deglycosylation, indicating a non-uniform distance between the two “joint” regions.29 We note that the CH2–CH3 interface is the binding site for the FcRn receptor, and the helix containing L317 and N318 is directly involved in the binding (pdb entry 1I1C),31 so the increased flexibility of this region may be partially responsible for the decreased binding affinity between FcRn and the antibody. However, for other antibody-binding receptors, such as FcγRI,32 the antibody glycans are reported to be directly involved in the binding of the receptor, indicating that the glycans may play versatile roles in modulating the antibody's structure and function.33
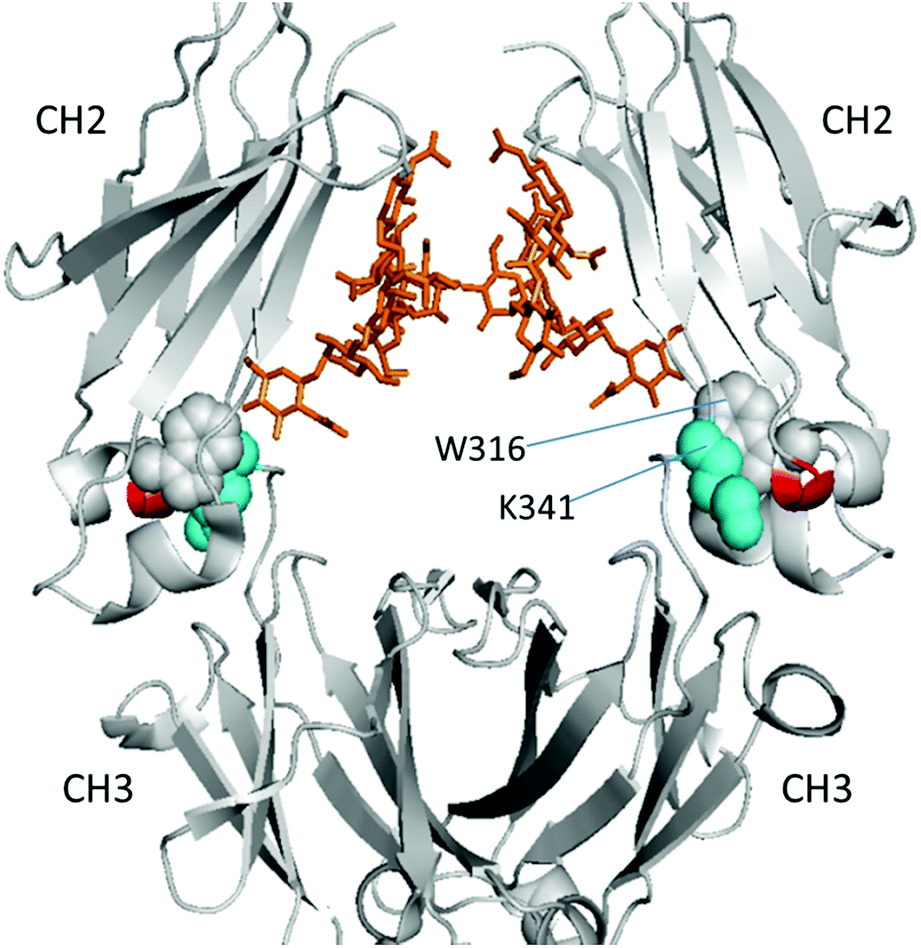 | ||
| Fig. 5 Location of affected residues (red) on the HER heavy chain (gray) (PDB entry 3D6G). Cyan spheres denote residue K341 from the loop and gray spheres denote W316 next to the two affected residues. Golden stick-diagrams show the two glycan chains attached to residue N300. | ||
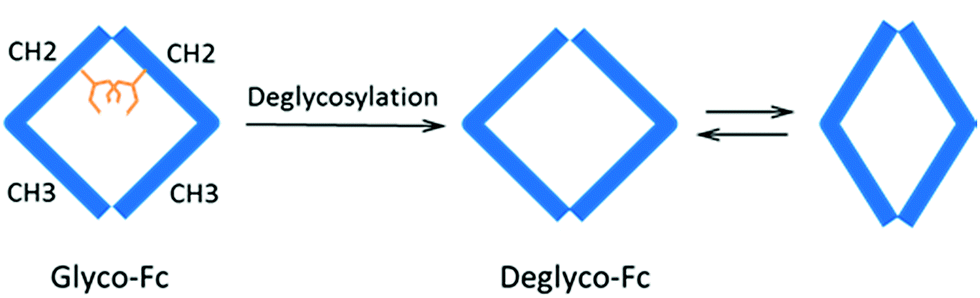 | ||
| Fig. 6 Proposed mechanism of deglycosylation effect on antibody. Removal of glycans may have enhanced the dynamic nature of Fc and the movement of CH2–CH3 interface through breathing-like motions. CH2 and CH3 represent the two IgG domains of the heavy chain, and the golden sticks denote glycans. | ||
Conclusion
Most structural studies on antibodies have used Fc or Fab fragments as surrogates for the whole antibody due to its size as well as its complex and dynamic nature. The middle-down HDX-MS approach reported in the current work provides a new means for whole antibody higher-order structural characterization with HDX information for 100% of the light chain and 95.3% of the heavy chain. When combined with online ETD fragmentation, deuteration information for individual amides can be obtained. Compared to the most widely used bottom-up HDX-MS,34–36 this hybrid middle-down approach inherits its advantage of basically no protein size limit through enabling pepsin to perform digestion in a fast but also specific manner under HDX quench conditions, which results in the production of only a number of fragments from 150 kDa antibodies. On the other hand, compared to top-down HDX-MS,9–13,17 the middle-down approach inherits its advantages of residue level spatial resolution and minimal back exchange through the use of online ETD fragmentation and HPLC separation at subzero temperature. Its spatial resolution may be further improved by performing online ETD on a newer-generation instrument where a faster MS2 scan rate can be achieved. As both the bottom-up and top-down HDX-MS continue to mature, we anticipate the current middle-down approach to be used as a competitive alternative method for antibody structural characterization such as antibody–receptor interaction, antibody paratope mapping, and antigen epitope mapping.The limited specific pepsin digestion due to the intrinsic disulfide bonds in the target antibody is the key to the generation of large protein segments, which are of a perfect size for further online dissociation using fragmentation mechanisms such as ETD. Since disulfide bonds occur frequently in human proteins, this method should not be limited to antibodies; it may also be applicable to other large proteins. We expect that this new middle-down HDX-MS technology will be widely used in the future for the accurate and comprehensive structural characterization of antibodies and for deciphering the structural dynamics of large proteins.
Acknowledgements
The authors would like to thank the Natural Sciences and Engineering Research Council of Canada (NSERC) for financial support. We are also grateful to Genome Canada and Genome BC for providing Science and Technology Innovation Centre funding and support for the University of Victoria-Genome BC Proteomics Centre, and we thank Carol E. Parker for helpful discussions and careful review of this manuscript.Notes and references
- K. Henzler-Wildman and D. Kern, Nature, 2007, 450, 964–972 CrossRef CAS PubMed.
- J. B. Munro, J. Gorman, X. C. Ma, Z. Zhou, J. Arthos, D. R. Burton, W. C. Koff, J. R. Courter, A. B. Smith, P. D. Kwong, S. C. Blanchard and W. Mothes, Science, 2014, 346, 759–763 CrossRef CAS PubMed.
- S. A. Berkowitz, J. R. Engen, J. R. Mazzeo and G. B. Jones, Nat. Rev. Drug Discovery, 2012, 11, 527–540 CrossRef CAS PubMed.
- L. Konermann, S. Vahidi and M. A. Sowole, Anal. Chem., 2014, 86, 213–232 CrossRef CAS PubMed.
- L. Konermann, J. Pan and Y. Liu, Chem. Soc. Rev., 2011, 40, 1224–1234 RSC.
- T. E. Wales and J. R. Engen, Mass Spectrom. Rev., 2006, 25, 158–170 CrossRef CAS PubMed.
- I. A. Kaltashov and S. J. Eyles, Mass Spectrom. Rev., 2002, 21, 37–71 CrossRef CAS PubMed.
- I. A. Kaltashov, C. E. Bobst and R. R. Abzalimov, Anal. Chem., 2009, 81, 7892–7899 CrossRef CAS PubMed.
- J. Pan, J. Han, C. H. Borchers and L. Konermann, J. Am. Chem. Soc., 2009, 131, 12801–12808 CrossRef CAS PubMed.
- S. Amon, M. B. Trelle, O. N. Jensen and T. J. D. Jørgensen, Anal. Chem., 2012, 84, 4467–4473 CrossRef CAS PubMed.
- G. B. Wang, R. R. Abzalimov, C. E. Bobst and I. A. Kaltashov, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20087–20092 CrossRef CAS PubMed.
- G. Wang and I. A. Kaltashov, Anal. Chem., 2014, 86, 7293 CrossRef CAS PubMed.
- J. Pan, S. Zhang, C. E. Parker and C. H. Borchers, J. Am. Chem. Soc., 2014, 136, 13065–13071 CrossRef CAS PubMed.
- Q. Zhang, L. N. Willison, P. Tripathi, S. K. Sathe, K. H. Roux, M. R. Emmett, G. T. Blakney, H. M. Zhang and A. G. Marshall, Anal. Chem., 2011, 83, 7129–7136 CrossRef CAS PubMed.
- D. Houde, Y. Peng, S. A. Berkowitz and J. R. Engen, Mol. Cell. Proteomics, 2010, 9, 1716–1728 CAS.
- P. F. Jensen, V. Larraillet, T. Schlothauer, H. Kettenberger, M. Hilger and K. D. Rand, Mol. Cell. Proteomics, 2015, 14, 148–161 CAS.
- J. Pan and C. H. Borchers, Proteomics, 2014, 14, 1249–1258 CrossRef CAS PubMed.
- X. Zhang, E. Y. T. Chien, M. J. Chalmers, B. D. Pascal, J. Gatchalian, R. C. Stevens and P. R. Griffin, Anal. Chem., 2010, 82, 1100–1108 CrossRef CAS PubMed.
- J. E. P. Syka, J. J. Coon, M. J. Schroeder, J. Shabanowitz and D. F. Hunt, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9528–9533 CrossRef CAS PubMed.
- M. Zehl, K. D. Rand, O. N. Jensen and T. J. D. Jørgensen, J. Am. Chem. Soc., 2008, 130, 17453–17459 CrossRef CAS PubMed.
- J. Pan, J. Han, C. H. Borchers and L. Konermann, J. Am. Chem. Soc., 2008, 130, 11574–11575 CrossRef CAS PubMed.
- C. Wu, J. C. Tran, L. Zamdborg, K. R. Durbin, M. X. Li, D. R. Ahlf, B. P. Early, P. M. Thomas, J. V. Sweedler and N. L. Kelleher, Nat. Methods, 2012, 9, 822–824 CrossRef CAS PubMed.
- L. Fornelli, D. Ayoub, K. Aizikov, A. Beck and Y. O. Tsybin, Anal. Chem., 2014, 86, 3005–3012 CrossRef CAS PubMed.
- H. Jaquet and J. J. Cebra, Biochem., 1965, 4, 954–963 CrossRef CAS.
- Y. Pang, W. H. Wang, G. E. Reid, D. F. Hunt and M. L. Bruening, Anal. Chem., 2015, 87, 10942–10949 CrossRef CAS PubMed.
- Y. O. Tsybin, L. Fornelli, C. Stoermer, M. Luebeck, J. Parra, S. Nallet, F. M. Wurm and R. Hartmer, Anal. Chem., 2011, 83, 8919–8927 CrossRef CAS PubMed.
- Y. Mao, S. G. Valeja, J. C. Rouse, C. L. Hendrickson and A. G. Marshall, Anal. Chem., 2013, 85, 4239–4246 CrossRef CAS PubMed.
- A. Beck, S. Sanglier-Cianferani and A. van Dorsselaer, Anal. Chem., 2012, 84, 4637–4646 CrossRef CAS PubMed.
- M. T. Kelliher, R. D. Jacks, M. S. Piraino and C. A. Southern, Mol. Immunol., 2014, 60, 103–108 CrossRef CAS PubMed.
- M. J. Borrok, S. T. Jung, T. H. Kang, A. F. Monzingo and G. Georgiou, ACS Chem. Biol., 2012, 7, 1596–1602 CrossRef CAS PubMed.
- W. L. Martin, A. P. West, L. Gan and P. J. Bjorkman, Mol. Cell, 2001, 7, 867–877 CrossRef CAS PubMed.
- J. Lu, J. Chu, Z. Zou, N. B. Hamacher, M. W. Rixon and P. D. Sun, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 833–838 CrossRef CAS PubMed.
- M. Dalziel, M. Crispin, C. N. Scanlan, N. Zitzmann and R. A. Dwek, Science, 2014, 343, 1235681 CrossRef PubMed.
- K. D. Rand, M. Zehl, O. N. Jensen and T. J. D. Jørgensen, Anal. Chem., 2009, 81, 5577–5584 CrossRef CAS PubMed.
- M. Rey, M. L. Yang, K. M. Burns, Y. P. Yu, S. P. Lees-Miller and D. C. Schriemer, Mol. Cell. Proteomics, 2013, 12, 464–472 CAS.
- Z. Y. Kan, B. T. Walters, L. Mayne and S. W. Englander, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16438–16443 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 to S5. See DOI: 10.1039/c5sc03420e |
| This journal is © The Royal Society of Chemistry 2016 |