
Chitosan-folate coated mesoporous silica nanoparticles as a smart and pH-sensitive system for curcumin delivery†‡
Mohammad Porgham Daryasaria,
Mohammad Reza Akhgara,
Fatemeh Mamashlib,
Bahareh Bigdelib and
Mehdi Khoobi*cd
aDepartment of Chemistry, Kerman Branch, Islamic Azad University, Kerman, Iran
bInstitute of Biochemistry and Biophysics, University of Tehran, Tehran, Iran
cPharmaceutical Sciences Research Center, Tehran University of Medical Sciences, Tehran, Iran
dDepartment of Pharmaceutical Biomaterials, Medical Biomaterials Research Center, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran. E-mail: m-khoobi@tums.ac.ir; Mehdi.khoobi@gmail.com; Fax: +98-21-66461178; Tel: +98-21-64121113
First published on 7th November 2016
Abstract
In this work, normal and large pore size mesoporous silica nanoparticles (NMSNs and LMSNs) were prepared by co-condensation method and coated with (3-aminopropyl)triethoxysilane (APTES) to prepare amine functionalized MSNs (MSN-NH2). They were then conjugated with succinic anhydride (SA) to obtain carboxylic acid functionalized MSNs (MSN-COOH). Curcumin (CUR) as a hydrophobic drug was loaded into the synthesized MSNs with two different pore sizes to compare the loading capacity and efficiency. The results showed that LMSNs had 2-fold larger loading efficiency and capacity for CUR than NMSNs. Chitosan (CS), as a pH-sensitive polymer, was also conjugated to folic acid (FA) as an active targeting agent and then coated on the surface of carboxylic acid enriched MSNs via electrostatic interaction. The MSNs were fully characterized by scanning electron microscopy (SEM), transmission electron microscopy (TEM), zeta potential analysis, dynamic light scattering (DLS), nitrogen adsorption/desorption analysis, thermo gravimetric analysis (TGA), XRD analysis, NMR and UV-visible spectroscopies. The mechanism of CUR release from CS-FA coated LMSNs was pH-sensitive and in vitro release modeling revealed that CUR is released via Korsmeyer–Peppas mechanism. No significant toxicity was observed for CUR free MSNs, while the CUR loaded MSNs inhibited proliferation of HeLa and NIH-3T3 cell lines, showing more cytotoxic effect on cancerous HeLa cells. Moreover, the selective cellular uptake of CUR loaded LMSNs-COOH@CS-FA by folate receptor-positive HeLa cells was assessed and confirmed. Hemocompatibility and protein corona of the target carrier were also studied to show negligible hemolytic activity and suitable protein–target LMSNs interactions.
Introduction
Cancer is the second leading cause of death in the world and has a great effect on global health. The global incidence of cancer, and the number of new cancer cases, is continuing to rise.1 Among the different approaches for cancer treatment, chemotherapeutics agents are still widely used as a practical strategy for the treatment of cancers. Common chemotherapeutic agents are often distinguished by short circulation time, nonspecific cell biodistribution and low delivery efficiency at the targeted tumor tissue which mainly arise from the hydrophobicity and degradability of the administered drug.2,3Nanotechnology has opened a new way to selectively engineer a myriad of different materials and remove all obstacles in front of the conventional formulations. The possibility of engineering nanomaterials with controllable size and morphology as well as programing their surface chemistry with the aid of a multitude of biological and medical functions provide a great opportunity for designed nanoplatforms to reach targeted tissue and selectively destroy cancer cells in the body.3–5
However, clinical application of conventional nanocarriers has been still limited due to the premature drug release before reaching to the target, uncontrollable rate of drug release and low local concentration, and inefficient cellular uptake.6,7
Among various DDSs, mesoporous silica nanoparticles (MSNs) stand out to be a promising candidate due to some of their conspicuous virtues, such as uniform particle sizes (80–500 nm), large surface areas (>1000 m2 g−1), high pore volumes (0.5–2.5 cm3 g−1), tunable pore diameters (1.3–30 nm), and capability of being modified with various functional groups in both exterior and interior surfaces independently. Drug encapsulation inside the pores followed by blocking the pore entrances with stimuli responsive agents can endow MSNs with the ability to precise control of release without leaching prior to reaching the targeted cells.
Various polymer coatings were exploited to modify the surface of MSNs and create a more advanced carrier with combined benefits of both components.8 Compared to the all applied stimulus-sensitive polymer, the pH sensitive polymers as more general and ideal coating agents have been regarded to control selective release of drug at tumor tissue. The pH of tumor environment is lower than normal tissue due to the high rate of glycolysis in cancer cells. This media can act as perfect trigger for selective release of anticancer drugs.9,10
Chitosan (CS) is a non-toxic biodegradable polycation with a high number of primary amino groups. These amine functional groups render cationic character to the polymer and are responsible for a range of significant features including in situ gelation, mucoadhesion, efflux pump inhibition, high cellular permeability as well as bioavailability for oral administration of drugs which make CS as an outstanding candidate in DDSs. CS can be swelled in cancerous tissues due to their acidic media and this property endows the polymer with the ability of discrimination between normal and cancerous cells for controlled drug release.11–16
On the other hand, active targeting can also efficiently increase nanoparticles (NPs) internalization through receptor-mediated endocytosis and improve the efficacy of their payloads.17–20 This ligand mediated targeting employ the affinity of the ligands on the surface of NPs to increase cellular uptake by the targeted cells receptors that overexpressed in the diseased tissues or cells.21–26 Folic acid (FA) as an inexpensive, water soluble and stable vitamin without adverse effect on normal cells and low immunogenic response has attracted a great deal of attention for active targeting.27 The over expression of the FA receptor in epithelial malignancies, such as colorectal, ovarian, and breast cancer cells in comparison with most normal cells make FA conjugates as facile and infallible strategy to promote the receptor-mediated endocytosis of nanoparticles.28–34 The vesicular trafficking of FA conjugates makes them able to move through many organelles and release efficiently their cargo into the cell cytoplasm.35
Curcumin (CUR) as an inexpensive drug and a natural polyphenol compound, is extracted from the herb Curcuma longa (turmeric), with a diverse range of therapeutic properties. The anti-proliferative property of CUR as well as its low toxicity, high dose tolerance, and safe profile make it as a suitable candidate for cancer therapy.36,37 However, the pharmaceutical preparations of CUR are restricted due to its low water solubility and poor bioavailability; the main challenges that can be improved through a proper DDS.38,39
So far, a few researches have been performed on the use of modified MSNs for CUR delivery. It has been demonstrated that the amino functionalized MCM-41 as hydrophilic and positively charged particles can comparatively control release of CUR and enhance endocytosis in cancer cells compared to the naked MCM-41.40 Kotcherlakota et al. prepared amino functionalized MSNs with different morphology, particle and pore sizes (KIT-6, MSU-2 and MCM-41) for CUR delivery. It was revealed that the encapsulation efficiency was enhanced by amino functionalized MCM-41 in comparison to other MSNs.41 In a recent study, Kim et al. have developed a CUR delivery system based on MCM-41 with coating layer of tannic acid–Fe(III) complex (MSN-TA), as a pH- and glutathione-responsive DDS. The results showed that CUR loaded MCM-41 induced higher cytotoxicity compared to pure CUR in the presence of MRC5 cells.42
In the light of the above mentioned results, in this work, we reported a facile strategy to prepare a pH-sensitive MSNs coated with FA conjugated CS as double responsive targeting DDS. MCM-41 in two different pore sizes was prepared and their CUR loading efficiency (CLE) and CUR loading capacity (CLC) were evaluated. The cytotoxic activity and cellular uptake of MSNs and CUR loaded MSNs were analyzed and compared with CUR in the presence of the folate receptor-positive HeLa cells as cancerous and folate receptor-negative NIH-3T3 cells, as normal cells. Hemolysis assay was also carried out to evaluate biosafety of MSNs on erythrocytes.
Experimental
Materials
Cetyltrimethylammonium bromide (CTAB), tetraethyl orthosilicate (TEOS), (3-aminopropyl)triethoxysilane (APTES), succinic anhydride (SA), N-hydroxysuccinimide (NHS), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), ammonium nitrate (NH4NO3), folic acid (FA), dimethyl sulfoxide (DMSO) and all other solvents and reagents were purchased from Merck (Darmstadt, Germany) and used without further purification. Curcumin (CUR) was from Merck (Darmstadt, Germany). The HeLa and NIH-3T3 cell lines were purchased from National Cell Bank of Iran (Pasteur Institute, Iran). The cells were incubated in RPMI 1640 (Gibco) media supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Gibco) and 200 μg mL−1 streptomycin (Jaberebn-Hayan, Iran) and 500 μg mL−1 penicillin (Sigma, USA) at 37 °C in humidified atmosphere and 5% CO2. Millipore Milli-Q® (Burlington, MA, USA) high purified water was used to make aqueous solutions.Synthesis of functionalized MSN nanoparticles
CTAB was removed using ionic exchange method.48 In summary, 1 g of prepared MSNs were dispersed in 100 mL of ethanol (95%) containing 1 g of NH4NO3, and refluxed for 6 h at 80 °C. The removed template MSNs were recovered by filtration; washed with ethanol and dried overnight under vacuum at 45 °C. To improve the efficiency of the process, the above procedure was repeated for two times.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm to separate the yellow precipitate from solution which was washed with deionized water and excess ethanol before freeze drying.
000 rpm to separate the yellow precipitate from solution which was washed with deionized water and excess ethanol before freeze drying.Ninhydrin assay
Determination of primary amines content on the amino modified samples was carried out by ninhydrin colorimetric assay.50,51 Briefly, 1 mL color reagent (10 g of Na2HPO4·12H2O, 6 g of KH2PO4, 0.5 g of ninhydrin in 100 mL of distilled water) was added into a dispersed solution of 3 mg of sample in 2 mL of distilled water, and the mixture was heated in the boiling water bath for 16 min and then cooled in the water bath at 20 °C for 20 min. Afterward 5 mL of diluting solution (2 g of KIO3 in 600 mL distilled water and 400 mL of ethanol 96%) was added to the mixture. The color of the solution turns to purple blue due to the reaction of the amino groups with ninhydrin. The yield of the reaction was measured by UV-visible spectrophotometer at 570 nm. The reaction of APTES with ninhydrin was applied for preparation of the calibration curve.Loading of CUR in MSNs
CUR was loaded into the MSN-COOH according to the reported method with some modifications.52 30 mg of dry MSN-COOH was added in 5 mL of CUR solution in dichloromethane (1 mg mL−1), and the mixture was sonicated for 5 min by ultrasonic bath to obtain a well dispersed suspension. After stirring for 48 h under light sealed conditions, the CUR loaded MSNs were collected by centrifugation and washed with deionized water. To calculate the CUR loading efficiency (CLE) and loading capacity (CLC), the residual CUR content in the supernatant was determined through plotting calibration curve of CUR standard solutions by UV-visible measurement at 420 nm. The CUR loading efficiency and capacity were calculated as follows:| CLE (%) = mass of loaded CUR in MSN/total mass of CUR added initially × 100 |
| CLC (%) = mass of loaded CUR in MSN/mass of MSN × 100. |
In vitro CUR release profile
The release profile of CUR was investigated as follow: Tween-80 0.1% (w/v) was added to the phosphate buffer saline (PBS) to facilitate the release and dissolution of CUR in aqueous phase and maintain a sink condition. 2 mg of the sample was immersed into the 3 mL PBS (pH 7.4 with 0.1% Tween-80) and (pH 5.5 with 0.1% Tween 80) tubes. The release assay was performed at 37 °C in dark using a shaking water bath at 100 rpm. Sampling was done at predetermined time points. In each time point, all release medium was taken out by centrifugation at 10000 rpm for 15 minutes and replaced with equivalent volume of fresh medium. The cumulative release profile of CUR was evaluated by UV-visible spectrophotometry (at 420 nm). The calibration curve of the released CUR concentration was plotted by given concentration of CUR standard solutions in the same condition.52,53 In vitro release kinetics modeling was analyzed by KinetDS 3 rev 2010 software.
Cellular uptake analysis
Flow cytometric analysis was investigated for cellular uptake of CUR loaded MSNs. Owing to the intrinsic green fluorescence of CUR, cellular uptake of CUR and CUR-loaded MSNs was studied by flow cytometry. NIH-3T3 and HeLa cells were grown in 6-well plates (40000 cells per well) up to 80% confluency. Cells were then treated with freshly prepared serum-free medium (pH 6) containing 100 μg mL−1 of CUR (in the form of free CUR or CUR loaded MSN-COOH, LMSN-COOH@CS or LMSN-COOH@CS-FA). After 30 min incubation, the cells were trypsinized, washed with PBS three times, and resuspended in 1 mL PBS.54 CyFlow Space (Parpec, Germany) flow cytometer was used to examine cellular uptake and FloMax software was used to analyze the data.
MTT cell viability assay
Cytotoxicity of the drug loaded MSNs was determined by MTT (3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay against NIH-3T3 fibroblasts and HeLa cells as normal and tumor cells, respectively. NIH-3T3 and HeLa cells were separately seeded in 96-well plates at the density of 7 × 103 cells per well and culture medium of RPMI (200 μL). After 24 h incubation to allow cells to attach, the media were replaced with fresh media containing LMSN-COOH, LMSN-COOH@CS-FA, LMSN-COOH-CUR@CS-FA (prepared in deionized water), and CUR (prepared in DMSO with safety concentration of 0.1%) at a concentration range of 0.1–100 μg mL−1 and in a total volume of 200 μL. The plates were then further incubated at 37 °C for 72 h. 100 μL of PBS containing 5 mg mL−1 MTT (Sigma, USA) was then added to each well. After cells incubation for an additional 4 h, 100 μL of DMSO was added to each well. The absorbance of each well was read on Bio Tek microplate reader (USA) at a test wavelength of 570 nm and a reference wavelength of 630 nm. Cell viability was calculated based on the following formula:55| Number of viable cells = (Abs of sample × 100)/(Abs of control). |
Hemolysis assay
For hemolysis assay, the human red blood cells (HRBCs) were obtained according to the reported procedure.37,56 Briefly, the fresh human blood was stabilized and treated with EDTA to remove the plasma as supernatant by centrifugation at 2000 rpm for 10 min and refined by successive rinsing with PBS buffer (pH 7.4). The suspension of HRBCs was diluted 10 times with PBS buffer (pH 7.4), and then 200 μL of HRBCs suspension was added to 800 μL of each sample with different concentration (1.95–1000 μg mL−1). In the case of positive control, 200 μL of HRBCs suspension was added to 800 μL Triton X100 (2% v/v), and for negative control 200 μL of HRBCs suspension was added to 800 μL of PBS buffer (pH 7.4). Afterwards, all of the samples were incubated for 2 h at room temperature by moderate shaking. Finally, the samples were centrifuged at 10000 rpm for 2 min, and the absorbance of supernatant (hemoglobin) was measured by UV-visible spectrophotometer at 541 nm. The hemolytic activity percentages of the different samples were calculated as follows:| Hemolysis% = (Abssample − Absctrl−/Absctrl+ − Absctrl−) × 100. |
Preparation of protein corona
The protein coated MSNs were prepared by incubating the MSNs with plasma proteins at protein concentrations of 10% (simulation of an in vitro milieu) and 100% (simulation of an in vivo milieu) for 1 h under slow stirring at 37 °C (human body temperature) to allow proteins to condense onto the surface of MSNs. Plasma proteins coated MSNs (at concentration of 10%) were prepared by adding 100 μL of MSNs (1 mg mL−1) to stock solution of 100 μL plasma protein in 800 μL PBS (pH = 7.4), and the above sample at the concentration of 100% was prepared by adding 100 μL of MSNs (1 mg mL−1) to stock solution of 900 μL plasma protein to confirm complete surface coverage. After incubation, samples were centrifuged for 3 times at the following conditions: (1) at 11000 rpm for 30 min where the supernatant was removed and replaced with 500 μL PBS, (2) and (3) at the same condition of the first step while the samples were centrifuged for 20 min. Washing steps were performed to remove the loosely attached proteins from the surface of the MSNs and leave only the strongly bound “hard corona” proteins attached to the MSNs.57–59 Separation of protein samples was done by SDS-PAGE to investigate hard corona protein. For this purpose, 50 μL of each sample was mixed with 15 μL of sample buffer [Tris-base, pH 6.8, SDS 10% (w/v), bromophenol blue (10% w/v), glycerol (20% w/v), 2-mercaptoethanol (10% w/v)] and immersed in water bath at 100 °C for 15 min. Equal volumes of the samples were loaded on 12% acrylamide SDS-PAGE, run for 4 h at 120 V, 20–30 A. Silver nitrate staining method was used to demonstrate proteins in gel. Gel densitometry was analyzed by Gel Analyzer 2010 software.
Characterization
In order to verify the successful functionalization of MSNs, Fourier transform infrared spectroscopy (FTIR) were taken by FT-IR Magna 550, Nicolet using the KBr plates. NMR experiment was recorded on Avance III ultrasheild spectrometer manufactured by Bruker at a field strength of 11.7 tesla (500 MHz) and spectroscopy data are collected using TopSpin software. Powder XRD patterns were collected on STOE Theta–theta Powder Diffraction System, radiation: 1.54060 Cu, generator: 40 kV, 40 mA. Surface area analyzer (Quantachrome NOVA Automated Gas Sorption System, 2000e) was used to calculate nitrogen adsorption isotherm, specific surface area (by BET method) and pore size distribution curve (by BJH method). Thermal decomposition experiments were carried out by thermogravimetric analysis (TGA) on Thermogravimetric analyzer METTLER TOLEDO from room temperature to 800 °C under nitrogen flow. UV-visible spectra measurements were acquired using UV-visible Jasco-530 spectrophotometer in the range of 220–800 nm. Hydrodynamic size distribution and zeta potential were determined by using a ZEN3600 Zetasizer (Malvern Instruments, UK). The morphology of the samples was investigated by transmission electron microscope (HRTEM, Philips CM30, 300 kV, Netherlands) and field emission scanning electron microscope (FE-SEM, Tescan/Mira, Czech Republic).Results and discussion
This study mainly focused on the preparation of active and passive targeted MSNs, and investigation of the effect of pore size on the loading efficiency and surface coating on the mechanism of the drug release, and biocompatibility. Scheme 1 shows the sequence steps for the preparation of the target MSNs and drug loading. MSNs were prepared and functionalized with APTES followed by SA to obtain carboxylic acid functionalized MSN-COOH. CUR was then loaded into the pores of MSNs after template removing using ionic exchange method. CUR loading efficiency and capacity were assessed for both LMSNs and NMSNs. Since the best results were achieved for LMSNs, the next modification was done for LMSNs. CS was conjugated with FA and then coated on the surface of LMSNs via electrostatic interaction to provide an active and passive targeted CUR delivery system. All samples were fully characterized and evaluated in the viewpoint of their cytotoxicity, blood compatibility, and plasma proteins interaction as well as CUR release at different pH.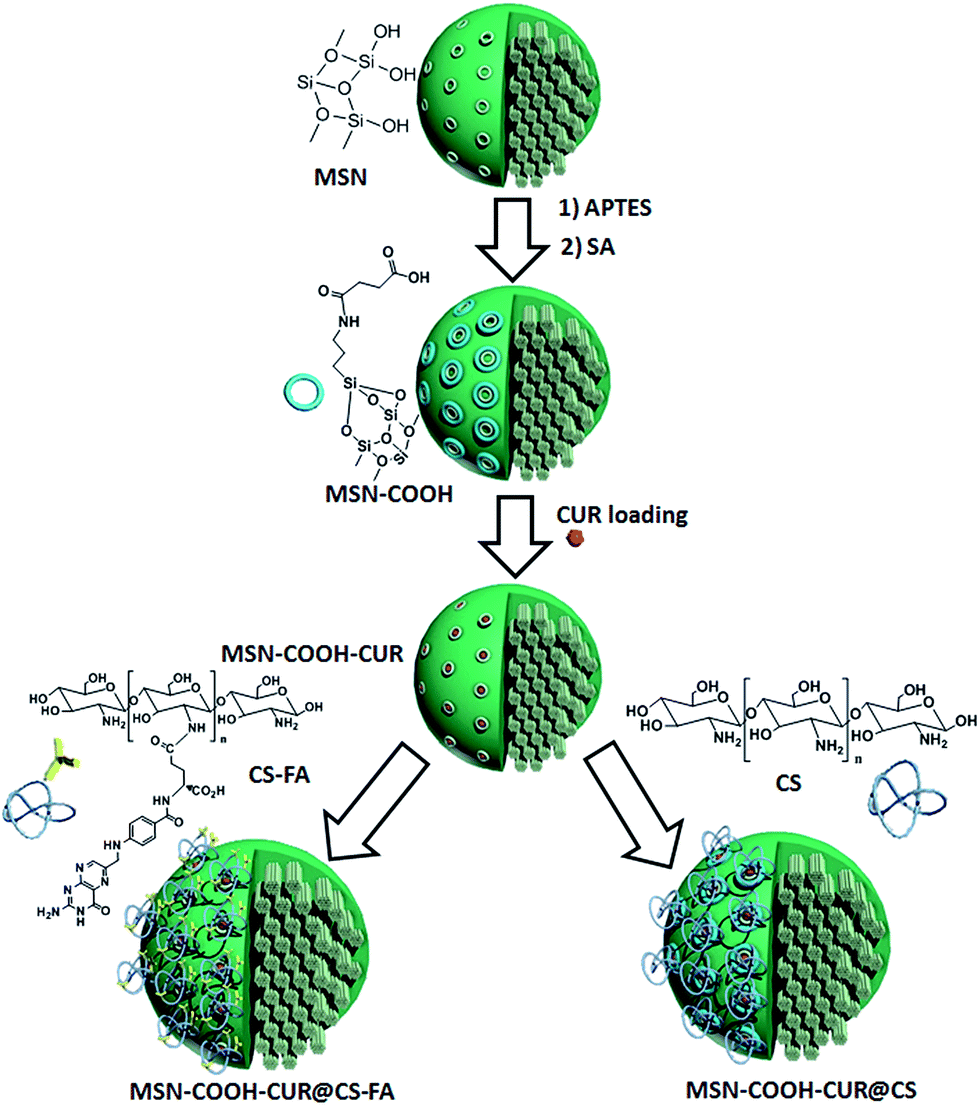 | ||
| Scheme 1 MSNs functionalization and CUR loading. | ||
Characterization of MSNs
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations (νCO) of the new carboxyl acid groups. Also, the intensity of the band at around 1640 cm−1 was obviously increased which may be assigned to the stretching vibrations of amide bonds (Fig. S2, see ESI‡). The FTIR spectrum of CUR exhibited a sharp peak at 3490 cm−1 and a broad peak at about 3400 cm−1 relating to the OH groups of CUR. The bands appeared at around 1629 and 1600 cm−1 are attributed to the stretching vibration of CO bonds and CC aromatic rings. All other bands were in agreement with the FT-IR spectrum of CUR. The FT-IR spectrum of the CUR loaded MSNs showed the combined characteristic peaks of CUR and MSNs (Fig. 1).
O stretching vibrations (νCO) of the new carboxyl acid groups. Also, the intensity of the band at around 1640 cm−1 was obviously increased which may be assigned to the stretching vibrations of amide bonds (Fig. S2, see ESI‡). The FTIR spectrum of CUR exhibited a sharp peak at 3490 cm−1 and a broad peak at about 3400 cm−1 relating to the OH groups of CUR. The bands appeared at around 1629 and 1600 cm−1 are attributed to the stretching vibration of CO bonds and CC aromatic rings. All other bands were in agreement with the FT-IR spectrum of CUR. The FT-IR spectrum of the CUR loaded MSNs showed the combined characteristic peaks of CUR and MSNs (Fig. 1).
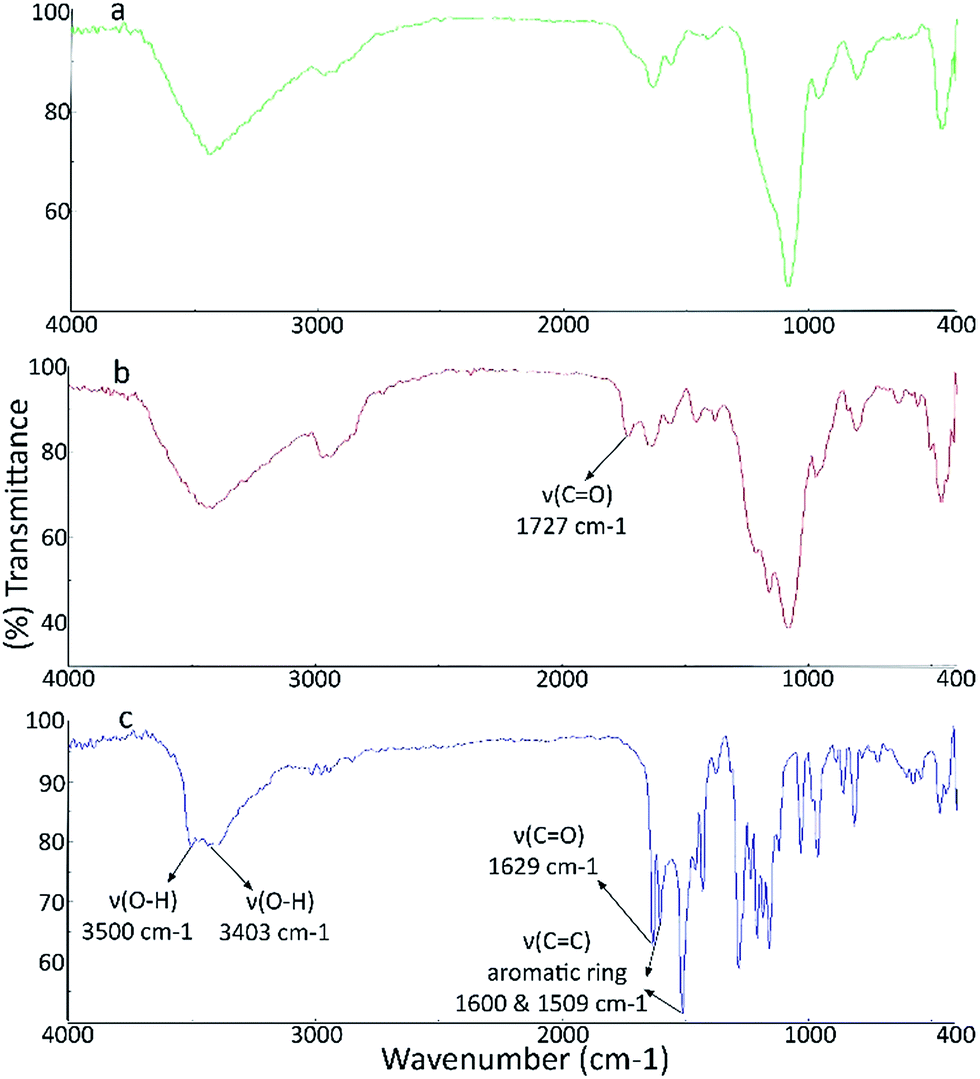 | ||
| Fig. 1 FT-IR spectra of LMSN-COOH@CS-FA (a), LMSN-COOH-CUR@CS-FA (b) and CUR (c). | ||
 | ||
| Fig. 2 Morphological characterization of MSNs: FE-SEM images of LMSNs-removed (a), LMSN-COOH@CS-FA (b); TEM image of LMSNs-removed (c) and LMSN-COOH@CS-FA (d). | ||
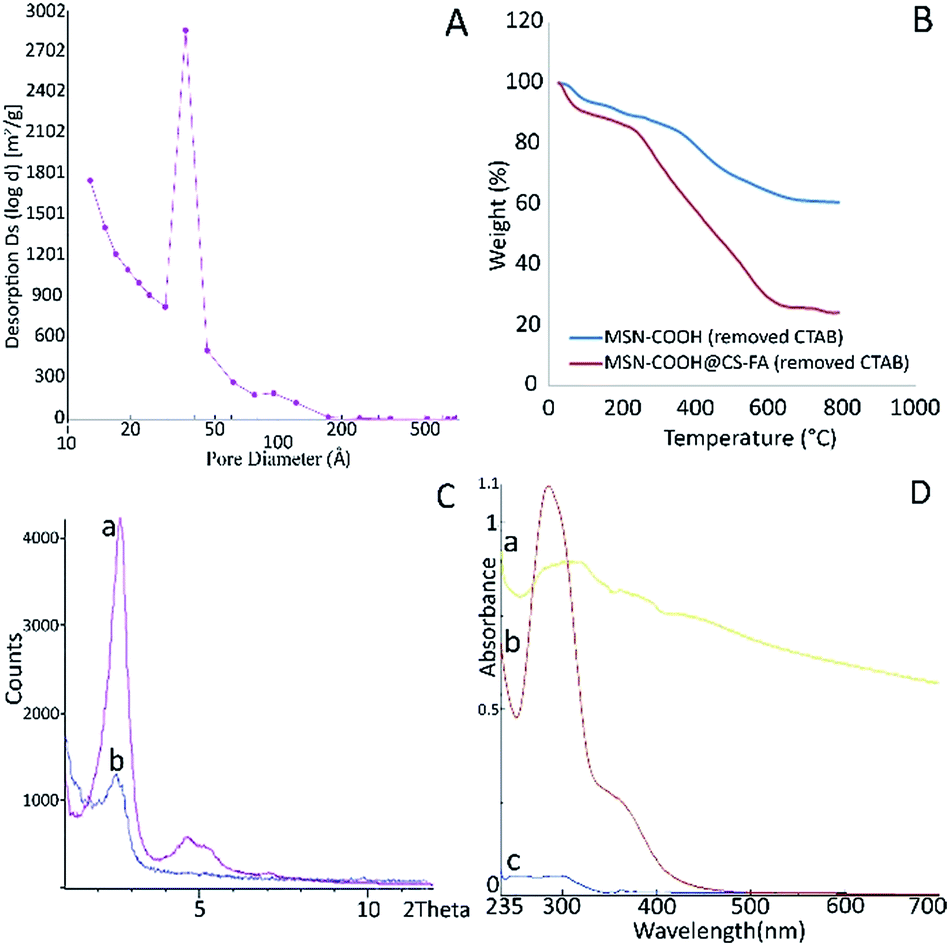 | ||
| Fig. 3 BJH pattern of LMSN-removed (A); TGA curves of LMSN-COOH and LMSN-COOH@CS-FA (B); XRD pattern of (a) LMSN and (b) LMSN-COOH@CS-FA after the removal of template (C) and UV-visible spectrum of (a) FA, (b) CS-FA and (c) CS (D). | ||
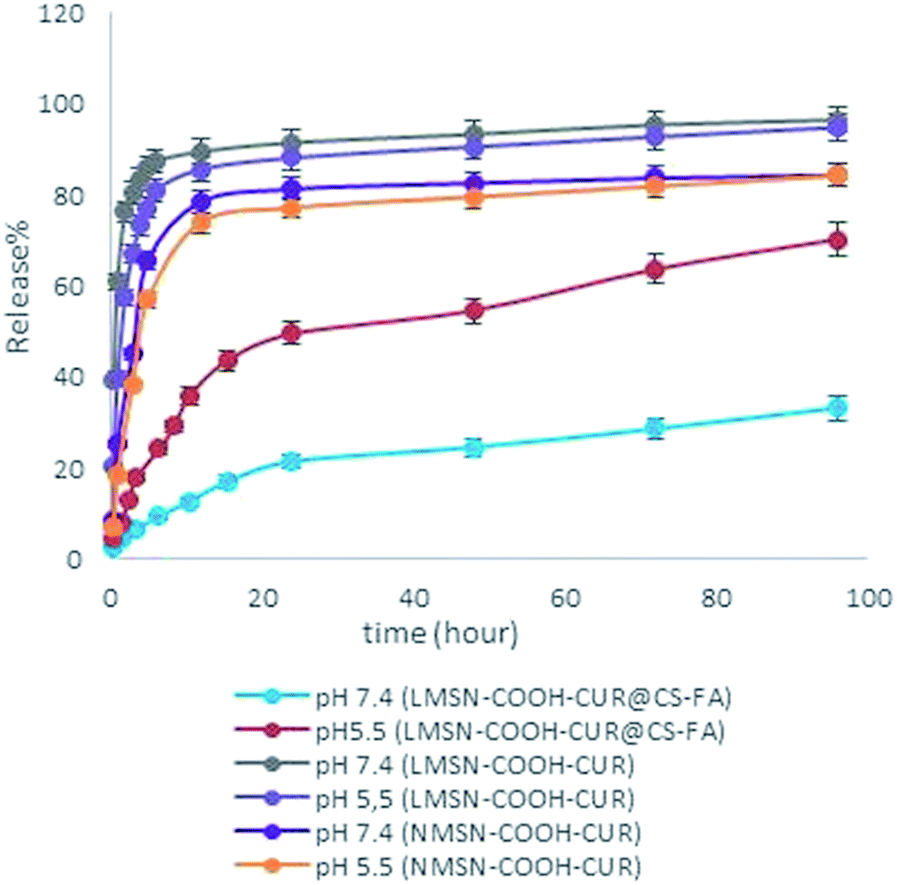 | ||
| Fig. 4 Release profiles of CUR from LMSNs and NMSNs. | ||
In sum, the prepared DDS demonstrated a pH-sensitive and sustained CUR release behavior which is of crucial importance for selective and efficient treatment of diverse types of cancers. For further studies, the in vitro release modeling was performed to predict the release profile kinetics by most used mathematical models, zero-order, first-order and Korsmeyer–Peppas.70 The results were listed in Table S4.‡ In order to fit of a model equation, best mathematical model was chosen by using the coefficient of determination (R2). According to the R2 results, release data were found to be adjusted to Korsmeyer–Peppas model. This model is generally used to evaluate the release profile mechanism particularly when more than one parameter or not well known mechanism involved.71 In order to determine release mechanism, n value in Korsmeyer–Peppas model, consist of n ≤ 0.45 for Fick diffusion mechanism and 0.45 < n ≤ 1.0 for mass transfer following non-Fickian or anomalous diffusion model were determined.70,71
As mentioned above, CUR release from LMSN-COOH-CUR@CS-FA at pH 5.5 and 7.4 seems to be controlled by non-Fickian diffusion. In non-Fickian diffusion mechanism, the kinetic of CUR release is controlled by both diffusion and polymer swelling, while the release kinetic of LMSN-COOH-CUR and NMSN-COOH-CUR at pH 5.5 and 7.4 seems to be described by Fickian diffusion and system acts as a simple barrier.72 In sum, good correlation could be observed between experimental and linear regression data.
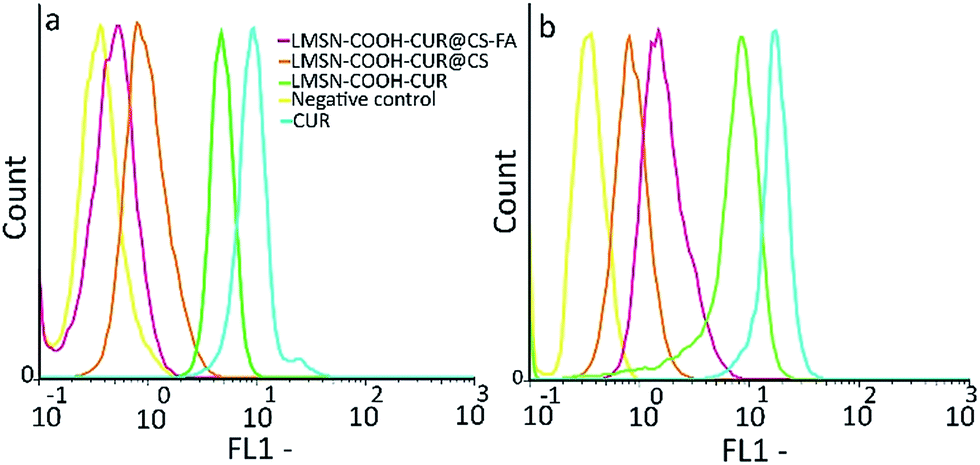 | ||
| Fig. 5 Flow cytometric analysis of CUR, LMSN-COOH-CUR, LMSN-COOH-CUR@CS, and LMSN-COOH-CUR@CS-FA cellular uptake in (a) NIH-3T3 and (b) HeLa cells. | ||
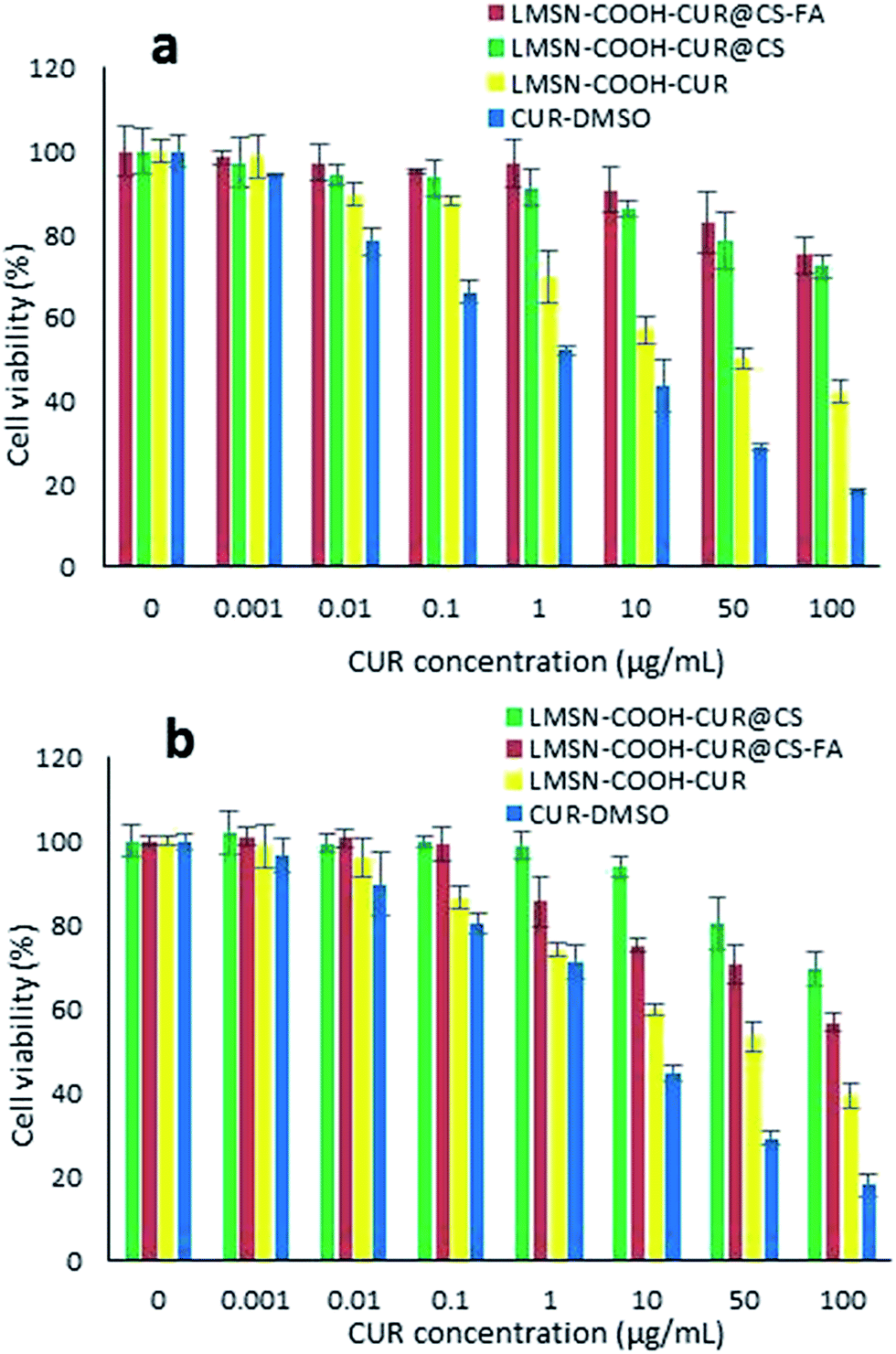 | ||
| Fig. 6 Cytotoxicity of free and CUR loaded nanoparticles measured by MTT assay in NIH-3T3 (a) and HeLa (b) cells after 72 h incubation. Data is expressed as mean ± S.D. (n = 3). | ||
In comparison, LMSN-COOH-CUR exhibited significantly cytotoxicity as compared to CS and CS-FA coated MSNs which could be related to the high CUR release from LMSN-COOH-CUR (Fig. 4). In fact, the absence of CS and CS-FA coatings leads to prompt CUR release from MSNs and therefore more free diffusion of free CUR through phospholipid cell membrane (Fig. 6 and S13, S14‡).67,77 Moreover, slow and sustained CUR release from CS and CS-FA coated MSNs could be another reason for low cytotoxicity of LMSN-COOH-CUR@CS and LMSN-COOH-CUR@CS-FA in both cell lines as compared to free CUR.77,78
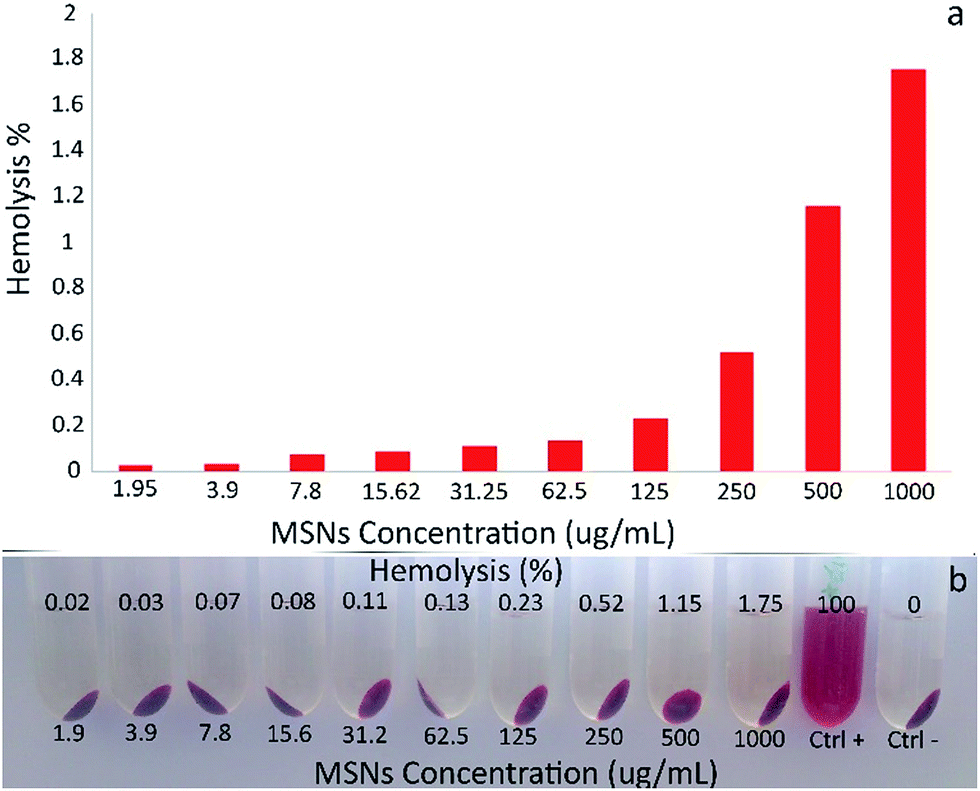 | ||
| Fig. 7 Hemolytic activity and hemolysis percentage of LMSN-COOH-CUR@CS-FA (a), Photographs of RBCs treated with LMSN-COOH-CUR@CS-FA at different concentrations (b). | ||
Conclusion
There has been a considerable interest to design and develop passive and active targeting DDSs with a novel approach focused on enhancing aqueous solubility of hydrophobic drugs and improving the drug loading capacity, therapeutic efficacy, and sustained release. To achieve this goal, herein we introduced LMSNs coated with FA conjugated CS as a smart nanocarrier for targeted delivery of CUR as model hydrophobic anticancer drug. The results showed an increase in loading capacity, pH dependent and sustained in vitro release profile, and biocompatibility. Our findings exhibited that CUR free LMSNs were safe for normal cell lines, and folate receptor-positive HeLa cell line was more sensitive to the antiproliferative effect of CUR as a result of higher cellular uptake of CS-FA coated LMSNs by HeLa cells than folate receptor-negative NIH-3T3 cell line. These results confirmed selective targeting and successful delivery of CUR by the designed MSNs. These findings suggest LMSN-COOH-CUR@CS-FA as a promising candidate for targeted hydrophobic anticancer delivery. Study on biodistribution of the carrier and its tumor shrinking power via in vivo analyses could be the objects of future researches.Acknowledgements
This study was supported by a grant from the Research Council of Tehran University of Medical Sciences (Grant no. 30429) and from the Iran National Science Foundation (INSF; Grant no. 92037263).References
- A. Jemal, R. Siegel, E. Ward, Y. Hao, J. Xu and M. J. Thun, Ca-Cancer J. Clin., 2009, 59, 225–249 CrossRef PubMed.
- K.-N. Yang, C.-Q. Zhang, W. Wang, P. C. Wang, J.-P. Zhou and X.-J. Liang, Cancer Biol. Med., 2014, 11, 34–43 CAS.
- N. T. Phuoc, J. F. Quinn, M. R. Whittaker and T. P. Davis, Polym. Chem., 2016, 7, 4295–4312 RSC.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. Chan, Nature Reviews Materials, 2016, 1, 16014 CrossRef.
- N. T. Tran, Z. Jia, N. P. Truong, M. A. Cooper and M. J. Monteiro, Biomacromolecules, 2013, 14, 3463–3471 CrossRef CAS PubMed.
- M. Talelli, M. Iman, A. K. Varkouhi, C. J. Rijcken, R. M. Schiffelers, T. Etrych, K. Ulbrich, C. F. van Nostrum, T. Lammers and G. Storm, Biomaterials, 2010, 31, 7797–7804 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- S. B. Hartono, N. T. Phuoc, M. Yu, Z. Jia, M. J. Monteiro, S. Qiao and C. Yu, J. Mater. Chem. B, 2014, 2, 718–726 RSC.
- M. Stubbs, P. M. McSheehy, J. R. Griffiths and C. L. Bashford, Mol. Med. Today, 2000, 6, 15–19 CrossRef CAS PubMed.
- J. Liu, Y. Huang, A. Kumar, A. Tan, S. Jin, A. Mozhi and X.-J. Liang, Biotechnol. Adv., 2014, 32, 693–710 CrossRef CAS PubMed.
- P. C. Berscht, B. Nies, A. Liebendörfer and J. Kreuter, Biomaterials, 1994, 15, 593–600 CrossRef CAS PubMed.
- M. Thanou, J. Verhoef and H. Junginger, Adv. Drug Delivery Rev., 2001, 50, S91–S101 CrossRef CAS PubMed.
- M. Thanou, J. Verhoef and H. Junginger, Adv. Drug Delivery Rev., 2001, 52, 117–126 CrossRef CAS PubMed.
- S. A. Agnihotri, N. N. Mallikarjuna and T. M. Aminabhavi, J. Controlled Release, 2004, 100, 5–28 CrossRef CAS PubMed.
- X. Qu, A. Wirsén and A. C. Albertsson, J. Appl. Polym. Sci., 1999, 74, 3186–3192 CrossRef CAS.
- A. Popat, J. Liu, G. Q. M. Lu and S. Z. Qiao, J. Mater. Chem., 2012, 22, 11173–11178 RSC.
- J. Hrkach, D. Von Hoff, M. M. Ali, E. Andrianova, J. Auer, T. Campbell, D. De Witt, M. Figa, M. Figueiredo and A. Horhota, Sci. Transl. Med., 2012, 4, 128 Search PubMed.
- D. B. Kirpotin, D. C. Drummond, Y. Shao, M. R. Shalaby, K. Hong, U. B. Nielsen, J. D. Marks, C. C. Benz and J. W. Park, Cancer Res., 2006, 66, 6732–6740 CrossRef CAS PubMed.
- D. W. Bartlett, H. Su, I. J. Hildebrandt, W. A. Weber and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15549–15554 CrossRef CAS PubMed.
- O. C. Farokhzad, J. Cheng, B. A. Teply, I. Sherifi, S. Jon, P. W. Kantoff, J. P. Richie and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6315–6320 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- N. Kamaly, Z. Xiao, P. M. Valencia, A. F. Radovic-Moreno and O. C. Farokhzad, Chem. Soc. Rev., 2012, 41, 2971–3010 RSC.
- J. Shi, Z. Xiao, N. Kamaly and O. C. Farokhzad, Acc. Chem. Res., 2011, 44, 1123–1134 CrossRef CAS PubMed.
- Z. Cheng, A. Al Zaki, J. Z. Hui, V. R. Muzykantov and A. Tsourkas, Science, 2012, 338, 903–910 CrossRef CAS PubMed.
- A. Koshkaryev, R. Sawant, M. Deshpande and V. Torchilin, Adv. Drug Delivery Rev., 2013, 65, 24–35 CrossRef CAS PubMed.
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS PubMed.
- N. T. Tran, N. P. Truong, W. Gu, Z. Jia, M. A. Cooper and M. J. Monteiro, Biomacromolecules, 2013, 14, 495–502 CrossRef CAS PubMed.
- E. K. Park, S. B. Lee and Y. M. Lee, Biomaterials, 2005, 26, 1053–1061 CrossRef CAS PubMed.
- P. Chan, M. Kurisawa, J. E. Chung and Y.-Y. Yang, Biomaterials, 2007, 28, 540–549 CrossRef CAS PubMed.
- M. O. Oyewumi, R. A. Yokel, M. Jay, T. Coakley and R. J. Mumper, J. Controlled Release, 2004, 95, 613–626 CrossRef CAS PubMed.
- S. Mansouri, Y. Cuie, F. Winnik, Q. Shi, P. Lavigne, M. Benderdour, E. Beaumont and J. C. Fernandes, Biomaterials, 2006, 27, 2060–2065 CrossRef CAS PubMed.
- S.-Q. Liu, N. Wiradharma, S.-J. Gao, Y. W. Tong and Y.-Y. Yang, Biomaterials, 2007, 28, 1423–1433 CrossRef CAS PubMed.
- S. D. Weitman, R. H. Lark, L. R. Coney, D. W. Fort, V. Frasca, V. R. Zurawski and B. A. Kamen, Cancer Res., 1992, 52, 3396–3401 CAS.
- D. Dubé, M. Francis, J.-C. Leroux and F. M. Winnik, Bioconjugate Chem., 2002, 13, 685–692 CrossRef.
- S.-J. Yang, F.-H. Lin, K.-C. Tsai, M.-F. Wei, H.-M. Tsai, J.-M. Wong and M.-J. Shieh, Bioconjugate Chem., 2010, 21, 679–689 CrossRef CAS PubMed.
- A. B. Kunnumakkara, P. Anand and B. B. Aggarwal, Cancer Lett., 2008, 269, 199–225 CrossRef CAS PubMed.
- M. Akrami, M. Khoobi, M. Khalilvand-Sedagheh, I. Haririan, A. Bahador, M. A. Faramarzi, S. Rezaei, H. A. Javar, F. Salehi and S. K. Ardestani, RSC Adv., 2015, 5, 88096–88107 RSC.
- E. I. Paramera, S. J. Konteles and V. T. Karathanos, Food Chem., 2011, 125, 913–922 CrossRef CAS.
- S. S. Bansal, M. Goel, F. Aqil, M. V. Vadhanam and R. C. Gupta, Cancer Prev. Res., 2011, 4, 1158–1171 CrossRef CAS PubMed.
- S. Jambhrunkar, Z. Qu, A. Popat, J. Yang, O. Noonan, L. Acauan, Y. Ahmad Nor, C. Yu and S. Karmakar, Mol. Pharmaceutics, 2014, 11, 3642–3655 CrossRef CAS PubMed.
- R. Kotcherlakota, A. K. Barui, S. Prashar, M. Fajardo, D. Briones, A. Rodríguez-Diéguez, C. R. Patra and S. Gómez-Ruiz, Biomater. Sci., 2016, 4, 448–459 RSC.
- S. Kim, S. Philippot, S. Fontanay, R. E. Duval, E. Lamouroux, N. Canilho and A. Pasc, RSC Adv., 2015, 5, 90550–90558 RSC.
- H. Tang, J. Guo, Y. Sun, B. Chang, Q. Ren and W. Yang, Int. J. Pharm., 2011, 421, 388–396 CrossRef CAS PubMed.
- J. Zheng, X. Tian, Y. Sun, D. Lu and W. Yang, Int. J. Pharm., 2013, 450, 296–303 CrossRef CAS PubMed.
- B. Lindlar, A. Kogelbauer, P. J. Kooyman and R. Prins, Microporous Mesoporous Mater., 2001, 44, 89–94 CrossRef.
- J. Zhang, M. Niemelä, J. Westermarck and J. M. Rosenholm, Dalton Trans., 2014, 43, 4115–4126 RSC.
- L. Yuan, Q. Tang, D. Yang, J. Z. Zhang, F. Zhang and J. Hu, J. Phys. Chem. C, 2011, 115, 9926–9932 CAS.
- N. Lang and A. Tuel, Chem. Mater., 2004, 16, 1961–1966 CrossRef CAS.
- M. Gulfam and B. G. Chung, Macromol. Res., 2014, 22, 412–417 CrossRef CAS.
- B. Chertok, A. E. David and V. C. Yang, Biomaterials, 2010, 31, 6317–6324 CrossRef CAS PubMed.
- A. Szegedi, M. Popova, I. Goshev, S. Klebert and J. Mihaly, J. Solid State Chem., 2012, 194, 257–263 CrossRef CAS.
- J. Shen, Q. He, Y. Gao, J. Shi and Y. Li, Nanoscale, 2011, 3, 4314–4322 RSC.
- M. M. Yallapu, S. F. Othman, E. T. Curtis, N. A. Bauer, N. Chauhan, D. Kumar, M. Jaggi and S. C. Chauhan, Int. J. Nanomed., 2012, 7, 1761–1779 CAS.
- S. Alam, J. J. Panda and V. S. Chauhan, Int. J. Nanomed., 2012, 7, 4207–4222 CAS.
- N. M. Khoram, B. Bigdeli, A. Nikoofar and B. Goliaei, Int. J. Breast Cancer, 2016, 19, 18–25 CrossRef PubMed.
- J. Li, L. Zheng, H. Cai, W. Sun, M. Shen, G. Zhang and X. Shi, Biomaterials, 2013, 34, 8382–8392 CrossRef CAS PubMed.
- S. Mirsadeghi, R. Dinarvand, M. H. Ghahremani, M. R. Hormozi-Nezhad, Z. Mahmoudi, M. J. Hajipour, F. Atyabi, M. Ghavami and M. Mahmoudi, Nanoscale, 2015, 7, 5004–5013 RSC.
- M. Mahmoudi, I. Lynch, M. R. Ejtehadi, M. P. Monopoli, F. B. Bombelli and S. Laurent, Chem. Rev., 2011, 111, 5610–5637 CrossRef CAS PubMed.
- M. Mahmoudi, M. P. Monopoli, M. Rezaei, I. Lynch, F. Bertoli, J. J. McManus and K. A. Dawson, ChemBioChem, 2013, 14, 568–572 CrossRef CAS PubMed.
- H. Jin, M. B. Ansari and S.-E. Park, Chem. Commun., 2011, 47, 7482–7484 RSC.
- H. Song, C. Su, W. Cui, B. Zhu, L. Liu, Z. Chen and L. Zhao, BioMed Res. Int., 2013, 2013, 723158 Search PubMed.
- J. Ji, P. Zuo and Y.-L. Wang, Nanoscale Res. Lett., 2015, 10, 1–8 CrossRef PubMed.
- J. Zhang, X. Li, J. M. Rosenholm and H.-c. Gu, J. Colloid Interface Sci., 2011, 361, 16–24 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- L. Jia, J. Shen, Z. Li, D. Zhang, Q. Zhang, C. Duan, G. Liu, D. Zheng, Y. Liu and X. Tian, Int. J. Pharm., 2012, 439, 81–91 CrossRef CAS PubMed.
- Y. Xiao, H. Hong, A. Javadi, J. W. Engle, W. Xu, Y. Yang, Y. Zhang, T. E. Barnhart, W. Cai and S. Gong, Biomaterials, 2012, 33, 3071–3082 CrossRef CAS PubMed.
- A. Pourjavadi and Z. M. Tehrani, Int. J. Polym. Mater. Polym. Biomater., 2014, 63, 692–697 CrossRef CAS.
- V. Dodane, M. A. Khan and J. R. Merwin, Int. J. Pharm., 1999, 182, 21–32 CrossRef CAS PubMed.
- M. Torkpur-Biglarianzadeh and M. Salami-Kalajahi, RSC Adv., 2015, 5, 29653–29662 RSC.
- P. Costa and J. M. S. Lobo, Eur. J. Pharm. Sci., 2001, 13, 123–133 CrossRef CAS PubMed.
- A. R. Khare and N. A. Peppas, Biomaterials, 1995, 16, 559–567 CrossRef CAS PubMed.
- Y. Gao, Y. Li, Y. Li, L. Yuan, Y. Zhou, J. Li, L. Zhao, C. Zhang, X. Li and Y. Liu, Nanoscale, 2015, 7, 597–612 RSC.
- A. Kunwar, A. Barik, B. Mishra, K. Rathinasamy, R. Pandey and K. Priyadarsini, Biochim. Biophys. Acta, Gen. Subj., 2008, 1780, 673–679 CrossRef CAS PubMed.
- S. Honary and F. Zahir, Trop. J. Pharm. Res., 2013, 12, 255–264 Search PubMed.
- D. Feng, Y. Song, W. Shi, X. Li and H. Ma, Anal. Chem., 2013, 85, 6530–6535 CrossRef CAS PubMed.
- L. Jiang, Z.-m. Gao, L. Ye, A.-y. Zhang and Z.-g. Feng, Biomater. Sci., 2013, 1, 1282–1291 RSC.
- S. Jambhrunkar, S. Karmakar, A. Popat, M. Yu and C. Yu, RSC Adv., 2014, 4, 709–712 RSC.
- C. C. Fleischer and C. K. Payne, Acc. Chem. Res., 2014, 47, 2651–2659 CrossRef CAS PubMed.
Footnotes |
| † This work is dedicated to the memory of Prof. Abbas Shafiee (1937–2016). |
| ‡ Electronic supplementary information (ESI) available: Scheme S1, Fig. S1–S13 and Tables S1–S4. See DOI: 10.1039/c6ra23182a |
| This journal is © The Royal Society of Chemistry 2016 |