
Enhancing mechanical performance of epoxy thermosets via designing a block copolymer to self-organize into “core–shell” nanostructure
Zhengguang Henga,
Zhong Zengb,
Bin Zhanga,
Yinfu Luoa,
Jiemin Luoa,
Yang Chen*a,
Huawei Zou*a and
Mei Lianga
aState Key Laboratory of Polymer Materials Engineering, Polymer Research Institute of Sichuan University, Sichuan University, Chengdu 610065, China. E-mail: cy3262276@163.com; hwzou@163.com; Fax: +86-28-85402465; Tel: +86-28-85408288
bSafety Environment Quality Surveillance and Inspection Research Institute of CNPC Chuanqing Drilling & Exploration Corporation, Chengdu 618300, China
First published on 9th August 2016
Abstract
A rigid-flexible amphiphilic pentablock copolymer, polystyrene-block-poly(ε-caprolactone)-block-polydimethylsiloxane-block-poly(ε-caprolactone)-block-polystyrene (PS-PCL-PDMS-PCL-PS, SLDLS), was designed. SLDLS block copolymers will self-organize into “core–shell” nanostructures in epoxy thermosets, with rigid PS as the “shell” and flexible PDMS as the “core”. Due to the incorporation of the “core–shell” nanostructures, the tensile strength and toughness of the epoxy composites were simultaneously improved and the storage modulus maintained. It is believed that designing specific structures in epoxy resins through self-organization could provide new methods for the preparation of advanced functional epoxy composites.
1. Introduction
Self-organization is a very common phenomenon in nature. Depending on the inherent characteristics, various structures with their corresponding properties could be obtained. A typical example of self-assembly is nacre, which is composed of an aragonite layer and elastic biopolymer layer. Because of the unique layer-by-layer structure, nacre exhibits flexible and stable features.1 Over the past decades, materials scientists have aspired to exploit nature's assembly principles to create artificial materials, with hierarchical structures and tailored properties, for the fabrication of functional devices.As a most important class of thermosets resins, epoxy resins (EP) are widely utilized in many applications (i.e., aerospace, automotive industry etc.) owing to their excellent mechanical performance, resistance to chemicals and low shrinkage during curing. However, undesirable properties, such as their inherently brittle and poor damping property, adversely affected most of the physical and mechanical properties, which limited their applications in high performance composites.
Block copolymers have attracted much research interest because they could self-organize into different nanostructures in the selective solvent. For example, in selective solvent, because polymer surfactants could self-assemble into micelles or vesicles, they were widespread studied in the field of drug carrier. Similarly, when epoxy served as a selective solvent, block copolymers could also self-organize into nanostructures with various morphologies, such as spherical, spheres on spheres, cylindrical and so on.2–4 However, research on obtaining the desired properties by designing block copolymers and the nanostructures in epoxy is rarely targeted.5
As one of the most widely utilized thermosets, epoxy resins have various advantages, while its inherent brittleness is an unavoidable issue. “Core–shell” nanoparticles have been proved to be a classical method to tough epoxy.6–8 These particles consist of a soft rubbery core with a rigid shell around it. Poly(methyl methacrylate), which is compatible with the epoxy matrix, is usually used as shell material, soft materials (polyurethane, siloxane, butadiene etc.) are chosen as core materials. Giannakopoulos9 reported that the fracture energy of composite containing 15 wt% core–shell rubber (CSR) particles was increased to 860 J m−2 compared with the unmodified epoxy (77 J m−2). Chen's work also shown CSR particles increased the fracture toughness significantly at room temperature and cryogenic temperatures.9 However, both of them indicated the Young's modulus and tensile strength of the epoxy polymer would reduce because of the addition of the CSR particles. Generally, it is shown that the classical “core–shell” rubber particles could significantly improve the toughness9–11 while the strength and modulus were inevitably decreased. Thus, rigid particles were incorporated to cope with this disadvantage.12,13 The dispersity of the modifiers, however, is still a problem. In this context, nanostructures with rigid “shell” and flexible “core” were prepared to maximize the advantages of classical “core–shell” structure.
Herein, a novel kind of rigid-flexible amphiphilic block copolymer, polystyrene-block-poly(ε-caprolactone)-block-polydimethylsiloxane-block-poly(ε-caprolactone)-block-polystyrene (PS-PCL-PDMS-PCL-PS, SLDLS), was designed and incorporated into epoxy thermosets. Compared with those “core–shell” nanoparticles mentioned above, except for the soft-core (PDMS) and the rigid shell (PS), we introduce a new PCL subchain, which is miscible with the epoxy matrix, to optimize the interactions between the matrix and the modifier. The “soft core–rigid shell” nanostructures, where the rigid PS as the “shell” and the flexible PDMS as the “core”, were self-organized in the selective solvent epoxy through the mechanisms of self-assembly and reaction-induced microphase separation during the curing reaction in our study. Besides, the interfacial bonding could obviously improve due to the appearance of miscible PCL subchain. Hence, it is reasonable to obtain a new family of epoxy thermosets, which the storage modulus, maintained, the tensile strength and toughness were simultaneously improved.
2. Materials
The hydroxyl-terminated polydimethylsiloxane (HTPDMS) was kindly supplied by Shin-Etsu Chemical Co., Ltd., Japan. Before use, it was dried by azeotropic distillation with anhydrous toluene. The monomer of ε-caprolactone (ε-CL) (Aladdin, 99%) was dried over calcium hydride (CaH2) and distilled under decreased pressure prior to use. Stannous octanoate [Sn(Oct)2] was purchased from Aladdin Co. and used as the catalyst. N,N,N′,N′,N′-Pentamethyldiethylenetriamine (PMDETA), 2-bromoisobutyryl bromide and copper(I) bromide (CuBr) were purchased from Aldrich Co. Prior to use, CuBr was purified by stirring in glacial acetic acid overnight, filtered off, washed with ethanol, and then dried in a vacuum oven at 60 °C for 24 h. Styrene was purchased from Chemical Reagent Factory of Kelong and washed with an aqueous solution of sodium hydroxide (5 wt%) three times and then with water until neutralization. It was also dried by azeotropic distillation with anhydrous toluene prior to use. 4-Dimethylaminopyridine (DMAP) was purchased from Nanjing Tianhua Reagent Co., China, and recrystallized in toluene at 80 °C. Epoxy resins used in this work are diglycidyl ether of bisphenol A-based (DGEBA) epoxy resin E-51, which was obtained from Jiangsu Wuxi Resin Plant, China. 3,3′-Dichloro-4,4′-diamino diphenyl methane (MOCA), which was purchased from Changshan beier Co., China, is used as curing agent. All other solvent were used as received.2.1 Synthesis of PCL-b-PDMS-b-PCL (LDL) triblock copolymer
Poly(ε-caprolactone)-block-polydimethylsiloxane-block-poly(ε-caprolactone) triblock copolymer (PCL-b-PDMS-b-PCL, LDL) was synthesized via the ring-opening polymerization (ROP) of ε-caprolactone (ε-CL) in the presence of hydroxyl-terminated polydimethylsiloxane (HTPDMS). Stannous octanoate [Sn(Oct)2] was used as the catalyst. Typically, HTPDMS (25.404 g, 10.11 mmol) and ε-CL (80.88 g, 709.44 mmol) were charged to a 250 ml pre-dried round-bottom Schlenk flask equipped with a magnetic stirrer then Sn(Oct)2 was added at the ratio of 1/1000 (w/w) with respect to ε-CL using a syringe. The flask was connected to a standard Schlenk line system and the freeze–pump–thaw cycle was repeated for three times to eliminate moisture. Then the flask was immersed into a thermostatted oil bath at 120 °C to initiate the ring-opening polymerization. After the polymerization was carried out for 36 h, the system was cooled to room temperature and the crude product was dissolved in tetrahydrofuran (THF). The solution was slowly dropped into a great amount of petroleum ether to afford the precipitates, repeated several times until white solids were obtain. The product was dried in a vacuum oven at 40 °C until a constant weight was obtained with a yield of 93.8%.2.2 Synthesis of PS-b-PCL-b-PDMS-b-PCL-b-PS (SLDLS) pentablock copolymer
The macromolecular initiator was first prepared by following the literature method.14,15 LDL (50 g, 4.76 mmol), DMAP (3.491 g, 28.58 mmol) and triethylamine (TEA, 2.64 mL, 19.05 mmol) were dissolved in 100 mL of CH2Cl2 and the solution was cooled to 0 °C. 2-Bromo-isobutyryl bromide (BiBB, 5.91 mL, 47.63 mmol) in 50 mL of CH2Cl2 was slowly injected dropwise for about 2 h. After the addition was completed, the temperature was allowed to rise to room temperature. The mixture reacted for another 24 h at room temperature under stirring. Then the solution was removed in a rotary evaporator. The crude macroinitiator was dropped into an excessive amount of cold methanol to afford the precipitates, recrystallized for 3 times, and dried in a vacuum oven at 40 °C for 48 h.Then above macroinitiator was used as the macroinitiator of atom transfer radical polymerization for styrene to obtain the PS-b-PCL-b-PDMS-b-PCL-b-PS pentablock copolymer. General, to a 250 mL pre-dried round-bottom Schlenk flask, macroinitiator (46.8 g, 4.457 mmol) was first dried by azeotropic distillation with anhydrous toluene, then CuBr (1 g, 6.971 mmol), PMDETA (1.483 mL, 7.077 mmol), and styrene (72.8 g, 700 mmol) were charged. The reactive system was degassed via three freeze–pump–thaw cycles and then immersed in a thermostatted oil bath at 110 °C. After 16 h, the Schlenk flask was exposed to air, cooled to room temperature and further dissolved in THF. Then the solution was passed over a column of neutral alumina, concentrated and dropped into an excessive amount of cold methanol to afford the precipitates. The product was dried in a vacuum oven at 40 °C until a constant weight was obtained with a yield of 59.9%. The process of synthesis for SLDLS block copolymer is summarized in Fig. 1.
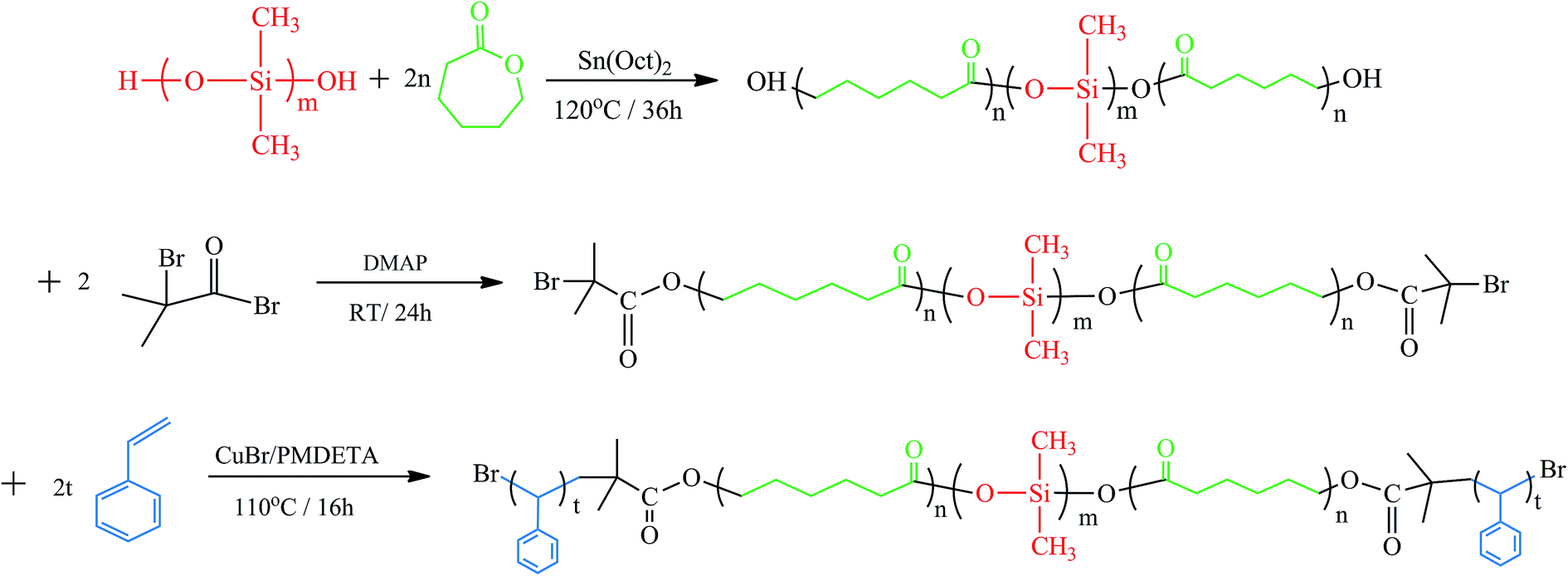 | ||
| Fig. 1 The process of synthesis for PS-b-PCL-b-PDMS-b-PCL-b-PS pentablock copolymer. | ||
2.3 Preparation of nanostructured epoxy resin
The desired amount of PS-b-PCL-b-PDMS-b-PCL-b-PS pentablock copolymer and DGEBA were mixed at 120 °C with vigorous stirring until the mixtures became homogenous. Then the curing agent MOCA was added to system with continuously vigorous stirring. The mixtures were degassed under vacuum, poured into Teflon molds and subjected to the thermal curing at 150 °C for 2 h plus 180 °C for 2 h for post curing. The thermosets containing PS-b-PCL-b-PDMS-b-PCL-b-PS pentablock copolymer up to 20 wt% were prepared.2.4 Measurement and characterization
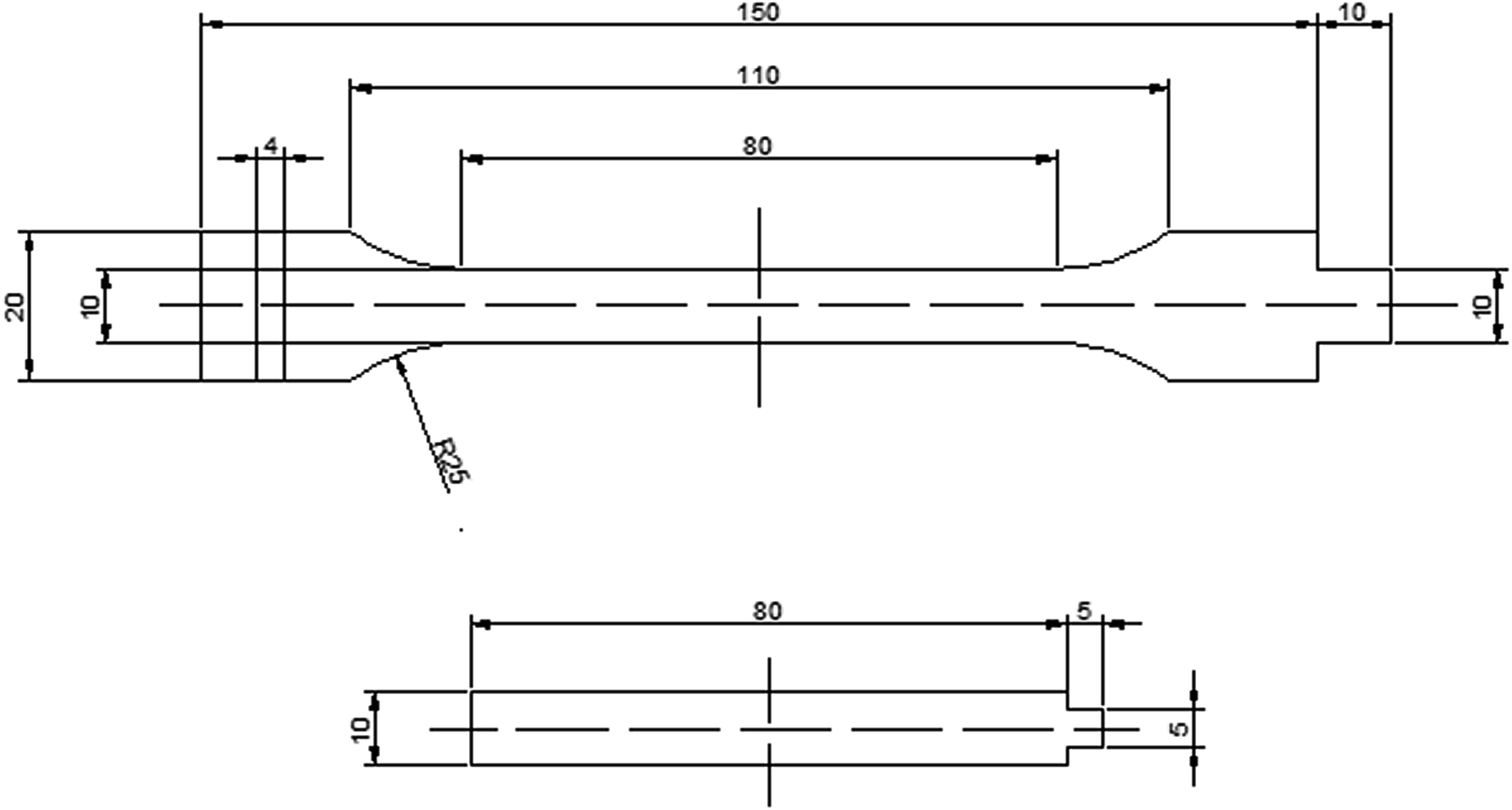 | ||
| Scheme 1 The dimensions of the tensile specimens. | ||
3. Results and discussion
3.1 Synthesis of PCL-b-PDMS-b-PCL triblock copolymer
Fig. 2 presented the 1H NMR spectrum of the PCL-b-PDMS-b-PCL triblock copolymer. The signal at 0.07–0.09 was ascribed to [Si(CH3)2], which is the characteristic group of PDMS. Meanwhile, the signals at 1.35–1.41, 1.60–1.68, 2.28–2.38, 4.04–4.07 were also observed in the 1H-NMR spectrum, which were attributed to the methylene protons of PCL, [OCOCH2CH2CH2CH2CH2], [OCOCH2CH2CH2CH2CH2], [OCOCH2(CH2)4], [OCO(CH2)4CH2], respectively. The results indicate that the resulting polymer combined the structural features from both PCL and PDMS. The triblock copolymer was also subjected to gel permeation chromatography (GPC) to measure the molecular weight, and the GPC curves were shown in Fig. 3. The GPC curves displayed a unimodal distribution of molecular weight, and the molecular weight of PDMS and the triblock copolymer were determined to be Mn = 3123 and 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 551 g mol−1, respectively. The results of 1H NMR and GPC indicated that the PCL-b-PDMS-b-PCL triblock copolymer was successfully obtained.
551 g mol−1, respectively. The results of 1H NMR and GPC indicated that the PCL-b-PDMS-b-PCL triblock copolymer was successfully obtained.
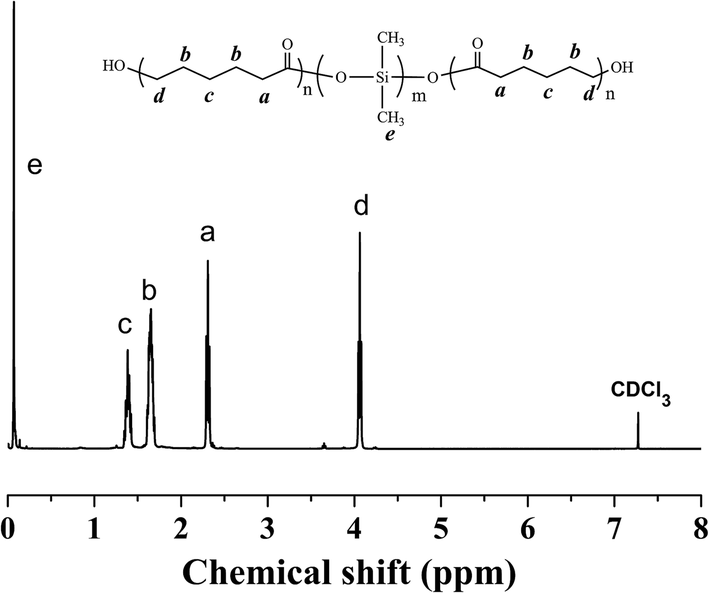 | ||
| Fig. 2 1H NMR spectrum of the PCL-b-PDMS-b-PCL triblock copolymer. | ||
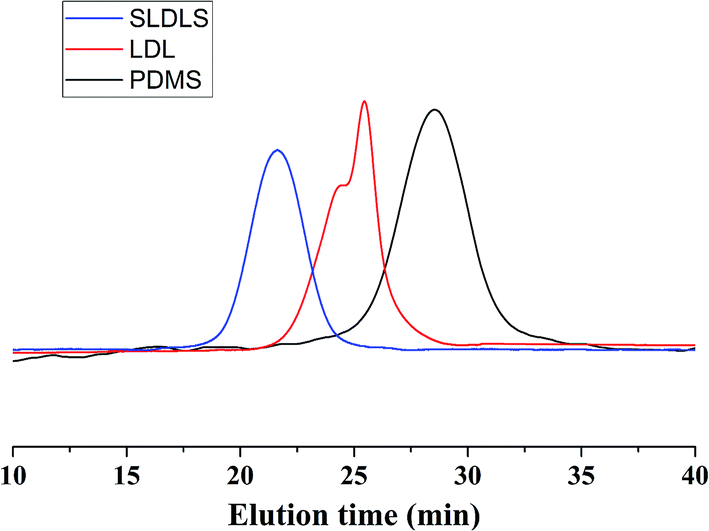 | ||
| Fig. 3 GPC curves of PDMS, PCL-b-PDMS-b-PCL and PS-b-PCL-b-PDMS-b-PCL-b-PS block copolymers. | ||
3.2 Synthesis of PS-b-PCL-b-PDMS-b-PCL-b-PS pentablock copolymer
The 1H NMR spectrum of the PS-b-PCL-b-PDMS-b-PCL-b-PS pentablock copolymer was shown in Fig. 4. In addition to the above peaks appeared in the 1H NMR spectrum of the PCL-b-PDMS-b-PCL triblock copolymer, some new signals were observed. The signals at 6.30–7.30 and 1.29–2.12 ppm are ascribed to the phenyl protons and methylene/methine from styrene repeating units, respectively. The pentablock copolymer was also measured by GPC (see Fig. 3), the result shown that GPC curve displayed a unimodal distribution of molecular weight, and the molecular weight was determined to be Mn = 48227 g mol−1. Both 1H NMR and GPC indicate that the PS-b-PCL-b-PDMS-b-PCL-b-PS pentablock copolymer was successfully obtained.
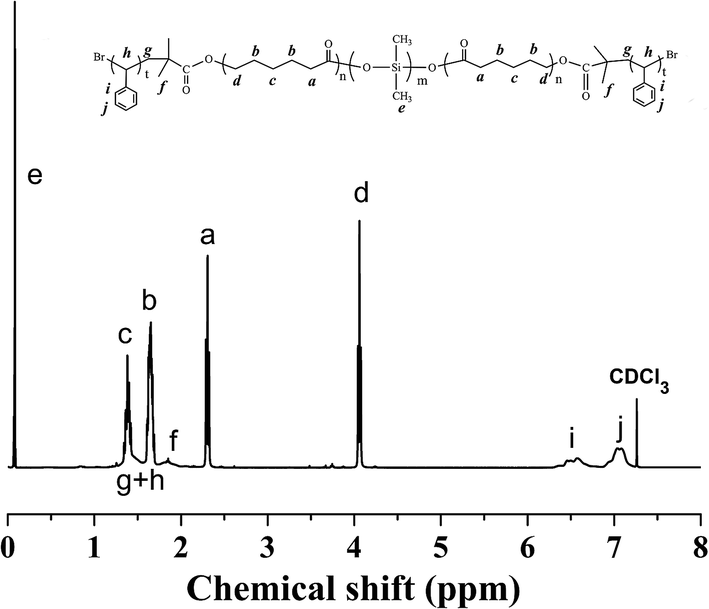 | ||
| Fig. 4 1H NMR spectrum of the PS-b-PCL-b-PDMS-b-PCL-b-PS pentablock copolymer. | ||
3.3 Nanostructures in epoxy thermosets containing PS-b-PCL-b-PDMS-b-PCL-b-PS
As mentioned above, the SLDLS pentablock copolymer was synthesized via a combination of the ring-opening polymerization and atom transfer radical polymerization. Then it was incorporated in to epoxy to prepare the nanostructured thermosets. After curing, all the specimens were homogenous and transparent, which indicated that no macroscopic phase separation occurred.The nanostructures were further measured by transmission electron microscopy (TEM). The TEM images of the epoxy thermosets containing 10 wt% and 20 wt% SLDLS pentablock copolymer are shown in Fig. 5. In order to increase the electron density contrast, the ultrathin sections of the epoxy thermosets was stained with RuO4. In this case, PDMS nanodomains remained almost unaffected whereas PS nanodomains were stained. It is seen that SLDLS pentablock copolymers were homogeneously dispersed into the continuous epoxy matrix for both of the thermosets containing 10 wt% and 20 wt% (Fig. 5a and b). Besides, in the higher magnification TEM image, all the spherical nanodomains displayed a typical “core–shell” structure, which is composed of the gray “core” and the black “shell” (Fig. 5a′ and b′). It is proposed that the outer “shell” is attributable to PS phase whereas the “core” to PDMS phase. And the diameter of the “core–shell” structure was about ∼150 nm.
 | ||
| Fig. 5 The TEM images of the epoxy thermosets containing 10 wt% (a and a′) and 20 wt% (b and b′) PS-b-PCL-b-PDMS-b-PCL-b-PS pentablock copolymer. | ||
3.4 The formation mechanism of nanostructures
The formation of nanostructures in thermosets containing amphiphilic block copolymers could follow self-assembly16–18 and/or reaction induced microphase separation (RIMPS).19–22 In self-assembly approach, nanostructures were formed via self-assembly of the block copolymers prior to curing and further fixed through subsequent curing reaction. For the mechanism of RIMPS, all the copolymer blocks are miscible with the thermosets. The nanostructures were not created until the curing reaction was performed with sufficiently high conversion.In the present study, SLDLS pentablock copolymer was constituted by PDMS, PCL and PS subchains. Own to the big difference in solubility parameter between epoxy and PDMS, PDMS was immiscible with epoxy before and after curing reaction. As for PCL subchain, because of the intermolecular specific interactions (e.g., hydrogen-bonding) between PCL and epoxy matrix, it remained miscible with epoxy after curing,23 which is consistent with the results of DMA (Fig. 7). It has been known that the blends of PS and epoxy displayed an upper critical solution temperature (UCST) behavior, PS blocks were miscible with epoxy prior to curing.22 While during the process of the subsequent curing reaction, reaction-induced phase separation would occur. Therefore, in this work, both self-assembly and reaction-induced microphase separation mechanisms were concurrently involved with the formation of the nanostructures in the thermosets.
The formation of nanostructures could speculate as below, PDMS subchain was first self-assembled into spherical prior to curing, then the preformed PDMS sphere acted as the templates of the reaction-induced microphase separation of PS subchain to form “core–shell” nanostructures, where the rigid PS as the “shell” and the flexible PDMS as the “core” (see Fig. 6). This is also verified by the results of TEM shown in Fig. 5.
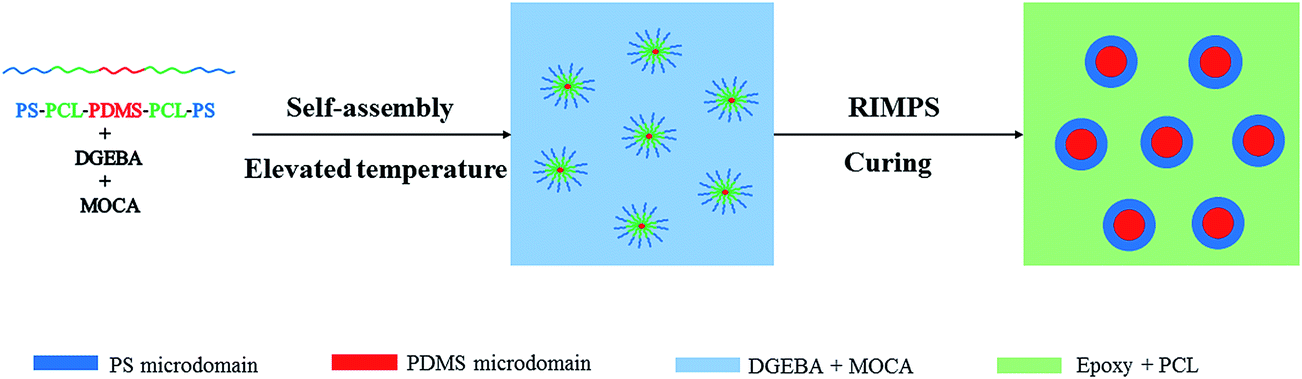 | ||
| Fig. 6 The formation of nanostructures in the epoxy thermosets containing PS-b-PCL-b-PDMS-b-PCL-b-PS pentablock copolymer. | ||
3.5 Dynamic mechanical properties
The nanostructured composites containing SLDLS pentablock copolymer were also subjected to dynamic mechanical analysis in Fig. 7. For the neat epoxy, apart from the α transition, the thermosets exhibited the secondary transitions (viz. β-relaxation) at 68.8 °C and −55.6 °C, respectively.24 The former is attributed predominantly to the motion of diphenyl groups in amine-crosslinked epoxy, while the latter to that of hydroxyl ether structural units. It is known that the Tg of PCL are approximately −65 °C, however, this transition was not observed for the thermosets containing 5 wt% and 20 wt% SLDLS. Thus, it is concluded that PCL subchains that connected with PDMS/PS through covalent bond were remain fixed in the epoxy network after curing.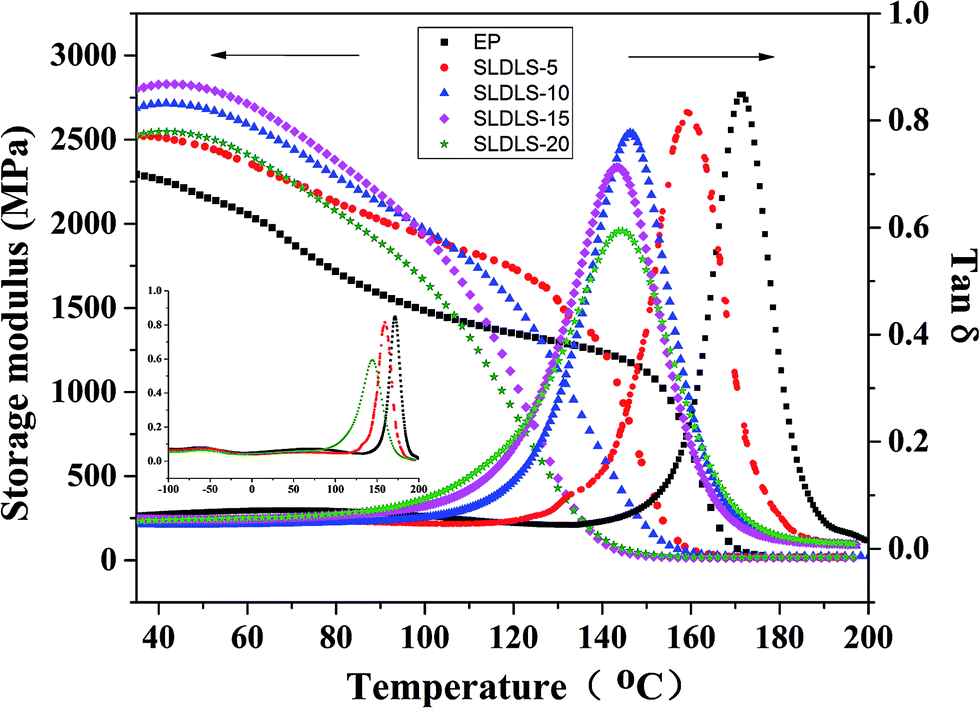 | ||
| Fig. 7 Dynamic mechanical analysis of the composites containing PS-b-PCL-b-PDMS-b-PCL-b-PS pentablock copolymer. | ||
Adding the triblock copolymer into the thermosets, the damping temperature range of the composites were broaden from 24.6 °C to 42.7 °C (Table 1), which suggested that the pentablock copolymers were well interpenetrated into the epoxy network and thereby enhanced the damping characteristics of the composites.25 Besides, this implied that the incorporation of the pentablock copolymers enhanced the energy dissipation capacity of the epoxy matrix. Owning to the incorporation of PDMS subchains with excellent damping property, soft PDMS phase would dissipate more energy than the matrix. Because soft PDMS was connected with PS through the PCL subchains, when the composites were subjected to the outer forces, PDMS subchains would be limited by the restrictions of the PS subchains, which could also dissipate more energy. Meanwhile, PCL remained miscible with epoxy, which play role of plasticizers. Thus the damping temperature range become broader compared with the neat epoxy.
| Sample code | EP | SLDLS-5 | SLDLS-10 | SLDLS-15 | SLDLS-20 |
|---|---|---|---|---|---|
| Tg (°C) | 170.7 | 158.6 | 145.3 | 143.4 | 144.0 |
| Damping temperature in range tanδ > 0.2 (°C) |
157.7–182.3 | 143.3–172.8 | 126.1–162.1 | 120.8–160.0 | 119.4–162.1 |
It also observed storage modulus was greater for the modified epoxy than the neat at room temperature indicating that the modulus of the composites were not compromised by the incorporation of the pentablock copolymer. Meanwhile, the Tgs of the epoxy phase decreased slightly for the modified composites due to the plasticization effect of miscible PCL subchains in the epoxy matrix, which has a good agreement with the ref. 20.
3.6 Tensile properties
It is known that the morphology of the separated phases is a key factor to determine the properties of the composites. The stress–strain curves of the epoxy thermosets containing SLDLS pentablock copolymer were shown in Fig. 8. The tensile strength and elongation at break of neat epoxy were 57.6 MPa and 3.0%, respectively (Table 2). When the “core–shell” nanostructures was incorporated, the tensile strength and toughness of the epoxy composites were simultaneously improved. And the maximum values were obtained when the content was 20 wt%, 75.1 MPa and 6.0%, respectively.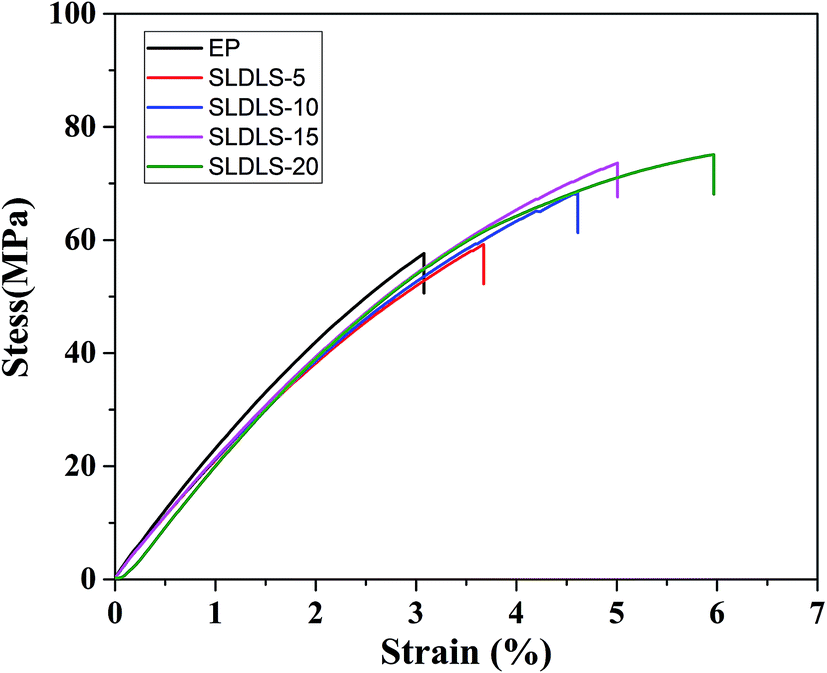 | ||
| Fig. 8 The stress–strain curves of the epoxy thermosets containing PS-b-PCL-b-PDMS-b-PCL-b-PS pentablock copolymer. | ||
| Sample code | EP | SLDLS-5 | SLDLS-10 | SLDLS-15 | SLDLS-20 |
|---|---|---|---|---|---|
| Tensile strength (MPa) | 57.6 ± 3.9 | 59.2 ± 3.9 | 68.3 ± 0.53 | 73.6 ± 4.9 | 75.1 ± 0.37 |
| Elongation at break (%) | 3.0 | 3.7 | 4.6 | 5.0 | 6.0 |
The tensile fracture surfaces of neat epoxy and composites containing 10 wt%, 15 wt% and 20 wt% SLDLS block copolymers were shown in Fig. 9. The neat epoxy fracture surfaces revealed a mostly smooth fracture surface, representative of brittle failure (Fig. 9a). With the introduction of the SLDLS pentablock copolymer, the fracture surface became rougher. Owing to the appearance of “soft core–rigid shell” nanostructures, the interfacial bonding between the pentablock copolymers and epoxy matrix was obviously improved. Besides, PCL subchains that connected with PDMS/PS through covalent bond were remain fixed in the epoxy network after curing, which was further optimized the interactions between the matrix and the modifier, and would further dissipate more energy than the matrix during stretching. Hence, mechanical properties of the composites were improved.
 | ||
| Fig. 9 The tensile fracture surfaces of neat epoxy (a) and composites containing 10 wt% (b), 15 wt% (c) and 20 wt% (d) PS-b-PCL-b-PDMS-b-PCL-b-PS pentablock copolymer. | ||
4. Conclusions
In summary, SLDLS pentablock copolymer was designed and synthesized via the combination of the ring-opening polymerization and atom transfer radical polymerization. Own to the difference of every blocks of SLDLS and epoxy matrix, SLDLS pentablock copolymer could self-organize into “core–shell” nanostructure in epoxy through the mechanism of self-assembly and reaction-induced microphase separation. The mechanical results showed that these self-organized “core–shell” nanostructures could optimize the advantages of classical “core–shell” rubber particles, meanwhile, the tensile strength, storage modulus and damping temperature rang were simultaneously improved compared with the neat epoxy.References
- X. Li, W.-C. Chang, Y. J. Chao, R. Wang and M. Chang, Nano Lett., 2004, 4, 613–617 CrossRef CAS.
- H. Cong, L. Li and S. Zheng, Polymer, 2015, 80, 146–158 CrossRef CAS.
- S. Ritzenthaler, F. Court, L. David, E. Girard-Reydet, L. Leibler and J. Pascault, Macromolecules, 2002, 35, 6245–6254 CrossRef CAS.
- H. Cong, L. Li and S. Zheng, Polymer, 2014, 55, 1190–1201 CrossRef CAS.
- Z. Heng, Y. Chen, H. Zou and M. Liang, RSC Adv., 2015, 5, 42362–42368 RSC.
- E. D. Bain, D. B. Knorr Jr, A. D. Richardson, K. A. Masser, J. Yu and J. L. Lenhart, J. Mater. Sci., 2016, 51, 2347–2370 CrossRef CAS.
- G. Giannakopoulos, K. Masania and A. Taylor, J. Mater. Sci., 2011, 46, 327–338 CrossRef CAS.
- K. Xiao and L. Ye, Polym. Eng. Sci., 2000, 40, 70–81 CAS.
- J. Chen, A. Kinloch, S. Sprenger and A. Taylor, Polymer, 2013, 54, 4276–4289 CrossRef CAS.
- K. Gam, M. Miyamoto, R. Nishimura and H. Sue, Polym. Eng. Sci., 2003, 43, 1635–1645 CAS.
- D. Quan and A. Ivankovic, Polymer, 2015, 66, 16–28 CrossRef CAS.
- H.-Y. Liu, G. Wang and Y.-W. Mai, Compos. Sci. Technol., 2012, 72, 1530–1538 CrossRef CAS.
- H. Y. Liu, G. T. Wang, Y. W. Mai and Y. Zeng, Composites, Part B, 2011, 42, 2170–2175 CrossRef.
- Q. Yan and Y. Zhao, J. Am. Chem. Soc., 2013, 135, 16300–16303 CrossRef CAS PubMed.
- K. Jankova, X. Chen, J. Kops and W. Batsberg, Macromolecules, 1998, 31, 538–541 CrossRef CAS.
- M. A. Hillmyer, P. M. Lipic, D. A. Hajduk, K. Almdal and F. S. Bates, J. Am. Chem. Soc., 1997, 119, 2749–2750 CrossRef CAS.
- P. M. Lipic, F. S. Bates and M. A. Hillmyer, J. Am. Chem. Soc., 1998, 120, 8963–8970 CrossRef CAS.
- M. Blanco, M. López, G. Kortaberria and I. Mondragon, Polym. Int., 2010, 59, 523–528 CrossRef CAS.
- Z. Xu and S. Zheng, Macromolecules, 2007, 40, 2548–2558 CrossRef CAS.
- R. Yu, S. Zheng, X. Li and J. Wang, Macromolecules, 2012, 45, 9155–9168 CrossRef CAS.
- F. Meng, S. Zheng, H. Li, Q. Liang and T. Liu, Macromolecules, 2006, 39, 5072–5080 CrossRef CAS.
- D. Hu and S. Zheng, Eur. Polym. J., 2009, 45, 3326–3338 CrossRef CAS.
- W. Fan, L. Wang and S. Zheng, Macromolecules, 2010, 43, 10600–10611 CrossRef CAS.
- B.-S. Kim and P. T. Mather, Macromolecules, 2002, 35, 8378–8384 CrossRef CAS.
- J. Liu, H.-J. Sue, Z. J. Thompson, F. S. Bates, M. Dettloff, G. Jacob, N. Verghese and H. Pham, Macromolecules, 2008, 41, 7616–7624 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |