
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Using steric bulk for selective recognition; blocking the binding site to differentiate guests†
Ryan N.
Robson
and
Frederick M.
Pfeffer
*
Centre for Chemistry and Biotechnology, School of Life and Environmental Sciences, Deakin University, Waurn Ponds, Victoria 3216, Australia. E-mail: fred.pfeffer@deakin.edu.au
First published on 16th June 2016
Abstract
Selectivity is demonstrated in a supramolecular host:guest system using a receptor with a non-linear binding site. For the “open” receptor 1 strong binding for both flexible and rigid guests was observed. Receptor 2, with a “blocked” binding site, also bound flexible guests effectively but its affinity for rigid guests was 50 fold lower.
The concepts of preorganisation and complementarity are central to supramolecular chemistry.1,2 It is accepted that through the careful application of these concepts that a host can be constructed with high selectivity for a specific guest.2
In Nature recognition is also achieved using non-linear binding sites, for example, the well-known thiazolidinedione insulin sensitizer rosiglitazone (marketed as Avandia) adopts a “U” shape when bound to its target the peroxisome proliferator-activated receptor (PPARγ).3 As long as the guest can adopt a low energy conformation to accommodate a “twisted” or “hindered” binding site then strong interactions, and in turn biological activity, can be achieved. The pharmaceutical guest rosiglitazone can assume the required non-linear conformation due to judiciously placed flexible methylene and ethylene links. Not all ligands, substrates, and even pharmaceutical guests can adopt such a conformation and in this way enhanced selectivity can be achieved.
In supramolecular chemistry a great deal of attention has been devoted to designing and synthesising perfectly matched (preorganised and complementary) host:guest systems. The use of hosts with non-linear binding sites/clefts to elicit selectivity amongst guests is as yet not well explored.
Herein we report two new hosts 1 and 2 (Fig. 1) for the binding of dicarboxylates. Host 2 was designed to favour the binding of flexible guests and its structure comprises a bisurea cleft that is centrally functionalised with a blocking unit that prevents linear “end-to-end” binding. While the synthesis of both 1 and 2 was broadly based on previous polynorbornyl bis-thiourea systems,4 a unique central unit was required that could be further functionalised. Hedaya diene 3 (Scheme 1), a diester synthesised by a tandem Diels–Alder reaction,5 was identified as ideally suited for further modification.6
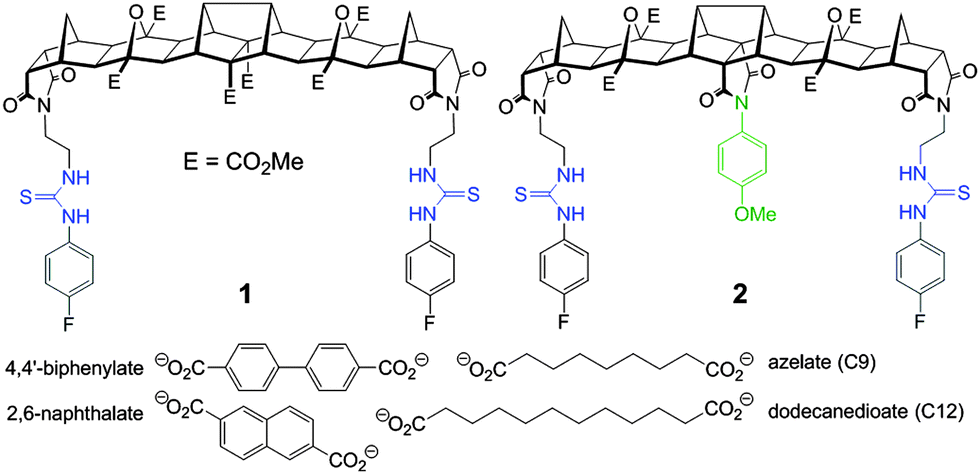 | ||
| Fig. 1 Structure of fused polynorbornane hosts 1 and 2 highlighting the sterically restricted cleft of host 2. Also shown are the four dicarboxylate guests used in this study. | ||
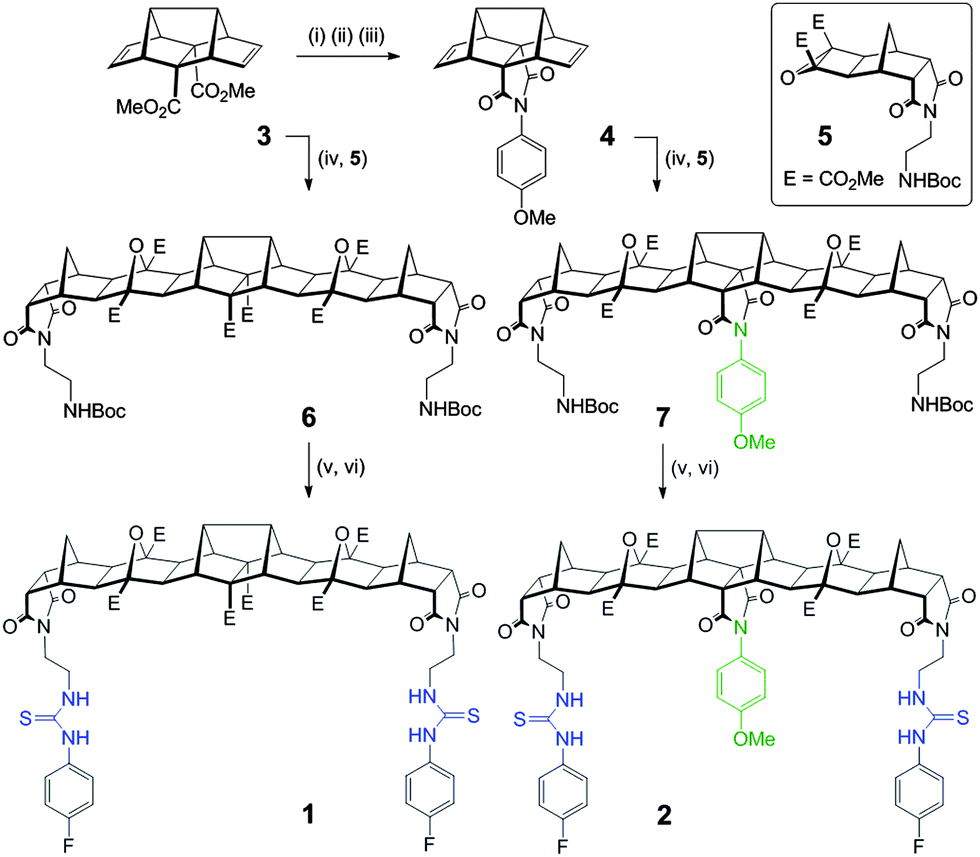 | ||
| Scheme 1 Synthesis of new hosts 1 and 2. Reagents and conditions: (i) KOH, CH3OH, 60 °C, 4 h, 89% (ii) Ac2O, CH2Cl2, 40 °C, 4 h, 99% (iii) 4-methoxyaniline, PhCH3, 150 °C (μw), 30 min, 21% (iv) 5 (2 equiv.), 150 °C (μw), 10 min, 65% for 6, 71% for 7 (v) 20% TFA in CH2Cl2, 4 h (vi) 4-fluorophenylisothiocyanate, NEt(iPr)2, CHCl3, 24 h, 77% for 1, 73% for 2. | ||
Saponification of diene 3 to the diacid was followed by dehydration (using Ac2O) to the corresponding anhydride. Reaction of the anhydride with 4-methoxyaniline gave the desired centrally functionalised imide 4 in 21% yield (Scheme 1, see ESI,† for a detailed description of all synthetic procedures). The microwave mediated 1,3 dipolar cycloaddition7 between either 3 or 4 and the carbonyl ylide generated from cyclobutane epoxide 58 afforded the fused polynorbornane frameworks 6 and 7 (65% and 71% yield respectively). A two-step protocol involving removal of the protecting groups (TFA/CH2Cl2) then immediate reaction with 4-fluorophenylisothiocyanate gave the new thiourea hosts 1 and 2 in 77% and 73% yield respectively. The new compounds were fully characterised using 1H, 13C and 19F NMR spectroscopy and for 2 the appearance of two sets of AB pairs in the 1H spectrum clearly indicated the central and end functionalised para substituted aromatics. Due to the primarily aliphatic nature of the frameworks the region of the spectrum containing the urea N–H resonances (δ = 9.53 & 7.67 ppm for 1 and δ = 9.58 & 7.71 ppm for 2) was free from other resonances making them ideally suited for 1H NMR titration experiments to evaluate binding.
Binding studies were performed using both the rigid dicarboxylates (2,6-naphthalate and 4,4′-biphenylate, Fig. 1) as well as flexible dicarboxylates of similar length (azelate (C9) and dodecanedioate (C12), Fig. 1). All 1H NMR titrations were performed in d6-DMSO at a host concentration of 2.5 μM. Binding constants (see Table 1) were determined using global fitting (Bindfit software).9 All isotherms with fitplots are provided in the ESI.†
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K1:1)a for hosts 1 and 2 with both rigid and flexible guests
K1:1)a for hosts 1 and 2 with both rigid and flexible guests
For new bisurea host 1 binding of both the rigid and flexible guests was comparable (Table 1). For example host 1 bound both naphthalate and azelate (C9) in a strong 1:1 fashion (logK = 3.7 and 4.3 respectively, Table 1). The results clearly indicate that the new bisurea host is well suited to the binding of larger dicarboxylates. These results also clearly reinforce the fact that for preorganised bisurea clefts that do not possess some form of additional means of binding site discrimination little selectivity is achievable between guests of similar length.
The binding behaviour of the new hosts 1 and 2 to both flexible guests was near identical. Indeed, both 1 and 2 bound azelate (C9) equally effectively and the binding isotherms (Fig. 2) and in turn the binding constants (logK = 4.3 and logK = 4.5 respectively) for this anion were near identical indicating that strong binding was taking place regardless of the central methoxyphenyl group (Fig. 3).
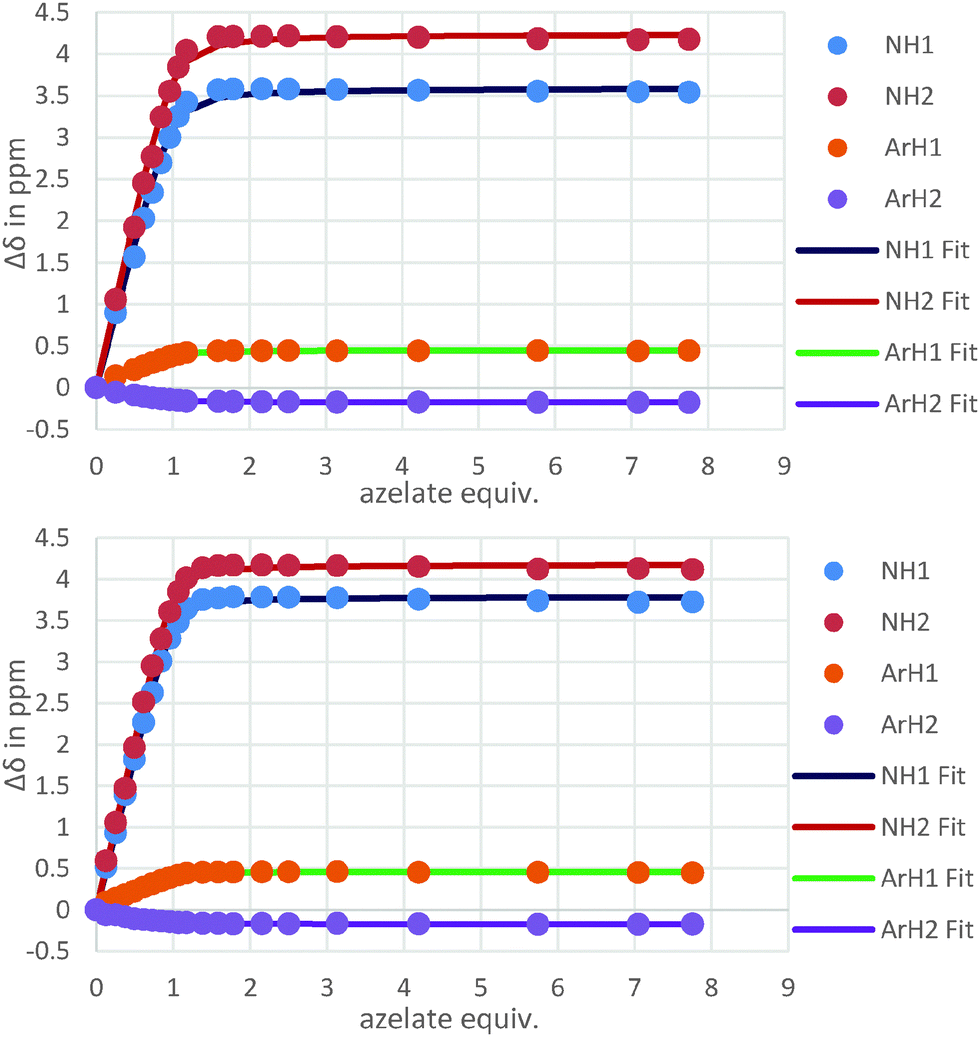 | ||
| Fig. 2 Isotherms resulting from the 1H NMR titrations of hosts 1 and 2 with azelate (C9). | ||
 | ||
| Fig. 3 Proposed binding conformation of hosts 1 and 2 with azelate (C9). | ||
While the central methoxyphenyl group in 2 had little influence on the binding of flexible guests it had significant influence on the binding of rigid dicarboxylates of similar length. Indeed, compared to host 1 the binding of the same guest was an order of magnitude less potent; for example the binding of 2 to 2,6-naphthalate was modest (logK = 2.8) whereas the unencumbered host 1 bound this guest much more effectively (logK = 3.7).
For host 2 the discrimination of flexible versus rigid guests was highlighted in the binding of azelate (C9) versus the similarly sized 2,6-naphthalate (logK = 4.5 versus logK = 2.8, Fig. 4). The difference in binding constants amounts to 50 fold selectivity for the flexible guest over the rigid.
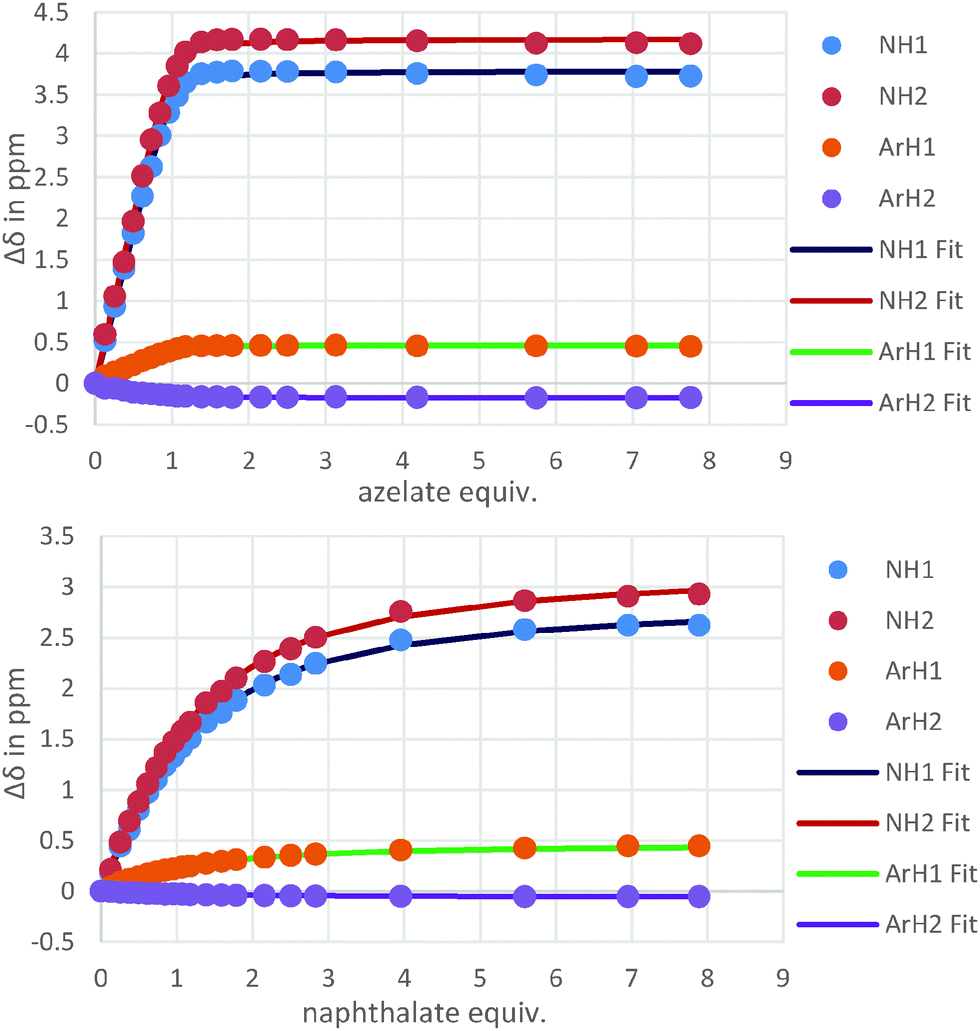 | ||
| Fig. 4 Isotherms resulting from the 1H NMR titrations of hosts 2 with azelate (C9) (top) and naphthalate (bottom) showing the clear difference in binding behaviour. | ||
Features of both host 2 and the guest make this possible. First, for the host, with ethylene linkers to the thiourea groups, a reorientation of the H bond donors to face away from the central cleft is possible and as such a non-linear binding site is readily created. Second and most importantly, guests such as azelate are highly flexible and as such can adopt a low energy “U” shape conformation that can interact with the host regardless of the central unit. Due to the central group of 2, the rigid guests, despite their appropriate size, cannot form strong linear hydrogen bonds with the thiourea groups (Fig. 5).
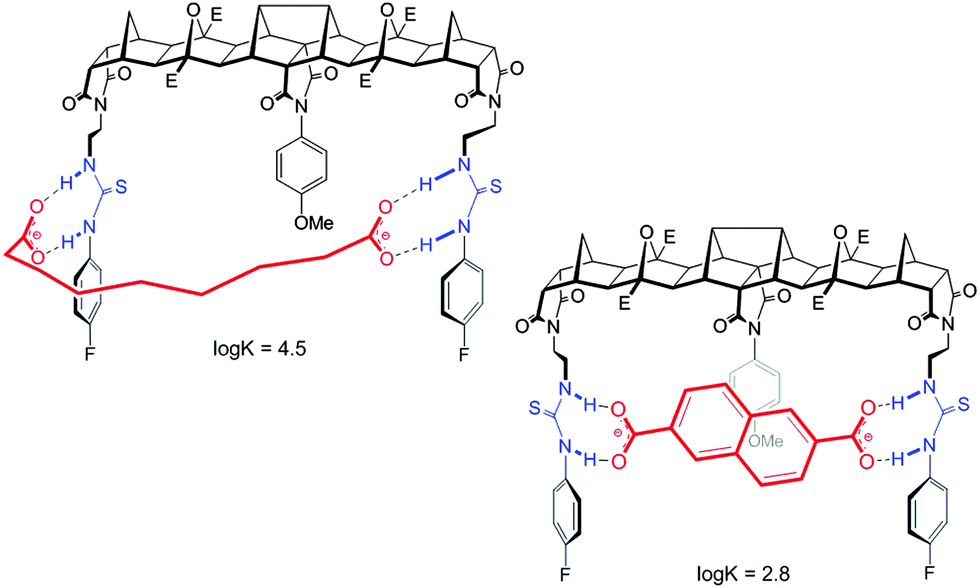 | ||
| Fig. 5 Proposed binding of host 2 with azelate (C9) and naphthalate. | ||
In conclusion we have successfully demonstrated that selectivity in supramolecular recognition can be achieved using a steric block to discriminate against guests that cannot adopt the “U” shape geometry required for strong binding. Such a general strategy is likely to be particularly useful in the binding of larger, typically more challenging substrates.
FMP thanks the Australian Research Council (ARCDP0140100227) and Centre for Chemistry and Biotechnology for financial support. The authors would also like to acknowledge the Australian Research Council for funding the Magnetic Resonance Facility at Deakin through LIEF grant LE110100141.
Notes and references
- Anion Receptor Chemistry, ed. J. L. Sessler, P. A. Gale and W.-S. Cho, The Royal Society of Chemistry, Cambridge, 2006 Search PubMed; Supramolecular Chemistry: from molecules to nanomaterials, ed. J. W. Steed and P. A. Gale, John Wiley and Sons, Chichester, 2012 Search PubMed.
- D. J. Cram, Science, 1988, 240, 760–767 CrossRef CAS PubMed; D. Cram, Angew. Chem., Int. Ed., 1988, 27, 1009–1020 CrossRef; K. P. McDonald, Y. Hua, S. Lee and A. H. Flood, Chem. Commun., 2012, 48, 5065–5075 RSC; H. T. Ngo, X. J. Liu and K. A. Jolliffe, Chem. Soc. Rev., 2012, 41, 4928–4965 RSC; E. M. Boyle, S. Comby, J. K. Molloy and T. Gunnlaugsson, J. Org. Chem., 2013, 78, 8312–8319 CrossRef PubMed; P. A. Gale, N. Busschaert, C. J. E. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205–241 RSC; P. Rios, T. S. Carter, T. J. Mooibroek, M. P. Crump, M. Lisbjerg, M. Pittelkow, N. T. Supekar, G.-J. Boons and A. P. Davis, Angew. Chem., Int. Ed., 2016, 55, 3387–3392 CrossRef PubMed.
- R. T. Nolte, G. B. Wisely, S. Westin, J. E. Cobb, M. H. Lambert, R. Kurokawa, M. G. Rosenfeldk, T. M. Willson, C. K. Glass and M. V. Milburn, Nature, 1998, 395, 137–143 CrossRef CAS PubMed; H. E. Xu, M. H. Lambert, V. G. Montana, K. D. Plunket, L. B. Moore, J. L. Collins, J. A. Oplinger, S. A. Kliewer, R. T. Gampe Jr., D. D. McKee, J. T. Moore and T. M. Willson, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 13919–13924 CrossRef PubMed.
- A. J. Lowe and F. M. Pfeffer, Chem. Commun., 2008, 1871–1873 RSC; A. J. Lowe and F. M. Pfeffer, Org. Biomol. Chem., 2009, 7, 4233–4240 Search PubMed; A. J. Lowe, B. M. Long and F. M. Pfeffer, J. Org. Chem., 2012, 77, 8507–8517 CrossRef CAS PubMed; A. J. Lowe, B. M. Long and F. M. Pfeffer, Chem. Commun., 2013, 49, 3376–3388 RSC; B. M. Long and F. M. Pfeffer, Chem. – Asian J., 2014, 9, 1091–1098 CrossRef PubMed.
- D. McNeil, B. R. Vogt, J. J. Sudol, S. Theodoropulos and E. Hedaya, J. Am. Chem. Soc., 1974, 96, 4673–4674 CrossRef CAS; L. A. Paquette, M. J. Wyvratt, H. C. Berk and R. E. Moerck, J. Am. Chem. Soc., 1978, 100, 5845–5855 CrossRef; R. T. Taylor, M. W. Pelter and L. A. Paquette, Organic Syntheses, Coll., 1993, vol. 8, p. 298 Search PubMed; R. T. Taylor, M. W. Pelter and L. A. Paquette, Organic Syntheses, Coll., 1990, vol. 68, p. 198 Search PubMed.
- M. Abou-Gharbia, U. R. Patel, M. B. Webb, J. A. Moyer, T. H. Andree and E. A. Muth, J. Med. Chem., 1988, 31, 1382–1392 CrossRef CAS PubMed; M. Golic, M. R. Johnston, D. Margetic, A. C. Schultz and R. N. Warrener, Aust. J. Chem., 2006, 59, 899–914 CrossRef; M. D. Duque, C. Ma, E. Torres, J. Wang, L. Naesens, J. Juarez-Jimenez, P. Camps, F. Javier Luque, W. F. DeGrado, R. A. Lamb, L. H. Pinto and S. Vazquez, J. Med. Chem., 2011, 54, 2646–2657 CrossRef PubMed; M. D. Johnstone, E. K. Schwarze, G. H. Clever and F. M. Pfeffer, Chem. – Eur. J., 2015, 21, 3948–3955 CrossRef PubMed.
- R. C. Foitzik, A. J. Lowe and F. M. Pfeffer, Tetrahedron Lett., 2009, 50, 2583–2584 CrossRef CAS.
- F. M. Pfeffer, T. Gunnlaugsson, P. Jensen and P. E. Kruger, Org. Lett., 2005, 7, 5357–5360 CrossRef CAS PubMed; F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Org. Biomol. Chem., 2007, 5, 1894–1902 Search PubMed.
- http://supramolecular.org, accessed 15th May, 2016; P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC; A. J. Lowe, F. M. Pfeffer and P. Thordarson, Supramol. Chem., 2012, 24, 585–594 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, 1H, 13C and 19F NMR spectra of compounds, as well as all titration isotherms. See DOI: 10.1039/c6cc04549a |
| This journal is © The Royal Society of Chemistry 2016 |