
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
M12L8 metallo-supramolecular cube with cyclotriguaiacylene-type ligand: spontaneous resolution of cube and its constituent host ligand†
Jonathan M.
Fowler
,
Flora L.
Thorp-Greenwood
,
Stuart L.
Warriner
,
Charlotte E.
Willans
and
Michaele J.
Hardie
*
School of Chemistry, University of Leeds, Woodhouse Lane, Leeds LS2 9JT, UK. E-mail: m.j.hardie@leeds.ac.uk
First published on 22nd June 2016
Abstract
The racemic ligand (±)-tris-(4-methylthiazolyl)cyclotriguaiacylene forms a homochiral crystalline Ag12L8 cube with spontaneous resolution. The ligand itself likewise crystallises in a chirally pure fashion in two clathrate complexes. Ag12L8 is the first example of a cyclotriguaiacylene-type coordination cube and a rare example of a M12L8-type metallo-cube.
Metallo-cages, also referred to as coordination cages, are three-dimensional, hollow architectures that often closely resemble Platonic or Archimedean solids. Investigation of their self-assembly, and applications as nano-scale host assemblies is a highly active area.1 One approach to a cube assembly is linking together eight corner units in an orthogonal manner. Most examples of such edge-directed coordination cage cubes have been M8L12 cages where the metal supplies the corner piece.2 An alternative design is a M12L8 assembly where the metal represents the cube edge and the corner piece is a ligand, though examples are much rarer.3 Cyclotriveratrylene (CTV) and its analogues have a bowl-shape tribenzo[a,d,g]-cyclononatriene core with the arene faces approximately orthogonal to one another which suggests that CTV-type ligands would be an ideal corner piece for an edge-directed cage. Indeed this concept has been demonstrated by Warmuth who reported an assembly where eight aldehyde-functionalised cyclotriguaiacylene (CTG) analogues were linked into a cube through reversible imine bond formation with twelve diaminobenzene units.4 Despite this organic precedent, directly analogous metallo-supramolecular examples with CTV or CTG-type ligands are not known. These ligand types do form an array of metallo-cages,5–8 but the closest examples to a cube are M6L8 stella octangula structures of octahedral symmetry,7 and a topologically complex self-entangled Pd4L4 cube.8 Likewise building tectons based on a conformationally-locked tribenzotriquinacene-core (akin to a bridge-head CTV) can be assembled into cube structures,9 but the only 3D metallo-cage examples of these ligands are M3L2 assemblies with trigonal bipyramidal shape.10 We report here the first example of a M12L8 cube assembled from a CTG-type ligand, noting CTG-ligands are chiral and this cube displays both chiral self-sorting in its assembly and in its crystallisation into two conglomerate clathrate complexes.
The racemic ligand (±)-tris-(4-methylthiazolyl)cyclotriguaiacylene L was synthesised in 80% yield through reaction of 4-(chloromethyl)thiazole hydrochloride with cyclotriguaiacylene in the presence of base, Scheme 1. The 1H NMR spectrum is characteristic of the C3-symmetric bowl-shaped cavitand with the bridging endo and exo methylene protons of the tribenzo[a,d,g]-cyclononatriene core appearing as doublets at 4.71 and 3.49 ppm respectively. The structures of two clathrate complexes of L, namely L·4(H2O) and L·3(CH3NO2), were determined by single crystal X-ray techniques.‡ Each complex crystallises in a chiral trigonal/hexagonal space group with an asymmetric unit composed of solvent molecules and one third of an L ligand, with the ligand having crystallographic three-fold symmetry. For both complexes, the thiazole-methyl groups of the ligand extending outwards in-plane with the CTG-arene faces, at Cthiazole–CH2–O–Caryl torsion angle ca. 179°, however the rotation of the thiazole is distinct for the two complexes, Fig. 1.
 | ||
| Scheme 1 Synthetic route to (±)-tris-(4-methylthiazolyl)cyclotriguaiacylene. | ||
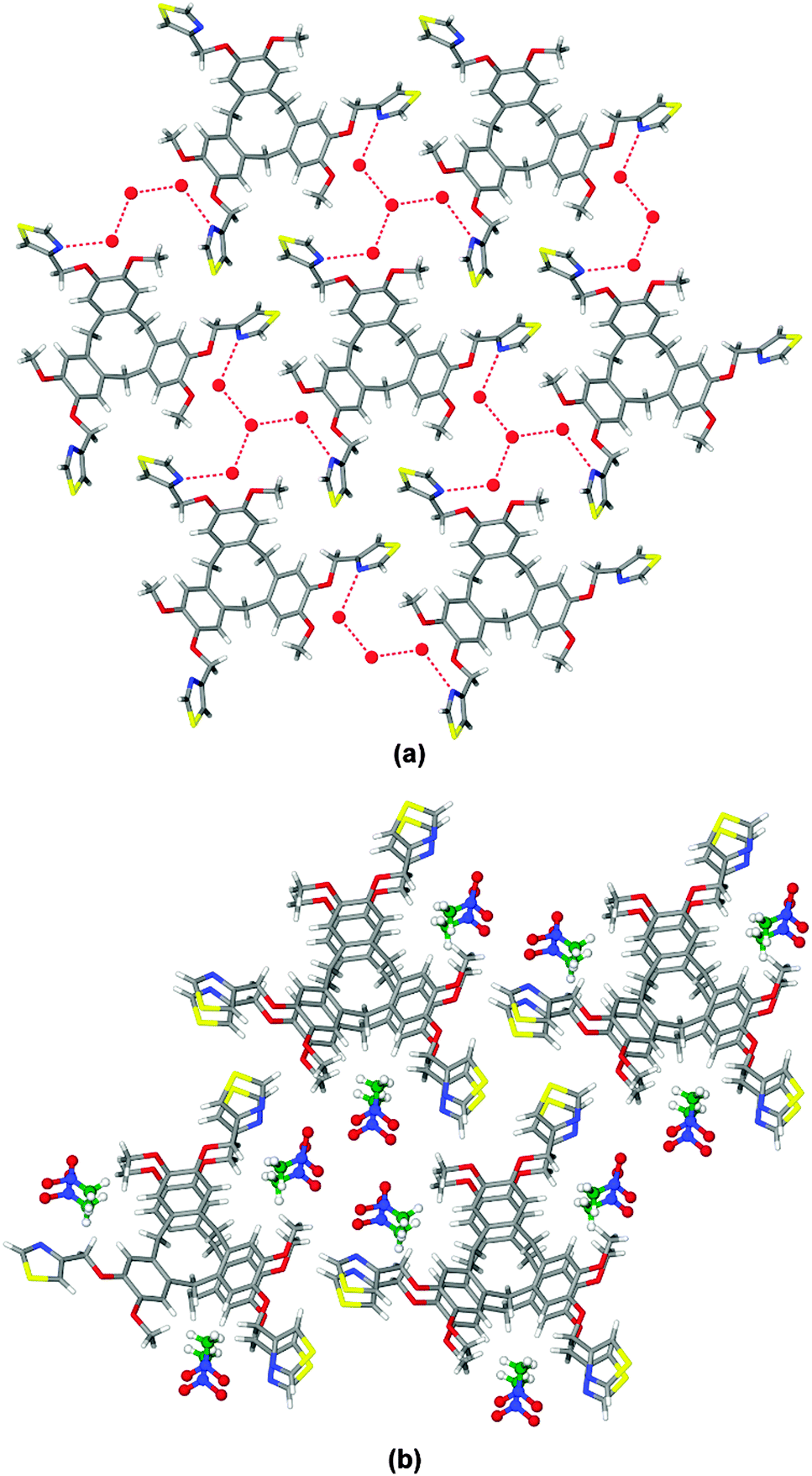 | ||
| Fig. 1 Clathrate complexes of L. (a) 2D hydrogen bonded network from crystal structure of L·4(H2O) viewed down c axis. Water shown as spheres, N⋯O and O⋯O separations are 2.895 and 2.918 Å respectively. (b) Packing diagram of L·3(CH3NO2) with CH3NO2 shown in ball-and-stick. | ||
In L·4(H2O) there are two types of water molecules with one sited on a three-fold rotation axis. A network of hydrogen bonds forms between the thiazole groups and waters of crystallisation (Fig. 1a) such that a tetrameric cluster of water molecules connects to three L ligands, and vice versa to form a 2D network with 63 topology. In the overall lattice layers of networks pack in an ABAB arrangement with a rotated bowl-in-bowl stacking of the cavitand ligands (see ESI†).
Bowl-in-bowl stacking of ligands also occurs in complex L·3(CH3NO2), however here the ligands are perfectly aligned with a separation of 4.4 Å, Fig. 1b. The CH3NO2 guest molecules occupy lattice positions within the structure. Both clathrate complexes exhibit spontaneous resolution as all L ligands within each structure are the same enantiomer. Spontaneous resolution on crystallisation is known as conglomerate formation.
Vapour diffusion of diethyl ether into a dimethylformamide (DMF) solution of L and AgX (X = ReO4−, BF4−) gave crystals of the complexes [Ag12(L)8]·12ReO4·n(DMF) 1 and [Ag12(L)8]·12BF4·n(DMF) 2.§ The structures of complexes 1 and 2 were determined by single crystal crystallography and are isostructural, being solved in the chiral cubic space group F432.‡ The asymmetric unit of complex 1 comprises one third of ligand L, a Ag(I) cation on a 2-fold axis and fragments of ReO4− anions, one on a 3-fold axis and the other, disordered and with low occupancy, sited on a 4-fold axis.§
As for the ligand structures, ligand L in 1 has crystallographic 3-fold symmetry, however with a quite different conformation where the thiazole-methyl groups are folded inwards above the cavitand bowl, at Cthiazole–CH2–O–Caryl torsion angle −83.6°. As expected for a thiazole, only the nitrogen heterocyclic atoms act as donors to the metal, with each thiazole binding to a separate Ag(I) cation. The Ag(I) cations have near linear coordination to thiazole groups of two L ligands at Ag–N distance 2.151(15) Å and N–Ag–N angle 175.9(8)°. Each L1 ligand thus bridges between three metals and each metal links two L1 ligands. These linkages result in a discrete [Ag12L8]12+ cubic cage with the L1 ligands at the corners, Fig. 2a. The Ag(I) cations are located at the internal rather than external edge of the cube due to the conformation adopted by the thiazole-methyl groups. The S-heteroatoms of each thiazole moiety are located at the faces of the cube, and the space-filling view, Fig. 2b, shows small windows into the cubes which are bounded by these S atoms (S⋯S distances across diagonal of window is ca. 6 Å). Space-filling also shows that Ag(I) centres are well within the cube, and thus protected from additional coordination exo to the cage. The Ag(I) cations are arranged in a cuboctahedron with Ag⋯Ag separation of 8.71 Å. The overall size of the cube is ca. 27 Å across the body diagonal measured between centres of the –(CH2)3– cavitand ligands.
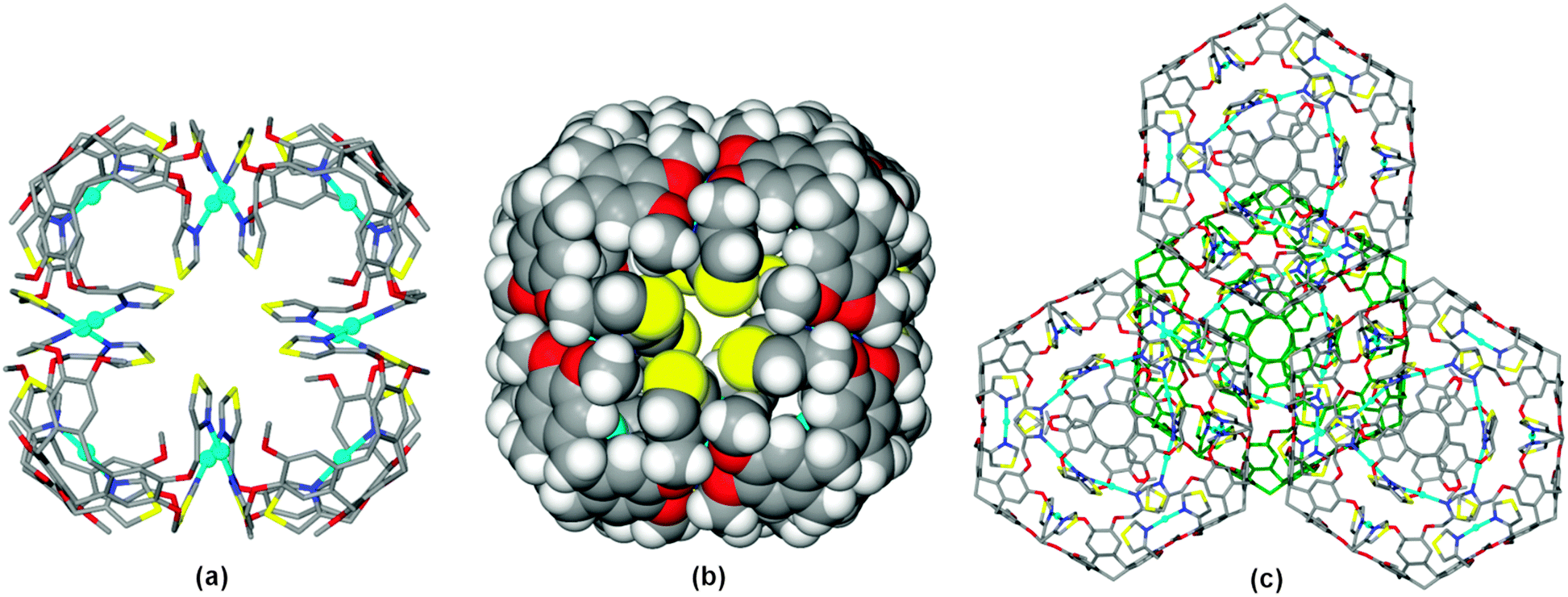 | ||
| Fig. 2 From the crystal structure of [Ag12(L)8]·12ReO4·n(DMF) 1 showing (a) the [Ag12(L)8]12+ cube; (b) space-filling view; (c) partial packing of cubes through π–π stacking interactions into a cubic close packed array. | ||
The eight L ligands within each [Ag12L8]12+ cube are the same ligand enantiomer, hence the cube displays homochiral recognition or chiral self-sorting. Homochiral self-sorting of a racemic mixture of enantiomeric ligands during metal-directed self-assembly processes has been previously reported, both for helicate systems,11 and also for cage-like assemblies,6–8,12 including with CTG-type ligands.6–8 Complex 1 displays a further level of spontaneous chiral resolution as it crystallises as a conglomerate where each crystal of 1 contains only one enantiomer of the chiral [Ag12L8]12+ cube. The bulk sample contains crystals of both enantiomers, and coincidentally the selected crystal studied for the isostructural complex 2 was of the opposite enantiomer to that selected for 1 (see ESI†). Spontaneous resolution is not common for metallo-supramolecular systems. Resolution of metallo-supramolecular species with achiral ligands and helical chirality at the metal has been reported for a M3L3 helicate13 and recently Rissanen and co-workers reported the first example for a 3D metal-linked coordination cage.14 A handful of examples have been reported for simpler CTG-type systems, namely a metalled hemi-cryptophane,15 a CTG-type coordination polymer,16 a trinuclear Re(I)3L complex17 as well as organic cage cryptophanes.18
There are extensive π–π stacking interactions between [Ag12(L)8]12+ cubes in the crystal lattice, with each arene of the tribenzo[a,d,g]-cyclononatriene ligand core forming a face-to-face interaction at centroid separation 3.81 Å. The cubes form a cubic close packed array, Fig. 2c, with π-stacking along the cube edges such that each cube interacts with twelve others.
There are two crystallographically distinct ReO4− positions in the crystal lattice of complex 1, one is fully occupied on a 3-fold axis and is external to the [Ag12L8]12+ cube, whilst the other has less than full occupancy, and is located on a 4-fold axis (hence is symmetry-disordered) and located inside the cube, aligned with the facial windows. The very high symmetry of the structure is reflected in the Russian Doll arrangement of prisms that can be identified, with the internal ReO4− anions forming an octahedron which is internal to the cuboctahedron of Ag(I) sites, itself part of the [Ag12L8]12+ cube, which is surrounded by a rhombicuboctahedron of external ReO4− anions, Fig. 3 and Fig. S19 (ESI†).
 | ||
| Fig. 3 Matryoshka (Russian Doll) arrangement of prisms-in-prisms of complex 1 with octahedron of endo-ReO4 sites shown in dark red; cuboctahedron of Ag(I) sites in light blue; cube of ligand positions in green; and rhombicuboctahedron of exo-ReO4 sites in purple. | ||
Thermogravimetric analysis of complexes 1 and 2 showed a gradual weight loss to ca. 190–200 °C of ca. 7–10% for 1 and 2 respectively (see ESI†), consistent with the loss of approximately 10 DMF solvent molecules from inside the [Ag12L8]12+ cubes from 1 and 12 DMF molecules from 2 which are solvation levels easily accommodated within the cube, with an estimated internal volume of ca. 5000 Å3 (excluding anions) using a 1.2 Å probe.19
Crystals of the cube can be re-dissolved in coordinating solvents such as DMSO but this leads to disassociation of the complex. The 1H NMR spectrum of L and AgBF4 in d7-DMF was very broad (see Fig. S9, ESI†), and ESI-MS of a DMF solution of complex 1 showed only m/z peaks corresponding to [AgL]+ and [AgL2]+ species (Fig. S10, ESI†).
In summary, the racemic ligand (±)-tris-(4-methylthiazolyl)cyclotriguaiacylene crystallises in a chiral fashion as a hydrate, and Ag(I) complexes of the same ligand form a Ag12L8 cube structure that shows dual levels of chiral sorting, with both chiral self-sorting of the cubes and spontaneous resolution of the chiral cubes on crystallisation for both ReO4− and BF4− salts.
This work was supported by the EPSRC through equipment grant EP/K039202/1, through an EPSRC PhD studentship (JMF, DTG-2014), and the Leverhulme Trust (RPG-2014-148). We thank Algy Kazlauciunas for EDX and TGA measurements, and Tanya Marinko-Covell and Stephen Boyer for microanalysis. Data accessibility: data supporting this study are available in supplementary information and at http://doi.org/10.5518/87.
Notes and references
- For recent reviews, see: L. Chen, Q. Chen, M. Wu, F. Jiang and M. Hong, Acc. Chem. Res., 2015, 48, 201 CrossRef CAS PubMed; S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419 RSC; J. J. Henkelis and M. J. Hardie, Chem. Commun., 2015, 51, 11929 RSC; M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848 RSC; T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734 CrossRef PubMed; K. Harris and M. Fujita, Chem. Commun., 2013, 49, 6703 RSC; M. D. Ward, Chem. Commun., 2009, 4487 RSC.
- For example: C. Browne, S. Brenet, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 1944 CrossRef CAS PubMed; I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, L. Russo, W. Clegg and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 15167 CrossRef PubMed; J.-Y. Xu, X. Qiao, H.-B. Song, S.-P. Yan, D.-Z. Liao, G. Song, Y. Journaux and J. Cano, Chem. Commun., 2008, 6414 RSC; Y. Liu, V. Kravtsov, R. D. Walsh, P. Poddar, H. Srikanth and M. Eddaoudi, Chem. Commun., 2004, 2807 Search PubMed; Z. R. Bell, L. P. Harding and M. D. Ward, Chem. Commun., 2003, 2432 RSC; S. Roche, C. Haslam, S. L. Heath and J. A. Thomas, Chem. Commun., 1998, 1681 RSC.
- M. Wang, C. Wang, X.-Q. Hao, X. Li, T. J. Vaughn, Y.-Y. Zhang, Y. Yu, Z.-Y. Li, M.-P. Song, H.-B. Yang and X. Li, J. Am. Chem. Soc., 2014, 136, 10499 CrossRef CAS PubMed; C. Wang, X.-Q. Hao, M. Wang, C. Guo, B. Xu, E. N. Tan, Y.-Y. Zhang, Y. Yu, Z.-Y. Li, H.-B. Yang, M.-P. Song and X. Li, Chem. Sci., 2014, 5, 1221 RSC; B. F. Abrahams, S. J. Egan and R. Robson, J. Am. Chem. Soc., 1999, 121, 3535 CrossRef.
- D. Xu and R. Warmuth, J. Am. Chem. Soc., 2008, 130, 7520 CrossRef CAS PubMed.
- J. J. Henkelis, C. J. Carruthers, S. E. Chambers, R. Clowes, A. I. Cooper, J. Fisher and M. J. Hardie, J. Am. Chem. Soc., 2014, 136, 14393 CrossRef CAS PubMed; B. F. Abrahams, B. A. Boughton, N. J. FitzGerald, J. L. Holmes and R. Robson, Chem. Commun., 2011, 47, 7404 RSC; T. K. Ronson, H. Nowell, A. Westcott and M. J. Hardie, Chem. Commun., 2011, 47, 176 RSC; B. F. Abrahams, N. J. FitzGerald and R. Robson, Angew. Chem., Int. Ed., 2010, 49, 2896 CrossRef PubMed; A. Westcott, J. Fisher, L. P. Harding, P. Rizkallah and M. J. Hardie, J. Am. Chem. Soc., 2008, 130, 2950 CrossRef PubMed; Z. Zhong, A. Ikeda, S. Shinkai, S. Sakamoto and K. Yamaguchi, Org. Lett., 2001, 3, 1085 CrossRef PubMed.
- A. Schaly, Y. Rousselin, J.-C. Chambron, E. Aubert and E. Espinosa, Eur. J. Inorg. Chem., 2016, 832 CrossRef CAS; J. J. Henkelis, T. K. Ronson, L. P. Harding and M. J. Hardie, Chem. Commun., 2011, 47, 6560 RSC; C. J. Sumby, J. Fisher, T. J. Prior and M. J. Hardie, Chem. – Eur. J., 2006, 12, 2945 CrossRef PubMed.
- J. J. Henkelis, J. Fisher, S. L. Warriner and M. J. Hardie, Chem. – Eur. J., 2014, 20, 4117 CrossRef CAS PubMed; T. K. Ronson, J. Fisher, L. P. Harding and M. J. Hardie, Angew. Chem., Int. Ed., 2007, 46, 9086 CrossRef PubMed.
- T. K. Ronson, J. Fisher, L. P. Harding, P. J. Rizkallah, J. E. Warren and M. J. Hardie, Nat. Chem., 2009, 1, 212 CrossRef CAS PubMed.
- S. Klotzbach and F. Beuerle, Angew. Chem., Int. Ed., 2015, 54, 10356 CrossRef CAS PubMed; J. Strübe, B. Neumann, H.-G. Stammler and D. Kuck, Chem. – Eur. J., 2009, 15, 2256 CrossRef PubMed.
- J. Wei, Z.-M. Li, X.-J. Jin, X.-J. Yao, X.-P. Cao, H.-F. Chow and D. Kuck, Chem. – Asian J., 2015, 10, 1150 CrossRef CAS PubMed.
- O. Gidron, M. Jirásek, N. Trapp, M.-O. Ebert, X. Zhang and F. Diederich, J. Am. Chem. Soc., 2015, 137, 12502 CrossRef CAS PubMed; C. Gütz, R. Hovorka, N. Struch, J. Bunzen, G. Meyer-Eppler, Z.-W. Qu, S. Grimme, F. Topíc, K. Rissanen, M. Cetina, M. Engeser and A. Lützen, J. Am. Chem. Soc., 2014, 136, 11830 CrossRef PubMed; M. Boiocchi and L. Fabbrizzi, Chem. Soc. Rev., 2014, 43, 1835 RSC; M. Huntin, C. J. Cramer, L. Gagliardi, A. R. M. Shahi, G. Bernardinelli, R. Cerny and J. R. Nitschke, J. Am. Chem. Soc., 2007, 129, 8774 CrossRef PubMed; S. G. Telfer, T. Sato, R. Kuroda, J. Lefebvre and D. B. Leznoff, Inorg. Chem., 2004, 43, 421 CrossRef PubMed.
- S. A. Boer and D. R. Turner, Chem. Commun., 2015, 51, 17375 RSC; L.-L. Yan, C.-H. Tan, G.-L. Zhang, L.-P. Zhou, J.-C. Bünzli and Q.-F. Sun, J. Am. Chem. Soc., 2015, 137, 8550 CrossRef CAS PubMed; C. Gütz, R. Hovorka, G. Schnakenburg and A. Lützen, Chem. – Eur. J., 2013, 19, 10890 CrossRef PubMed; C. Maeda, T. Kamada and A. Osuka, Coord. Chem. Rev., 2007, 251, 2743 CrossRef.
- R. Krämer, J.-M. Lehn, A. de Cian and J. Fischer, Angew. Chem., Int. Ed. Engl., 1993, 32, 703 CrossRef.
- P. Bonakdarzadeh, F. Pan, E. Kalenius, O. Jurček and K. Rissanen, Angew. Chem., Int. Ed., 2015, 54, 14890 CrossRef CAS PubMed.
- Y. Makita, K. Sugimoto, K. Furuyoshi, K. Ikeda, T. Fujita, S.-I. Fujiwara and A. Ogawa, Supramol. Chem., 2011, 23, 269 CrossRef CAS.
- J. J. Henkelis, S. A. Barnett, L. P. Harding and M. J. Hardie, Inorg. Chem., 2012, 51, 10657 CrossRef CAS PubMed.
- F. L. Thorp-Greenwood, V. E. Pritchard, M. P. Coogan and M. J. Hardie, Organometallics, 2016, 35, 1632 CrossRef CAS.
- M. A. Little, J. Donkin, J. Fisher, M. A. Halcrow, J. Loder and M. J. Hardie, Angew. Chem., Int. Ed., 2012, 51, 764 CrossRef CAS PubMed; C. Givelet, J. Sun, D. Xu, T. J. Emge, A. Dhokte and R. Warmuth, Chem. Commun., 2011, 47, 4511 RSC.
- A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, A46, C34 Search PubMed.
- T. K. Ronson and M. J. Hardie, CrystEngComm, 2008, 10, 1731 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR, MS, IR spectra, TGA, EDX, pXRD, details of structure refinements and additional diagrams. CCDC 1470477–1470479 and 1478562. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc04130b |
| ‡ Crystal data. L·4(H2O): C36H41N3O10S3, Mr = 771.9, hexagonal, a = b = 14.5223(19) Å, c = 10.2701(12) Å, V = 1875.8(4) Å3, space group P63, Z = 2, λ = 1.54184 Å, θmax = 73.42°, 158 parameters, 1 restraint, R1 = 0.0886 (for 1394 data I > 2σ(I)), wR2 = 0.2598 (all 2239 data), Flack parameter = −0.07(7). CCDC 1470477. L·3(CH3NO2): C39H42N6O12S3, Mr = 882.97, trigonal (hexagonal), a = b = 28.138(3) Å, c = 4.4445(5) Å, V = 3047.4(5) Å3, space group R3, Z = 3, λ = 1.54184 Å, θmax = 73.82°, 183 parameters, 1 restraint, R1 = 0.0719 (for 1635 data I > 2σ(I)), wR2 = 1873 (all 2150 data), Flack parameter = 0.08(5). CCDC 1478562. Complex 1: C288H264Ag12N24O96Re12S24, Mr = 9895.52, cubic, a = b = c = 35.7237(4) Å, V = 45590(9) Å3, space group F432, Z = 4, λ = 1.54184 Å, θmax = 73.74°, 168 parameters, 4 restraints, R1 = 0.0847 (for 2667 data I > 2σ(I)), wR2 = 0.2886 (all 3794 data), Flack parameter = 0.03(4). CCDC 1470478. Complex 2: C288H264Ag12B12F48N24O48S24, Mr = 7934.84, cubic, a = b = c = 35.4548(7) Å, V = 44568(1) Å3, space group F432, Z = 4, λ = 1.54184 Å, θmax = 73.64°, 152 parameters, 42 restraints, R1 = 0.0718 (for 1639 data I > 2σ(I)), wR2 = 0.2256 (all 2572 data), Flack parameter = 0.15(3). Squeeze was employed for complex 2.18 CCDC 1470479. |
| § Large crystals of complexes 1 and 2 can be obtained repeatedly in good yield in ca. 1 week from crude L which contains a slight HCl contamination. We have previously noted such contamination in a similar ligand synthesis.20 Use of highly pure L only gives a small number of very small crystals of 1 or 2 after a month alongside crystals of the ligand. EDX measurements on individual single crystals do not show Cl with Re, Ag and S the only heavy elements. |
| This journal is © The Royal Society of Chemistry 2016 |