
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bead-like structures and self-assembled monolayers from 2,6-dipyrazolylpyridines and their iron(II) complexes†
Laurynas
Pukenas
a,
Florence
Benn
a,
Edmund
Lovell
a,
Amedeo
Santoro
b,
Laurence J.
Kershaw Cook
b,
Malcolm A.
Halcrow
*b and
Stephen D.
Evans
*a
aSchool of Physics and Astronomy, E. C. Stoner Building, University of Leeds, Leeds, LS2 9JT, UK. E-mail: s.d.evans@leeds.ac.uk; Fax: +44 113 343 3900; Tel: +44 113 343 3852
bSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK. E-mail: m.a.halcrow@leeds.ac.uk; Fax: +44 113 343 6565; Tel: +44 113 343 6506
First published on 27th May 2015
Abstract
Drop-casting acetone solutions of [Fe(bpp)2][BF4]2 (bpp = 2,6-di[pyrazol-1-yl]pyridine) onto a HOPG surface affords unusual chain-of-beads nanostructures. The beads in each chain are similar in size, with diameters in the range of 2–6 nm and heights of up to 10 Å, which is consistent with them containing between 10–50 molecules of the compound. The beads can be classified into two types, which exhibit different conduction regimes by current-imaging tunnelling spectroscopy (CITS) which appear to correlate with their positions in the chains, and may correspond to molecules containing high-spin and low-spin iron centres. Similarly drop-cast films of the complex on a gold surface contain the intact [Fe(bpp)2][BF4]2 compound by XPS. 4-Mercapto-2,6-di[pyrazol-1-yl]pyridine undergoes substantial decomposition when deposited on gold, forming elemental sulfur, but 4-(N-thiomorpholinyl)-2,6-di[pyrazol-1-yl]pyridine successfully forms SAMs on a gold surface by XPS and ellipsometry.
Introduction
Spin-crossover compounds, which undergo a transition between electronic spin states under the influence of temperature, pressure or light, are a versatile form of molecular switch.1–5 They are molecular complexes or coordination polymers of first row transition ions with a d4–d7 electron count, and an intermediate ligand field that makes their high-spin and low-spin states close in energy. The phenomenon is common in iron(II),6 iron(III)7 and cobalt(II)8 chemistry, although examples containing other transition ions are also well known. The spin-crossover event in a solid material leads to concomitant changes to a number of its physical properties, including its magnetic moment,9 colour,9 electrical resistance10,11 and dielectric constant.12 It can also lead to unusual thermal expansion behaviour around the temperature of the transition.13Much recent effort has been devoted to investigating spin-transition compounds in nanoscale materials.14–16 Nanoparticle preparations and films of ≥50 nm in diameter generally show comparable spin-state switching properties to the corresponding bulk materials. At smaller thicknesses the switching performance is influenced by inactive metal centres at the surface of the nanostructures. This usually leads to an attenuation of the switching performance as the size is reduced, although a recovery in switching cooperativity was recently detected in 2–3 nm particles which was attributed to the rigidity of the particle surface.17
Most of these studies have been performed using coordination polymer materials,18 and the nanochemistry of molecular spin-crossover compounds is less advanced. Best studied is [Fe(H2Bpz)2(phen)] (pz = pyrazolyl, phen = 1,10-phenanthroline), which has been fabricated into homogeneous films on a number of surfaces by vacuum deposition, at thicknesses ranking from 102 nm to sub-monolayer coverage.19–25 Isolated molecules of [Fe(H2Bpz)2(phen)] on Au(111) can exist in either their high- or low-spin states by STM, but appear to be unable to switch between the two24,25 (in some cases, this could reflect partial decomposition of the molecules on the substrate22). In contrast monolayer, bilayer or multi-layer films can undergo the expected spin-state switching under a thermal, optical or charge stimulus, although the conditions leading to the onset of switching in the films have varied between studies. Functional, polycrystalline films and surface patterns of other molecular spin-crossover switches have also been produced by vacuum deposition,26–28 spin-coating,10,29 drop-casting30,31 and lithographic methods.32,33 In contrast, self-assembled monolayers (SAMs) of spin-crossover complexes have not yet been reported, although progress towards that end has been achieved using a different type of molecular spin-state switch.34
Much of our work has focussed on the [Fe(bpp)2]2+ family of complexes, where bpp is 2,6-di(pyrazol-1-yl)pyridine or a derivative of it (Scheme 1).35 The synthetic flexibility of the bpp ligand allows a variety of substituents to be appended to the complex molecule, while retaining its spin-crossover functionality.35,36 Derivatives of [Fe(bpp)2]2+ have been fabricated into surface nanostructures10,30,31,33 and single-molecule junctions.37 Of particular note is a report from Grohmann et al., who drop-cast [Fe(L1)2][BF4]2 onto HOPG.30 This afforded unusual “chain-of-beads” structures on the graphite surface, each bead having a diameter of 2 nm by scanning-tunnelling microscopy (STM). That corresponds to a small cluster of molecules if the beads contain the intact complex. Individual beads gave different I/V response curves by current imaging tunnelling spectroscopy (CITS), which were proposed to arise from molecules in their high- and low-spin states. Some beads were observed to switch between the two forms over a period of minutes, under a constant bias, in an apparently random manner.38 Comparable data from two salts of the unsubstituted analogue [Fe(bpp)2]2+ were also briefly described by the same group.38
 | ||
| Scheme 1 The structure of the [Fe(bpp)2]2+ family of complexes, and the bpp ligand derivatives discussed in this work. | ||
We report here two investigations of the surface chemistry of [Fe(bpp)2]2+ derivatives. First is a study of drop-casting [Fe(bpp)2][BF4]239 on HOPG, which has afforded comparable surface structures to those of [Fe(L1)2][BF4]2.30 In addition to STM and CITS data, we also report the first XPS measurements that support the chemical composition of the drop-cast nanostructures. Second, is an investigation of SAMs formed from two bpp derivatives containing sulfur substituents, L2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 40 and the new ligand L3.
40 and the new ligand L3.
Results and discussion
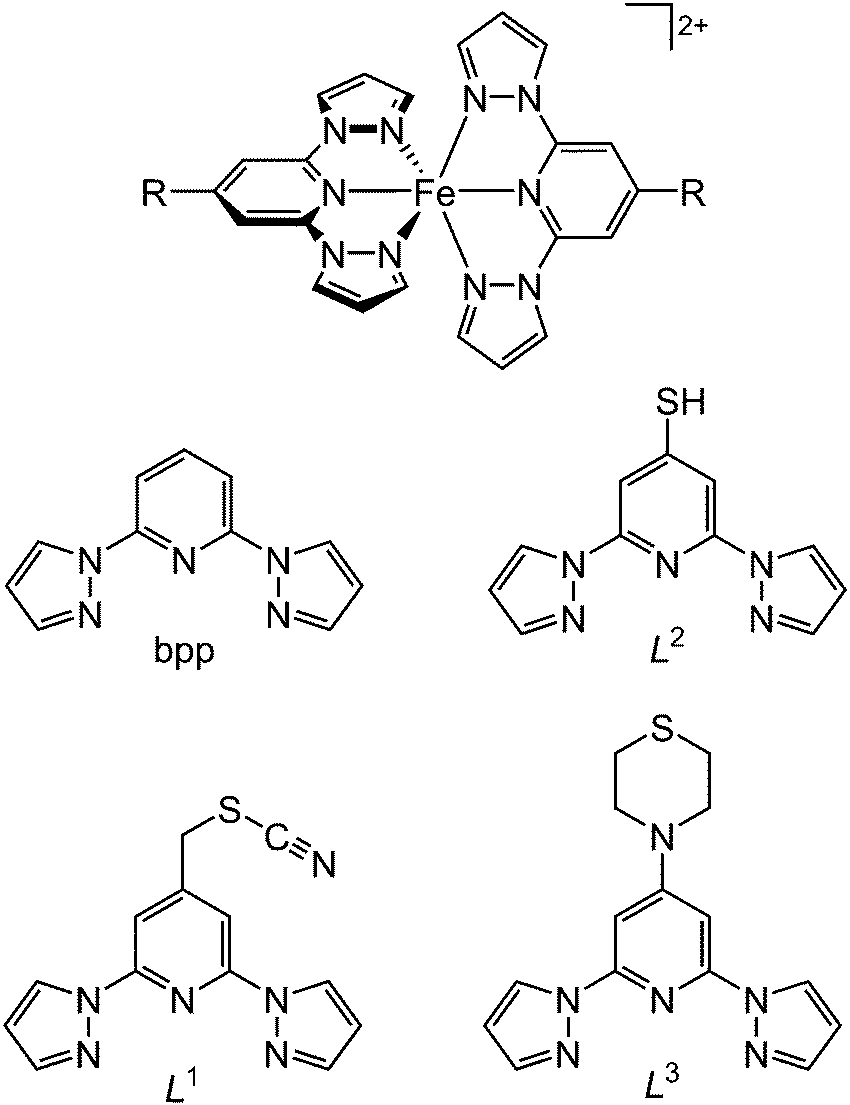
Acetone solutions of [Fe(bpp)2][BF4]239 were drop-cast onto a freshly cleaved HOPG surface, and the resultant nanostructures were examined by STM. The drop-casting procedure was found to consistently lead to “chain of beads” structures on the graphite surface (Fig. 1). The chains were up to 1–2 μm in length, and while the bead structures in each chain were reasonably homogeneous, there were some differences between the beads in different chains. The diameters of beads in different sets varied between 2–6 nm, and their heights ranged from 4 Å up to as much as 10 Å (Fig. 1). Peak-to-peak separations of between 5 nm and 12 nm were evident in different chains, although beads were sometimes touching or sometimes well-separated in the chains depending on the bead size (Fig. 2).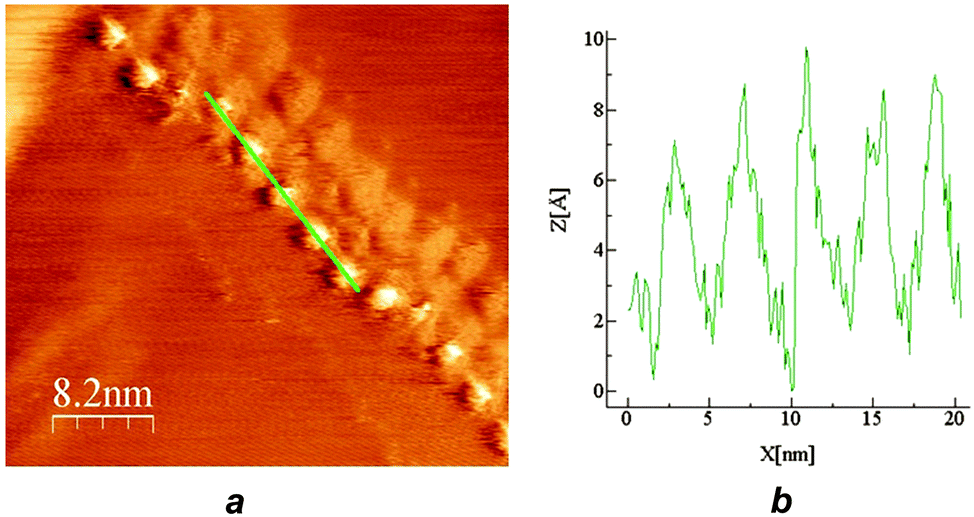 | ||
| Fig. 1 (a) STM image of a representative chain of beads from [Fe(bpp)2][BF4]2 drop cast onto HOPG, with widths of 2.5 nm and heights of 7 Å. (b) Profile over the five beads highlighted in (a). | ||
 | ||
| Fig. 2 STM images at three different scales of the same set of beads. The underlying graphite lattice can clearly be seen in (c). These beads are comparable in size to those in Fig. 1, but are more widely spaced. | ||
Other recurring features in the deposited complexes include chains traversing across substrate step edges, and chains that change direction at distinct, consistent angles (ESI†). However, different types of structure were observed at the ends of chains, including an abrupt termination of a chain associated with a sequence of HOPG step edges, and a more gradual petering out of the beads on an apparently featureless region of the surface (ESI†). Although the beads superficially resemble moiré patterns on the HOPG surface,41 the bead structures are clearly distinguishable from moiré patterns by STM and by CITS (see below), in images that contain both features (ESI†).
Several different chains-of-beads were scanned using CITS at 290 K and 203 K where, based on the properties of the bulk complex, they would be expected to be in the high-spin and low-spin state respectively.39 In general, there is a marked difference between the spectroscopy of the beads and the substrate, with some beads being darker (type I) and some brighter in current (type II).38 The difference between type I and type II beads is also evident in the I/V response, since type II beads exhibit a larger current than type I (Fig. 3). At room temperature this difference was only evident at larger biases of >±0.15 V, but improved sensitivity between the beads at lower biases was observed at 203 K. In both the images and the I/V curves, the difference between the types of beads was more pronounced at negative than at positive bias (Fig. 3 and ESI†).
 | ||
| Fig. 3 Left: (a) an STM image taken at 203 K, containing a mixture of type I and type II beads, with corresponding spectroscopic current maps (b) to (d) at different negative biases. Right: I–V curves of the substrate (black line), type I beads (average of ten beads; red circles), type II beads (average of 6 beads; blue squares), and a bead observed to possibly switch at negative bias in the STM images (type III; black triangles). A figure showing the error ranges of these I/V data is in the ESI.† | ||
One interpretation of these data would be that type I and type II beads contain complex molecules in their high-spin and low-spin states.30,38 However, since type II beads were observed more frequently in the vicinity of kinks or other discontinuities in the chains (Fig. 3), the conduction properties of the beads may also be influenced by their relationship to their nearest neighbours. Nearly all beads retained the same type I or type II conduction behaviour when the same chain was measured at different biases. However, the “type III” bead in Fig. 3 appears to switch at 203 K, from type I to type II as the bias was decreased in the spectroscopic current maps. This apparent switching is less clear cut in the I/V curves from the same set of beads, however (Fig. 3).
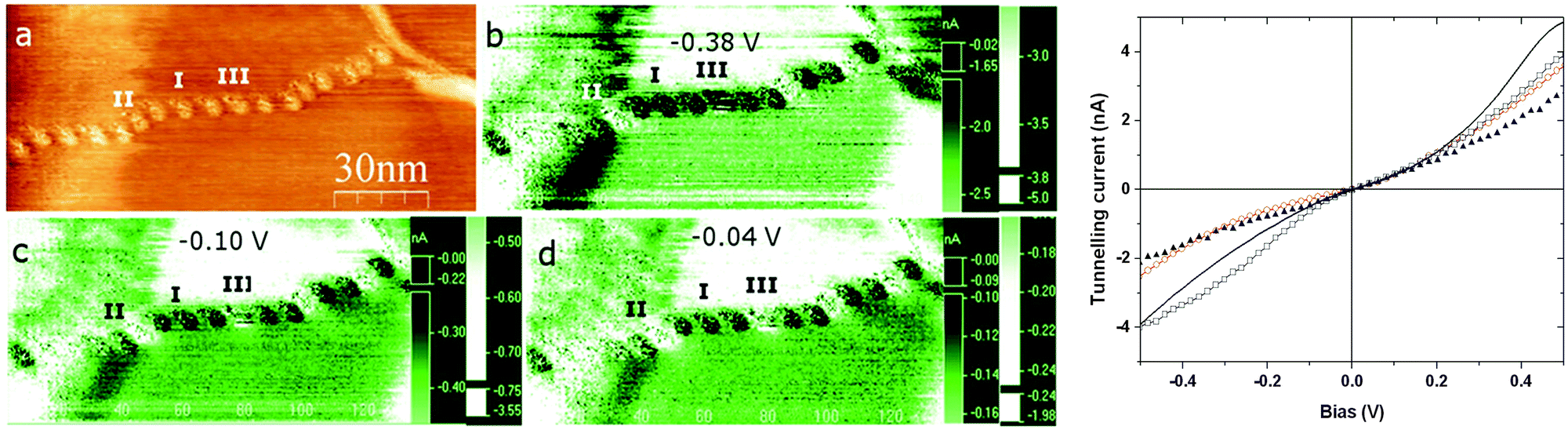
Since the crystallographic dimensions of the [Fe(bpp)2]2+ cation are 13 × 13 × 14 Å, with a molecular volume of ca. 620 Å3 at 290 K, the beads are too large to correspond to an isolated molecule on the HOPG surface. By volume, the typical bead structure contains 10–50 molecules of [Fe(bpp)2]2+ with accompanying anions, assuming the beads contain the intact complex molecule. To address that question, XPS measurements were performed on nm films of [Fe(bpp)2][BF4]2, drop-cast from acetone solution onto Au(111). The resultant photoelectron scan at 293 K clearly showed the presence of C, N, B, F and Fe (Fig. 4); the Fe:C:N composition is 1:27:10, in reasonable agreement with the theoretical values (1:22:10), while the B:F ratio is 1:3 (predicted 1:4). The C and N 1s peaks were both clearly split into multiple components. Binding energies of the fitted carbon peaks at 284.5 eV, 285.4 eV and 287.1 eV (Fig. 4b) are comparable to those reported for a metal-free bpp derivative, while the two N peaks at 399.7 eV and 401.3 eV (which have an approximate 2:3 ratio, Fig. 4c) can be attributed to the pyrrolic and pyridinic N atom environments in the bpp ligand, respectively.42 Although the conditions of the STM and XPS experiments involve different substrates and film thicknesses, these data provide clear evidence that [Fe(bpp)2][BF4]2 can retain its structural integrity when drop-cast from acetone.
 | ||
| Fig. 4 XPS spectra of (a) Fe 2p1/2 and 2p3/2, (b) C 1s, (c) N 1s, (d) F 1s and (e) B 1s, in films of [Fe(bpp)2][BF4]2 drop-cast onto Au(111). The carbon region consist of three components, as expected for pyrazole and pyridine, however, the weakest peak is shifted by 1 eV to a higher binding energy than expected. Experimental data are represented by empty circles, while the solid lines correspond to envelopes of fitted components shown in filled squares, dashed and dotted lines. | ||
The Fe 2p1/2 and 2p3/2 XPS emissions exhibit high-energy satellite peaks, reflecting 3d → ligand MLCT excitations during the electron emission.27,43 The pronounced nature of these satellites is characteristic of the complex being high-spin at the temperature of measurement.44 However, cycling the XPS samples about the spin-transition temperature of the compound (T1/2 = 261 K) did not lead to the 1–2 eV reduction in the 2p3/2 binding energy, or the weakening of the satellite peaks, that would be characteristic of spin-crossover (ESI†). While that could reflect decomposition of the compound in the X-ray beam,45 an aspect of the chemistry of [Fe(bpp)2]2+ derivatives that would also explain this observation is discussed below.
With a view to producing SAMs of [Fe(bpp)2]2+-type centres, L240 was deposited onto gold-coated glass by drop-casting and by SAM formation. XPS data from both types of L2 layers demonstrated the presence of C, N, S and, unexpectedly, a small amount of O. However, the relative abundance of each element did not match the empirical formula of the ligand, with higher-than-expected quantities of sulfur compared to the other elements (tabulated in the ESI†). This discrepancy was more pronounced in the SAMs than in the drop-cast samples, where the L2 layers were thicker and formed more rapidly. The S 2p XPS profiles were more complex than expected, and demonstrated the presence of elemental sulfur as well as bound, unbound and partially oxidised thiol residues, in varying ratios (ESI†). Similarly, the N 1s emission demonstrated the presence of the intact bpp moiety in drop-cast films of L2, showing two peaks in a 2:3 ratio as before. However, in the SAMs of L2 the ratio of these components was closer to 1:1, indicating that the bpp moiety may have fragmented on the surface under these conditions. Finally, the thickness of the deposited layers also varied widely between experiments, at between 3–34 Å by ellipsometry.
We interpret these data as indicating the partial decomposition of L2 at the gold surface by cleavage of the C–S bond, leading to deposition of elemental sulfur. Similar observations have also been made from SAMs derives from 4-mercaptopyridine, which decompose to elemental sulfur on a gold surface over a period of hours in aqueous solution, and more rapidly when dissolved in ethanol.46 The precise mechanism behind the instability of Au/L2 surfaces is unclear, but the N 1s region indicates that at least a significant part of L2 is not intact on the surface, and suggests presence of bpp-derived byproducts from the C–S cleavage process.
In the light of these results, the new ligand L3 was prepared, as a derivative of bpp with a remote sulfur-containing tether group that should be more robust on a gold substrate (Fig. 5 and ESI†). SAMs derived from L3 were indeed more promising, in giving an elemental composition that is close to the formula of the ligand, C:N:S = 17.1:3.9:1 (expected ratio 15:6:1). While SAMs produced from methanol, acetonitrile and chloroform all gave broadly consistent data, the best results were obtained using methanol as solvent. The carbon 1s spectrum shows similar binding energies and fitted component ratios as [Fe(bpp)2][BF4]2 (ESI†). The S 2p XPS profile shows two duplets with 2p3/2 peaks located at 161.9 eV and 163.5 eV (Fig. 6). Although these are commonly assigned to bound and unbound thiol residues, respectively, that does not indicate a poorly formed SAM; both duplets are also characteristic for monolayers of thiane derivatives on gold.47 The N 1s peaks at 401.5 eV and 399.7 eV have an approximate 1:2 area ratio that is close to the empirical formula of the ligand (Fig. 6). Ellipsometry data gave consistent thicknesses of 8–9 ± 1 Å for different SAMs of L3. While that is shorter than the crystallographic length of the extended L3 molecule (ca. 13 Å), that thickness would be consistent with 20–30% lower surface coverage than a well-packed alkanethiol SAM, or a binding mode where the L3 thiomorpholine group lies flat on the Au surface according to molecular models (ESI†). For comparison, the thickness of a well-ordered SAM of a different bpp derivative with an alkanethiol tether was measured at 9.7 Å, albeit by a different technique.42
Incubating SAMs of L3 in acetonitrile solutions of Fe[ClO4]2 did not lead to incorporation of iron into the monolayer, according to XPS data which were identical to those from the initial SAM. Similarly, incubating methanol or acetonitrile solutions of the pre-formed complex [Fe(L3)2][ClO4]2 (ESI†) on gold afforded layers of L3 only, with no iron being present. Evidently, the surface-bound L3 ligand binds iron too weakly to form spin-crossover SAMs, either because of the geometry of the adsorbed ligand or on electronic grounds. Since salts of [Fe(L3)2]2+ are high-spin at room temperature (ESI†), the iron centres in surface-bound L3–Fe complexes should be labile towards ligand displacement reactions which would explain the observed weak interaction between the metal and the SAM.
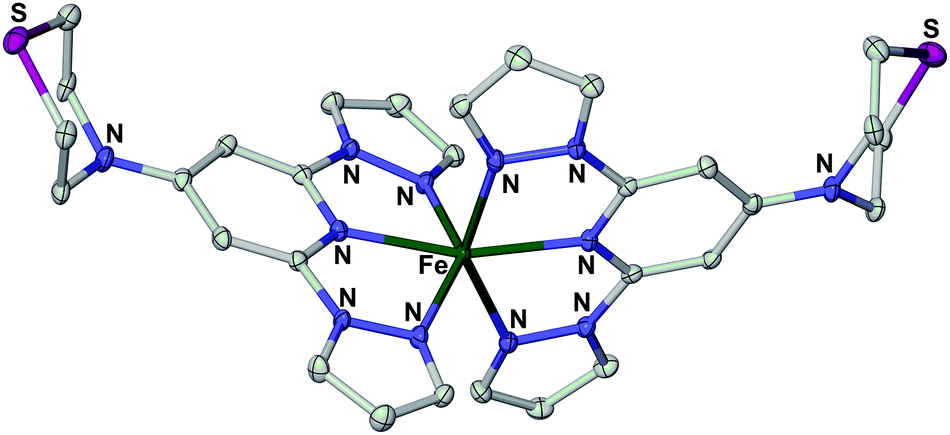 | ||
| Fig. 5 View of the complex dication in the single crystal X-ray structure of [Fe(L3)2][BF4]2·H2O. Displacement ellipsoids are at the 50% probability level, and H atoms have been omitted for clarity. Experimental details and metric parameters from the crystal structures of three salts of [Fe(L3)2]2+ are in the ESI.† | ||
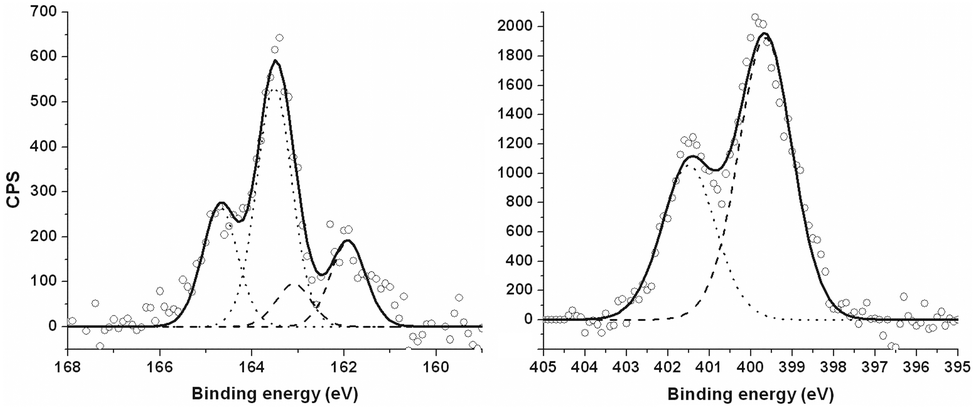 | ||
| Fig. 6 High-resolution S 2p3/2 (left) and N 1s (right) XPS data from SAMs of L3, produced from methanol solution. Empty circles represent experimental data, dashed and dotted lines correspond to bound and unbound thiols, respectively, in S 2p region (left) and two distinct environments of nitrogen atoms in N 1s spectrum (right). Solid lines show envelopes of the fitted peaks. | ||
Conclusions
We have reproduced the unusual results reported by Grohmann et al., of the formation of “chain of beads” nanostructures upon drop-casting derivatives of [Fe(bpp)2]2+ (Scheme 1) onto HOPG.30,38 Since similar structures are obtained using the parent compound [Fe(bpp)2][BF4]2, the sulfur-containing substituents in [Fe(L1)2][BF4]2 clearly have no role in formation of the bead structures. Although the composition of the beads could not be probed directly, XPS data from [Fe(bpp)2][BF4]2 drop-cast onto a gold surface strongly imply that the complex retains its integrity during the drop-casting process.‡ That being the case, the beads (which vary in size between chains) are of appropriate size to contain 10–50 molecules of the compound. Different beads in the same chain exhibit one of two conduction regimes by CITS, which appears to correlate with their proximity to discontinuities in the chain structures. Their different conduction properties might indicate that these beads contain molecules in the high-spin and low-spin states.10,11,38 However, with rare exceptions (Fig. 3), no evidence for switching of the beads' conduction properties under increasing bias was observed, that would indicate charge-induced spin-crossover.The apparent absence of conduction switching in the beads does not invalidate their assignment as high- and low-spin states of the compound. Recent work has indicated that iron(II) complexes which exhibit spin-crossover in the bulk can be either high-spin or low-spin on a surface, but may remain trapped in their spin-states under increasing bias or changing temperature in nanostructures below a certain size regime. The threshold size for the recovery of spin-crossover has varied between studies,24,25,28 although coverages approaching a full monolayer are required to induce spin-state switching in at least some cases. Hence, the inactivity of 2–6 nm beads of [Fe(bpp)2][BF4]2 towards switching under a bias is not unreasonable.
Another factor that could also be relevant to these data, which is unique to the [Fe(bpp)2]2+ spin-crossover system, is the tendency of the high-spin compounds to adopt an unusual angular Jahn–Teller distortion that inhibits spin-crossover in the solid state.35 For example, the three salts of [Fe(L3)2]2+ that we have examined all exhibit this distorted structure by X-ray crystallography, and are not spin-crossover active as a result (Fig. 6 and ESI†). While [Fe(bpp)2][BF4]2 does not suffer from this distortion as a bulk material, and thus exhibits spin-crossover,39 some other salts of the [Fe(bpp)2]2+ cation do show it in the crystalline state.48 The adoption of the distorted structure by [Fe(bpp)2]2+ following drop-casting could also contribute to the lack of switching in the bead structures, and to the compound remaining high-spin below 260 K in the XPS data.
While SAMs of L3 were achieved, attempts to metallate them with iron salts were unsuccessful. This contrasts with closely related derivatives of [Fe(terpy)2]2+ (terpy = 2,2′:6′,2′′-terpyridine), which can be assembled into SAMs and other surface nanostructures.49 An obvious difference relates to the spin states of the two classes of compounds: [Fe(terpy)2]2+ derivatives are low-spin and kinetically inert at room temperature, while [Fe(bpp)2]2+ centres are predominantly high-spin and more reactive towards ligand substitution and solvolysis. We are currently pursuing SAMs based on alternative spin-crossover iron centres, that are less reactive in their high-spin forms and thus more likely to produce switchable surface nanostructures.
Experimental
The complex [Fe(bpp)2][BF4]2 was synthesised by the literature method.39 The ligand L2 was prepared by a modification of our published procedure,40 to avoid contamination by an iodine-containing byproduct (ESI†). The synthesis of the new ligand 4-(N-thiomorpholinyl)-2,6-di(pyrazol-1-yl)pyridine (L3) and its iron complex salts, and crystal structures and magnetic susceptibility data from the complexes, are also described in the ESI.†Samples of [Fe(bpp)2][BF4]2 for STM measurements were prepared by drop-casting 10−7 M solutions of the complex onto freshly cleaved HOPG. The surface was left to dry before loading into UHV chamber. Gold-coated microscope slides produced by our previously reported procedure50 were typically immersed into 1 mM methanol, acetonitrile or chloroform solution of the compounds and left over the night to form SAMs. The samples were removed from the solutions, rinsed with copious amounts of corresponding solvent, dried with a nitrogen stream, rinsed with Milli-Q water, and dried again.
STM measurements were performed with Omicron Variable Temperature UHV STM system at a base pressure of 10−9–10−11 mbar using Pt/Ir (80:20) tips mechanically cut from 0.25 mm wire. Tunnelling current was typically set between 0.1 nA and 0.8 nA with applied bias of 0.1 V to 0.2 V. CITS data was acquired by sweeping voltage between −0.7 V and +0.7 V at every pixel of the topography images. STM data were processed using the Scala Pro and WSxM programs.51
High-resolution XPS spectra were acquired with a Thermo Electron Corporation ESCA Lab 250 at 20 eV pass energy with 0.2 eV resolution and processed with CasaXPS software. Binding energies for SAMs were calibrated using the Au 4f7/2 peak at 83.9 eV. However, spectra of the drop-cast films, which showed weak gold signals, were referenced to the N 1s peak at the lower binding energy of 399.7 eV, to maintain consistency across the samples. The S 2p regions were fitted with duplets of 2p3/2 and 2p1/2 peaks with a 2:1 area ratio, respectively, and separation of 1.18 eV in binding energy.
Temperature readings for the STM measurements were recorded with the thermocouple located in the cooling block, which was coupled to the sample. However, the thermocouple for the XPS measurements was in direct contact with the sample.
The thicknesses of the SAMs was measured with a Jobin-Yvon UVISEL spectroscopic ellipsometer at 70° angle of incidence and the wavelength was varied between 300 and 800 nm in 5 nm steps. Data were modelled and fitted with a simple three-layer system using DeltaPsi2 software. A clean gold substrate was used to obtain values for the base layer, while Cauchy approximation with an assumed refractive index of ninf = 1.45 was used to model the SAM layers.
Other measurements
Elemental microanalyses were performed by the University of Leeds School of Chemistry microanalytical service. Electrospray mass spectra (ESMS) were obtained on a Bruker MicroTOF spectrometer, from MeCN feed solutions. 1H NMR spectra employed a Bruker DPX300 spectrometer, operating at 300.2 MHz. Variable temperature magnetic susceptibility measurements were performed on a Quantum Design SQUID/VSM magnetometer, in an applied field of 5000 G. A diamagnetic correction for the sample was estimated from Pascal's constants;52 a diamagnetic correction for the sample holder was also measured separately, and applied to the data.Acknowledgements
This work was funded by EPSRC grant EP/I014039/1, and by the University of Leeds.Notes and references
- Spin Crossover in Transition Metal Compounds I-III, ed. P. Gütlich and H. A. Goodwin, Top. Curr. Chem., 2004, pp. 233–235 Search PubMed.
- Spin-Crossover Materials – Properties and Applications, ed. M. A. Halcrow, John Wiley & Sons, Chichester, 2013, p. 568 Search PubMed.
- A. Bousseksou, G. Molnár, L. Salmon and W. Nicolazzi, Chem. Soc. Rev., 2011, 40, 3313 RSC.
- M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119 RSC.
- P. Gütlich, Eur. J. Inorg. Chem., 2013, 581 CrossRef PubMed; P. Gütlich, A. B. Gaspar and Y. Garcia, Beilstein J. Org. Chem., 2013, 9, 342 CrossRef PubMed.
- M. A. Halcrow, Polyhedron, 2007, 26, 3523 CrossRef CAS PubMed.
- M. Nihei, T. Shiga, Y. Maeda and H. Oshio, Coord. Chem. Rev., 2007, 251, 2606 CrossRef CAS PubMed.
- S. Hayami, Y. Komatsu, T. Shimizu, H. Kamihata and Y. H. Lee, Coord. Chem. Rev., 2011, 255, 1981 CrossRef CAS PubMed.
- O. Kahn, J. Kröber and C. Jay, Adv. Mater., 1992, 4, 718 CrossRef CAS PubMed.
- M. Matsuda and H. Tajima, Chem. Lett., 2007, 36, 700 CrossRef CAS.
- A. Rotaru, I. A. Gural'skiy, G. Molnár, L. Salmon, P. Demont and A. Bousseksou, Chem. Commun., 2013, 48, 4163 Search PubMed.
- A. Bousseksou, G. Molnár, P. Demont and J. Menegotto, J. Mater. Chem., 2003, 13, 2069 RSC; S. Bonhommeau, T. Guillon, L. M. L. Daku, P. Demont, J. S. Costa, J.-F. Létard, G. Molnár and A. Bousseksou, Angew. Chem., Int. Ed., 2006, 45, 1625 CrossRef CAS PubMed.
- H. J. Shepherd, T. Palamarciuc, P. Rosa, P. Guionneau, G. Molnár, J.-F. Létard and A. Bousseksou, Angew. Chem., Int. Ed., 2012, 51, 3910 CrossRef CAS PubMed; H. J. Shepherd, I. A. Gural'skiy, C. M. Quintero, S. Tricard, L. Salmon, G. Molnár and A. Bousseksou, Nat. Commun., 2013, 4, 2607 Search PubMed.
- H. J. Shepherd, G. Molnár, W. Nicolazzi, L. Salmon and A. Bousseksou, Eur. J. Inorg. Chem., 2013, 653 CrossRef CAS PubMed; G. Molnár, L. Salmon, W. Nicolazzi, F. Terki and A. Bousseksou, J. Mater. Chem. C, 2014, 2, 1360 RSC.
- M. Cavallini, Phys. Chem. Chem. Phys., 2012, 14, 11867 RSC.
- P. N. Martinho, C. Rajnak and M. Ruben, in Spin-Crossover Materials – Properties and Applications, ed. M. A. Halcrow, John Wiley & Sons, Chichester, 2013, ch. 14, pp. 375–404 Search PubMed.
- H. Peng, S. Tricard, G. Félix, G. Molnár, W. Nicolazzi, L. Salmon and A. Bousseksou, Angew. Chem., Int. Ed., 2014, 53, 10894 CrossRef CAS PubMed.
- M. C. Muñoz and J. A. Real, Coord. Chem. Rev., 2011, 255, 2068 CrossRef CAS PubMed; O. Roubeau, Chem. – Eur. J., 2012, 18, 15230 CrossRef PubMed.
- T. Miyamachi, M. Gruber, V. Davesne, M. Bowen, S. Boukari, L. Joly, F. Scheurer, G. Rogez, T. K. Yamada, P. Ohresser, E. Beaurepaire and W. Wulfhekel, Nat. Commun., 2012, 3, 938 CrossRef PubMed.
- T. G. Gopakumar F. Matino, H. Naggert, A. Bannwarth and F. Tuczek, and R. Berndt, Angew. Chem., Int. Ed., 2012, 51, 6262 CrossRef PubMed.
- H. Naggert, A. Bannwarth, S. Chemnitz, T. von Hofe, E. Quandt and F. Tuczek, Dalton Trans., 2011, 40, 6364 RSC.
- T. G. Gopakumar, M. Bernien, H. Naggert, F. Matino, C. F. Hermanns, A. Bannwarth, S. Mühlenberend, A. Krüger, D. Krüger, F. Nickel, W. Walter, R. Berndt, W. Kuch and F. Tuczek, Chem. – Eur. J., 2013, 19, 15702 CrossRef CAS PubMed.
- T. Palamarciuc, J. C. Oberg, F. El Hallak, C. F. Hirjibehedin, M. Serri, S. Heutz, J.-F. Létard and P. Rosa, J. Mater. Chem., 2012, 22, 9690 RSC.
- B. Warner, J. C. Oberg, T. G. Gill, F. El Hallak, C. F. Hirjibehedin, M. Serri, S. Heutz, M.-A. Arrio, P. Sainctavit, M. Mannini, G. Poneti, R. Sessoli and P. Rosa, J. Phys. Chem. Lett., 2013, 4, 1546 CrossRef CAS.
- A. Pronschinske, Y. Chen, G. F. Lewis, D. A. Shultz, A. Calzolari, M. Buongiorno Nardelli and D. B. Dougherty, Nano Lett., 2013, 13, 1429 Search PubMed; A. Pronschinske, R. C. Bruce, G. Lewis, Y. Chen, A. Calzolari, M. Buongiorno-Nardelli, D. A. Shultz, W. You and D. B. Dougherty, Chem. Commun., 2013, 49, 10446 RSC.
- S. Shi, G. Schmerber, J. Arabski, J.-B. Beaufrand, D. J. Kim, S. Boukari, M. Bowen, N. T. Kemp, N. Viart, G. Rogez, E. Beaurepaire, H. Aubriet, J. Petersen, C. Becker and D. Ruch, Appl. Phys. Lett., 2009, 95, 043303 CrossRef PubMed.
- T. Mahfoud, G. Molnár, S. Cobo, L. Salmon, C. Thibault, C. Vieu, P. Demont and A. Bousseksou, Appl. Phys. Lett., 2011, 99, 053307 CrossRef PubMed.
- M. Bernien, D. Wiedemann, C. F. Hermanns, A. Krüger, D. Rolf, W. Kroener, P. Müller, A. Grohmann and W. Kuch, J. Phys. Chem. Lett., 2012, 3, 3431 CrossRef CAS.
- A. D Naik, M. M. Dîrtu and Y. Garcia, J. Phys.: Conf. Ser., 2010, 217, 012032 CrossRef.
- M. S. Alam, M. Stocker, K. Gieb, P. Müller, M. Haryono, K. Student and A. Grohmann, Angew. Chem., Int. Ed., 2010, 49, 1159 CrossRef CAS PubMed.
- M. Cavallini, I. Bergenti, S. Milita, J. C. Kengne, D. Gentili, G. Ruani, I. Salitros, V. Meded and M. Ruben, Langmuir, 2011, 27, 4076 CrossRef CAS PubMed.
- M. Cavallini, I. Bergenti, S. Milita, G. Ruani, I. Salitros, Z.-R. Qu, R. Chandrasekar and M. Ruben, Angew. Chem., Int. Ed., 2008, 47, 8596 CrossRef CAS PubMed.
- S. Basak, P. Hui and R. Chandrasekar, Chem. Mater., 2013, 25, 3408 CrossRef CAS.
- S. Bin-Salamon, S. Brewer, S. Franzen, D. L. Feldheim, S. Lappi and D. A. Shultz, J. Am. Chem. Soc., 2005, 127, 5328 CrossRef CAS PubMed; G. Poneti, L. Poggini, M. M. B. Cortigiani, L. Sorace, E. Otero, P. Sainctavit, A. Magnani, R. Sessoli and A. Dei, Chem. Sci., 2015, 6, 2268 RSC.
- M. A. Halcrow, Coord. Chem. Rev., 2009, 253, 2493 CrossRef CAS PubMed; L. J. Kershaw Cook, R. Mohammed, G. Sherborne, T. D. Roberts, S. Alvarez and M. A. Halcrow, Coord. Chem. Rev., 2015, 289–290, 2 CrossRef PubMed.
- M. A. Halcrow, New J. Chem., 2014, 38, 1868 RSC.
- D. Secker, S. Wagner, S. Ballmann, R. Härtle, M. Thoss and H. B. Weber, Phys. Rev. Lett., 2011, 106, 136807 CrossRef; V. Meded, A. Bagrets, K. Fink, R. Chandrasekar, M. Ruben, F. Evers, A. Bernand-Mantel, J. S. Seldenthuis, A. Beukman and H. S. J. van der Zant, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 245415 CrossRef.
- A. Grohmann, M. Haryono, K. Student, P. Müller and M. Stocker, Eur. J. Inorg. Chem., 2013, 662 CrossRef CAS PubMed.
- J. M. Holland, J. A. McAllister, Z. Lu, C. A. Kilner, M. Thornton-Pett and M. A. Halcrow, Chem. Commun., 2001, 577 RSC.
- L. J. Kershaw Cook, J. Fisher, L. P. Harding and M. A. Halcrow, Dalton Trans., 2015, 44, 9417 RSC.
- C. Liu, H. Chang and A. J. Bard, Langmuir, 1991, 7, 1138 CrossRef CAS; T. M. Bernhardt, B. Kaiser and K. Rademann, Surf. Sci., 1998, 408, 86 CrossRef.
- C. Shen, M. Haryono, A. Grohmann, M. Buck, T. Weidner, N. Ballav and M. Zharnikov, Langmuir, 2008, 24, 12883 CrossRef CAS PubMed.
- S. G. Chiuzbaian, M. Neumann, O. Waldmann, B. Schneider, I. Bernt and R. W. Saalfrank, Surf. Sci., 2001, 482–485, 1272 CrossRef CAS; T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441 CrossRef PubMed.
- L. N. Mazalov, I. P. Asanov and V. A. Varnek, J. Electron Spectrosc. Relat. Phenom., 1998, 96, 209 CrossRef CAS.
- M. S. Lazarus, M. A. Hoselton and T. S. Chou, Inorg. Chem., 1977, 16, 2549 CrossRef CAS.
- E. A. Ramírez, E. Cortés, A. A. Rubert, P. Carro, G. Benítez, M. E. Vela and R. C. Salvarezza, Langmuir, 2012, 28, 6839 CrossRef PubMed.
- M. C. Lennartz, M. Baumert, S. Karthäuser, M. Albrecht and R. Waser, Langmuir, 2011, 27, 10312 CrossRef CAS PubMed.
- J. M. Holland, J. A. McAllister, C. A. Kilner, M. Thornton-Pett, A. J. Bridgeman and M. Halcrow, J. Chem. Soc., Dalton Trans., 2002, 548 RSC; J. Elhaïk, D. J. Evans, C. A. Kilner and M. A. Halcrow, Dalton Trans., 2005, 1693 RSC; J. Elhaïk, C. A. Kilner and M. A. Halcrow, Dalton Trans., 2006, 823 RSC.
- S. Liatard, J. Chauvin, D. Jouvenot, F. Loiseau and A. Deronzier, J. Phys. Chem. C, 2013, 117, 20431 CAS; R. Sakamoto, K.-H. Wu, R. Matsuoka, H. Maeda and H. Nishihara, Chem. Soc. Rev., 2015 10.1039/c5cs00081e.
- Z. H. Al-Lawati, R. J. Bushby and S. D. Evans, J. Phys. Chem. C, 2013, 117, 7533 CAS.
- I. Horcas, R. Fernandez, J. M. Gomez-Rodriguez, J. Colchero, J. Gomez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS PubMed.
- C. J. O'Connor, Prog. Inorg. Chem., 1982, 29, 203 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterisation data for L2, L3 and salts of [Fe(L3)2]2+; experimental details, figures and tables for crystal structures of [Fe(L3)2]X2 (X− = BF4−, ClO4− and CF3SO3−); additional STM images, XPS and ellipsometry data. CCDC 1058749–1058751. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5tc01233c |
| ‡ 30 nm films of [Fe(bpp)2][BF4]2 have also been produced by spin-coating acetonitrile solutions of the complex onto a glass substrate. While XPS data were not reported, the spin-state properties of the thin films closely resemble bulk samples of the same compound.10 |
| This journal is © The Royal Society of Chemistry 2015 |