
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioconjugation of hydrogen-bonded organic semiconductors with functional proteins†
E. D.
Głowacki
*a,
R. R.
Tangorra
*b,
H.
Coskun
a,
D.
Farka
a,
A.
Operamolla
bc,
Y.
Kanbur
a,
F.
Milano
d,
L.
Giotta
d,
G. M.
Farinola
b and
N. S.
Sariciftci
a
aLinz Institute for Organic Solar Cells (LIOS), Physical Chemistry, Johannes Kepler University, Linz, Austria. E-mail: eric_daniel.glowacki@jku.at
bDipartimento di Chimica, Università degli studi di Bari Aldo Moro, Via Orabona, 4, 70126 Bari, Italy. E-mail: roberto.tangorra@uniba.it
cIstituto di Chimica dei Composti Organometallici (ICCOM), Consiglio Nazionale delle Ricerche, Via Orabona, 4, 70126, Bari, Italy
dIstituto per i Processi Chimico Fisici (IPCF), Consiglio Nazionale delle Ricerche, Via Orabona, 4, 70126 Bari, Italy
First published on 29th May 2015
Abstract
We demonstrate the direct bioconjugation of hydrogen-bonded organic semiconductors with two different complex functional proteins in an aqueous environment. The representative semiconductors are epindolidione and quinacridone, materials used in devices in the form of vacuum-evaporated polycrystalline films. First, these molecules in thin films react spontaneously with N-hydroxysuccinimide functionalized linkers: disuccinimidyl suberate and succinimidyl biotinate. The suberate linker is then used to covalently bind the Rhodobacter sphaeroides reaction centre (RC), the key photoenzyme for conversion of light into electrical charges in photosynthetic bacteria. Similarly, the biotin linker is used to bridge streptavidin to the surface of the hydrogen-bonded semiconductor film. Multiple-reflection infrared spectroscopy, water contact angle measurements, and atomic force microscopy are used to verify surface functionalization. The presence and functional integrity of the immobilized proteins are demonstrated by specific experiments: a charge recombination kinetics assay in the case of the RC, and photoluminescence measurements for quantum dot-labelled streptavidin. As key results of our work, we have shown that upon bioconjugation, the semiconductors preserve their favourable electrical properties: as evidenced by photoconductor devices operating under water sensitized by the RC, and thin film transistor measurements before and after bioconjugation. These are enabling steps for using hydrogen-bonded semiconductors as platforms for multifunctional bioelectronics devices.
Introduction
Soft organic electronic materials are emerging as candidates for next-generation bioelectronics devices. Due to low-toxicity, biocompatibility, low elastic modulus, and potential low-cost, they are envisioned to be applicable where other traditional semiconductors simply cannot be applied.1–3 For bioelectronics applications such as sensing and actuation, drug delivery, and ionic/electronic signal transduction, functionalization with bioactive molecules is critical to achieve specific interfacing with the desired biosystem.4–7 Such a chemical modification of organic semiconductors has proven to be problematic, as the introduction of various functions disturbs the intra- or intermolecular conjugation necessary for charge transport. From the biological macromolecule side, interaction with non-physiological environments can compromise the structure necessary for a fully functional hybrid architecture.Two successful approaches have been proven, where the semiconducting molecules are chemically derivatized prior to device fabrication8,9 or more commonly, semiconductor layers are modified following device fabrication with the help of various functional interlayers.4,5,10–13 This introduces issues related to additional material processing steps that often plague multilayered structures and that sum up to the worsening of the semiconductor performance parameters, in terms of mobility and operational stability, often observed in the required physiological environment.
H-bonded pigments are a class of organic colorants, which features many natural-origin molecules that have been used for centuries, as well as numerous mass-produced industrial synthetic compounds used in applications as various as outdoor paints, cosmetics, and printing inks.14,15 Their widespread use in the dye and pigment industry is motivated by three favourable properties: low-cost production, excellent stability, and low toxicity.16–18 Many of these materials, including the ones used here, belong to the lowest toxicity category rating for commercial colorants, meaning they are considered less hazardous than even water-soluble food dyes.18 Recently, hydrogen-bonded pigments have emerged as promising organic semiconductors, with epindolidione (EPI) and quinacridone (QNC) (Fig. 1) demonstrating hole mobility in the range of 0.1–1 cm2 V−1 s−1 and outstanding operational stability in both air and in aqueous environments with pH 3–10.10,19,20 This latter finding is motivating for deploying these materials in applications requiring direct interfacing with biological “wet” environments. Finally, the N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functional groups characterizing hydrogen-bonded pigments are chemical handles that are in principle amenable for direct bioconjugation.21,22 In this paper we describe two-step bioconjugation chemistry on hydrogen-bonded pigment films for immobilization of protein monolayers on their surface.
O functional groups characterizing hydrogen-bonded pigments are chemical handles that are in principle amenable for direct bioconjugation.21,22 In this paper we describe two-step bioconjugation chemistry on hydrogen-bonded pigment films for immobilization of protein monolayers on their surface.
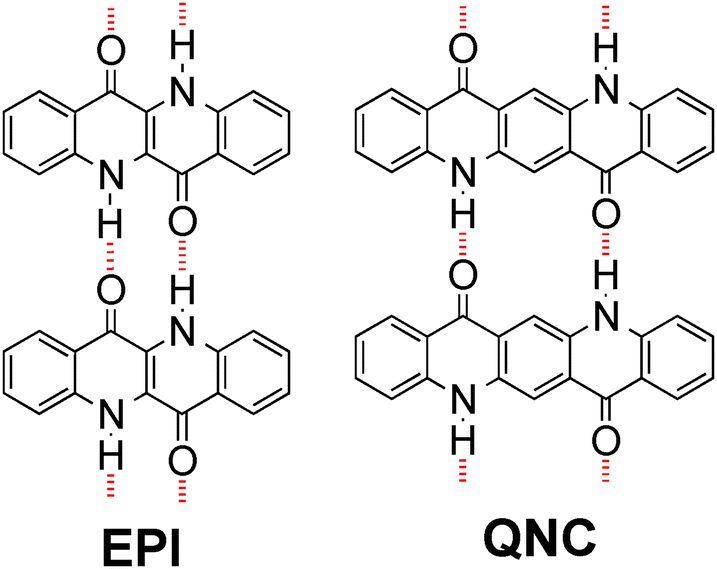 | ||
| Fig. 1 Molecular structures of hydrogen-bonded pigments epindolidione (EPI) and quinacridone (QNC). Red dashed lines indicate intermolecular hydrogen-bonding. | ||
Our bioconjugation protocol, besides being general and straightforward, is demonstrated to preserve the semiconducting properties of the pigment while maintaining the functions of the proteins. In the first step, an aqueous solution of a flexible aliphatic linker is kept in contact with the hydrogen-bonded pigment film. The N-succinimidyl ester (NHS) function on the linker is introduced to selectively address amine nucleophiles on the pigments. The experimental data collected and discussed below suggest that the N–H group on the pigment can react with the linker, likely producing an amide linkage and an N-hydroxysuccinate as a leaving group (Fig. 2 and 3). In the second step, the pigment-linker adduct (Fig. 2, inset) functions as a hook for a given biomacromolecule. We selected two different proteins for surface bioconjugation: Rhodobacter sphaeroides R26 photosynthetic reaction centre (RC) (Fig. 2) and streptavidin (Fig. 3).
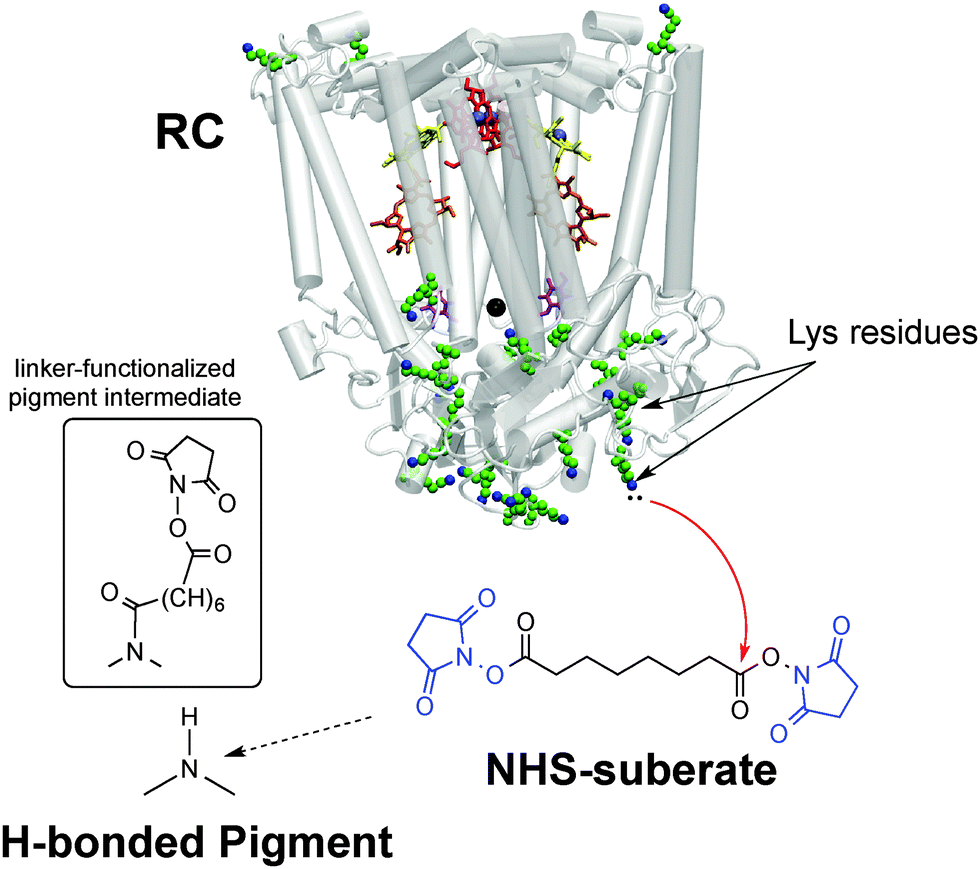 | ||
| Fig. 2 Bioconjugation procedure for the Rhodobacter sphaeroides photosynthetic reaction centre (RC) to hydrogen-bonded pigments using the disuccinimidyl suberate linker, with the NHS leaving groups shown in blue. The crystal structure23 of the RC is shown, highlighting the 22 lysine residues available for conjugation. The inset shows the presumed product of the reaction of the hydrogen-bonded pigment with the NHS-terminated linker – one NHS group leaves and the other remains on the other side of the linker. | ||
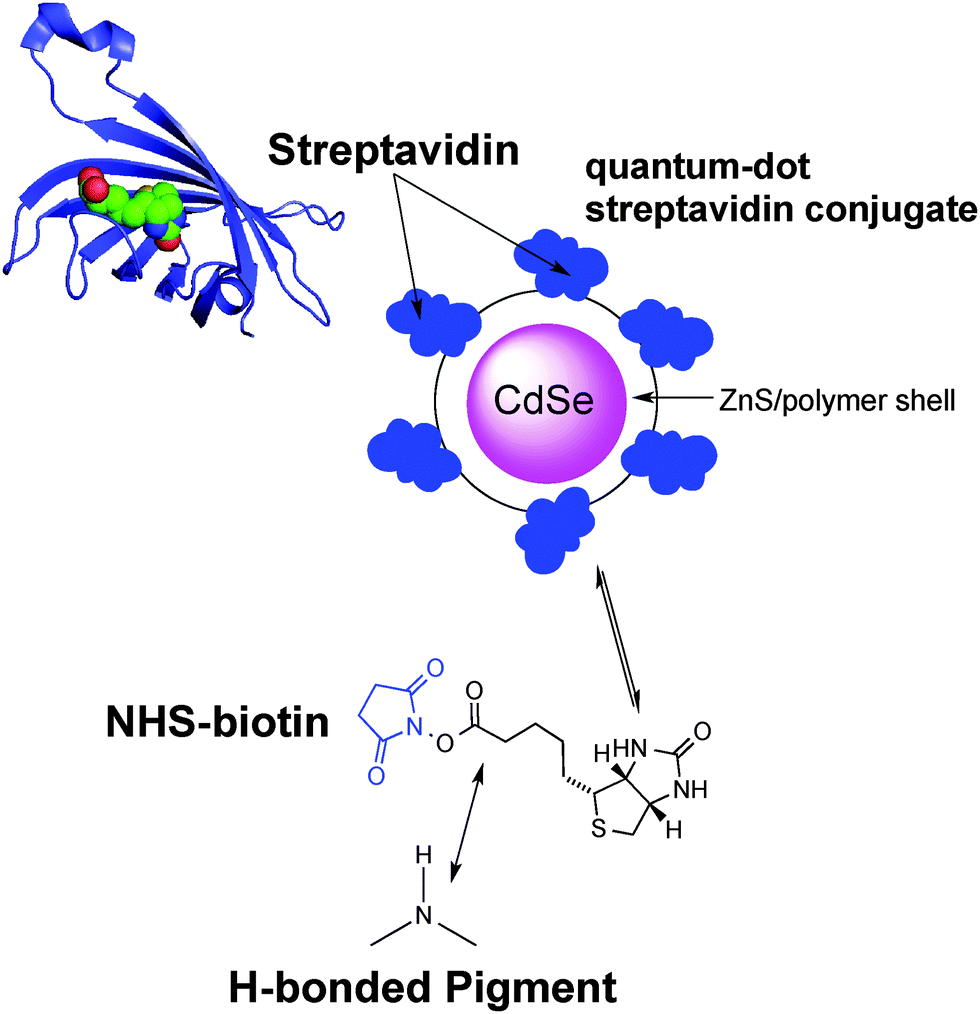 | ||
| Fig. 3 The structure of the monomeric streptavidin protein with a bound biotin molecule; along with the qdot-705 luminescence label structure. The NHS–biotin linker is used to biotinylate the hydrogen-bonded semiconductor surface, affording specific binding sites for streptavidin. | ||
Both are well-studied complex functional proteins. The RC is a photoenzyme that can be easily bioconjugated by reaction of the N-succinimidyl activated ester with its lysine amino acid residues24 and the biotin–streptavidin couple is an archetypical noncovalent lock-and-key binding pair used in numerous biochemical procedures.
We explore if the hydrogen-bonded semiconductor films retain appropriate semiconducting properties following bioconjugation, by investigating the performances of an aqueous-environment RC-sensitized photoconductor, and of field-effect transistor devices before and after surface-modification of the semiconductor layer with the suberate or biotin linkers. These experiments show that both the semiconducting properties of the pigments and the biological functionality of the proteins are preserved. These results suggest that hydrogen-bonded pigments are promising for further developments of multifunctional bioelectronics devices, especially when operation in direct contact with aqueous biological environments is desired, and that selective and mild bioconjugation chemistry is an elegant way to achieve a biohybrid architecture without compromising the protein or semiconductor partners.
Results and discussion
Bioconjugation of the Rhodobacter sphaeroides RC with the suberate (SUB) linker
The photosynthetic RC (Fig. 2) is a membrane-spanning protein, acting as a photoconverter in the photosynthetic process. Nine cofactors are found in the protein scaffold: two ubiquinone-10 (UQ10) molecules (QA, QB), an iron ion, two bacteriopheophytins (BPhe) and four bacteriochlorophylls (BChl), two of which form a functional dimer (D). Upon photon absorption, with an efficiency close to unity, one electron sitting on D is excited and shuttled to the electron acceptor quinone QA and finally to quinone QB; in the absence of external reductants, a charge recombination reaction occurs in a timescale from milliseconds up to seconds.23 In the RC, 22 lysines (Lys) are found, nine of which are available for bioconjugation,24 hence these aminoacid residues are involved in the bioconjugation reaction with the linker-modified surface. Optical transitions associated with the chlorin pigments (BChl and BPhe) give distinct and well-assigned UV-Vis signals; however, at the level of few RC layers, these could not be strong enough to be detected. We relied on a multi-reflection mode FTIR technique, using the amide I and amide II diagnostic signals at 1655 and 1545 cm−1 respectively, as probes for the presence of RC on the pigment film surface. The IR spectra (Fig. 4) of H-bonded semiconductors deposited on ITO-coated glass slides, shown in (a), gave distinctive peaks corresponding to C–C bonds in the aromatic skeleton rings convoluted with those related to CO and N–H functional groups present in both quinacridone and epindolidione backbones (peak assignment in the ESI† S1).
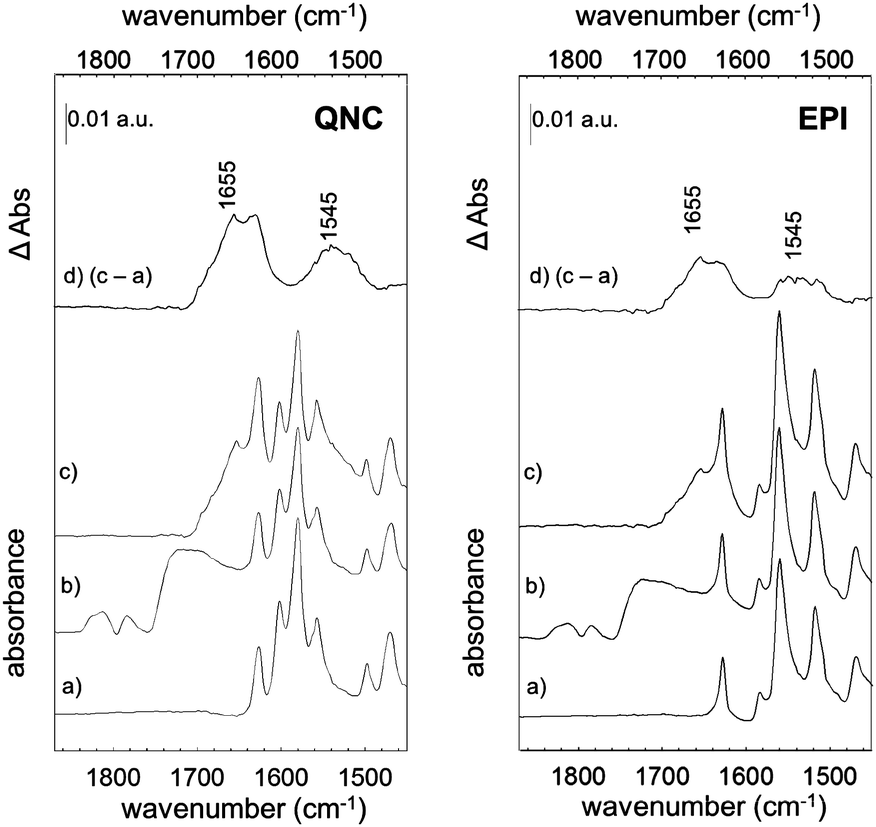 | ||
| Fig. 4 RC bioconjugation to H-bonded semiconductors traced by FTIR spectra of RC–quinacridone (left) and RC–epindolidione hybrid systems (right): (a) the pristine organic semiconductors; (b) SUB linked to semiconductors; (c) RC bioconjugated to semiconductor surfaces by the SUB linker. (d) Difference spectrum between (c) and (a). | ||
After reaction with SUB (Fig. 4b), the appearance of three pronounced bands at 1814, 1784 and 1725 cm−1 is clearly evident in both QNC and EPI films, corresponding to the stretching vibration (ν) of the two CO in the succinimidyl ring and to the CO ester, respectively. These three bands, arising from the SUB linker, suggest the successful modification of the organic semiconductor surfaces. An important question is whether the NH function on the hydrogen-bonded semiconductor initiates nucleophilic attack on the NHS-functionalized molecule, resulting in an amide linkage (Fig. 2, inset). Deconvolution of the broad SUB peak centered at 1725 cm−1 shows that it is composed of two peaks, one corresponding to the CO on the succinimidyl ring, and the second peak, which we assigned to the amide CO (see Fig. S1.2 of ESI†). In the last bioconjugation step (Fig. 4c), after RC incubation, two additional signals appear at 1655 and 1545 cm−1 (amide I and amide II bands of proteins), concomitantly with the disappearance of SUB signals observed above. In Fig. 4d, the absorption difference spectra between traces (c) and (a) are shown, in which the arising of amide I and II modes strongly indicates the effective amide coupling between RC and SUB. Furthermore, the similarity of the collected mode positions to the reported native RC signals25 also suggests that RC retains the essential secondary structure after bioconjugation reaction. However, more conclusive experiments have been carried out to assess the protein integrity and functionality, as discussed in the following.
Evidence supporting the effective RC bioconjugation to organic semiconductor materials can be provided by recording the FTIR spectra of RC bioconjugated samples and RC physisorbed ones after each washing step with phosphate-buffered saline, PBS (Fig. 5). For bioconjugated RC, no absorption differences were detected after two consecutive rinses in buffer solution (Fig. 5a and b). Conversely, for physisorbed RC systems, no signals from the protein were detected already after the first buffer rinsing, ruling out any aspecific interaction with the hydrogen-bonded pigment surfaces.
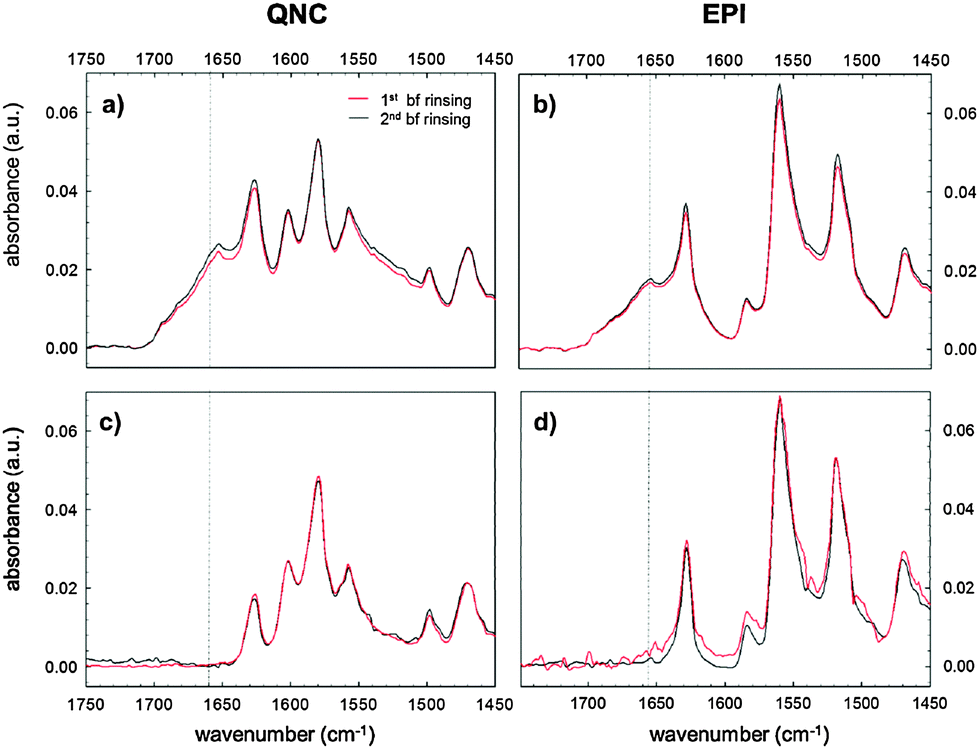 | ||
| Fig. 5 FTIR spectra after subsequent washing steps of bioconjugated (a) and (b) and physisorbed (c) and (d) RC samples both with quinacridone and epindolidione substrates. Washing was performed with PBS, pH 7.2. | ||
The analysis of the washing solutions, performed using the cytochrome assay described in detail in the ESI† S2, further supports the effective covalent binding of RC to the substrates. In the absence of SUB, all RC is desorbed from the surface with the first washing. With SUB-modified surfaces, some RC is still present in the washing solution after the second washing for QNC and even after the third washing for EPI.
Atomic force microscopy (AFM) furnishes a powerful tool for monitoring surface modifications. The occurrence of bioconjugation on QNC or EPI surfaces with RCs was followed step by step by acquisition of surface topographies for each sample before and after each surface treatment step. These results were supplemented by recording changes in the water contact angle. These values are reported together with surface roughnesses in Table 1. AFM images of evaporated films of QNC and EPI are shown in Fig. 6a and d, respectively. Both evaporated films characteristically exhibit well-defined crystallites of size on the order of a few hundred nanometers, with EPI having larger grains. QNC forms distinctive rods with a length of 200–300 nm and a width of 10–20 nm; EPI crystallites are larger, with 60–80 nm width and more than 300 nm length. QNC and EPI aggregates are considered responsible for their peculiar solid-state semiconducting features. This aggregation profile is still recognizable in the topographies acquired after overnight treatment with an aqueous solution of SUB (middle panels, Fig. 6b and e for QNC and EPI respectively), demonstrating that treatment with such a solution does not disrupt surface organization. Along with SUB functionalization, the root mean square roughness of EPI changes from 2.9 ± 0.3 nm for the untreated sample to 2.8 ± 0.5 nm for the SUB-functionalized sample. Likewise, QNC show a decrease in surface roughness from 3.4 ± 0.3 nm for the untreated sample to 3.2 ± 0.3 nm for the SUB-functionalized sample. In parallel, both samples show a decrease in contact angle: while untreated QNC and EPI are relatively hydrophobic (89° and 85° respectively), more hydrophilic values are found after SUB treatment (59° and 74°, respectively). All these findings together support the hypothesis that SUB, possessing a linear and flexible aliphatic skeleton, can densely pack on the surface of both QNC and EPI, exploiting the free pendant NH functionalities of the semiconductor for covalent immobilization and leaving numerous hydrophilic N-hydroxysuccinimidyl units protruding from the surface.
| Untreated EPI | EPI@SUB | EPI@SUB@RC | Untreated QNC | QNC@SUB | QNC@SUB@RC | |
|---|---|---|---|---|---|---|
| Roughness RMS [nm] | 2.9 ± 0.3 | 2.8 ± 0.5 | 4.6 ± 0.7 | 3.4 ± 0.3 | 3.2 ± 0.3 | 7.2 ± 0.6 |
| Contact angle [°] | 86 | 74 | <10 | 89 | 59 | <10 |
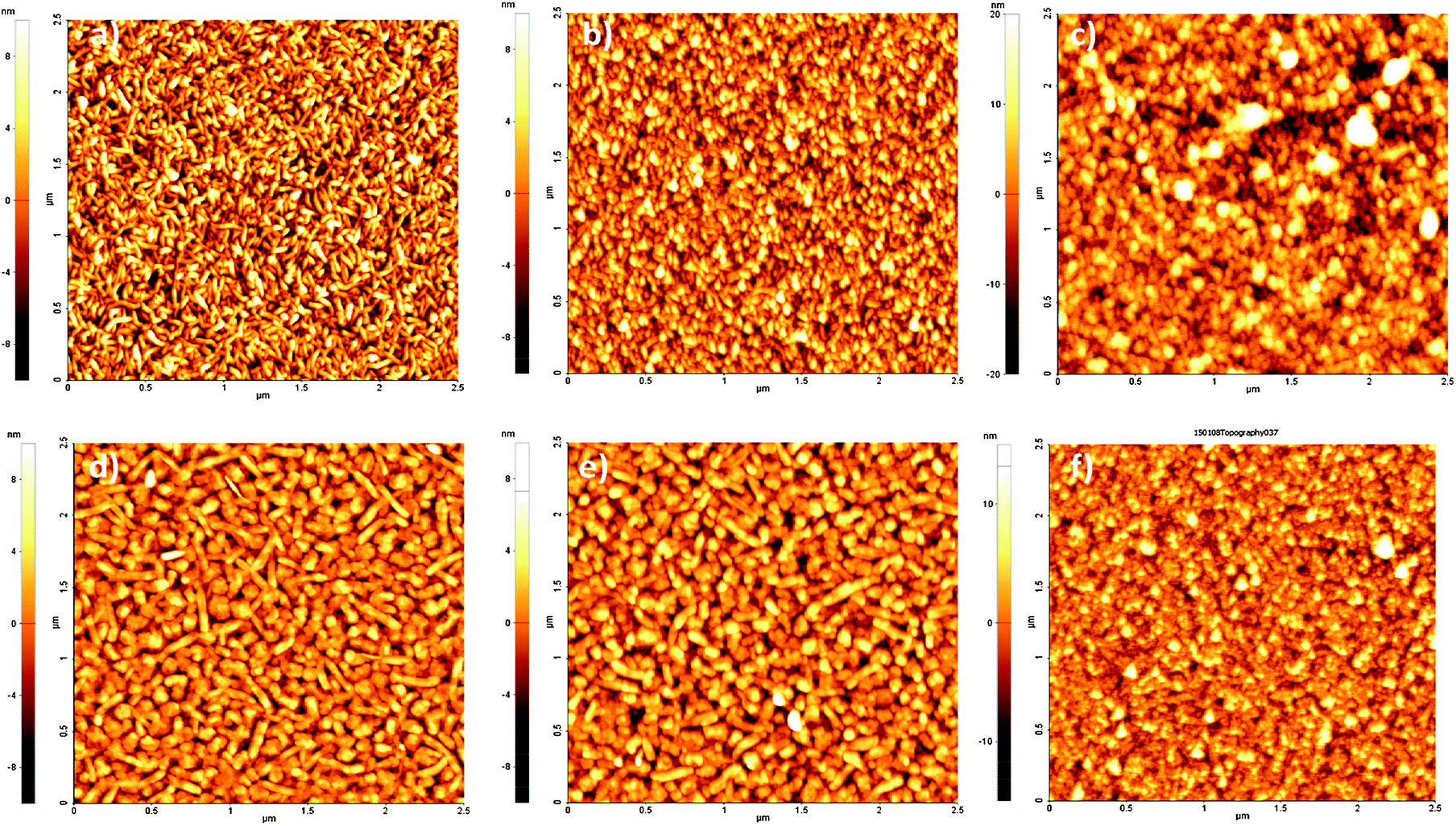 | ||
| Fig. 6 Tapping-mode 2.5 × 2.5 μm AFM topographies of films of QNC (top panels) and EPI (bottom panels): (a, d) pristine evaporated films; (b, e) SUB functionalized films; (c, f) bacterial RC on SUB functionalized films after washing with buffer solution. | ||
After incubation for four hours with the RC solution, EPI and QNC films in AFM appear to be completely covered with globular particles and no clear structure from the semiconductors is distinguishable (Fig. S3a and b in ESI†). Washing these samples four times with phosphate-buffered saline (PBS) and finally distilled water removes physisorbed proteins and yields a nanostructure with relatively well-defined globules superimposed on the crystalline substrate, as shown in Fig. 6c and f. In these topographies regular globular structures of soft materials cover the surface, corresponding to an increased RMS (4.6 ± 0.7 nm in the case of EPI and 7.2 ± 0.6 nm in the case of QNC). The height distribution increases markedly for the RC-modified samples. Histograms of height distributions for each topography are shown in Fig. S4 and S5 of the ESI.† These AFM results clearly evidence that the semiconductor surfaces retain the RC protein on the surface following washing.
The recombination time for the photoexcited charge-separated states on the RC can be followed spectrophotometrically using a pump–probe technique.26,27 We utilized this method to assess protein integrity once it is anchored on the semiconductor surface. The fitting of the experimental data (Fig. 7) with a monoexponential curve (A(t) = A0·e−kt) enabled determination of a kinetic constant, k, of 10 s−1 in accordance with the literature values associated with the recombination kinetics from the quinone QA.28 This result indicates that the loosely-bound secondary quinone QB has been completely removed during the bioconjugation process. However the signal amplitude is compatible with a fully active monolayer of protein, indicating that neither the bioconjugation reaction nor the close proximity to the organic semiconductor surface hinders the RC ability to generate a photoinduced electron–hole pair. This is a fundamental preliminary requirement for developing a hybrid device achieving effective electron transfer interactions between the RC and the H-bonded organic semiconductor.
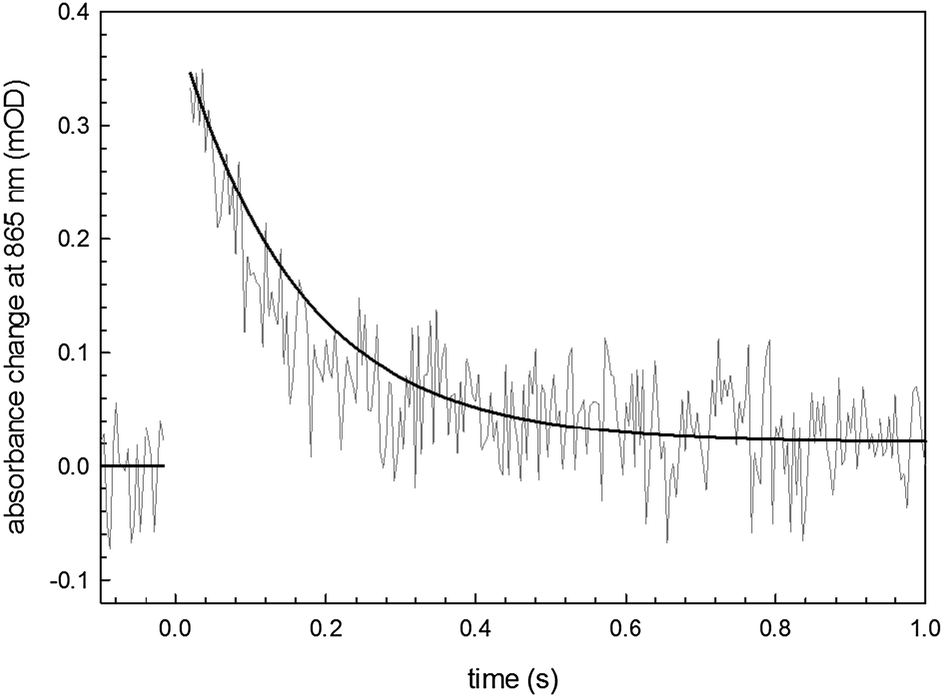 | ||
| Fig. 7 Transient absorbance of the SUB-bioconjugated RC on EPI with a monoexponential fitting curve. | ||
RC-sensitized photoconductive detector in an aqueous environment
The idea of harvesting charges photogenerated by the RC has been demonstrated in various electrochemical devices where the RC was immobilized on both metallic29,30 or semiconducting electrodes.31 For semiconductor devices, the RC can be exploited as a photosensitizer, absorbing efficiently in the red region of the spectrum. We followed this principle to sensitize photoconductivity in films of EPI using the SUB-immobilized RC. EPI was chosen for these experiments since it has a band gap of 2.4 eV; therefore red light can be used to selectively excite the RC in regions where the EPI does not exhibit any photoconductivity by itself. Moreover, recently it was reported that EPI transistors operate stably while in direct contact with aqueous solutions with various ionic strengths and pH values between 3 and 10.20 This property enables testing for RC-sensitized photoconductivity effects in wet environments. For these experiments, a simple resistor device was fabricated as shown in Fig. 8a. A pair of Cr/Au electrodes is evaporated onto glass, followed by a silanization treatment with OTS in order to prevent delamination of the EPI film during water exposure. A 40 nm-thick EPI film is then evaporated through a shadow mask in such a way as to cover the interdigitated area while leaving contact pads on the sides exposed. A poly(dimethyl siloxane), PDMS, block with a molded circular hole is then used to create a reservoir of the aqueous solution over the active EPI region.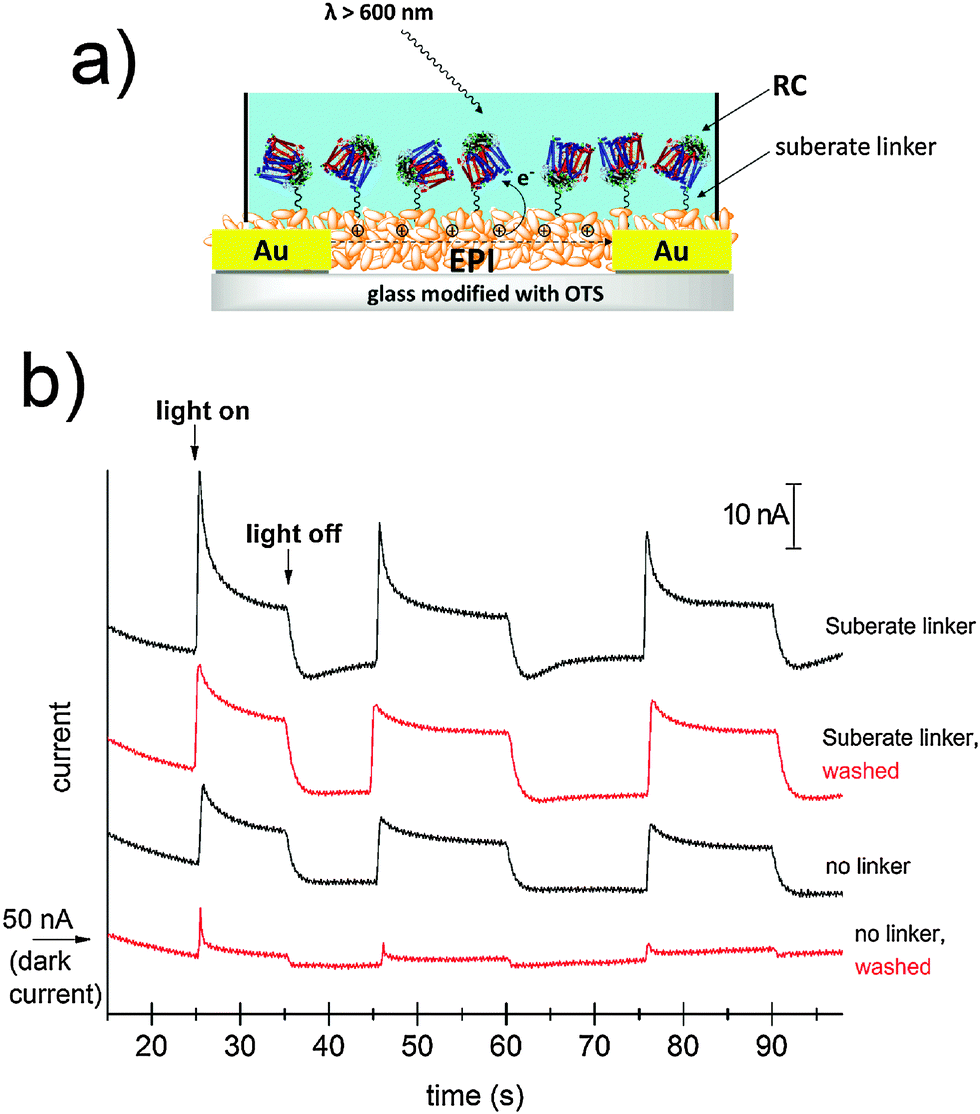 | ||
| Fig. 8 (a) Schematic of the EPI photoconductor sample showing the SUB-anchored RC on the surface of the EPI crystallites. The aqueous solution is maintained over the active area of the device with the help of a PDMS block. (b) Current transients obtained for the EPI photoconductor devices with a bias voltage of 0.5V. A ≈ 50 nA dark-current characterized all devices. | ||
The EPI devices measured under PBS solution showed dark currents of ≈50 nA under a bias of 500 mV. No photoconductivity was observed upon illumination by a tungsten lamp spectrally restricted with a filter to give λ > 600 nm. EPI samples were then prepared with and without SUB linker treatment, followed by 4 hour incubation with the RC, as described in the above section. Samples were then rinsed with 18 MΩ water, a fresh PBS solution was added to the reservoir, and photocurrent was measured (Fig. 8b). Samples with the linker were found to show a higher photoresponse than those without the linker. Importantly, following a second washing step with PBS, unmodified EPI samples lost the photoconductivity response, while those modified with SUB retained photoconductivity. We interpret this as evidence that the SUB linker indeed covalently attaches the RC to the EPI molecules on the surface of the film. Possible mechanisms behind photosensitization can be postulated. Photoinduced oxidation of EPI by the long-lived hole on the D+, can be hypothesized to be a likely process. A first consideration is that electron transport in EPI under oxygenated conditions does not occur due to the high-lying LUMO level, so under ambient conditions such EPI-based resistors can be regarded as hole-only devices. To better understand the mechanism we carried out photoelectrochemistry with EPI modified with RC operating as the working electrode (Fig. S6–S9, ESI†). At potentials more negative than +200 mV vs. Ag/AgCl, cathodic photocurrent is observed. Under illumination that selectively excites the RC, only at potentials in excess of +350 mV a small anodic photocurrent appeared. This means that the photocurrent electron flow is from the EPI to the RC, and does not reverse until sufficiently high positive potentials are applied to the EPI electrode. Based on the results from photoelectrochemistry that show electron flow from the EPI to the RC, and the EPI p-type character, we hypothesize that a likely mechanism for photoconductivity is photoinduced hole injection from the RC into the EPI.
Bioconjugation of streptavidin with biotin
Biotin (Vitamin B7) is a well-established functional molecule in biochemistry due to its strong and highly specific interaction with streptavidin. The streptavidin–biotin “lock and key” pair is used in many biochemical assays and imaging technologies.21,32,33 For these reasons, we explored the bioconjugation of the succinimidyl biotinate linker (B7) onto EPI and QNC, as shown in Fig. 3. In Fig. 9, IR spectra of EPI and QNC after B7 linker incubation and after washing with PBS solution are compared to IR spectra of pristine B7 and EPI and QNC alone. In the IR spectrum of pure B7 (traces marked ‘b’ in Fig. 9), the signals at 1817, 1790, 1749 and 1730 cm−1 can be related to the two carbonyl functional groups on the succinimidyl ring and to the ester carbonyl, while the peak at 1700 cm−1 is due to the stretching of biotin ureido carbonyl.34 After B7 incubation, a new band appeared centered at about 1660 cm−1, in correspondence to the absorption of the amide bond, while no signals at 1817 and 1790 were detected, suggesting the complete reaction of the NHS ring. Furthermore, the broad shape of the peak at 1660 cm−1 can be deconvoluted into three bands: one at 1660 belonging to the pigment, while the peak at 1715 cm−1 corresponds to the ureido carbonyl stretching, which is not involved in the possible covalent binding and, finally, the signal at 1650 cm−1 is due to the amide binding (ESI,† Fig. S10). These data strongly suggest that the B7 is covalently bound to the nitrogen atom of the pigment. However, after the PBS washing step, the IR features belonging to B7 completely disappeared (traces marked ‘d’ in Fig. 9). This disappearance of peaks could suggest that the B7 is removed from the surface during washing; however it has been reported previously in other immobilization experiments of B7 that these peaks disappear following aqueous washing steps despite the fact that the linker is still there, evidenced by the persistence of other distinctive peaks originating from the B7. This has been attributed to shielding or other changes in the local environment effecting the vibrational properties of the ureido ring.34,35 In our case, no appreciable difference is detected between traces a and d, since the spectral region of interest is covered by the pigment signals; however the effectiveness of bioconjugation is supported by the morphology changes recorded by AFM and luminescence assays discussed in the following.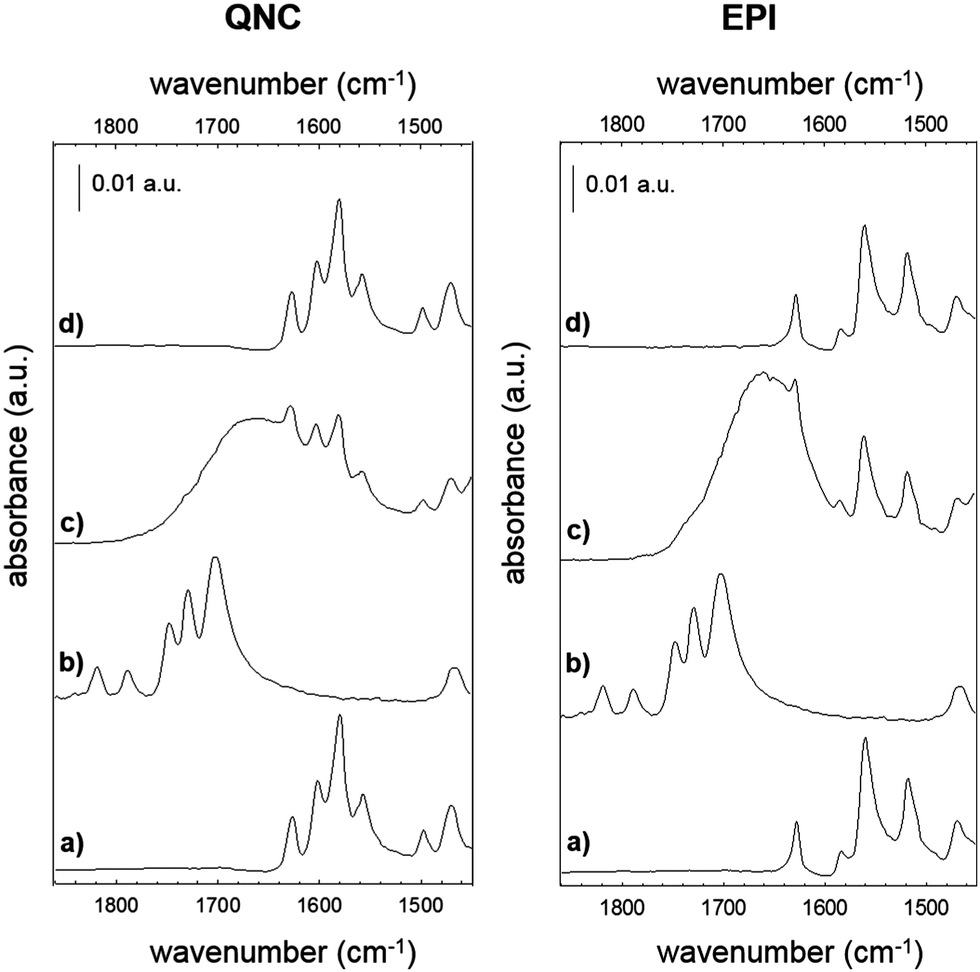 | ||
| Fig. 9 Reflection FTIR spectra of QNC and EPI films before and after B7 incubation. (a) Pristine hydrogen-bonded pigment film, (b) pristine B7, (c) pigment films incubated with B7, (d) after incubation with B7, following washing. | ||
AFM analysis was conducted for EPI and QNC films with B7 functionalization and after incubation with streptavidin (Fig. 10). As in the case of the SUB linker, B7 functionalization leads to a change in roughness and masking of the original distinctive crystallite morphology of the films (Fig. 10a and c; ESI,† Fig. S11–S14). An additional evidence for successful functionalization of the EPI and QNC films with B7 is a decrease in water contact angle (Table 2). Streptavidin is much smaller than the RC protein, and we could not find morphology changes that are unambiguously attributable to streptavidin, though it is known that this protein can exhibit aggregates of 100 nm in size when deposited on flat surfaces.36 However on QNC (Fig. 10b) aggregates of the same dimension can be found and on EPI (Fig. 10d) it can be seen that the surface is covered with globular structures of ∼30 nm in size, which are not found in unfunctionalized EPI (see also Fig. S15 in the ESI,† for a height profile evidencing the presence of smaller aggregates). Based on the following discussion on luminescent probe-labelled streptavidin, it is apparent that conjugation of streptavidin proceeds to a higher degree on EPI than on QNC.
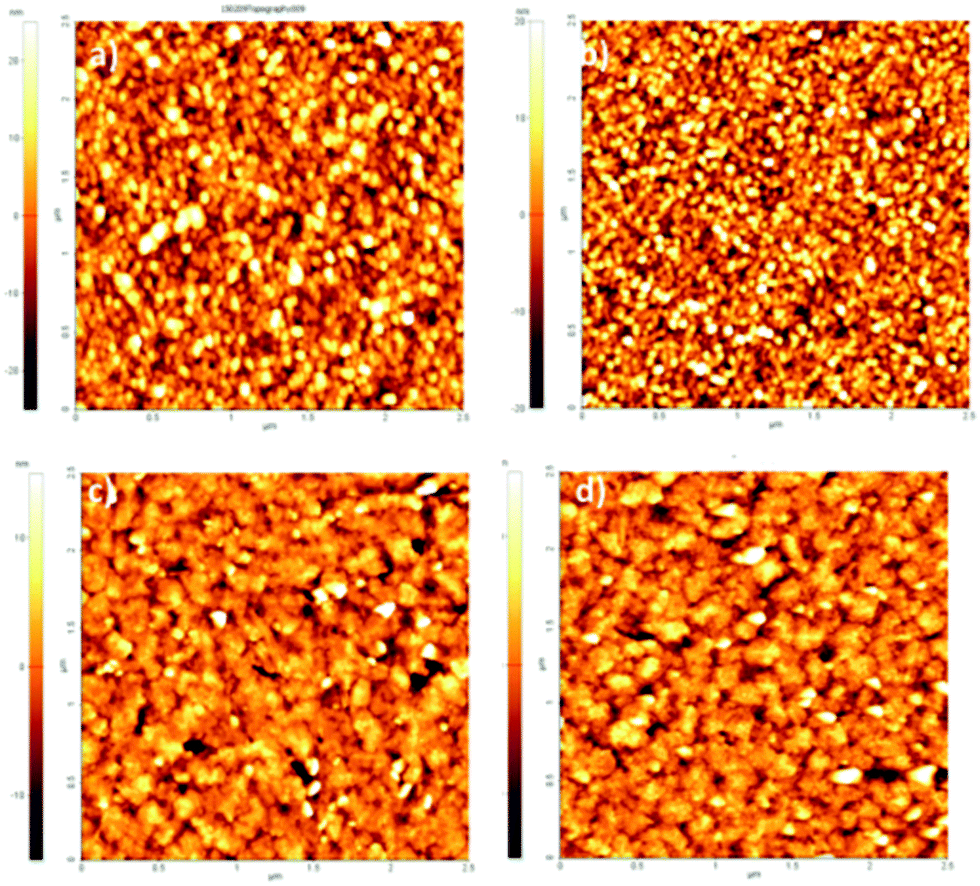 | ||
| Fig. 10 2.5 × 2.5 μm AFM topographies of (a) QNC functionalized with B7; (b) QNC with B7 and streptavidin; (c) EPI with B7; (d) B7 and streptavidin. | ||
| Untreated EPI | EPI@B7 | EPI@B7@SA | Untreated QNC | QNC@B7 | QNC@B7@SA | |
|---|---|---|---|---|---|---|
| RMS [nm] | 2.9 ± 0.3 | 4.4 ± 0.4 | 4.8 ± 0.4 | 3.4 ± 0.3 | 8.9 ± 0.6 | 6.8 ± 0.6 |
| Contact angle [°] | 86 | 71 | <10 | 89 | 70 | <10 |
A convenient way to probe the functionalization of EPI and QNC surfaces with B7 is to use luminophore-labeled streptavidin and measure photoluminescence, comparing B7-functionalized and unfunctionalized samples before and after washing steps. It follows that successful B7 functionalization would lead to higher luminescence than unfunctionalized samples, and the luminescence should be retained even after washing. To conduct this assay, as shown in Fig. 3, we used a commercial fluorescent probe consisting of CdSe/ZnS nanocrystals functionalized with streptavidin proteins on the surface (Life Technologies qdot705). This particular label was chosen because of the photoluminescence peak at 705 nm, which is sufficiently bathochromic of the absorbance of QNC and EPI and therefore could be selectively excited at wavelengths (>640 nm) where the pigment semiconductors do not absorb and energy transfer and quenching effects can be excluded. As a control sample, aminopropyl silanized (APS) glass (structure in inset of Fig. 11a) was used, both with and without functionalization with B7. As shown in Fig. 11a, only B7-modified samples showed luminescence from the qdot705 probe following a single washing with PBS solution. This indicates that little or no non-specific binding of the streptavidin on the APS surface occurs. EPI and QNC samples, in contrast, showed a significant amount of luminescence both with and without B7 modification, with more subtle differences than the qdot705 probes on the APS-modified glass. Over the course of four washing cycles, luminescence diminished, but never completely. An example of luminescence for an EPI sample, both with and without B7 modification, is shown in Fig. 11b. To obtain data of statistical significance, measurements over 16 samples for each assay were done, and the results of the integrated luminescence originating from EPI and QNC are shown in Fig. 11c and d. For comparison, we used the non-hydrogen bonded analogues tetracene and pentacene, respectively. All samples showed nonspecific binding affinity with the streptavidin-qdot705, though the hydrogen-bonded materials demonstrated more luminescence, i.e. better binding, overall. The other trend that emerges is that B7-modified hydrogen-bonded semiconductors always have the most luminescence after the initial streptavidin-qdot705 incubation and first wash, though subsequent washes easily diminish this luminescence. The other interesting point is that the streptavidin protein appears to have fairly good affinity for all four organic semiconductor films, and especially the hydrogen-bonded materials. For comparison, the amine-terminated APS-functionalized glass (Fig. 11a) lost nearly all luminescence after an initial washing step, while the luminescent-labelled streptavidin was clearly present on the hydrogen-bonded pigments after several washes. Based on the small difference in apparent streptavidin binding between B7-modified and pristine surfaces, some effort was made to explore different functionalization protocols to increase the amount of B7 binding. All procedures featured the B7-NHS dissolved in 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O:DMSO, with various ionic strength of the solution adjusted by the phosphate–buffer system and both with and without non-nucleophilic base catalysts like pyridine and dimethylaminopyridine which are known to deprotonate the NH function on hydrogen-bonded pigments and therefore increase their nucleophilicity.37 Ultrasonication to increase reactivity was also attempted. All procedures yielded results within the standard deviation of the data shown in Fig. 11c and d. From the luminescence and FTIR data, it is clear that biotinylation occurs to some degree, but not necessarily that it is stable. Nevertheless, the data for B7 support the conclusion that, as with SUB, reaction of the hydrogen-bonded pigments with NHS-functionalized linkers is a viable approach.
:1 H2O:DMSO, with various ionic strength of the solution adjusted by the phosphate–buffer system and both with and without non-nucleophilic base catalysts like pyridine and dimethylaminopyridine which are known to deprotonate the NH function on hydrogen-bonded pigments and therefore increase their nucleophilicity.37 Ultrasonication to increase reactivity was also attempted. All procedures yielded results within the standard deviation of the data shown in Fig. 11c and d. From the luminescence and FTIR data, it is clear that biotinylation occurs to some degree, but not necessarily that it is stable. Nevertheless, the data for B7 support the conclusion that, as with SUB, reaction of the hydrogen-bonded pigments with NHS-functionalized linkers is a viable approach.
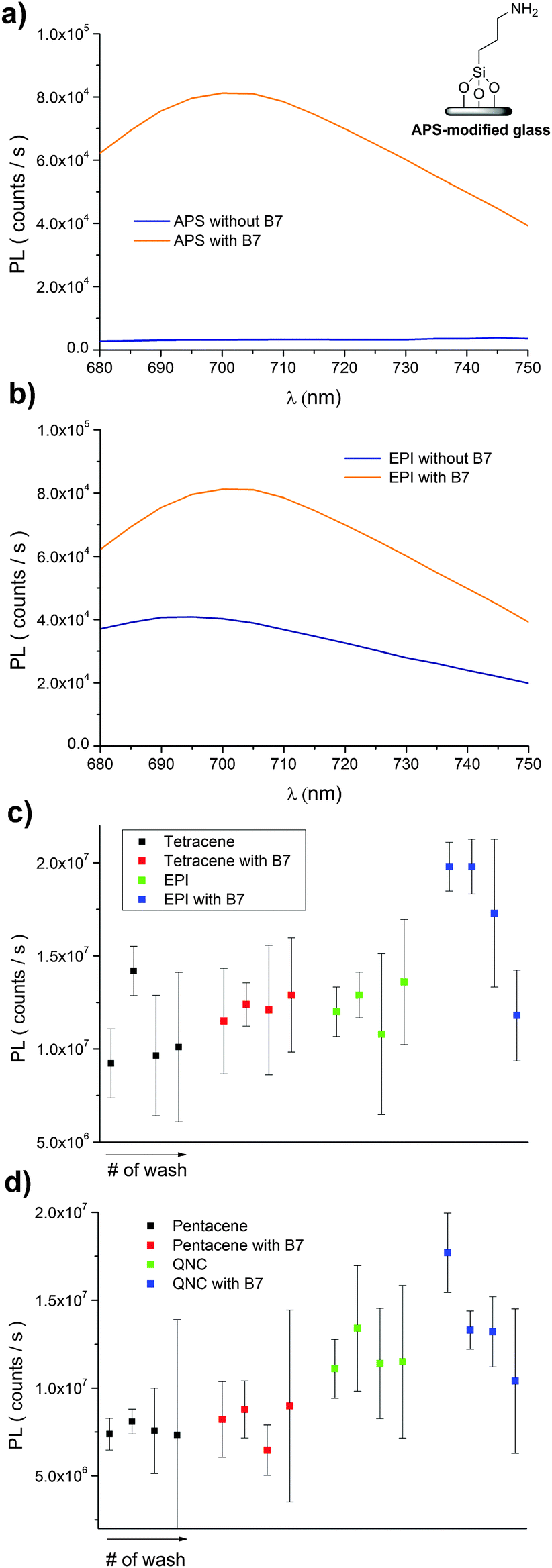 | ||
| Fig. 11 Comparison of luminescence originating from streptavidin-functionalized CdSe/Zns quantum dots incubated on B7-treated versus non-treated substrates, over the course of four subsequent washing steps. (a) “Control” sample of APS-functionalized glass with and without B7 modification. Only B7-modified APS samples show the 705 nm-peak qdot luminescence. (b) An example comparison of photoluminescence of EPI incubated with qdot705, both with and without B7-modification. (c) Integrated luminescence comparison of samples of tetracene and EPI, with and without B7. (d) Samples of pentacene and QNC, with and without B7. | ||
Surface-modified field-effect transistors
An important practical consideration is how bioconjugation might affect the hydrogen-bonded pigment in an active semiconductor device. Examples in the literature show that organic semiconductors often suffer severe decrease in mobility following derivatization with biomolecules.8 Therefore we tested how the surface functionalization protocols with SUB and B7, and their respective protein pairs, affect the performance of field-effect transistors (device structure shown in Fig. 12a). These FETs are made using a bottom-gate/top-contact geometry with the polycrystalline EPI surface in direct contact with the aqueous environment, and are typically operated in the linear regime with low VSD values between 0.1–0.5 V and VG values not exceeding 3–3.5 V in order to avoid any electrochemical reactions and parasitic currents. The gold source–drain contacts are not passivated in any way. Fig. 12b shows linear-regime transfer curves of EPI FETs before and after SUB functionalization. These are measured in air on samples that are nominally dry. Mobility decreases in the case of SUB from 0.1 cm2 V−1 s−1 to 0.06 cm2 V−1 s−1 after reaction; however devices remain stable in this state over measurement for at least 5 days. Incubation with RC solutions, however, leads to immediate degradation of the FET, with no measurable modulation remaining after the start of incubation. We attribute this to the presence of a highly polar LDAO surfactant in the RC solution, since test measurements with LDAO in water had an identical effect. In the case of B7-functionalized devices, mobility was unaffected following functionalization (Fig. 12c) and measurement of devices in dry air. In the case of both linkers, the off current increases to around 100 pA and the threshold voltage decreases after functionalization. Nevertheless, as found previously20 for such EPI FET structures, the performance remains stable at this level over hundreds of measurement cycles. We observed significant differences in response of linear-mode FETs to streptavidin for functionalized versus unfunctionalized control devices (Fig. 12d and e). FETs without B7, upon exposure to streptavidin solution, demonstrate a threshold voltage decrease of around 400 mV relative to the same device measured in PBS (Fig. 12d). “On” current is not affected. Devices modified with B7 show a substantial decrease in “on” current immediately when B7 incubation is begun, and a pronounced hysteresis (Fig. 12e). After 10 minutes, the transfer curves stabilize with a lower hysteresis and threshold voltage shift by 1 V to a higher gate voltage. Exposure of these devices to streptavidin then leads to a marked drop in the threshold voltage, by about 1 V, and an increase in current. The threshold voltage is higher by about 500 mV relative to unfunctionalized FETs incubated with streptavidin. This characteristic is retained after washing the devices with PBS solution and drying. It can be concluded that B7 modification does change the way the devices respond to streptavidin. Importantly, based on these initial results, it is clear that FETs based on EPI can continue to operate during and after surface functionalization and thus that they can be a suitable platform for bioelectronics applications. Mechanistic factors behind the observed changes in FET electrical characteristics, for example changes in the threshold voltage, remain to be fully understood.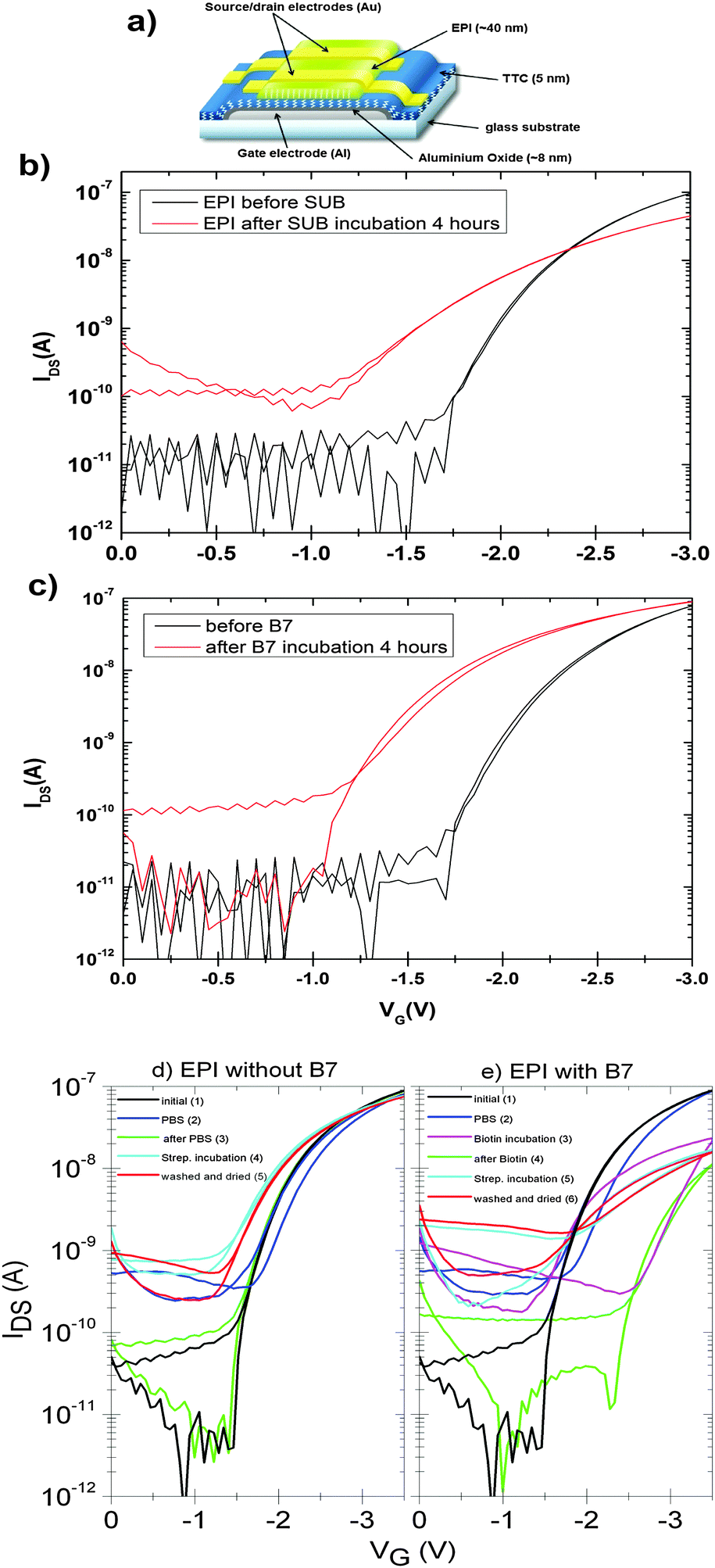 | ||
| Fig. 12 (a) FET structure schematic of low-voltage EPI devices. Anodized aluminium oxide passivated with TTC functions as a high-capacitance hybrid gate dielectric. Linear-mode transfer characteristics of low-voltage FETs: (b) EPI FETs measured before and after SUB functionalization, VSD = −500 mV. (c) EPI FETs measured before and after B7 functionalization, VSD = 500 mV. (d) EPI devices measured initially, in dry air (1), in PBS solution (2), (3) washed with water and dried, (4) during incubation with streptavidin in PBS solution, and finally washed and dried (5). (e) The same type of device as shown in (d), however with an additional incubation step with B7. A VSD of −500 mV was used in all measurements. | ||
Experimental
Materials
Epindolidione was synthesized and purified as previously reported.20 The quinacridone powder was purchased as Pigment Violet 19 from Kremer Pigmente and purified by temperature gradient sublimation. N-Succinimidyl biotinate (TCI) and disuccinimidyl suberate (Thermo Scientific) were used as-received. DMSO, decylubiquinone, lauryldimethylamine oxide, horse heart cytochrome c, and potassium phosphate were from Sigma. Phosphate-buffered saline from Alfa Aesar was used. (3-Aminopropyl)triethoxysilane (APS) was purchased from Alfa Aesar (98%) and used as-received. Modification of glass surfaces with APS was accomplished by a vapor-phase treatment, heating a vial of APS to 70 °C in a sealed, dry container with the glass substrates. Indium tin oxide (ITO) coated glass slides were used as the reflective substrate for FTIR experiments. Glass substrates were silanized with n-octyltrichlorosilane (OTS) by sealing O2 plasma cleaned glass in a closed chamber with a reservoir of OTS heated to 90 °C for 12 h. Samples were then rinsed with toluene and isopropanol, and sonicated in toluene, and finally rinsed with isopropanol and distilled water. Silanization of ITO substrates was performed in the identical way, except instead of n-octyltrichlorosilane, n-octyltriethoxysilane was used. In all cases, silanized hydrophobic substrates were used to avoid any delamination of the pigment films upon prolonged exposure to water. RC isolation from Rhodobacter sphaeroides strain R26 was performed following a standard procedure.38 The reaction centre purity and integrity were checked by the absorbance ratio A280/A802 and A760/A865, which were kept below 1.3 and 1, respectively.:1 v/v to give a concentration of 0.15 mM. This solution was applied directly on top of the surface to be functionalized and allowed to incubate in a sealed chamber with high humidity. After a period of 4 or 12 hours, as specified, the drop was removed with a pipette and the surface was rinsed under a stream of distilled water. For experiments with streptavidin alone, streptavidin obtained from IBA GmbH (Göttingen, Germany) was used. For luminescence experiments, streptavidin-qdot705 conjugate, purchased from Life Technologies, was diluted to a concentration of 50 μM in PBS and samples were incubated in this solution for 20 minutes to achieve full coverage of the surface. Longer incubation times did not yield higher luminescence. Following incubation, samples were rinsed with deionized water. Subsequent washing steps were performed with PBS followed by deionized water.
Infrared spectroscopy characterization
Mid-infrared spectra were acquired using a Perkin Elmer Spectrum One FTIR spectrometer equipped with a DTGS (deuterated tryglicine sulfate) detector. The spectral resolution used for all measurements was 4 cm−1. A variable specular reflection device designed specifically for thin film analyses (AmplifIR, Smiths Detection) was employed for IR measurements both on bioconjugated and physisorbed protein-modified semiconductors onto ITO coated glass slides. The background spectrum was acquired using a plain ITO slide.Surface morphology
Atomic force microscopy topographies were taken using a Park XE-100 SPM system microscope. Images were acquired in the non-contact tapping mode using tips (Type PPP-NCHR) on a cantilever of 125 μm length, about 330 kHz resonance frequency, 42 N m−1 nominal force constant and <10 nm guaranteed tip curvature radius. Surface areas were sampled with a scan rate of 1 Hz. Topographies were analyzed using the software XEI (Park System Corporation, version 1.8.0).RC kinetic measurements
Charge recombination reactions were monitored with a kinetic spectrophotometer of local design,39 by following the absorbance change at 865 nm over time. RC-modified semiconductors on ITO slides were placed at 45° angle with respect to the measuring beam and excited using a saturating flash light. 128 individual traces were averaged and recorded on a Digital Oscilloscope (Tektronics TDS-3200).Conclusions
Surface reactivity for decorating the surfaces of organic semiconductor films with functional biomolecules is an enabling step for realizing various bioelectronics devices. In this work, it has been demonstrated that simple bioconjugation procedures involving N-succinimidyl-functionalized molecules, well-established in biotechnological applications, can be used directly to modify hydrogen-bonded pigment semiconductor films. Infrared vibrational spectroscopy suggests that the NH group on the hydrogen-bonded pigment acts as a nucleophile, producing an amide linkage to the NHS-functionalized molecule. The examples shown in this work involving attachment of a bacterial photosynthetic reaction centre demonstrate that the semiconductor–biomolecule conjugate preserves the functions of both components, as evidenced by assays probing the reaction centre integrity and the appearance of sensitized photoconductivity, for example. In the case of the bacterial RC, physisorption of the protein can easily be reversed by washing with buffer solution, while the modification of the semiconductor with the suberate linker leads to samples that are resilient to washing, strongly indicating that true chemisorption occurs; as previously surmised based on the FTIR results. Streptavidin, on the other hand, displays a high degree of nonspecific binding to the semiconductor surface, but its interaction with the surface enhances the presence of the biotin linker. The aqueous-based functionalization of OFETs with suberate and biotin linkers does modify their electrical properties, however the devices continue to operate with similar mobility. Our findings of facile bioconjugation of H-bonded semiconducting pigments suggest that these materials are suitable for biocompatible electronic devices.Acknowledgements
We are grateful for support by the Austrian Science Foundation, FWF, within the Wittgenstein Prize of N. S. Sariciftci Solare Energie Umwandlung Z222-N19 and the Translational Research Project TRP 294-N19 “Indigo: From ancient dye to modern high-performance organic electronics circuits.” Y. Kanbur is grateful to TUBITAK-BIDED for the International Post-Doctoral Research Fellowship. This work was also financially supported by Ministero dell'Istruzione, dell'Università e della Ricerca (MIUR), Research Project of National Interest (prot. 2010C4R8M8 and 2012A4Z2R, Project AQUA-SOL); Regione Puglia “Progetto Reti di Laboratori Pubblici di Ricerca WAFITECH Prog. Cod. 09”; MIUR and DiTECH (PON 02_00563_3316357 Molecular Nanotechnology for Health and Environment MAAT). We thank G. Romanazzi for supplying the epindolidione material and R. Hasslacher for contact angle measurements. The authors thank Dr Massimo Trotta and Alessandra Antonucci for useful discussions.References
- M. D. Angione, R. Pilolli, S. Cotrone, M. Magliulo, A. Mallardi, G. Palazzo, L. Sabbatini, D. Fine, A. Dodabalapur, N. Cioffi and L. Torsi, Mater. Today, 2011, 14, 424–433 CrossRef CAS.
- M. Irimia-Vladu, E. D. Głowacki, G. Voss, S. Bauer and N. S. Sariciftci, Mater. Today, 2012, 15, 340–346 CrossRef CAS.
- J. Rivnay, R. M. Owens and G. G. Malliaras, Chem. Mater., 2014, 26, 679–685 CrossRef CAS.
- H. U. Khan, M. E. Roberts, O. Johnson, R. Förch, W. Knoll and Z. Bao, Adv. Mater., 2010, 22, 4452–4456 CrossRef CAS PubMed.
- M. L. Hammock, A. N. Sokolov, R. M. Stoltenberg, B. D. Naab and Z. Bao, ACS Nano, 2012, 6, 3100–3108 CrossRef CAS PubMed.
- T. Cramer, A. Campana, F. Leonardi, S. Casalini, A. Kyndiah, M. Murgia and F. Biscarini, J. Mater. Chem. B, 2013, 1, 3728–3741 RSC.
- L. Torsi, M. Magliulo, K. Manoli and G. Palazzo, Chem. Soc. Rev., 2013, 42, 8612–8628 RSC.
- L. Torsi, G. M. Farinola, F. Marinelli, M. C. Tanese, O. H. Omar, L. Valli, F. Babudri, F. Palmisano, P. G. Zambonin and F. Naso, Nat. Mater., 2008, 7, 412–417 CrossRef CAS PubMed.
- W. Yoon, S. H. Lee, O. S. Kwon, H. S. Song, E. H. Oh, T. H. Park and J. Jang, Angew. Chem., Int. Ed., 2009, 48, 2755–2758 CrossRef PubMed.
- E. D. Głowacki, M. Irimia-Vladu, M. Kaltenbrunner, J. Gąsiorowski, M. S. White, U. Monkowius, G. Romanazzi, G. P. Suranna, P. Mastrorilli, T. Sekitani, S. Bauer, T. Someya, L. Torsi and N. S. Sariciftci, Adv. Mater., 2013, 25, 1563–1569 CrossRef PubMed.
- G. Palazzo, D. De Tullio, M. Magliulo, A. Mallardi, F. Intranuovo, M. Y. Mulla, P. Favia, I. Vikholm-Lundin and L. Torsi, Adv. Mater., 2015, 27, 911–916 CrossRef CAS PubMed.
- M. Muskovich and C. J. Bettinger, Adv. Healthcare Mater., 2012, 1, 248–266 CrossRef CAS PubMed.
- F. Buth, A. Donner, M. Sachsenhauser, M. Stutzmann and J. A. Garrido, Adv. Mater., 2012, 24, 4511–4517 CrossRef CAS PubMed.
- H. Zollinger, Color Chemistry. Syntheses, Properties and Applications of Organic Dyes and Pigments, Wiley-VCH, Weinheim, 3rd edn, 2003 Search PubMed.
- High Performance Pigments, ed. E. B. Faulkner and R. J. Schwartz, Wiley-VCH, Weinheim, 2nd edn, 2009 Search PubMed.
- W. Herbst and K. Hunger, Industrial Organic Pigments, Wiley-VCH, Weinheim, 3rd edn, 2004 Search PubMed.
- K. Hunger and W. Herbst, Ullmann's Encycllopedia Industrial Chemistry, 2012, vol. 27, p. 379 Search PubMed.
- K. Hunger, Rev. Prog. Color. Relat. Top., 2005, 35, 76–89 CrossRef CAS PubMed.
- E. D. Głowacki, G. Voss and N. S. Sariciftci, Adv. Mater., 2013, 25, 6783–6800 CrossRef PubMed.
- E. D. Głowacki, G. Romanazzi, C. Yumusak, H. Coskun, U. Monkowius, G. Voss, M. Burian, R. T. Lechner, N. Demitri, G. J. Redhammer, N. Sünger, G. P. Suranna and S. Sariciftci, Adv. Funct. Mater., 2015, 25, 776–787 CrossRef PubMed.
- N. Stephanopoulos and M. B. Francis, Nat. Chem. Biol., 2011, 7, 876–884 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- G. Feher, J. P. Allen, M. Y. Okamura and D. C. Rees, Nature, 1989, 339, 111–116 CrossRef CAS PubMed.
- F. Milano, R. R. Tangorra, O. Hassan Omar, R. Ragni, A. Operamolla, A. Agostiano, G. M. Farinola and M. Trotta, Angew. Chem., Int. Ed., 2012, 51, 11019–11023 CrossRef CAS PubMed.
- P. R. Rich and M. Iwaki, Mol. BioSyst., 2007, 3, 398–407 RSC.
- L. Nagy, F. Milano, M. Dorogi, A. Agostiano, G. Laczkó, K. Szebényi, G. Váró, M. Trotta and P. Maróti, Biochemistry, 2004, 43, 12913–12923 CrossRef CAS PubMed.
- F. Milano, L. Gerencser, A. Agostiano, L. Nagy, M. Trotta and P. Maroti, J. Phys. Chem. B, 2007, 111, 4261–4270 CrossRef CAS PubMed.
- F. Mavelli, M. Trotta, F. Ciriaco, A. Agostiano, L. Giotta, F. Italiano and F. Milano, Eur. Biophys. J., 2014, 43, 301–315 CrossRef CAS PubMed.
- H. Yaghoubi, Z. Li, D. Jun, E. Lafalce, X. Jiang, R. Schlaf, J. T. Beatty and A. Takshi, J. Phys. Chem. C, 2014, 118, 23509–23518 CAS.
- M.-J. den Hollander, J. G. Magis, P. Fuchsenberger, T. J. Aartsma, M. R. Jones and R. N. Frese, Langmuir, 2011, 27, 10282–10294 CrossRef CAS PubMed.
- Y. Kim, S. A. Shin, J. Lee, K. D. Yang and K. T. Nam, Nanotechnology, 2014, 25, 342001 CrossRef PubMed.
- P. C. Weber, D. H. Ohlendorf, J. J. Wendoloski and F. R. Salemme, Science, 1989, 243, 85–88 CAS.
- G. T. Hermanson, Bioconjugate Techniques, Elsevier, Amsterdam, 2nd edn, 2008 Search PubMed.
- Biointerface Characterization by Advanced IR Spectroscopy, ed. C. M. Pradier and Y. J. Chabal, Elsevier, Amsterdam, 1st edn, 2011 Search PubMed.
- Functionalization of Semiconductor Surfaces, ed. F. Tao and S. L. Bernasek, Wiley, Hoboken, 2012 Search PubMed.
- M. D. Angione, S. Cotrone, M. Magliulo, A. Mallardi, D. Altamura, C. Giannini, N. Cioffi, L. Sabbatini, E. Fratini, P. Baglioni, G. Scamarcio, G. Palazzo and L. Torsi, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6429–6434 CrossRef CAS PubMed.
- E. D. Głowacki, G. Voss, K. Demirak, M. Havlicek, N. Sunger, A. C. Okur, U. Monkowius, J. Gąsiorowski, L. Leonat and N. S. Sariciftci, Chem. Commun., 2013, 49, 6063–6065 RSC.
- R. A. Isaacson, F. Lendzian, E. C. Abresch, W. Lubitz and G. Feher, Biophys. J., 1995, 69, 311–322 CrossRef CAS.
- F. Ciriaco, R. R. Tangorra, A. Antonucci, L. Giotta, A. Agostiano, M. Trotta and F. Milano, Eur. Biophys. J., 2015, 44, 183–192 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of FTIR and AFM measurements, and cyclic voltammetry of bioconjugated RC. See DOI: 10.1039/c5tc00556f |
| This journal is © The Royal Society of Chemistry 2015 |