
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photostable phosphorescent polymer nanospheres for high sensitivity detection†
A. N.
Edwards‡§
,
M.
Yamazaki‡
,
S. H.
Krishnadasan
,
T. W.
Phillips
 ,
L.
Rowlands
,
R.
Jourdain
,
A. M.
Nightingale¶
* and
J. C.
de Mello
*
,
L.
Rowlands
,
R.
Jourdain
,
A. M.
Nightingale¶
* and
J. C.
de Mello
*
Department of Chemistry, Imperial College London, Exhibition Road, South Kensington, London SW7 2AY, UK. E-mail: A.Nightingale@soton.ac.uk; j.demello@imperial.ac.uk
First published on 11th June 2015
Abstract
Time-gated interrogation of phosphorescent labels allows high sensitivity detection of analytes by excluding background fluorescence and stray excitation light. Here we report a method for preparing phosphorescent polymer nanospheres with optical properties specifically designed to enable time-gated detection with both organic and inorganic light-emitting diodes and photodiodes. The nanospheres were formed by co-precipitating the phosphorescent dye ruthenium diphenyl phenanthroline with the amphiphilic statistical co-polymer poly(styrene-co-maleic acid). To minimize aggregation and ensure effective encapsulation against water and oxygen ingress, the polymer chains were cross-linked with the small-molecule cross-linker bisphenol A diglycidyl ether, resulting in near-monodisperse nanospheres with excellent resilience against aggregation and quenching. The nanospheres exhibited long phosphorescence lifetimes of 5.3 ± 0.1 μs, high photoluminescence quantum efficiencies of 33 ± 5%, and excellent stability under UV irradiation. Using a blue light-emitting diode as a light-source and a silicon photodiode as a detector, a low detection limit of 4 μg ml−1 was achieved. The suitability of the nanospheres for bioanalysis was demonstrated by surface functionalising them with biotin to yield a simple biolabel for streptavidin.
Introduction
Time-gated phosphorescence is a powerful method for detecting the presence of trace analytes.1 By using a pulsed excitation source and measuring emission from a phosphorescent sample during the off-stage of the excitation, it is possible to eliminate spurious contributions from stray excitation light or background fluorescence. Consequently, time-gated detection is ideal for analysing biological media where scattering macromolecules and luminescent species abound. Whilst time-gating is usually considered to be a high-end detection method, implementation costs can be greatly reduced by judicious choice of phosphor. By selecting a material that absorbs above 400 nm and emits below 1100 nm with a radiative lifetime ≳1 μs, it is possible to perform time-gated detection using commodity light-emitting diodes (LEDs) and photodiodes in conjunction with low cost detection electronics. This opens up many opportunities in the field of point-of-care diagnostics, where a strong demand exists for sensitive detection techniques that can be implemented at the cost of a few dollars.Ruthenium complexes are near-ideal phosphors for low cost time-gated detection as they absorb strongly in the blue (λabs ∼ 460 nm) and emit efficiently in the red (λem ∼ 600 nm) with radiative lifetimes of several microseconds.2 Advantageously, these characteristics are compatible with excitation by both inorganic LEDs and organic LEDs (OLEDs), and with detection by both inorganic and organic photo-diodes.|| For bio-labelling applications, ruthenium dyes must be protected from exposure to oxygen and other emission quenchers by encapsulation in a protective glass or polymer matrix.6–10 The use of polymers for encapsulation is particularly advantageous since the size of the nanospheres can be readily tuned by controlling the polymer chain length, while inclusion of appropriate functional groups can be used to tailor the nanosphere surface for the attachment of bioactive molecules such as antibodies.
The most common method for preparing polymer nanospheres is emulsion polymerisation,11–13 in which small droplets of monomer are dispersed in a continuous phase of water prior to polymerisation. This method requires the monomer droplets to be stabilized by surfactants, which are often difficult to remove from the final polymer particles and can adversely affect their biological function.11 Consequently methods that avoid the use of surfactants are preferable for biolabelling. The simplest of the surfactant-free methods is the water-induced precipitation technique introduced by Fessi et al.,14 which yields sub-micron polymer particles in high yield. The method entails the co-dissolution of a lipophilic chromophore and an amphiphilic polymer in a small quantity of a water-miscible organic solvent, followed by the dropwise addition of excess water while vigorously stirring. The polymer is chosen to be insoluble in the water–organic solvent mixture and, at a sufficiently high water content, will precipitate from solution as a fine suspension of nanospheres, with hydrophilic polymer groups at the surface and dye molecules trapped within the hydrophobic interior.
Using ruthenium diphenyl phenanthroline [Ru(dpp)3Cl2] as the lipophilic dye, a co-polymer of acrylonitrile and acrylic acid as the host matrix, and N,N-dimethyl-formamide (DMF) as the organic solvent, Kürner et al. applied the precipitation method to the preparation of phosphorescent nanospheres with low susceptibility to oxygen quenching and high quantum efficiencies of up to 43%.11 Song et al. subsequently showed that crosslinking the polymer chains within the particles helped to protect the enclosed dye against quenching by oxygen.12 They dispersed Pt(II) and Pd(II) porphyrins in nanospheres formed from two polymers with crosslinkable moieties, poly(vinyl chloride-co-ethyl acetate-co-maleic acid) and poly(vinyl chloride-co-vinyl acetate-co-epoxy monomer). Crosslinking was induced by heating, yielding final crosslinked nanospheres with superior photostability to equivalent non-crosslinked nanospheres (due to the better exclusion of oxygen and other emission quenchers).12
As an alternative to using multiple polymers with cross-reactive functional groups, we report here the formation of crosslinked nanospheres using a small-molecule crosslinking agent, bisphenol A diglycidyl ether (BPA), in conjunction with a single amphiphilic polymer. Small molecule linkers have been shown to allow effective polymer chain crosslinking15–18 while at the same time reducing processing complexity by allowing nanosphere formation from a single constituent polymer. In particular, the size, morphology and surface chemistry of the nanospheres are controlled by a single material rather than a blend of polymers, thus avoiding the risk of composition-dependent variability in nanosphere properties. Below we describe a simple and reliable method for the preparation of easily functionalised high performance phosphorescent nanospheres from a single statistical co-polymer host, poly(styrene-co-maleic acid) (PS-co-MA) in combination with a BPA crosslinker and Ru(dpp)3Cl2 dye.
Results and discussion
To form the phosphorescent nanospheres, Ru(dpp)3Cl2 dye molecules and BPA crosslinkers were encapsulated within PS-co-MA nanospheres using the co-precipitation method, followed by thermally induced crosslinking of the polymer chains by the BPA. The chemical structures of the constituent materials are shown in Fig. 1. Ru(dpp)3Cl2 dye was chosen because of its favourable optical properties, commercial availability and proven efficacy when incorporated into polymer nanospheres.11 PS-co-MA was chosen as the host matrix since the carboxylic acid groups within the maleic acid component allow easy crosslinking and provide suitable surface groups for the attachment of biomolecules.19 The polystyrene co-constituent of the copolymer provides a good hydrophobic environment for the exclusion of oxygen and polar solvents, while the phenyl groups can be expected to further aid encapsulation of the dye molecule through π–π interactions with the phenyl groups on dpp. BPA was chosen as the small-molecule crosslinker because it is an inexpensive commercially available material with epoxide groups that can couple to the carboxylic acid groups of PS-co-MA; in addition, its phenyl rings ensure it is sufficiently hydrophobic to be incorporated into the interior of the nanospheres during precipitation.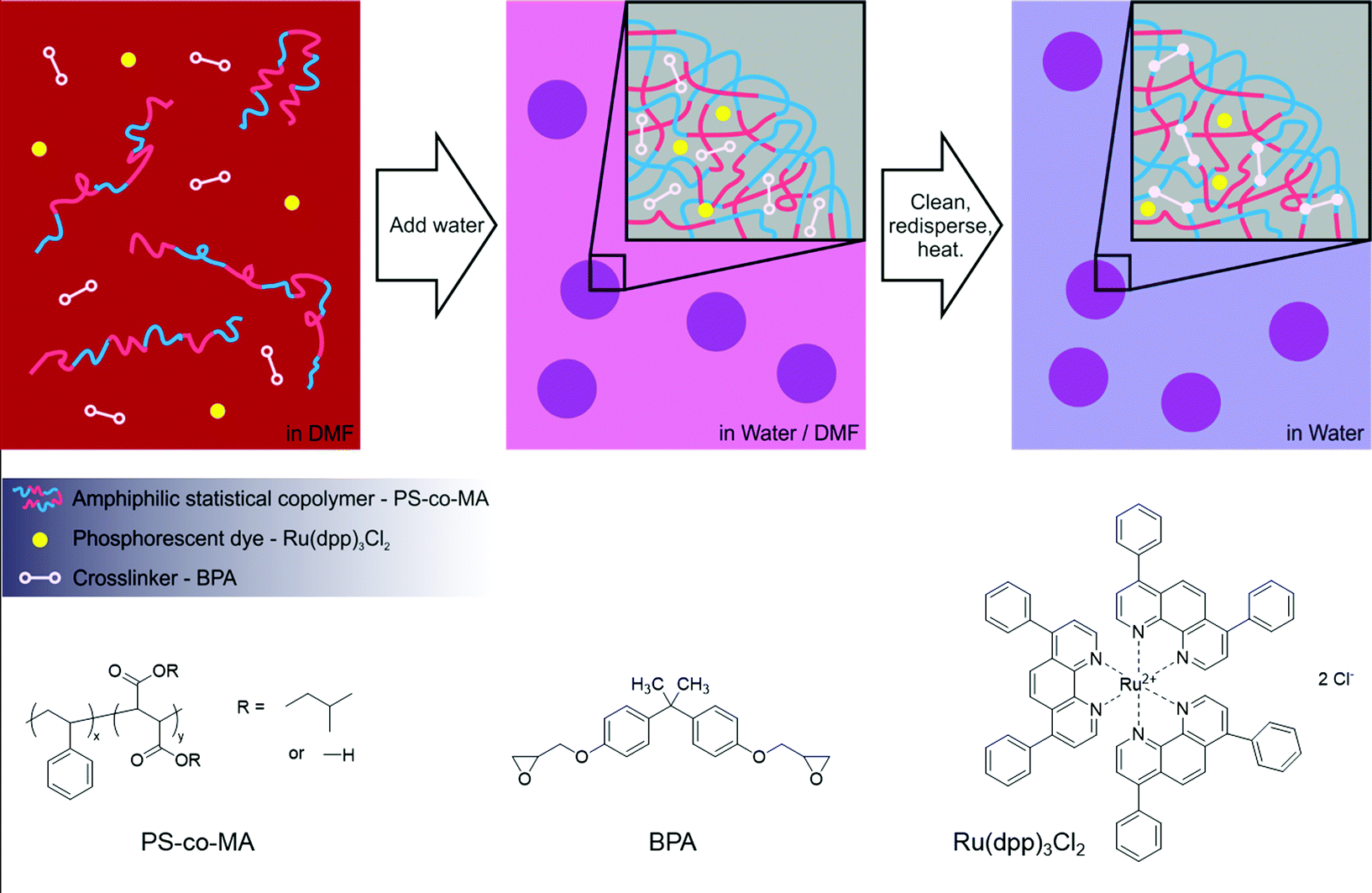 | ||
| Fig. 1 Schematic illustrating the co-precipitation/crosslinking process used to prepare the phosphorescent polymer nanospheres (top), and the molecular structures of the constituent materials (bottom). | ||
The general method for the co-precipitation of the nanospheres is illustrated in Fig. 1. PS-co-MA, Ru(dpp)3Cl2 and BPA were dissolved in (water-miscible) DMF at a relative loading of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:25 by mass. A syringe pump was used to slowly add an approximately five-fold excess of distilled water to the stirred solution. The solution soon turned turbid due to the formation of the (highly scattering) nanospheres. Stirring was continued for several minutes after completion of the water addition. The nanospheres were then cleaned three times by centrifugation, removal of the supernatant, and redispersal in deionised water. The dispersed nanospheres were crosslinked by heating at 85 °C overnight to yield a turbid light-orange dispersion and then finally cleaned with three more centrifugation/isolation/redispersal steps. (The stability of the cross-linked nanospheres was confirmed by adding a 30-fold excess of DMF to a sample of as-prepared nanospheres (in water). Following the addition, the resulting mixture remained turbid, indicating the continued presence of intact DMF-insoluble nanospheres).
:1:25 by mass. A syringe pump was used to slowly add an approximately five-fold excess of distilled water to the stirred solution. The solution soon turned turbid due to the formation of the (highly scattering) nanospheres. Stirring was continued for several minutes after completion of the water addition. The nanospheres were then cleaned three times by centrifugation, removal of the supernatant, and redispersal in deionised water. The dispersed nanospheres were crosslinked by heating at 85 °C overnight to yield a turbid light-orange dispersion and then finally cleaned with three more centrifugation/isolation/redispersal steps. (The stability of the cross-linked nanospheres was confirmed by adding a 30-fold excess of DMF to a sample of as-prepared nanospheres (in water). Following the addition, the resulting mixture remained turbid, indicating the continued presence of intact DMF-insoluble nanospheres).
Examination of the nanospheres by scanning electron microscopy (SEM) showed them to have uniform spherical shapes with diameters of 300 ± 120 nm and minimal coalescence (see Fig. 2a). Dynamic light scattering measurements in water were broadly consistent with this value, giving a hydrodynamic diameter of 480 ± 90 nm. Fig. 2b shows emission and excitation spectra for the nanospheres in water – the former being obtained at an excitation wavelength of 450 nm and the latter at the peak emission wavelength of 600 nm. (Note, a reliable absorption spectrum could not be obtained due to the strong optical scattering). The spectra exhibited the expected mirror symmetry between emission and excitation, and closely resembled the corresponding spectra for the free dye in de-aerated ethanol** (see Fig. S1, ESI†), indicating that incorporation of the dye into the nanospheres had little effect on the molecular conformation. Using time-correlated single photon counting, the phosphorescence lifetime of the nanosphere solution in aerated water was determined to be 5.5 ± 0.1 μs (see Fig. S2a, ESI†), close to the literature value of 6.3 ± 0.4 μs for the free dye in de-aerated water,20 and much higher than its value of 0.8 ± 0.1 μs in aerated water, see Fig. S2b (ESI†) and ref. 21. The photoluminescence quantum efficiency (PLQE) of the nanosphere solution was determined to be 33 ± 5% using an integrating sphere,22 which compares favourably with a reported value of 35 ± 4% for the free dye in de-aerated water.20 The ability to maintain long phosphorescence lifetimes and high quantum yields relative to the free dye in oxygen-free conditions shows that the incorporation of Ru(dpp)3Cl2 in the polymer host does not compromise its optical properties, with the interior of the PS-co-MA nanospheres providing a protective quencher-free environment.
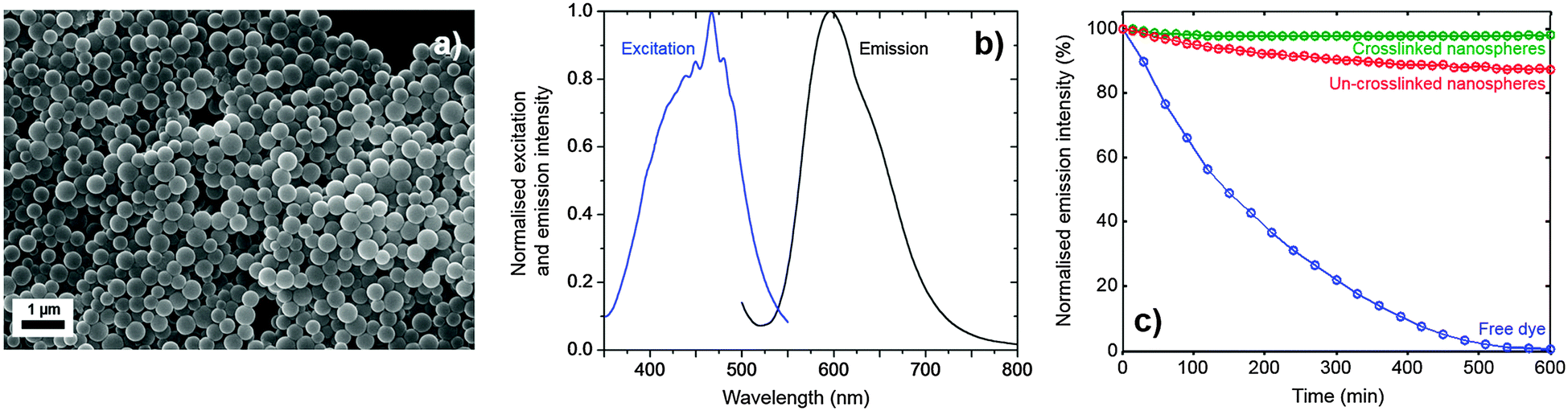 | ||
| Fig. 2 (a) Representative SEM image showing fully formed and crosslinked nanospheres, ∼300 nm in diameter prepared from water. (b) Normalised photoluminescence excitation and emission spectra for the crosslinked nanospheres in water. (c) Normalised emission intensity versus time for crosslinked and non-crosslinked nanospheres in water under constant excitation at 450 nm. Also shown is equivalent data for the free dye in de-aerated ethanol. | ||
The photostability of crosslinked and non-crosslinked nanosphere solutions was compared by continuously exciting at 465 nm (the approximate absorption peak of Ru(dpp)3Cl2) and measuring the peak intensity over time. Fig. 2c shows results for 0.1 mg ml−1 polymer nanospheres in water, with and without crosslinking. Also shown for comparison are equivalent results for the free dye in de-aerated ethanol (8.6 mM). The free dye underwent rapid quenching, losing 25% of its emission intensity in the first hour and virtually all of its emission intensity over the ten hour duration of the measurement. The dye in non-crosslinked PS-co-MA nanospheres fared better, retaining ∼88% of its initial intensity after ten hours. The dye in crosslinked PS-co-MA nanospheres fared better still, retaining virtually all (∼98%) of its intensity after ten hours. The excellent stability of the crosslinked dye-doped nanospheres confirms the rigorously oxygen- and quencher-free conditions within the nanosphere, and highlights the importance of crosslinking to achieve adequate encapsulation.
The optical properties of the Ru-doped nanospheres make them excellent candidates for low-cost high-sensitivity time-gated detection as they can be excited by blue LEDs and OLEDs and their emission can be detected using Si and organic photodiodes. In addition, the long emission lifetime allows time-gated detection to be employed without recourse to fast and costly instrumentation. To assess their viability as labels for time-gated detection, aqueous dispersions of the crosslinked Ru(dpp)3Cl2-doped nanospheres were measured at a range of concentrations between 6.5 μg ml−1 and 130 mg ml−1, using a simple homemade time-gated detection system. The optical system comprised a 1 mm path-length cuvette, fitted on opposing sides with a 470 nm blue inorganic LED (E1L31-3B0A2, Toyoda Gosei, Japan) and a silicon photodiode (S5972, Hamamatsu, Japan).
The LED was driven using a 20 mA, 20 kHz pulsed current source with a pulse duration of 6.2 μs. The output from the photodiode was passed to a precision switched-integrator transimpedance amplifier (IVC102) with an internal feedback capacitance of 100 pF. Using a microcontroller, the integrating amplifier was configured to remain in the reset state until 3.3 μs after the LED had extinguished, providing sufficient time for the LED to completely switch off (and for fluorescence from background contaminants to decay to zero). The amplifier was then released from the reset state and allowed to integrate for a period of 40 μs – several multiples of the Ru(dpp)3Cl2 lifetime – before the reset was re-enabled. This process was repeated at a repetition rate of 20 kHz, and the periodic output of the integrating amplifier was passed to the voltage-input of a digital lock-in amplifier, yielding a signal that was proportional to the average phosphorescence intensity over the 40 μs detection window. For the purposes of the current work, an SR830 digital lock-in amplifier from Stanford Research Instruments was used for signal acquisition, but a low-cost alternative could have equally been used (see for example ref. 23). For each of the nanosphere solutions, corrected signals were obtained by subtracting a reference signal obtained from a measurement of (dye-free) water. The corrected data, shown in Fig. 3, exhibited excellent linearity over a concentration range spanning four orders of magnitude. Defining the limit of detection λ as the concentration of analyte that would give a corrected signal equal to three times the standard deviation of the blank (σ = 2.5 nV), a value λ = 3.8 μg ml−1 was obtained, equivalent to a Ru(dpp)3Cl2 concentration of 30 ng ml−1 or <30 nM. It is important to note that this low detection limit was obtained using a face-on arrangement of the light-source and photodetector – the most convenient arrangement for use in miniaturised point-of-care diagnostic devices. This arrangement is extremely challenging for conventional fluorescence detection as it requires the use of rigorous optical filtering to prevent flooding of the detector by unabsorbed excitation light (see ref. 24 and 25). The use of time-gated detection elegantly avoids the need for optical filters, reducing the component count, whilst still enabling the use of a simple face-on optical arrangement.
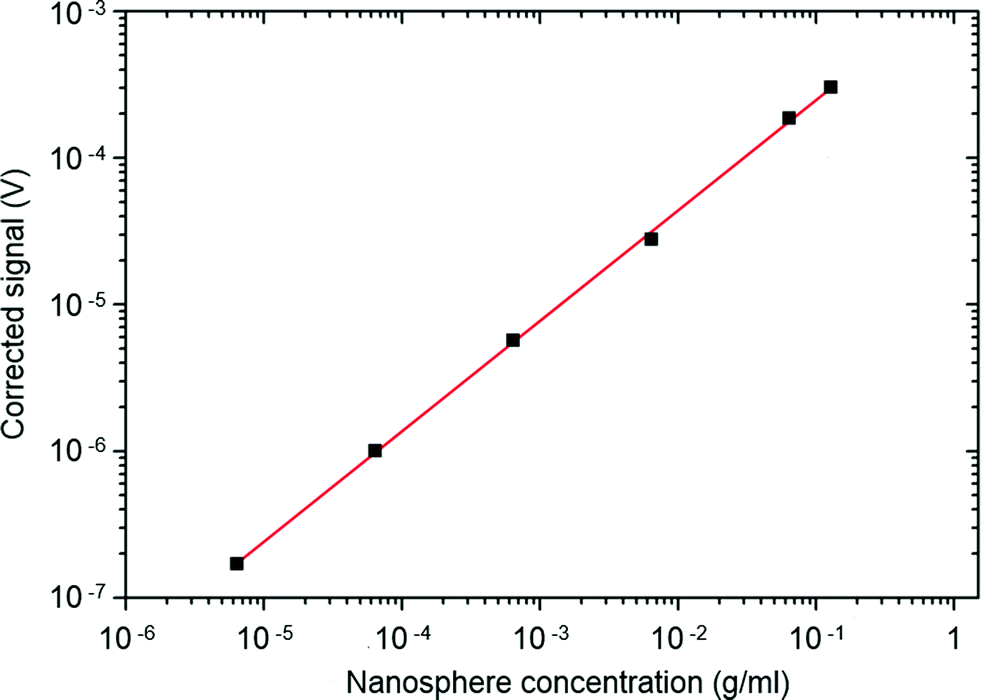 | ||
| Fig. 3 Time-gated signal intensity versus nanosphere concentration for crosslinked dye-doped nanospheres dispersed in water. | ||
To assess their suitability for use in bioassays, the feasibility of functionalising the surface of the nanospheres with a chemically selective binding group was assessed. As discussed above, PS-co-MA was selected as the host polymer in part because it was expected to provide a surface that would be rich in carboxylic acid groups that permit surface functionalisation by a wide range of biomolecules.26 This was confirmed by conductometric titration,27–29 which indicated that the number of surface carboxylic acid groups was ∼300000 per particle (i.e. approximately one per square nanometer, see ESI†).
Biotin was selected for surface functionalisation due to its strong affinity to streptavidin, which can be exploited in a wide range of bioassays.30 The biotinylation procedure is described in detail in the experimental section but, briefly, it entailed the covalent attachment of amine-PEG-biotin (APB) to the nanosphere surface via a 1-ethyl-3-(3-dimethyl-aminopropyl) carbodiimide hydrochloride (EDC) crosslinker,31 followed by cleaning-with and dispersal-in phosphate buffered saline (PBS). The result of this procedure is biotin molecules that are tethered to the nanosphere surface via a short polyethylene glycol (PEG) chain, which ensures sufficient separation between the biotin and the nanosphere surface for subsequent biotin–streptavidin conjugation.
After cleaning with PBS, the presence of the PEG-biotin on the nanosphere surface was confirmed by Fourier Transform Infrared (FTIR) spectroscopy. Fig. 4a shows the FTIR “fingerprint” region of the nanospheres after biotinylation and cleaning, along with comparative spectra for unfunctionalised nanospheres and APB. The APB spectrum showed a strong response at ∼1080 cm−1 (C–O stretch) associated with the PEG chain, a strong response at ∼1650 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch) due to the ketone in the biotin residue, plus several small intervening features which can be tentatively assigned to C–H and N–H bending modes. Prior to biotinylation the nanospheres exhibited only a single peak at 1650 cm−1 due to the carbonyl stretch of the carboxylic acid groups in the PS-co-MA. After biotinylation and cleaning, however, the nanosphere spectrum exhibited new features that were co-located with those of pure APB, consistent with successful functionalisation.
O stretch) due to the ketone in the biotin residue, plus several small intervening features which can be tentatively assigned to C–H and N–H bending modes. Prior to biotinylation the nanospheres exhibited only a single peak at 1650 cm−1 due to the carbonyl stretch of the carboxylic acid groups in the PS-co-MA. After biotinylation and cleaning, however, the nanosphere spectrum exhibited new features that were co-located with those of pure APB, consistent with successful functionalisation.
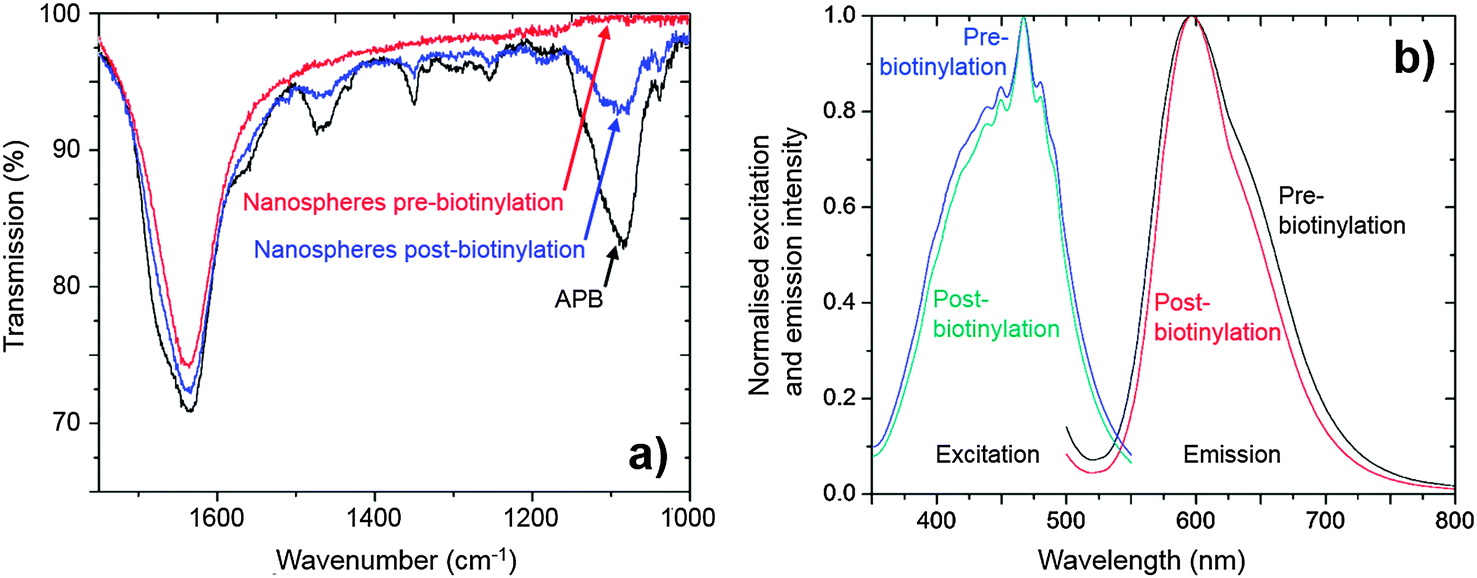 | ||
| Fig. 4 (a) FTIR spectra for nanospheres after biotinylation and cleaning (blue). Also shown for comparison are the spectra of the selected biotin derivative (APB, black) and the unfunctionalised nanospheres (red) prior to the biotinylation procedure. (b) Normalised photoluminescence excitation and emission spectra of nanospheres before and after biotinylation. In each case minimal changes in peak shape and location are seen. | ||
The biotinylation process had negligible effect on the optical properties of the nanospheres. The phosphorescence lifetime was measured to be 5.3 ± 0.1 μs before and 5.4 ± 0.1 μs afterwards, while the PLQE was 32.8 ± 5% beforehand and 33.2 ± 5% afterwards. Nor were any changes seen in the position and shape of the excitation and emission spectra (see Fig. 4b).†† Hence, despite significant chemical changes at the surface of the nanospheres, the optical properties of the Ru(dpp)3Cl2 dye within the nanosphere remained unaffected by the biotinylation procedure, confirming the effectiveness of the cross-linked PS-co-MA nanospheres as encapsulants.
To demonstrate the nanospheres could be applied to quantitative analysis, a simple assay was devised to quantify APB. The assay method entailed binding the APB to a fixed quantity of phosphorescent nanospheres, which were then extracted from the solution, re-dispersed, and quantified by time-gated detection (see Experimental for full details). Extraction was carried out magnetically by conjugating the APB-coated phosphorescent nanospheres to streptavidin-functionalised superparamagnetic microbeads, using a magnet to retain the beads (plus attached nanospheres) while the supernatant was removed.32 The upper set of images in Fig. 5a show the accumulation of magnetic beads (plus attached nanospheres) close to the pole of a permanent magnet, with the bulk solution becoming less turbid as it is depleted of scattering nanospheres. (The lower set of images shows the result of carrying out the same procedure in the absence of streptavidin-functionalised microbeads, with the bulk solution remaining uniformly turbid).
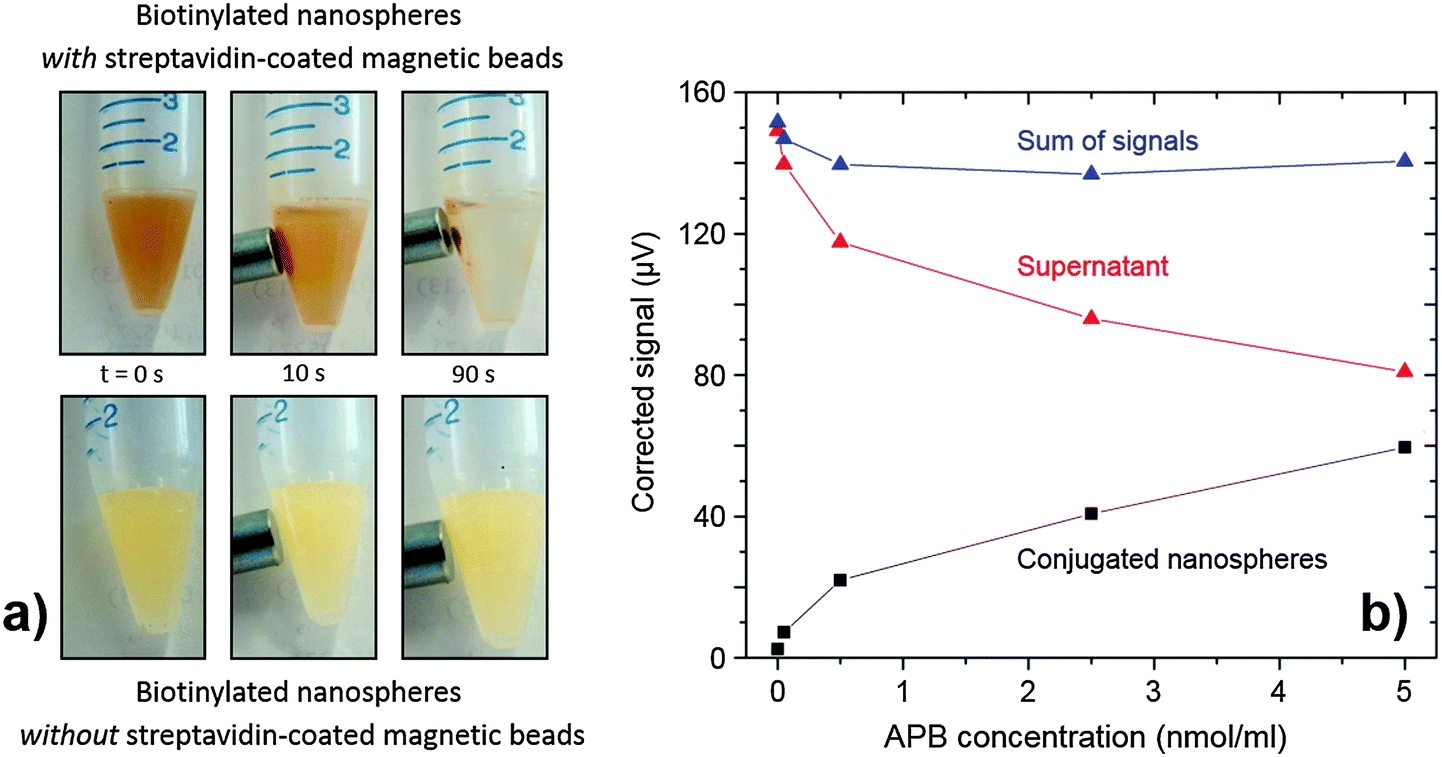 | ||
| Fig. 5 (a) Top: photographic images showing the extraction of biotinylated nanospheres using streptavidin-coated magnetic beads and a neodymium magnet; over 90 s, the solution clarified and material was seen to accumulate on the inside of the vial near the magnet. Bottom: a control test using the same procedure but without the streptavidin-coated magnetic beads showed no signs of separation. (b) The phosphorescent signal obtained from magnetically extracted conjugated nanospheres (black squares) redispersed in PBS, the remaining supernatant (red triangles) and the sum of the two signals (blue triangles), using different concentrations of APB during nanosphere biotinylation. | ||
Conjugation to the microbeads caused no significant changes to the optical properties of the biotinylated nanospheres: the shapes of the excitation and emission spectra remained unchanged (Fig. S3, ESI†), and negligible changes were seen in the measured phosphorescence lifetimes (5.4 μs before versus 5.3 μs after) and PLQEs (33.2 ± 5% before versus 35.8 ± 5% after), further confirming the effectiveness of the PS-co-MA/BPA nanospheres as encapsulants for the dye molecules.
Fig. 5b shows the measured signal I versus APB loading Q (black markers). Empirically, the response follows a sublinear power law (I ∝ Q0.45), consistent with multiple analytes attaching to a single nanosphere: at low concentrations of APB, each additional APB molecule is expected to attach to a new nanosphere but, as the APB concentration increases, it becomes increasingly likely that additional APB will attach to nanospheres that already host APB molecules, thereby decreasing the assay sensitivity. (This effect could be avoided by using smaller nanospheres to sterically suppress the occurrence of multiple attachments.) Nonetheless the assay is highly sensitive at lower concentrations, with a calculated limit of detection for APB of 0.011 nmol ml−1. (This compares favourably with typical limits of detection for commercial fluorescent label based biotin assays, e.g. 0.040 nmol ml−1 for a kit from Molecular Probes33).
The supernatant solution removed from the first wash contains the residual nanospheres that have not undergone biotinylation. These nanospheres were cleaned via three centrifugation–isolation–re-suspension cycles and then measured using the time-gated detection system. The resulting phosphorescence signal versus APB concentration is plotted in red in Fig. 5b. As expected, the red markers are the approximate mirror image of the black markers, with the sum of the two being approximately flat over the full concentration range. (The slight observed drop in the sum with increasing Q is attributable to some loss of phosphorescent nanospheres from the supernatant during the cleaning procedure). Hence, it is evident that the assay may be carried out by measuring either the magnetically extracted nanospheres or the supernatant – the latter perhaps being preferable in terms of reduced processing steps. Beyond the proof-of-principle assay reported here, the nanospheres should prove amenable to a broad range of bioassays due to their carboxylic acid-rich surface, which permits easy covalent attachment of a variety of different ligands.16
Conclusion
We have reported phosphorescent biolabels suitable for use in low-cost time-gated detection using either inorganic or organic light-emitting diodes and photodiodes. The biolabels consisted of dye-loaded poly(styrene-co-maleic acid) nanospheres cross-linked with bisphenol A diglycidyl ether. Ru(dpp)3Cl2-containing nanospheres had long phosphorescence lifetimes of 5.3 ± 0.1 μs, high photoluminescence quantum efficiencies of 33 ± 5%, and exhibited good resilience to quenching under UV irradiation. Using time-gated detection with a blue LED and Si photodiode in a face-on arrangement, the nanospheres were detectable down to low concentrations of ∼4 μg ml−1. Importantly, the nanospheres could be readily biofunctionalised via surface carboxylic acid groups without compromising their optical properties. This property was exploited to produce a simple proof-of-principle assay to quantify amine-PEG-biotin, using streptavidin-coated superparamagnetic microspheres to separate biotinylated and non-biotinylated nanospheres. In summary, the crosslinked PS-co-MA/BPA nanospheres are robust, versatile phosphorescent labels that allow sensitive low-cost time-gated detection, and are likely to prove attractive for a broad range of bioanalytical applications.Experimental
Formation of crosslinked nanospheres
100 mg of statistical co-polymer PS-co-MA (Sigma Aldrich, MW 180000 PS:MA ratio 1:<1) and 25 mg of BPA were dissolved in 20 ml DMF using a glass-coated magnetic stirrer bar rotating at 300 rpm. The quantity of BPA used was empirically determined to be the smallest amount that would ensure monodisperse non-aggregated nanospheres, while also providing effective protection of the dye against photobleaching. 1 mg of Ru(dpp)3Cl2 dissolved in 5 ml DMF was added to this solution, imparting a deep orange hue to the (previously colourless) solution.‡‡ 120 ml of distilled water was loaded into a syringe and injected into the solution at a rate of 2 ml min−1, causing a visual change from clear to cloudy after ∼5 min due to the formation of (highly scattering) nanospheres. The water addition was continued for a further 55 min until the full 120 ml had been delivered, resulting in a H2O:DMF volume ratio of approximately 5:1. The mixture was left to stir for a further ten minutes, after which it was shaken, centrifuged at 3000 rpm for 15 minutes, and separated from the supernatant, and topped up with 100 ml water. The first supernatant was a pale yellow colour, indicating that most of the dye had been encapsulated in the nanospheres. This washing procedure was repeated twice to eliminate residual DMF that might otherwise induce nanosphere coalescence. The solution was then heated overnight at 85 °C to induce crosslinking of the PS-co-MA and the BPA. The beads were then cleaned again by three centrifuge/supernatant-removal/redispersion steps. Solutions of the crosslinked nanospheres were stored in the dark at −20 °C.
Biotinylation of nanospheres
Phosphorescent nanospheres were dispersed in 0.1 M 2-(N-morpholino) ethane-sulfonic acid (MES) at a loading of 10 mg ml−1. 100 μl of a 10 mM solution of the 1-ethyl-3-(3-dimethyl-aminopropyl) carbodiimide hydrochloride (EDC) crosslinker in MES buffer and 1.1 mg of N-hydroxysulfosuccinimide (which helps prevent rehydrolysis of the carboxylic acid groups after EDC addition) were added to 1 ml samples of the nanospheres in MES, and the resulting mixture was shaken vigorously at room temperature for 15 min. Varying amounts of APB (EZ-Link amine-PEG8-biotin, Thermo Scientific) in 1 ml MES solution were added to the mixture before shaking vigorously for a further two hours. The biotinylated nanospheres were then centrifuged three times with 0.1 M phosphate-buffered saline (PBS) at pH 7.2 to stop the reaction and to remove any unreacted reagents. No aggregation was observed when working in PBS, either during the biotinylation procedure or in the course of carrying out the assay experiments.Assay for quantifying APB
Five 1 ml nanosphere solutions were prepared in PBS at a nanosphere concentration of 0.1 g ml−1, and each one was biotinylated with a different amount of APB in the range 0 to 5 nmol using the carbodiimide-based method outlined above. The biotinylated nanospheres were thoroughly washed in pH 7, 0.1 M PBS and re-suspended in 1 ml PBS. 100 μl of 2.5 nmol ml−1 magnetic microbead solution (Dynabeads MyOne Streptavidin C1, 10 mg ml−1 bead suspended in PBS pH 7.4, Invitrogen) was added to each of the phosphorescent nanosphere solutions and shaken for two hours. A neodymium magnet was then brought into contact with the lower outside surface of each sample container and held in place for five minutes, causing the bead–sphere conjugates to accumulate on the side of the container. The supernatant containing the non-biotinylated nanospheres was carefully removed with a pipette and retained. The magnetic beads left behind were re-suspended in 1 ml of fresh PBS solution. This process was repeated three times, and the solid was finally redispersed in a 1.0 mm quartz cuvette with 1 ml of 0.1 M PBS solution, ready for analysis using the time-gated detection system.Photoluminescence and absorption spectra
Spectra were obtained using a Jobin-Yvon Fluoromax-2 fluorimeter. A photodiode in the fluorimeter was used to monitor the transmitted light through the cuvettes to obtain absorption spectra.Time-correlated single photon counting
Time-resolved photoluminescence measurements were performed using a time-correlated single photon counting system (Horiba Jobin Yvon, Fluorocube). A 467 nm LED laser diode was used as the excitation source and the emission was detected at 610 nm. The obtained decay curves were fitted using a mono-exponential function of the form: I = Aexp(−t/τ) + I0.
Determination of photoluminescence quantum efficiency
Photoluminescence quantum efficiency (PLQE) measurements were performed using an integrating sphere (see ref. 22). Approximately 60 mg ml−1 of phosphorescent nanospheres was used for all measurements and 5 nmol of APB was used for the conjugates of the magnetic beads and the biotinylated nanospheres. 1 mm quartz cuvettes were used for all measurements.SEM measurements
A dilute suspension of beads was dropped onto a freshly cleaved ruby muscovite mica substrate (Agar Scientific G250-3). Samples were allowed to dry by evaporation before being attached to pin stubs with carbon tape. The samples were sputter coated with a 15 nm layer of chromium and had a silver contact painted on. The samples were dried under vacuum overnight before being analysed using a LEO Gemini 1525 FEGSEM.Dynamic light scattering measurement
DLS measurements were performed on a ZetaSizer ZS instrument (Malvern Instruments). Dilute aqueous suspensions of beads were sonicated for 3 minutes before being transferred to polystyrene cuvettes for analysis.Conductometric titration
A conductivity meter (EC-con 6, RMS, UK) was first calibrated using a hydrochloric acid reference solution. 114.5 mg of the dye-doped nanospheres was dispersed in 49 ml of distilled water, and 1 ml of 0.05 M sodium chloride solution was added. Following Pohl et al.,29 titrations of the nanospheres were carried out by adding 10 μl of 0.05 M sodium hydroxide solution at 1 min intervals with continuous stirring, noting the concentration at which the conductance reached its minimum value. This procedure was repeated three times using independent batches of polymer nanospheres.References
- A. Thibon and V. C. Pierre, J. Am. Chem. Soc., 2009, 131, 434–435 CrossRef CAS PubMed.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. Vonzelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- O. Hofmann, P. Miller, P. Sullivan, T. S. Jones, J. C. deMello, D. D. C. Bradley and A. J. deMello, Sens. Actuators, B, 2005, 106, 878–884 CrossRef CAS PubMed.
- O. Hofmann, X. Wang, J. C. deMello, D. D. C. Bradley and A. J. deMello, Lab Chip, 2005, 5, 863–868 RSC.
- J. Huang and J. C. deMello, in Organic Electronics in Sensors and Biotechnology, ed. R. Shinar and J. Shinar, McGraw-Hill Professional, 2009 Search PubMed.
- T. Behnke, C. Wuerth, K. Hoffmann, M. Huebner, U. Panne and U. Resch-Genger, J. Fluoresc., 2011, 21, 937–944 CrossRef CAS PubMed.
- S. M. Borisov, T. Mayr, A. A. Karasyov, I. Klimant, P. Chojnacki, C. Moser, S. Nagl, M. Schaeferling, M. I. Stich, A. S. Kocincova and O. S. Wolfbeis, in Fluorescence of Supermolecules, Polymers, and Nanosystems, ed. M. N. BerberanSantos, 2008, vol. 4, pp. 431–463 Search PubMed.
- S. M. Borisov, A. S. Vasylevska, C. Krause and O. S. Wolfbeis, Adv. Funct. Mater., 2006, 16, 1536–1542 CrossRef CAS PubMed.
- A. Gomez-Hens, J. M. Fernandez-Romero and M. P. Aguilar-Caballos, TrAC, Trends Anal. Chem., 2008, 27, 394–406 CrossRef CAS PubMed.
- A. S. Kocincova, S. M. Borisov, C. Krause and O. S. Wolfbeis, Anal. Chem., 2007, 79, 8486–8493 CrossRef CAS PubMed.
- J. M. Kürner, I. Klimant, C. Krause, H. Preu, W. Kunz and O. S. Wolfbeis, Bioconjugate Chem., 2001, 12, 883–889 CrossRef PubMed.
- X. Song, L. Huang and B. Wu, Anal. Chem., 2008, 80, 5501–5507 CrossRef CAS PubMed.
- J. F. Wang, P. A. G. Cormack, D. C. Sherrington and E. Khoshdel, Angew. Chem., Int. Ed., 2003, 42, 5336–5338 CrossRef CAS PubMed.
- H. Fessi, F. Puisieux and J. P. Devissaguet, Google Pat., publication number CA1293170 C, application number CA 555153, also published as DE3777793D1, EP0274961A1, EP0274961B1, US5049322, 1988 Search PubMed.
- Y. C. Hu and C. Y. Pan, Macromol. Rapid Commun., 2005, 26, 968–972 CrossRef CAS PubMed.
- M. J. Joralemon, K. S. Murthy, E. E. Remsen, M. L. Becker and K. L. Wooley, Biomacromolecules, 2004, 5, 903–913 CrossRef CAS PubMed.
- R. K. O’Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- Y. Wang, H. Du, L. Gao, H. Ni, X. Li, W. Zhu and Z. Shen, Polym. Chem., 2013, 4, 1657–1663 RSC.
- Q. Liu, Y. Li, S. Shen, X. Qiuguo, L. Chen, B. Liao, B. Ou and Y. Ding, J. Appl. Polym. Sci., 2011, 121, 654–659 CrossRef CAS PubMed.
- I. Klimant and O. S. Wolfbeis, Anal. Chem., 1995, 67, 3160–3166 CrossRef CAS.
- J. M. Kurner, PhD thesis, University of Regensburg, 2002.
- J. C. deMello, H. F. Wittmann and R. H. Friend, Adv. Mater., 1997, 9, 230–232 CrossRef CAS PubMed.
- S. K. Sengupta, J. M. Farnham and J. E. Whitten, J. Chem. Educ., 2005, 82, 1399 CrossRef CAS.
- M. Yamazaki, O. Hofmann, G. Ryu, X. Li, T. K. Lee, A. J. deMello and J. C. deMello, Lab Chip, 2011, 11, 1228–1233 RSC.
- M. Yamazaki, S. Krishnadasan, A. J. deMello and J. C. deMello, Lab Chip, 2012, 12, 4313–4320 RSC.
- A. N. Edwards, PhD thesis, Imperial College London, 2011.
- J. Hen, J. Colloid Interface Sci., 1974, 49, 425–432 CrossRef CAS.
- Z. L. Liu, Z. H. Ding, K. L. Yao, J. Tao, G. H. Du, Q. H. Lu, X. Wang, F. L. Gong and X. Chen, J. Magn. Magn. Mater., 2003, 265, 98–105 CrossRef CAS.
- H. A. Pohl, Anal. Chem., 1954, 26, 1614–1616 CrossRef CAS.
- E. P. Diamandis and T. K. Christopoulos, Clin. Chem., 1991, 37, 625–636 CAS.
- A. Gole and C. J. Murphy, Langmuir, 2005, 21, 10756–10762 CrossRef CAS PubMed.
- C. Smith, Nat. Methods, 2005, 2, 71–77 CrossRef CAS.
- Product Information: FluoReporter® Biotin Quantitation Assay Kit *for biotinylated nucleic acids* (F30755) by Invitrogen Detection Technologies, 2005.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tc00933b |
| ‡ These authors contributed equally to this work. |
| § Current address: Department of Materials Science and Engineering, University of Arizona, 4715 E Fort Lowell Road, Tucson, Arizona 85712, USA. |
| ¶ Current address: Faculty of Engineering and the Environment, University of Southampton, Highfield, Southampton SO17 1BJ, UK. |
| || Organic devices are of interest for use in low cost printed sensors for point of care diagnostics, see e.g.ref. 3 and 4. Typical OLEDS only emit strongly above ∼450 nm, while typical organic photodiodes are only sensitive to wavelengths below ∼750 nm and have slow response times in excess of 1 μs.5 Ruthenium dyes are therefore noteworthy as being one of the very few classes of dye molecules that enable sensitive phosphorescence detection using organic devices. |
| ** Note due to the poor solubility of Ru(dpp)3Cl2 in water, absorption and emission spectra were recorded in ethanol. |
| †† A slight reduction in signal intensity (∼8%) was observed for both the excitation and emission spectra, but this is most likely due to material loss during cleaning. |
| ‡‡ Note, a one percent weight ratio of dye to polymer was found to be optimum in terms of maximising the dye loading and ensuring effective encapsulation against oxygen; at substantially higher loadings, the nanospheres were found to be susceptible to photodegradation. |
| This journal is © The Royal Society of Chemistry 2015 |